

Abstract
Hydroxy fatty acids are critical lipid mediators involved in various pathophysiologic functions. We cloned and identified GPR31, a plasma membrane orphan G protein-coupled receptor that displays high affinity for the human 12-lipoxygenase-derived product 12-(S)-hydroxy-5,8,10,14-eicosatetraenoic acid (HETE). Thus, GPR31 is named 12-(S)-HETE receptor (12-HETER) in this study. The cloned 12-HETER demonstrated high affinity binding for 12-(S)-[3H]HETE (Kd = 4.8 ± 0.12 nm). Also, 12-(S)-HETE efficiently and selectively stimulated GTPγS coupling in the membranes of 12-HETER-transfected cells (EC50 = 0.28 ± 1.26 nm). Activating GTPγS coupling with 12-(S)-HETE proved to be both regio- and stereospecific. Also, 12-(S)-HETE/12-HETER interactions lead to activation of ERK1/2, MEK, and NFκB. Moreover, knocking down 12-HRTER specifically inhibited 12-(S)-HETE-stimulated cell invasion. Thus, 12-HETER represents the first identified high affinity receptor for the 12-(S)-HETE hydroxyl fatty acids.
Keywords: Eicosanoid, Eicosanoid Receptor, G Protein-coupled Receptors (GPCR), Receptors, Signal Transduction
Introduction
12-(S)-Hydroxy-5,8,10,14-eicosatetraenoic acid (12-(S)-HETE),3 a 12-lipoxygenase metabolite of arachidonic acid, has been demonstrated to evoke a wide variety of cellular responses (1). For example, 12-(S)-HETE has shown to have a prominent role in cytoskeleton remodeling so as to facilitate cell chemotaxis (2–4). Furthermore, addition of exogenous 12-(S)-HETE induces cells to secrete proteinases (5, 6) and vascular endothelial growth factor leading to an angiogenic response (7, 8). 12-(S)-HETE treatment of cancer cells also enhanced the expression of integrins (2, 9) and fibronectin (10), which prolong cell survival (11). In endothelial cells, 12-(S)-HETE induces the non-destructive retraction of monolayers (12) and promotes tumor cell adhesion (13). Also, the motility of isolated endothelial cells and tube formation is enhanced by 12-(S)-HETE treatment (14).
The diverse biological activities mediated by 12-(S)-HETE suggest that it functions as a critical signaling molecule in the regulation of physiological processes. Similar to the identified plasma membrane G protein-coupled receptors (GPCRs) for other eicosanoid (e.g. leukotrienes, prostaglandins, lipoxin), there is strong evidence implying that 12-(S)-HETE signaling involves an unidentified plasma membrane G protein-coupled receptor. Work in our laboratory demonstrated that activation of PKC-α by 12-(S)-HETE is through GPCR-mediated hydrolysis of inositol phospholipids (15). Our studies also suggest that 12-(S)-HETE stimulates the activities of a protein tyrosine phosphatase, the tyrosine kinase Src, and activates the PKC-ERK1/2 axis via an unidentified plasma membrane GPCR (16). Additionally, Hampson and Grimaldi (17) found that 12-(S)-HETE attenuates glutamate-induced calcium influx into neurons via a pertussis toxin-sensitive mechanism, suggesting that it acts via a G protein-coupled receptor. Collectively, these results suggest that the 12-(S)-HETE receptor is a G protein-coupled receptor.
We have conducted a computer-based database search for orphan GPCRs and developed an efficient technique to clone these GPCRs from PC3 human prostate cancer cells, a 12-(S)-HETE-responsive cell line (8, 14). We demonstrated that GPR31 is a high affinity GPCR for 12-(S)-HETE. Because GPR31 is deorphaned in the present study, it is renamed as 12-(S)-HETE receptor (12-HETER). The cloning, identification, and functional characterization of the membrane receptor for 12-(S)-HETE (i.e. 12-HETER) will provide significant insight into the molecular basis of the signaling and physiological functions mediated by 12-(S)-HETE.
EXPERIMENTAL PROCEDURES
Materials
Unlabeled 12-(S)-HETE, other HETEs, and 5-oxoETE were obtained from Cayman Chemical (Ann Arbor, MI). Monoclonal anti-β-actin antibody was purchased from Chemicon. Phospho-ERK antibody was from Cell Signaling Technology, Inc. (Danvers, MA). All other reagents were from Sigma unless specified.
Synthesis of 12-(S)-[3H]HETE
For the synthesis of 12-(S)-[3H]HETE, the purified platelet-type 12-lipoxygenase was used to catalyze the [3H]arachidonic acid ([3H]AA) into 12-(S)-[3H]HETE (supplemental Fig. S1). Briefly, bubble oxygen through 20 ml of phosphate buffer (pH 7.4) in 250 ml in an Erlenmeyer flask for 20 min at 30 °C. 250 μCi of [3H]AA (PerkinElmer, Boston, MA) was added to the oxygenated phosphate buffer. 50 μg of platelet-type 12-lipoxygenase was added, and the reaction was swirled vigorously for 5 min. The reaction was stopped by adding 60 mg of glutathione and 100 μl of glutathione peroxidase (10 units/ml, Sigma). The reaction mixture was further incubated at 30 °C for additional 5 min with continuous swirling. The mixture was then passed through a 6-ml LC-18 SPE column (Cat. 57054, Supelco). After washing the column with 10 ml of phosphate buffer, bound 12-(S)-[3H]HETE was eluted with 4 ml of methanol. Following evaporating, sample was reconstituted in 100 μl of 95% acetonitrile, and then injected into Waters 2695 HPLC monitored by the UV absorbance with the Waters 2998 photodiode array detector. Purified 12-(S)- [3H]HETE was collected in fractions eluted at 3.75–4.75 min (112 μCi/mmol, supplemental Fig. S1C).
Cell Lines
Chinese Hamster Ovary (CHO) cell, COS7 (Africa green monkey kidney) cells, human prostate carcinoma PC3, DU145, and PC-3M cells were obtained from the American Type Culture Collection (Manassas, VA) and maintained in a humidified atmosphere of 5% CO2 at 37 °C. Cells were routinely cultured in RPMI 1640 supplemented with 10% FBS (Invitrogen, Grand Island, NY), 2 mm l-glutamine, and 100 μg/ml penicillin-streptomycin.
Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR)
Total RNA was isolated using TRIzol reagent (Invitrogen) according to the manufacturer's protocol. cDNA was generated for PCR reactions using the superscript II Reverse Transcriptase (Invitrogen) and standard procedures.
Cloning the GPR31 cDNA from PC3 Cells
Specific oligonucleotide primers were designed based on GenBankTM accession number U65402: the sense and antisense primers, 5′-CACGG CCGGG TGATG CCATT CCCA-3′/5′-GTCAG GAATA GGAGT CTCTG GGGTT G-3′, were designed to amplify the full coding region of the human GPR31 cDNA as reported (18). The RT-PCR with these primers was performed on RNA isolated from human PC3 cells. Amplification was carried out for 30 cycles (30 s at 94 °C, 30 s at 53 °C and 2 min at 72 °C), followed by an additional 7 min at 72 °C, and resulted in a DNA fragment of ∼1 kb, which was cloned into pcDNA3.1/V5-His-TOPO vector (Invitrogen). The cloned cDNA sequence was confirmed by sequencing.
Transfection
CHO cells were seeded in 6-well plates (2 × 105 cells/well), and cultured in RPMI 1640 plus 10% fetal bovine serum at 37 °C for 20 h in a humidified atmosphere of 5% CO2. The cells were transfected with pcDNA3.1-GPR31 or other constructs with GenePorter reagent (Gene Therapy Systems, Inc) according to the manufacturer's protocol.
12-(S)-[3H]HETE Binding Assay
The binding assays were performed at room temperature with isolated cell membranes or cell monolayers grown in 24-well culture plates. For the membrane binding experiments, cell membranes were prepared as described by Yotomizo et al. (19). Binding assays were initiated by the addition of various concentrations of 12-(S)-[3H]HETE in the binding buffer (50 mm Tris-Cl, pH 7.4; 10 mm MgCl2; 10 mm NaCl, and 0.05% BSA). Membranes were incubated at room temperature with various concentrations of 12-(S)-[3H]HETE for total binding samples, and co-incubated with 12-(S)-[3H]HETE plus 1000-fold excess non-labeled 12-(S)-HETE for nonspecific binding samples. After 90 min of incubation, the sample was transferred onto Whatman GF/C glass fiber filter and was quickly rinsed with binding buffer to remove unincorporated counts. Filters were then placed in separate scintillation vials contain 2 ml of scintillation liquid, UltimaGold (PerkinElmer) and the radioactivity was determined by a liquid scintillation counter. For cell monolayer binding experiments, 1.5 × 105 cells were plated in each well of a 24-well plate. Confluent monolayers were washed twice with binding assay buffer (an equal volume mixture of RPMI 1640 and DMEM, 12.5 mm HEPES, pH 7.4). Binding assays were initiated by the addition of various concentrations of 12-(S)-[3H]HETE in the binding buffer. For total binding, the cells were incubated with various concentrations of 12-(S)-[3H]HETE. For the nonspecific binding, the cells were co-incubated in the binding buffer with 12-(S)-[3H]HETE plus 1000-fold non-labeled 12-(S)-HETE. After 90 min of incubation, the labeled buffer was removed, and cells were quickly rinsed three times with PBS. Cells were solubilized with 2% SDS (sodium dodecyl sulfate). Cell lysate was mixed with scintillation fluid, and radioactivity was determined with a liquid scintillation counter. Specific binding of 12-(S)-HETE was calculated by subtracting nonspecific binding from total binding.
Western Blot Analysis
Following ligand stimulation, protein samples (30 μg) were separated on 10% SDS-PAGE gels, transferred to membranes, and blocked as per standard methods. Dilutions of primary antibodies were: anti-phospho-ERK1/2, 1:1000; anti-phospho-MEK, 1:1000; anti-ERK1/2, 1:1000, anti-MEK, 1:1000; and mouse anti-actin 1:2500 in TBS-T with 5% low fat milk at 4 °C overnight. A peroxidase-conjugated goat anti-rabbit or anti-mouse IgG (1:2000) were subsequently used for detection and visualized by enhanced chemiluminescence (Amersham Biosciences).
[35S]GTPγS Coupling Assay
[35S]GTPγS binding assays (20) were performed with membrane preparations from GPR31 or pcDNA CHO cell transfectants. Membranes were diluted in an assay buffer (20 mm HEPES, pH 7.4, 100 mm NaCl, 10 mm MgCl2, 0.1% BSA, and 5 μm GDP) to a final protein concentration of 100 μg per assay point. Reactions were performed in a final volume of 200 μl in 96-well plates. [35S]GTPγS was added to each well to give a final concentration of 500 pm, and membranes were incubated for 60 min at 25 °C. The reactions were terminated by transferring to Whatman GF/C glass fiber filter and quickly rinsed with binding buffer to remove unincorporated counts. Bound radioactivity was measured using a liquid scintillation counter.
NFκB Promoter-driven Luciferase Assay
COS-7 cells (1 × 105 cells/well) were plated in 24-well culture plates for 24 h. Cells were transfected with GPR31 or pcDNA together with a pNFκB reporter plasmid (21). Forty-eight hours post-transfection, cells were serum starved overnight followed by treatment with or without 300 nm 12-(S)-HETE. Luciferase activity was measured after 6 h using the luciferase assay system (Promega) as we previously described (21).
shRNA-mediated Gene Knockdown
Four segments (nucleotide sequence: 411–430, 862–881, 1034–1053, and 1508–1527) of the GPR31 gene were chosen as target sequences using the Cenix algorithm (Ambion) and synthesized as the following shRNA templates: si-GPR31 #1: 5′-GATCC GGCCT TTTCT TTCAT TTTGT TCAAG AGACA AAATG AAAGA AAAGG CCTTT TTTGG AAA-3′; si-GPR31 #2: 5′-GATCC GGTCA ACCTG CTGTC TCCTT TCAAG AGAAG GAGAC AGCAG GTTGA CCTTT TTTGG AAA-3′; si-GPR31 #3: 5′-GATCC GCACT CTCCT GCCTT CAGTT TCAAG AGAAC TGAAG GCAGG AGAGT GCTTT TTTGG AAA-3′; and si-GPR31 #4: 5′-GATCC ATAAT AATTA CTCCT ACTTT TCAAG AGAAA GTAGG AGTAA TTATT ATTTT TTTGG AAA-3′. After subcloning into the pSilencer5.1-H1 Retroviral vector (Ambion), constructs were transfected into PC-3M cells using Effectene Transfection Reagent (Qiagen). Seventy-two hours later, transfected cells were selected with puromycin (1 μg/ml). The efficacy of GPR31 knockdown was measured by RT-PCR and real-time PCR.
Cell Invasion Assay
The invasive capacity of cells was determined by using the Neuro Probe A-series 96-well chamber with standard Framed Filters (8 μm pore size) (Neruro Probe, Gaithersburg, MD) following the manufacturer's protocol. PC3M stably transfected with si-GPR31 #2, #4 constructs, and control empty vector (#5 cells in the text) were grown to confluence in RPMI 1640 medium supplemented with 10% FBS. Cells were washed three times with PBS and cultured in serum-free medium for 12 h. Cell suspensions were prepared by trypsin, and resuspended in plain RPMI 1640 at 2 × 105 cells/ml. Standard Framed Filters were coated with matrigelTM (250 μg/ml, BD Bioscience, San Jose, CA) at 37 °C for 1 h and then air-dried. Cell suspensions (400 μl) were plated in the top chambers. Chemoattractants were added to the lower chambers. Cells were allowed to invade through matrigel for 8 h. Cells remaining on the upper surface of filters were removed by gently wiping with a cotton swab. Transmigrated cells were fixed with 4% paraformaldehyde and stained with 0.5% crystal violet for 30 min. After washing, the crystal violet dye was eluted with 10% acetic acid, and absorbance was measured at A595.
Data Analysis
The binding-saturation (non-linear regression) software of Prism 5 (GraphPad Software Inc.) was used to plot and analyze the binding data of 12-(S)-[3H]HETE. The dose-response-stimulation (log[agonist] versus response, non-linear regression) software of Prism 5 was used to calculate the EC50 values, plot and analyze the [35S]GTPγS coupling data. Student's t test was used for statistic analysis.
RESULTS
Radioligand Binding Assay Shows GPR31 Is a High Affinity 12-HETER
GPCRs are a large family of seven transmembrane proteins. On the basis of sequence comparisons, human GPCRs can be divided into four main groups (A, rhodopsin-like; B, secretin receptor family; C, metabotropic glutamate/pheromone; and others, adhesion, frizzled, taste type-2, etc) (22) that share conserved secondary domain structure, but not significant sequence homologies. Initially, we screened the GPCR database to search for GPCRs whose ligands are potentially bioactive lipids, by applying features derived from known lipid-bound GPCRs such as short N termini and the characteristics of ligand-binding pockets (22, 23). A total of 43 GPCRs that fall into the group A (rhodopsin-like) subfamily of GPCRs were identified. The phylogenetic tree of these GPCRs was constructed with the “All All Program” (the Computational Biochemistry Server at ETH Zürich) (Fig. 1A).
FIGURE 1.

Identification and cloning of GPR31. A, 43 candidate GPCRs have been identified as potential receptors for lipid ligands. The phylogenic tree was constructed based on the similarity level of their amino acid sequences. B, specific binding of 12-(S)- [3H]HETE in PC3 cell membranes. PC3 cell membranes were incubated with 0–50 nm of 12-(S)-[3H]HETE in the absence or presence of 50 μm non-radioactive 12-(S)-HETE. The specific binding was measured as described under “Experimental Procedures.” C, 16 of these candidate GPCRs were cloned from PC3 cells, and transiently transfected into CHO cells for 12-(S)- [3H]HETE (10 nm) binding assay. Note that transfection of GPR31 construct results in a 5.4-fold increase in 12-(S)-[3H]HETE binding.
PC3 human prostate cancer cells are responsive to 12-(S)-HETE treatment (8, 11, 14). Moreover, a radioligand binding assay with 12-(S)-[3H]HETE confirmed the presence of a 12-(S)-HETE receptor in PC3 cells (Fig. 1B). Among the 43 potential GPCRs, there were several well-characterized lipid-bound GPCRs (e.g. EDG1–8, EP1–4, etc) that were excluded from consideration. Of the remaining candidates, sixteen were detected in PC3 cells and subsequently cloned into the pcDNA3.1 expression vector for transient transfection of Chinese Hamster Ovary (CHO) cells. CHO cells do not have endogenous 12-(S)-[3H]HETE binding sites (Fig. 2A). The resulting transfected CHO cells were then evaluated for 12-(S)-[3H]HETE binding. As shown in Fig. 1C, the orphan receptor designated GPR31 (18) transiently expressed in CHO cells led to 5.4-fold higher levels of membrane-bound 12-(S)-[3H]HETE than the mock-transfected CHO cells (pcDNA vector). From the full-length sequence of this cDNA clone, we examined the hydropathy profile from the predicted secondary structure and found that the GPR31 cDNA encodes a 7 transmembrane receptor with 319 amino acids. As we predicted, this receptor belongs to the rhodopsin-like group A subfamily that binds small molecules, including eicosanoids (Fig. 1A).
FIGURE 2.

GPR31 is a high affinity receptor of 12-(S)-HETE. Membranes of parental (A) or CHO cells transiently transfected with pcDNA or GPR31 (B, C) were incubated with various concentrations of 12-(S)-[3H]HETE in the presence or absence of 1000-fold excess of non-radioactive 12-(S)-HETE. The specific binding was measured as described under “Experimental Procedures.” The Scatchard plot (B, insert) showed that the Kd of GPR31 is 4.87 nm. Note that there is no specific binding of 12-(S)-[3H]HETE in parental or pcDNA transiently transfected CHO cells. Also, note that the binding of 12-(S)-[3H]HETE is saturable (C). D, PC-3M cells were stably transfected with two different shRNA constructs for GPR31 receptors as indicated under “Experimental Procedures.” Note that #4 shRNA construct effectively knocked-down GPR31 expression (insert). Also note that there is no specific binding of 12-(S)-[3H]HETE in #4 shRNA-transfected cells. E, cell membranes of GPR31 transiently transfected CHO cells were incubated with 12-(S)-[3H]HETE (10 nm) and various competing eicosanoids (10 μm). Note that only 12-(S)-HETE can significantly replace the specific binding of 12-(S)-[3H]HETE (**, p < 0.01; t test). F, displacement of 12-(S)-[3H]HETE (10 nm) binding by various concentrations of non-radioactive 12-(S)-, 12-(R)-HETE in membranes of CHO cells stably expressing GPR31 receptors.
Next, plasma membranes of CHO cells transiently expressing GPR31 receptors were prepared as described by Yokomizo et al. (19). Radioligand binding assays were performed on these membranes with 12-(S)-[3H]HETE. The specific binding of 12-(S)-[3H]HETE to membranes of the GPR31-transfected cells is saturable (Fig. 2, B and C). Little or no specific 12-(S)-[3H]HETE binding was detected in the membranes of pcDNA control vector-transfected cells (Fig. 2B). Analysis of binding data revealed that cloned GPR31 has a Kd value of 4.8 ± 0.12 nm with Bmax value of 38.3 ± 0.23 pmol/mg protein. PC-3M cells express GPR31 receptors. Knockdown of GPR31 using shRNA diminished the specific binding of 12-(S)-[3H]HETE (Fig. 2D). Moreover, PC3 cells express ∼3-fold more GPR31 than Du145 prostate cancer cells. Accordingly, PC3 cells exhibit a corresponding increase in the specific binding activity of 12-(S)-[3H]HETE compared with Du145 cells (supplemental Fig. S2A). Previously, it was reported that the leukotriene B4 receptor, BLT2, has 12-(S)-HETE binding ability (22). To assess the specificity of binding, GPR31 and BLT2 receptors were expressed in CHO cells at similar levels and were compared for their affinities for 12-(S)-[3H]HETE. The results confirm the binding of 12-(S)-HETE to BLT2 receptors, albeit, the affinity of BLT2 receptor was much lower compared with GPR31 (supplemental Fig. S2B).
Moreover, non-radioactive 12-(S)-HETE effectively replaced 12-(S)-[3H]HETE bound to GPR31 receptors, whereas 12-(R)-HETE was unable to replace GPR31-bound 12-(S)-[3H]HETE (Fig. 2, E and F). This result suggests that binding of 12-(S)-HETE to GPR31 is stereospecific. The specificity of 12-(S)-[3H]HETE binding to GPR31 receptors were further examined by the ability of various eicosanoids to displace 12-(S)-[3H]HETE bound to the membrane fraction of CHO cells transfected with GPR31 construct. The results indicate that only the non-radiolabeled 12-(S)-HETE can significantly replace bound 12-(S)-[3H]HETE in the membrane from GPR31-transfected CHO cells (p < 0.01, t test) (Fig. 2E). This result strongly suggests that GPR31 is a high affinity receptor for 12-(S)-HETE, a monohydroxy fatty acid.
GTPγS Coupling Assay Indicates that GPR31 Is a High Affinity Receptor of 12-(S)-HETE
Next, we employed the GTPγS coupling assay (20) to confirm further whether the GPR31 receptor is a high affinity receptor of 12-(S)-HETE. Plasma membranes from CHO cells transiently transfected with GPR31 or pcDNA constructs were incubated with various concentrations of the structural analogs of 12-(S)-HETE in the presence of [35S]GTPγS. The EC50 of 12-(S)-HETE induced GTPγS coupling is 0.28 ± 1.26 nm in the membranes of GPR31 transfectants (Fig. 3A, and Table 1). In sharp contrast, the EC50 of 12-(S)-HETE induced GTPγS coupling in the membranes of pcDNA-transfected cells is ∼1 mm (Fig. 3A), which is most likely due to the nonspecific effect of the high concentration of lipid ligand. The stereoisomer, 12-(R)-HETE, was unable to stimulate specific coupling of GTPγS in membranes of either GPR31 or pcDNA-transfected cells (Fig. 3B), reflecting the stereospecificity of 12-(S)-HETE/GPR31 binding. The EC50 values of 15-(S)- or 5-(S)-HETE induced GTPγS coupling in membranes of GPR31 transfectants are 42.1 ± 31 and 385.7 ± 62 nm, respectively (Fig. 3, C and D, Table 1), which are approximately 2–3 orders of magnitude higher than that induced by 12-(S)-HETE. Moreover, similar EC50 values were observed in Hela and HEK293 cells transiently expressing GPR31 receptors (Fig. 3, E and F, Table 1). Together, these results suggest that 12-(S)-HETE is a regio- (positional-) specific ligand for GPR31.
FIGURE 3.

The stereo- and positional specificity of 12-(S)-HETE in GPR31-mediated GTPγS coupling. Plasma membranes from CHO (A–D), Hela (E), HEK293 (F) cells transiently transfected with GPR31 or pcDNA constructs were incubated with structural analogs of 12-(S)-HETE in the presence of [35S]GTPγS. The EC50 data for 12-(S)-HETE, 12-(R)-HETE, 15-(S)-HETE, and 5-(S)-HETE were summarized in Table 1.
TABLE 1.
EC50 of various agonists
The dose-response-stimulation (log[agonist] vs. response, non-linear regression) software of Prism 5 (GraphPad Software Inc) was employed to analyze the [35S]GTPγS coupling data and calculate the EC50 values. R-squared values of CHO/GPR31 cells treated with 12-(S)-HETE, 5-(S)-HETE, 15-(S)-HETE, and Hela or HEK293 cells treated with 12-(S)-HETE are: 0.9460, 0.9454, 0.8822, 0.9754, and 0.9840. n, numbers of experimental repeats. Each experiment has four determinations.
| Ligands | Cell | EC50 | n |
|---|---|---|---|
| 12-(S)-HETE | CHO/GPR31 | 0.28 ± 1.26 | (n = 6) |
| CHO/pcDNA | n.s.b.a | (n = 2) | |
| 12-(R)-HETE | CHO/GPR31 | n.s.b. | (n = 2) |
| CHO/pcDNA | n.s.b. | (n = 2) | |
| 5-(S)-HETE | CHO/GPR31 | 385.7 ± 62b | (n = 2) |
| CHO/pcDNA | n.s.b. | (n = 2) | |
| 15-(S)-HETE | CHO/GPR31 | 42.1 ± 31b | (n = 2) |
| CHO/pcDNA | n.s.b. | (n = 2) | |
| 12-(S)-HETE | Hela/GPR31 | 0.12 | (n = 1) |
| Hela/pcDNA | n.s.b. | (n = 1) | |
| 12-(S)-HETE | HEK293/GPR31 | 0.11 | (n = 1) |
| HEK293/pcDNA | n.s.b. | (n = 1) |
a n.s.b., no specific binding.
b p < 0.05, vs. 12-(S)-HETE-treated CHO/GPR31 cells, t-test.
12-(S)-HETE Activates MEK, ERK1/2, and NFκB via the GPR31 Receptor
It is known that treatment with 12-(S)-HETE leads to activation of signaling molecules such as extracellular signal-regulated kinase1/2 (ERK1/2), MEK, and NFκB (15, 16, 21). Therefore, we examined whether these 12-(S)-HETE-regulated signaling events are mediated by GPR31. As shown in Fig. 4A, treatment with 12-(S)-HETE resulted in ERK1/2 activation in CHO cells transiently transfected with HA-tagged and wild-type GPR31 constructs, whereas 12-(S)-HETE failed to activate ERK1/2 in parental or pcDNA vector-transfected CHO cells. Similarly, in COS-7 cells stably expressing GPR31, 12-(S)-HETE was able to significantly activate NFκB activity as measured by an NFκB promoter-driven luciferase assay (Fig. 4B). In contrast, 12-(S)-HETE was incapable of activating NFκB activity in COS-7 cells stably transfected with pcDNA control vector. 12-(S)-HETE dose-dependently (ranging from 1–300 nm) activated ERK1/2 and NFκB via GPR31 receptors (Fig. 4, C and D). In a control, 20-(S)-HETE (300 nm) was unable to activate ERK1/2 in GPR31-expressing cells (Fig. 4C). Moreover, 12-(S)-HETE/GPR31-mediated ERK1/2 activation was inhibited by pertussis toxin, suggesting that the involvement of the Gi/o heterotrimeric G proteins (Fig. 4C).
FIGURE 4.

GPR31 transduces 12-(S)-HETE signaling. A, CHO cells transiently transfected with GPR31, HA epitope tagged-GPR31 or pcDNA were treated with 12-(S)-HETE (300 nm). Cell lysates were immunoblotted with phosphospecific ERK1/2 antibody (upper panels). Lower panels, nitrocellulose membranes were reprobed with ERK1/2. B, COS-7 cells were stably transfected with pcDNA and pNFκB-Luc, or GPR31 and pNFκB-Luc. Luciferase activity was measured after 12-(S)-HETE (300 nm) treatment for 6 h. *, p < 0.05 (t test). C, CHO cells transiently transfected with GPR31 were treated with various concentrations of 12-(S)-HETE for 10 min in the presence or absence of pertussis toxin (PTx). ERK1/2 activation was measured. In a control, cells were treated with 300 nm 20-(S)-HETE. The ERK1/2 activation was quantitated by a densitometer (lower panel). D, NFκB activation was measured in COS-7 stably expressing GPR31 treated with various concentrations of 12-(S)-HETE. *, p < 0.05, versus no treatment control, t test. E, CHO cells were transiently transfected with GPR31, 5-oxoR, and HM74, together with pNFκB-Luc vector. Luciferase activity was measured after stimulation with or without 12-(S)-HETE. *, p < 0.05, versus GPR31-transfected cells without 12-(S)-HETE treatment, t test. F, PC3 cells stably transfected with pcDNA or GPR31 were treated with 12-(S)-HETE (300 nm). Upper panel, lysates was probed with phosphospecific MEK antibody; lower panel, membrane was reprobed with MEK antibody. G, left panel, RT-PCR (upper) and real-time PCR quantitation (lower) of GPR31 in PC-3M cells stably transfected with different shRNA constructs. Fold change was normalized to PC-3M cells transfected with the # 5 construct (control empty vector). Right panel, PC-3M cells stably transfected with #5 (control empty vector) were stimulated with various concentrations of 12-(S)-HETE for 10 min. The activation of MEK, indicated by the intensity of phospho-MEK Western blot, was quantitated by a densitometer (lower). H, MEK activation was measured in PC-3M cells stably transfected with si-GPR31 #2 or #4 constructs following 12-(S)-HETE stimulation.
There is a minimal increase of 12-(S)- [3H]HETE binding in CHO cells expressing 5-oxo-ETER (5-oxoR) and HM74 GPCRs (Fig. 1C). Therefore, we examined whether 5-oxoR and HM74 GPCRs can transduce the 12-(S)-HETE mediated signaling. As shown in Fig. 4E, treatment of 12-(S)-HETE was unable to activate NFκB in 5-oxoR and HM74-expressing cells. This result suggests that 5-oxoR and HM74 are not 12-(S)-HETE receptors. In addition, it should be noted that 12-(S)-HETE/GPR31 signaling axis was incapable of inducing the intracellular Ca+2 mobilization (supplemental Fig. S3).
PC3 cells exhibit high affinity binding for 12-(S)-[3H]HETE and express GPR31 (Fig. 1B, supplemental Fig. S2A). Consequently, MEK was activated in a time-dependent manner in PC3 cells transfected with pcDNA control vector following 12-(S)-HETE stimulation (Fig. 4F). The activation of MEK was markedly enhanced in PC3 cells stably overexpressing GPR31 (Fig. 4F). To evaluate the essential role of GPR31 in 12-(S)-HETE stimulated MEK activation, we employed several GPR31 shRNA constructs to knockdown GPR31 to different levels. As shown in Fig. 4G, the #4 shRNA efficiently knocked-down GPR31 in PC-3M cells, whereas knockdown with the #2 shRNA was inefficient. The level of GPR31 in #2-transfected cells is comparable to that of cells transfected with empty control vector (#5, Fig. 4G, left panel). 12-(S)-HETE dose-dependently stimulated MEK activation in PC-3M stably transfected with #5 empty control vector (right panel, Fig. 4G). Also, treatment with 12-(S)-HETE time-dependently activated MEK in cells stably expressing #2 si-GPR31 construct. In sharp contrast, MEK activation was completely abolished in cells stably expressing the #4 si-GPR31 construct. Collectively, these data indicate that 12-(S)-HETE-regulated activation of ERK1/2, NFκB, and MEK is specifically mediated by GPR31. Based on radioligand binding assay, GTPγS coupling, and signaling cascades activation, we firmly establish for the first time that GPR31 is a high affinity receptor of 12-(S)-HETE. Therefore, GPR31 is renamed as 12-HETE receptor (12-HETER) thereafter.
12-(S)-HETE/12-HETER Signaling Regulates Tumor Cell Invasion in Vitro
It is well established that 12-(S)-HETE plays a critical role in inflammation, tumor invasion, and metastasis (1–6, 11, 12). To investigate the role of 12-HETER in cell invasion, PC-3M cells expressing various levels of 12-HETER (Fig. 4G) were employed. Specifically, 12-HETER was efficiently knocked-down in si-GPR31#4 cells, whereas expression in si-GPR31#2 and si-GPR31#5 (empty vector control) cells was comparable to parental PC-3M cells. As shown in Fig. 5A, increasing doses of 12-(S)-HETE did stimulate the invasive capacity of both si-GPR#2 and si-GPR#5 cells in an in vitro invasion assay. In contrast si-GPR31#4 cells that no longer expressed 12-HETER were unresponsive to 12-(S)-HETE-induced invasion (Fig. 5A). The specificity of response to 12-(S)-HETE by the 12-HETER was demonstrated by the fact that invasion by the three PC-3M variants was nearly equivalent when stimulated with either EGF or 10% FBS (Fig. 5B). This result suggests that 12-(S)-HETE-stimulated tumor cell invasion is specifically mediated by the 12-HETER signaling.
FIGURE 5.
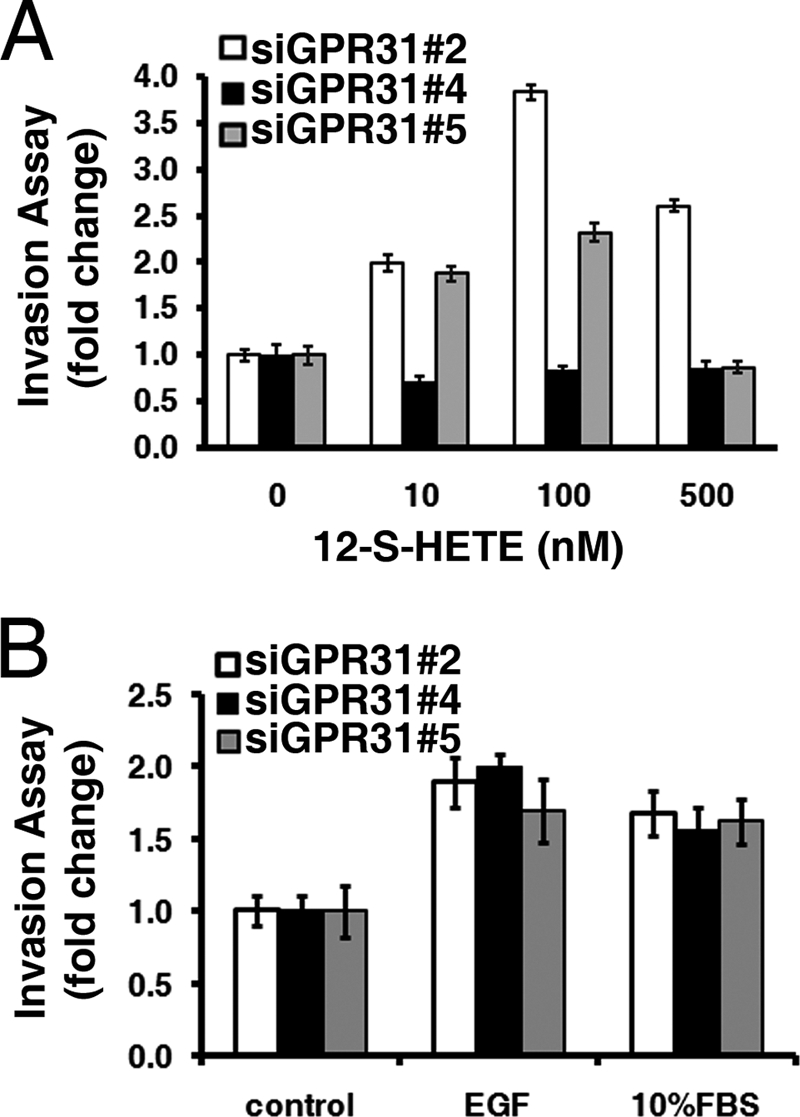
12-HETER mediates 12-(S)-HETE-stimulated cell invasion. A, invasion assays for PC-3M cells transfected with si-GPR31 #2, #4, or #5 (control empty vector) constructs in the presence or absence of various concentrations of 12-(S)-HETE. Note that knockdown of 12-HETER abolishes 12-(S)-HETE-stimulated cell invasion. B, invasiveness is indistinguishable between PC-3M cells transfected with si-GPR31 #2, #4, or #5 constructs, following EGF or 10% FBS stimulation.
DISCUSSION
As early as 1992, Arenberger et al. found that epidermal Langerhans cells of human skin possess specific binding for 12-(S)-HETE (Kd = 3.32 ± 0.45 nm) (22). To date, no molecular evidence for a 12-(S)-HETE receptor has been reported. Hammarström and co-workers (24–27) reported that murine Lewis lung carcinoma cells contain specific high affinity binding sites for 12-(S)-HETE. Their data showed that the binding sites have a cytosolic/nuclear localization and contain the heat shock proteins hsp70 and hsp90 as a high molecular weight cytosolic binding complex. The ligand binding subunit of this complex is an unidentified protein with an apparent molecular mass of ∼50 kDa. Also, their data suggested that the 50-kDa 12-(S)-HETE-binding protein is a nuclear receptor or co-activator protein. However, there has been no further evidence of the 50-kDa protein as a 12-(S)-HETE receptor.
Our laboratory found that the specific effect of 12-(S)-HETE on increasing tumor cell metastatic characteristics is positionally and stereochemically determined because the positional isomers 5-(S)-, 8-(S)-, 9-(S)-, 11-(S)-, and 15-(S)-HETE as well as the enantiomer 12-(R)-HETE failed to evoke any of the 12-(S)-HETE effects on tumor cells (28–30). Ligand binding experiments have revealed that high affinity (Kd = 1 nm) receptors of 12-(S)-HETE are present in several cell lines (15). The activation of MAPK signaling pathways by 12-(S)-HETE demonstrates that its receptor is G protein-coupled (15). In the present study, we have shown for the first time that the orphan GPR31 receptor is the high affinity receptor for 12-(S)-HETE receptor. Chinese hamster ovary (CHO) cells have no endogenous 12-(S)-HETE binding sites, as evidenced by the fact that there is no specific binding of 12-(S)- [3H]HETE in parental or pcDNA transfected CHO cells (Fig. 2, A and B). However, when the GPR31 cDNA construct was transfected into CHO cells, the transfected cells exhibited specific binding for 12-(S)-[3H]HETE with a Kd value of 4.87 nm (Fig. 2B), which is comparable to the affinity of 12-(S)-HETE receptor characterized previously (15, 28–31). Moreover, 12-(S)-HETE treatment specifically induced GTPγS coupling in membrane fractions of GPR31-transfected cells, but failed to induce GTPγS coupling in membrane fractions of pcDNA-transfected CHO cells (Fig. 3A). The 12-(S)-HETE effect on GTPγS coupling is regio- and stereospecific, as 15-(S)- and 5-(S)-HETE required two orders of magnitude greater concentration to induce GTPγS coupling and because 12-(R)-HETE is completely inactive (Fig. 3, Table 1). Furthermore, 12-(S)-HETE stimulated ERK1/2 and NFκB activation in GPR31-transfected CHO and COS7 cells. In contrast, there was no detectable ERK1/2 or NFκB activation in 12-(S)-HETE-treated mock-transfected cells (Fig. 4). Overexpression of GPR31 enhanced 12-(S)-HETE-stimulated MEK activation (Fig. 4F), whereas knockdown of GPR31 (Fig. 4H) abolished 12-(S)-HETE stimulated MEK activation (Fig. 4H) as well as invasive capability (Fig. 5). Collectively, these data indicate that we have identified the orphan receptor GPR31, for the first time, as a high affinity 12-(S)-HETE receptor (12-HETER).
Following the cloning and characterization of GPR31 as a high affinity receptor of 12-(S)-HETE, we initially performed an extensive analysis of array data deposited in the Gene Expression Omnibus (GEO) to examine the potential involvement of 12-HETER in various pathophysiological conditions. We observed that 12-HETER may be dysregulated in several diseases, including malignant megakaryocytes, arthritis, Alzheimer's disease, progressive B-cell chronic lymphocytic leukemia, diabetic nephropathy, high grade astrocytoma, prostate cancer, etc. Also, 12-(S)-HETE is an important signaling molecule which regulates various biological functions (1–14). For example, it has been shown that 12-(S)-HETE increases the invasiveness and metastatic potential in prostate tumors (8, 11, 12, 14) and is involved in the carcinogenesis of prostate tumors (11, 14, 32, 33). However, the pathophysiological roles of 12-(S)-HETE/12-HETER signaling await empirical determinations in the future. Therefore, further studies of the 12-HETER will provide mechanistic insights into 12-(S)-HETE-regulated signaling and functions, which may offer potential new therapeutics for a variety of diseases.
Supplementary Material
This work was supported, in whole or in part, by National Institutes of Health Grants CA029997, DoD W81XWH-06-1-0226 (to K. V. H.), and HL071071 (to M. J. L.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- 12-(S)-HETE
- 12-(S)-hydroxy-5,8,10,14-eicosatetraenoic acid
- GPCR
- G protein-coupled receptor
- GTPγS
- guanosine 5′-3-O-(thio)triphosphate
- 12-HETER
- 12-(S)-HETE receptor or GRP31
- AA
- arachidonic acid.
REFERENCES
- 1. Honn K. V., Tang D. G., Crissman J. D. (1992) Cancer Metastasis Rev. 11, 325–351 [DOI] [PubMed] [Google Scholar]
- 2. Chopra H., Timar J., Chen Y. Q., Rong X. H., Grossi I. M., Fitzgerald L. A., Taylor J. D., Honn K. V. (1991) Int. J. Cancer 49, 774–786 [DOI] [PubMed] [Google Scholar]
- 3. Tang D. G., Honn K. V. (1997) Adv. Exp. Med. Biol. 400A, 349–361 [DOI] [PubMed] [Google Scholar]
- 4. Timar J., Silletti S., Bazaz R., Raz A., Honn K. V. (1993) Int. J. Cancer 55, 1003–1010 [DOI] [PubMed] [Google Scholar]
- 5. Honn K. V., Timár J., Rozhin J., Bazaz R., Sameni M., Ziegler G., Sloane B. F. (1994) Exp. Cell Res. 214, 120–130 [DOI] [PubMed] [Google Scholar]
- 6. Liu X. H., Connolly J. M., Rose D. P. (1996) Clin. Exp. Metastasis 14, 145–152 [DOI] [PubMed] [Google Scholar]
- 7. Natarajan R., Bai W., Lanting L., Gonzales N., Nadler J. (1997) Am. J. Physiol. 273, H2224–H2231 [DOI] [PubMed] [Google Scholar]
- 8. Nie D., Krishnamoorthy S., Jin R., Tang K., Chen Y., Qiao Y., Zacharek A., Guo Y., Milanini J., Pages G., Honn K. V. (2006) J. Biol. Chem. 281, 18601–18609 [DOI] [PubMed] [Google Scholar]
- 9. Tang D. G., Diglio C. A., Bazaz R., Honn K. V. (1995) J. Cell Sci. 108, 2629–2644 [DOI] [PubMed] [Google Scholar]
- 10. Natarajan R., Gonzales N., Lanting L., Nadler J. (1994) Hypertension 23, I142–1147 [DOI] [PubMed] [Google Scholar]
- 11. Pidgeon G. P., Tang K., Cai Y. L., Piasentin E., Honn K. V. (2003) Cancer Res. 63, 4258–4267 [PubMed] [Google Scholar]
- 12. Honn K. V., Tang D. G., Gao X., Butovich I. A., Liu B., Timar J., Hagmann W. (1994) Cancer Metastasis Rev. 13, 365–396 [DOI] [PubMed] [Google Scholar]
- 13. Honn K. V., Tang D. G., Grossi I., Duniec Z. M., Timar J., Renaud C., Leithauser M., Blair I., Johnson C. R., Diglio C. A. (1994) Cancer Res. 54, 565–574 [PubMed] [Google Scholar]
- 14. Nie D., Hillman G. G., Geddes T., Tang K., Pierson C., Grignon D. J., Honn K. V. (1998) Cancer Res. 58, 4047–4051 [PubMed] [Google Scholar]
- 15. Liu B., Khan W. A., Hannun Y. A., Timar J., Taylor J. D., Lundy S., Butovich I., Honn K. V. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 9323–9327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Szekeres C. K., Tang K., Trikha M., Honn K. V. (2000) J. Biol. Chem. 275, 38831–38841 [DOI] [PubMed] [Google Scholar]
- 17. Hampson A. J., Grimaldi M. (2002) J. Neurosci. 22, 257–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zingoni A., Rocchi M., Storlazzi C. T., Bernardini G., Santoni A., Napolitano M. (1997) Genomics 42, 519–523 [DOI] [PubMed] [Google Scholar]
- 19. Yokomizo T., Kato K., Hagiya H., Izumi T., Shimizu T. (2001) J. Biol. Chem. 276, 12454–12459 [DOI] [PubMed] [Google Scholar]
- 20. Harrison C., Traynor J. R. (2003) Life Sci. 74, 489–508 [DOI] [PubMed] [Google Scholar]
- 21. Kandouz M., Nie D., Pidgeon G. P., Krishnamoorthy S., Maddipati K. R., Honn K. V. (2003) Prostaglandins Other Lipid Mediat. 71, 189–204 [DOI] [PubMed] [Google Scholar]
- 22. Bjarnadóttir T. K., Gloriam D. E., Hellstrand S. H., Kristiansson H., Fredriksson R., Schiöth H. B. (2006) Genomics 88, 263–273 [DOI] [PubMed] [Google Scholar]
- 23. Wettschureck N., Offermanns S. (2005) Physiol. Rev. 85, 1159–1204 [DOI] [PubMed] [Google Scholar]
- 24. Herbertsson H., Hammarström S. (1995) Biochim. Biophys. Acta 1244, 191–197 [DOI] [PubMed] [Google Scholar]
- 25. Herbertsson H., Kuhme T., Evertsson U., Wigren J., Hammarström S. (1998) J. Lipid Res. 39, 237–244 [PubMed] [Google Scholar]
- 26. Kurahashi Y., Herbertsson H., Söderström M., Rosenfeld M. G., Hammarström S. A. (2000) Proc. Natl. Acad. Sci. U.S.A. 97, 5779–5783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Herbertsson H., Kuhme T., Hammarstrom S. (1999) Arch. Biochem. Biophys. 367, 3–38 [DOI] [PubMed] [Google Scholar]
- 28. Grossi I. M., Fitzgerald L. A., Umbarger L. A., Nelson K. K., Diglio C. A., Taylor J. D., Honn K. V. (1989) Cancer Res. 49, 1029–1037 [PubMed] [Google Scholar]
- 29. Liu B., Timar J., Howlett J., Diglio C. A., Honn K. V. (1991) Cell Regul. 2, 1045–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Honn K. V., Nelson K. K., Renaud C., Bazaz R., Diglio C. A., Timar J. (1992) Prostaglandins 44, 413–429 [DOI] [PubMed] [Google Scholar]
- 31. Arenberger P., Kemény L., Rupec R., Bieber T., Ruzicka T. (1992) Eur. J. Immunol. 22, 2469–2472 [DOI] [PubMed] [Google Scholar]
- 32. Matsuyama M., Yoshimura R., Mitsuhashi M., Hase T., Tsuchida K., Takemoto Y., Kawahito Y., Sano H., Nakatani T. (2004) Int. J. Oncol 24, 821–827 [PubMed] [Google Scholar]
- 33. Shappell S. B., Olson S. J., Hannah S. E., Manning S., Roberts R. L., Masumori N., Jisaka M., Boeglin W. E., Vader V., Dave D. S., Shook M. F., Thomas T. Z., Funk C. D., Brash A. R., Matusik R. J. (2003) Cancer Res. 63, 2256–2267 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.