

Abstract
The CYTH superfamily of proteins is named after its two founding members, the CyaB adenylyl cyclase from Aeromonas hydrophila and the human 25-kDa thiamine triphosphatase. Because these proteins often form a closed β-barrel, they are also referred to as triphosphate tunnel metalloenzymes (TTM). Functionally, they are characterized by their ability to bind triphosphorylated substrates and divalent metal ions. These proteins exist in most organisms and catalyze different reactions depending on their origin. Here we investigate structural and catalytic properties of the recombinant TTM protein from Nitrosomonas europaea (NeuTTM), a 19-kDa protein. Crystallographic data show that it crystallizes as a dimer and that, in contrast to other TTM proteins, it has an open β-barrel structure. We demonstrate that NeuTTM is a highly specific inorganic triphosphatase, hydrolyzing tripolyphosphate (PPPi) with high catalytic efficiency in the presence of Mg2+. These data are supported by native mass spectrometry analysis showing that the enzyme binds PPPi (and Mg-PPPi) with high affinity (Kd < 1.5 μm), whereas it has a low affinity for ATP or thiamine triphosphate. In contrast to Aeromonas and Yersinia CyaB proteins, NeuTTM has no adenylyl cyclase activity, but it shares several properties with other enzymes of the CYTH superfamily, e.g. heat stability, alkaline pH optimum, and inhibition by Ca2+ and Zn2+ ions. We suggest a catalytic mechanism involving a catalytic dyad formed by Lys-52 and Tyr-28. The present data provide the first characterization of a new type of phosphohydrolase (unrelated to pyrophosphatases or exopolyphosphatases), able to hydrolyze inorganic triphosphate with high specificity.
Keywords: Bacterial Metabolism, Enzyme Structure, Mass Spectrometry (MS), Phosphatase, X-ray Crystallography, CYTH, Nitrosomonas, Triphosphatase, Triphosphate, Thiamine Triphosphate
Introduction
A large number of phosphohydrolases are able to hydrolyze triphosphorylated compounds, generally nucleoside triphosphates such as ATP or GTP. However, in 2002 we achieved the molecular characterization of a mammalian enzyme that specifically hydrolyzed thiamine triphosphate (ThTP),6 a compound unrelated to nucleotides (1). This 25-kDa soluble thiamine triphosphatase (ThTPase), which is involved in the regulation of cytosolic ThTP concentrations (2), has near absolute specificity for ThTP (it does not hydrolyze nucleotides) and a high catalytic efficiency. No sequence homology with other known mammalian proteins could be found.
Shortly thereafter, Iyer and Aravind (3) observed that the catalytic domains of human 25-kDa ThTPase (1) and CyaB adenylyl cyclase (AC2) from Aeromonas hydrophila (4) define a novel superfamily of domains that, according to these authors, should bind “organic phosphates.” This superfamily was called “CYTH” (CyaB-thiamine triphosphatase), and the presence of orthologs was demonstrated in all three superkingdoms of life. This suggested that CYTH is an ancient family of proteins and that a representative must have been present in the last universal common ancestor (LUCA) of all extant life forms. It was proposed (3) that this enzymatic domain might play a central role at the interface between nucleotide and polyphosphate metabolism, but this role remains largely undefined; in fact, the two structurally and enzymatically characterized members of the superfamily, AC2 and mammalian 25-kDa ThTPase, are likely to be secondary acquired activities, whereas other prokaryotic CYTH proteins would have performed more ancient and fundamental roles in organic phosphate and/or polyphosphate metabolism (3).
Using multiple alignments and secondary structure predictions, Iyer and Aravind (3) showed that the catalytic core of CYTH enzymes contained a novel α + β scaffold with six conserved acidic and four basic residues. At least four of the acidic residues (generally glutamate) are likely to chelate divalent cations required for catalysis, as is the case for adenylyl cyclases, DNA polymerases, and some phosphohydrolases (3, 5, 6).
More recently and independently of those bioinformatic studies, Shuman and co-workers (7) pointed out that RNA triphosphatases of fungi and protozoa exhibit striking similarities with bacterial and archaeal proteins of unknown function that belong to the CYTH superfamily. Although the primary structure conservation is low, the active site folds of the Cet1 RNA triphosphatase from Saccharomyces cerevisiae (8) and of several prokaryotic CYTH proteins are remarkably similar; in both cases, an eight-strand antiparallel β-barrel forms a topologically closed tunnel, and triphosphorylated substrates as well as divalent cation activators bind in the hydrophilic cavity. Therefore, Shuman and co-workers (7) called these proteins “triphosphate tunnel metalloenzymes” (TTM). They assumed that TTM was a larger superfamily including Cet1-like and CYTH proteins.
However, it is not established that all members of the CYTH superfamily exhibit the closed tunnel conformation. For instance, the 25-kDa mouse ThTPase has an open cleft structure when free in solution, whereas the enzyme-ThTP complex has a more closed, tunnel-like conformation (9). This suggests that the CYTH domain may be a versatile fold, both structurally and functionally.
Despite the presence of orthologs in most organisms, the general significance of CYTH enzymes remains a matter of conjecture. In unicellular eukaryotes, the essential role of RNA triphosphatases in RNA capping is clearly defined. But, like ThTPase activity in mammals, this is probably a secondary adaptation. In bacteria, two CYTH proteins, from A. hydrophila (4) and Yersinia pestis (10, 11) are considered as a particular class of adenylyl cyclases (class IV, CyaB), but this enzyme activity was found to be significant only under rather extreme conditions (50–60 °C and pH around 10). In addition, a more efficient class I adenylyl cyclase is also present in A. hydrophila (4) and Y. pestis (11). Archaeal and bacterial CYTH proteins have since been annotated as hypothetical (CyaB-like) adenylyl cyclases.
However, a bacterial CYTH ortholog from Clostridium thermocellum was recently shown to be completely devoid of adenylyl cyclase activity (12). The enzyme, which was characterized by Shuman and co-workers (12, 13) in considerable detail had a rather broad substrate specificity. It hydrolyzed guanosine 5′-tetraphosphate (Gp4), inorganic triphosphate (tripolyphosphate (PPPi)), and to a lesser extent nucleoside triphosphates and long-chain polyphosphates (12, 13).
In 2005 we obtained the three-dimensional crystal structure of the Nitrosomonas europaea CYTH ortholog (NeuTTM), the hypothetical protein NE1496 (Protein Data Bank code 2FBL); although labeled as a TTM (12), it exhibits a C-shaped cup conformation, or incomplete β-barrel.
Because of the structural similarities between NeuTTM and mouse 25-kDa ThTPase (open cleft rather than closed tunnel), we considered the possibility that ThTP might be a good substrate for NeuTTM. Indeed, in Escherichia coli, ThTP may be involved in responses to environmental stress (15), but no specific bacterial ThTPase has been characterized so far. However, the present data show that NeuTTM is actually devoid of significant ThTPase activity (as well as of adenylyl cyclase activity) but that NeuTTM has a strong preference and high affinity for PPPi. Furthermore, we show that two residues that are highly conserved in CYTH proteins (Lys-52 and Tyr-28) are important for catalysis. This is the first characterization of a specific tripolyphosphatase (PPPase), raising the question of the existence and possible role of PPPi in living cells.
EXPERIMENTAL PROCEDURES
Materials
Sodium tripolyphosphate (PPPi), sodium cyclic triphosphate (trimetaphosphate), polyphosphate (sodium phosphate glass, 65 ± 5 residues), ATP, ITP, GTP, and guanosine 5′-tetraphosphate (Gp4, Tris salt) were from Sigma. ThTP was synthesized and purified as previously described (16). Tetraphosphate (P4) (initially from Sigma) was a gift from Dr. Eric Oldfield, Dept. of Chemistry, University of Illinois, Urbana, IL. The pET15b vector for the expression of NeuTTM as a His-tag fusion protein was a gift from Dr. A. Savchenko, University of Toronto, Banting and Best Department of Medical Research, C. H. Best Institute, Toronto, Ontario, Canada).
Overexpression and Purification of NeuTTM
BL21(DE3) E. coli cells were transformed with a pET15b plasmid containing the sequence of NeuTTM with a His6 sequence at the N-terminal side of the gene. Transformed bacteria were grown overnight at 37 °C in 2 ml of LB medium containing 500 μg/ml ampicillin. Then the bacteria were cultured in 50–200 ml of 2× YT medium containing 250 μg/ml ampicillin until A600 reached 0.6–0.8. Expression of the fusion protein was induced with isopropyl β-d-1-thiogalactopyranoside (1 mm). After 3 h of incubation at 37 °C, the bacterial suspension was centrifuged 15 min at 8000 × g (4 °C). Bacteria were suspended in modified binding buffer (20 mm HEPES instead of phosphate buffer, 0.5 m NaCl, 30 mm imidazole, pH 7.5). After two cycles with a French press disruptor, the lysate was centrifuged 30 min at 50,000 × g. The tagged protein was purified on a FPLC system (ÄKTATM Purifier, Amersham Biosciences) coupled to a 5-ml HisTrapTM FF column (GE Healthcare) in the above-mentioned buffer. NeuTTM was then eluted in eluting buffer (20 mm HEPES, 0.5 m NaCl, 0.5 m imidazole, pH adjusted to 7.5 with 6 n HCl). Removal of the His tag was achieved by incubating 1 mg of the purified protein with 200 units of AcTEVTM protease (Invitrogen). After 2 h at room temperature, the mixture was purified on the HisTrap column to remove the released tag as well as the protease (also His-tagged). The purified untagged NeuTTM was recovered in the first fractions.
Crystallization, Data Collection, and Structure Solution
For crystallographic purposes, the NeuTTM gene was expressed as a selenomethionine-substituted protein using standard M9 high yield growth procedure according to the manufacturer's instructions (Shanghai Medicilon; catalogue number MD045004-50) with E. coli BL21(DE3) codon plus cells.
Crystals for the NeuTTM data collection were obtained with hanging drop vapor diffusion using a 24-well Linbro plate. 1 ml of well solution was added to the reservoir, and drops were made with 1 μl of well solution and 1 μl of protein solution. The crystals were grown at 20 °C using 3.5 m sodium formate as a precipitation agent and 0.1 m Bis-Tris propane buffer, pH 7.0.
The NeuTTM crystal was briefly dipped into a 5-μl drop of cryoprotectant solution (3.8 m sodium formate, 0.2 m NaCl, 4% sucrose, 4% glycerol, 4% ethylene glycol, and 0.1 m Bis-Tris propane buffer, pH 7.0) before flash-freezing in liquid nitrogen. Data collection was performed at the 19-ID beamline at the Advanced Photon Source, Argonne National Laboratory. One dataset was collected using 0.97959 Å wavelength. Data were indexed and processed with the HKL-2000 package (17).
Intensities were converted into structure factors, and 5% of the reflections were flagged for Rfree calculations using programs F2MTZ, Truncate, CAD, and Unique from the CCP4 package of programs (CCP4, 1994). The program BnP (18) was used for the determination of the selenium atom positions and phasing. ARP/wARP package (19) was used to build an initial model of the protein. Refinement and manual correction was performed using REFMAC5 (20) Version 5.5.0109 and Coot (21) Version 0.6. The final model included two protein chains (from residue 4 to residue 153 in chain A and from residue −1 to residue 153 in chain B), 3 molecules of ethylene glycol, 8 sodium ions, and 324 water molecules. The MOLPROBITY method (22) was used to analyze the Ramachandran plot and root mean square deviations of bond lengths, and angles were calculated from ideal values of Engh and Huber stereochemical parameters (23). Wilson B-factor was calculated using CTRUNCATE Version 1.0.11 (CCP4, 1994). The figures were created using PyMOL. The data collection and refinement statistics are shown in Table 1.
TABLE 1.
X-ray data collection and refinement statistics
Statistics for the highest resolution bin are in parentheses. r.m.s.d., root mean square deviation.
| Data collection | |
| Space group | P 32 2 1 |
| Unit cell (Å, °) | a = 52.28, b = 52.28, c = 252.48 |
| α = β = 90.0 γ = 120.0 | |
| Wavelength (Å) | 0.97959 |
| Temperature (K) | 100 |
| Resolution (Å) | 42.6-1.9 (1.95-1.9) |
| Observed reflections | 32,810 (2,358) |
| Rinta | 0.057 (0.560) |
| Average redundancy | 6.7 (4.9) |
| 〈I〉/〈σ(I)〉 | 20.5 (2.3) |
| Completeness, % | 99.7 (99.0) |
| Refinement | |
| R/Rfree | 0.213 (0.304)/0.270 (0.358) |
| Protein atoms | 2424 |
| Water molecules | 324 |
| Other atoms | 20 |
| r.m.s.d. from ideal bond length (Å)b | 0.02 |
| r.m.s.d. from ideal bond anglesb | 2.1° |
| Average B-factor for protein atoms (Å2) | 30.3 |
| Average B-factor for water molecules (Å2) | 39.8 |
| Ramachandran plot statistics (%)c | |
| Allowed | 100 |
| Favored | 98.3 |
| Outliers | 0 |
Native Electrospray Mass Spectrometry
Electrospray mass spectrometry experiments in native conditions (100 mm ammonium acetate aqueous solution, pH 6.5) were carried out on a Q-TOF Ultima Global mass spectrometer (Waters, Manchester, UK) with the electrospray source in the positive ion mode. The source tuning settings were as follows: capillary = 3 kV, source pirani pressure = 3.3 millibars, cone voltage = 40 V, source temperature = 40 °C, desolvation temperature = 60 °C, collision energy = 10 V. The RF lens 1 voltage was varied from 50 V (soft conditions to preserve the protein dimer) to 150 V (harder conditions to achieve a better desolvation and higher accuracy in the protein mass determination).
Molecular Sieve
A TSK column (G3000SW, 30 × 0.75 cm, 7 mm, Tosoh, Bioscience GmbH, 70567, Stuttgart, Germany) was used in 20 mm Hepes-Na (pH 6.8) and 200 mm NaCl at a flow rate of 0.5 ml/min.
Site-directed Mutagenesis of NeuTTM
Point mutations were introduced by the QuikChange method. The expression plasmid containing the coding sequence of NeuTTM was amplified with two mutagenic primers and Pfu DNA polymerase (Promega). Template DNA was removed by digestion with DpnI (Promega) for 2 h at 37 °C. The DNA was then introduced into E. coli cells, and single clones were isolated. The presence of the mutations was checked by DNA sequencing (Genotranscriptomics Platform, GIGA, University of Liège). The sequences of the oligonucleotides used are given in supplemental Table S1.
Determination of Phosphohydrolase Activities
The standard incubation medium (100 μl) contained 50 mm buffer, 5 mm MgCl2, 0.5 mm PPPi, and 20 μl of the enzyme at the adequate concentration. The mixture was incubated at 37 or 50 °C, and the reaction was stopped by the addition of 1 ml of phosphate reagent (24). The absorbance was read at 635 nm after 30 min and compared with a standard curve to estimate the released inorganic phosphate. When P4 was the substrate, the absorbance was read after only 15 min, as acid hydrolysis was rapid in the phosphate reagent. The buffers used for incubation at different pH values were: Na-MES (pH 6.0–7.0), Na-MOPS (pH 7.0–7.5), Na-HEPES (pH 7.5–8.0), Na-TAPS (pH 8.0–9.0), Na-CHES (pH 9.0–10.0), and Na-CAPS (pH 10.0–10.5).
Determination of In-gel Enzyme Activities
In-gel activities were measured as described (25). The gel was colored in phosphate precipitation reagent (1% w/v ammonium heptamolybdate, 1% w/v triethylamine, 1 n HNO3).
Determination of ATP, ADP, and cAMP
Purified NeuTTM was incubated under standard assay conditions with ATP (0.1 mm). The reaction was stopped by the addition of 12% trichloroacetic acid. After extraction of the acid with 3 × 1.5 ml of diethyl ether, nucleotides were determined by HPLC using UV detection (26).
Statistical Analyses
Data analysis was done with Prism 5 for Mac OS X (GraphPad Software, San Diego, CA) using nonlinear fitting to the Michaelis-Menten equation.
RESULTS
Cloning, Overexpression, and Purification of NeuTTM
The ORF for NeuTTM was subcloned in the pET15b vector, modified to allow the expression of N-terminal His-tag fusion proteins. The latter was purified as described under “Experimental Procedures.” The protein obtained after elution from the HisTrap column was essentially pure as judged by SDS-PAGE (supplemental Fig. S1). This poly-His fusion protein could be subsequently cleaved by TEV protease, which considerably increased the triphosphatase activity of the enzyme (see below). For crystallization, NeuTTM was expressed as selenomethionine-substituted protein.
Structural Properties of NeuTTM
The sequence and secondary structure elements are given in Fig. 1A. Crystallographic data show that NeuTTM, like 25-kDa ThTPase (9), has an open cleft structure in the absence of bound substrate (Fig. 1B). Several residues projecting into the cleft have been indicated. They seem to be important for substrate and metal binding and/or catalytic activity, as in the case of the closely related CthTTM, extensive mutational analysis (12) has revealed that most of these homologous residues are required for full PPPase activity. Four glutamate residues (Glu-4, -6, -114, -116, and possibly Glu-61 and -63) are supposed to be required for Mg2+ (or Mn2+) binding, whereas Arg-39 and -41 (and possibly Arg-87) probably interact with substrate phosphoryl groups. According to Keppetipola et al. (12), Lys-8 in CthTTM would form a hydrogen bond with a backbone carbonyl at the break of the C-terminal α4-α5 helix. However, the analysis of the crystal structure of NeuTTM leaves the existence of such a hydrogen bond in question. Although the distance between Lys-8NZ and Tyr-142-O is 2.9Å, the angle N…O-C is 117 degrees, which is rather far from the ideal geometry. Also, there is a water molecule (W429) located 3.5 Å away from the Tyr-142 carbonyl oxygen with the angle O…O-C of 172 degrees, which makes this water molecule a more likely partner for a hydrogen bond with the Tyr-142 carbonyl than Lys-8NZ.
FIGURE 1.

Structure of NeuTTM. A, amino acid sequence and secondary structure elements are shown. The residues conserved among known CYTH proteins (9) are highlighted by red boxes. B, a schematic representation of the NeuTTM monomer is shown. Side chains of residues probably involved in substrate and divalent cation binding and/or catalysis are rendered as sticks and are labeled. C, stabilization of the open β-barrel structure by hydrophobic interactions with α-turn 4 and α-helix 5 is shown. The NeuTTM monomer is shown in a schematic representation (gray). The residues forming a hydrophobic patch that stabilizes the protein core are shown in magenta sticks and labeled.
It seems that the β sheets are prevented from closing into a complete β-barrel by the insertion of a broken C-terminal helix (α4 and α5) into one end of the fold. Such a conformation is stabilized by hydrophobic interactions conferring a relatively rigid structure to NeuTTM (Fig. 1C).
Specificity and Kinetic Properties of Recombinant NeuTTM
In contrast to what we expected, only a slight ThTPase activity could be detected, indicating that, unlike its mammalian counterpart, NeuTTM is not a genuine ThTPase (Fig. 2).
FIGURE 2.
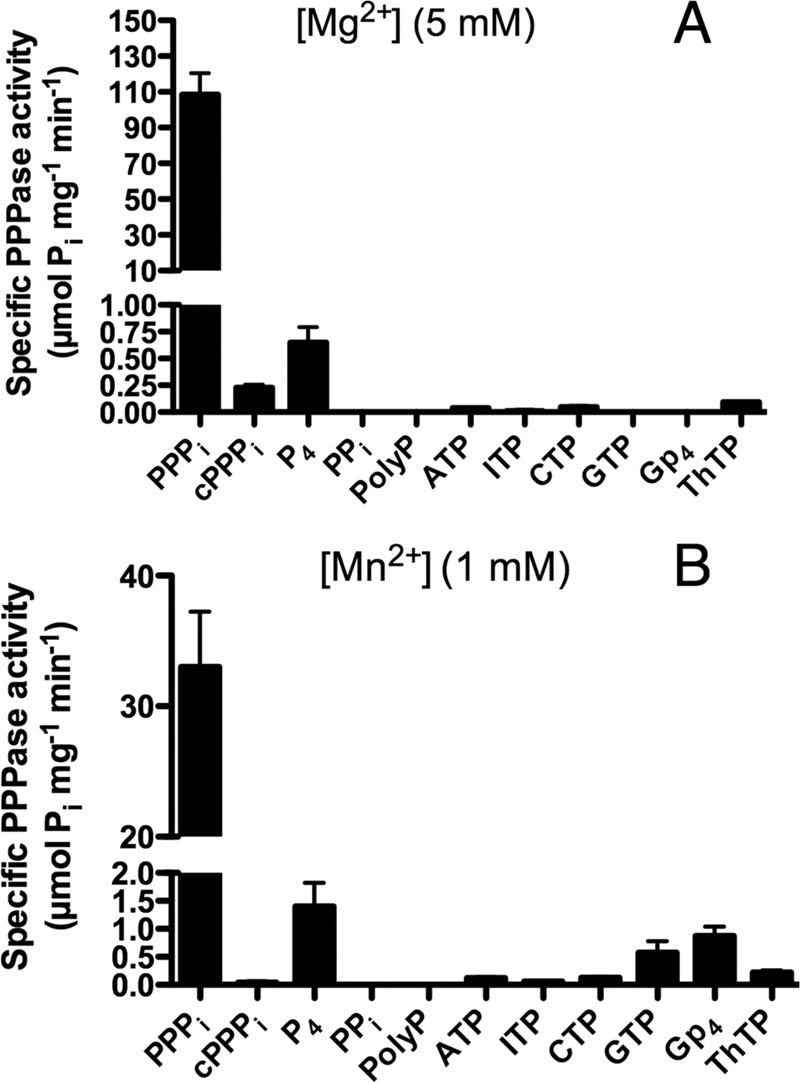
Substrate specificity of recombinant His-tagged non-mutated NeuTTM. The specificity was tested in the presence of 5 mm MgCl2 (pH 9.7) (A) or 1 mm MnCl2 (pH 10.4) (B). The incubation medium contained either 50 mm Na-CHES (pH 9.7) or 50 mm Na-CAPS (pH 10.4), 0.5 mm substrate, and 5 mm MgCl2 (pH 9.7) or 1 mm MnCl2 (pH 10.4). The temperature was 37 °C. Results are the mean ± S.D., n = 2–6. cPPPi, cyclic PPPi.
As ATP and other NTPs were hydrolyzed by a number of enzymes of the TTM family (7, 12), we tested these compounds as potential substrates for NeuTTM. Nucleoside triphosphatase activity was negligible in the presence of Mg2+, but a small activity was observed in the presence of Mn2+, in particular with GTP (Fig. 2).
By contrast to NTPs, PPPi was a very good substrate for NeuTTM with Mg2+ as activator (Fig. 2, Table 2). The enzyme had a strong preference for linear PPPi compared with cyclic PPPi (cyclic trimetaphosphate) and to the linear P4. The longer chain polyphosphate (containing 65 ± 5 phosphate residues) was not hydrolyzed either. Likewise, guanosine 5′-tetraphosphate (Gpppp or Gp4), which is a good substrate for CthTTM (13), was not significantly hydrolyzed. The NeuTTM adenylyl cyclase activity was also checked by incubating the enzyme with ATP plus MgCl2 or MnCl2 at different pH values, but in not cases did we detect the appearance of a cAMP peak by HPLC. Hence, NeuTTM appears to be devoid of adenylyl cyclase activity. Note that the closely related C. thermocellum CYTH protein (CthTTM) was also devoid of any such activity (12). So far the only bacterial CYTH proteins with significant (albeit low) adenylyl cyclase activity are the CyaB enzyme AC2 from A. hydrophila (4) and the homologous class IV adenylyl cyclase from Y. pestis (10, 11, 27).
TABLE 2.
Kinetic parameters for non-mutated (WT) and mutated recombinant NeuTTM
Data are the mean ± S.D.; n = 3–4.
| Enzyme prepraration | Substrate | Conditions of experiment | Kmapp | Vmax | kcat | Kcat/Km |
|---|---|---|---|---|---|---|
| μm | μmol min−1mg−1 | s−1 | s−1/M−1 | |||
| WT | PPPi | 50 °C, pH 9.7 | 40 ± 15 | 910 ± 70 | 288 | 7.2 106 |
| His-tagged | Mg2+ = 5 mm | |||||
| 37 °C, pH 9.7 | 21 ± 3 | 240 ± 30 | 76 | 3.6 106 | ||
| Mg2+ = 5 mm | ||||||
| 37 °C, pH 7.1 | 58 ± 5 | 60 ± 2 | 19 | 0.33 106 | ||
| Mg2+ = 5 mm | ||||||
| ATP | 50 °C, pH 8.1 | 800 ± 120 | 1.2 ± 0.2 | 0.36 | 0.45 103 | |
| Mn2+ = 10 mm | ||||||
| WT untagged | PPPi | 50 °C, pH 9.7m Mg2+ = 5 mm | 100 ± 20 | 25,000 ± 2,000 | 7900 | 79 106 |
| K8A | PPPi | 50 °C, pH 9.7 | 390 ± 30 | 2,800 ± 500 | 887 | 2.3 106 |
| His-tagged | Mg2+ = 5 mm | |||||
| PPPi | 37 °C, pH 9.7 | 280 ± 55 | 235 ± 30 | 74 | 0.19 106 | |
| Mg2+ = 5 mm | ||||||
| ATP | 50 °C, pH 10.1 | 1200 ± 500 | 12.5 ± 3.2 | 3.96 | 3.3 103 | |
| Mn2+ = 10 mm | ||||||
| K85A | PPPi | 50 °C, pH 9.7 | 720 ± 60 | 65 ± 7 | 21 | 29 103 |
| His-tagged | Mg2+ = 5 mm | |||||
| PPPi | 37 °C, pH 9.7 | 2600 ± 400 | 33 ± 3 | 10 | 4 103 | |
| Mg2+ = 5 mm | ||||||
| K52R | PPPi | 37 °C, pH 9.7 | 191 ± 8 | 3.1 ± 0.1 | 0.98 | 5.1 103 |
| His-tagged | Mg2+ = 5 mm |
General kinetic properties of NeuTTM PPPase are displayed in Fig. 3. The optimal temperature was 50–55 °C (Fig. 3A), and the optimum pH was around 9.7 (Fig. 3B). Thermal stability and alkaline pH optimum are characteristic features of several other enzymes of the CYTH superfamily, notably the founding members CyaB-AC2 (4) and human 25-kDa ThTPase (28).
FIGURE 3.
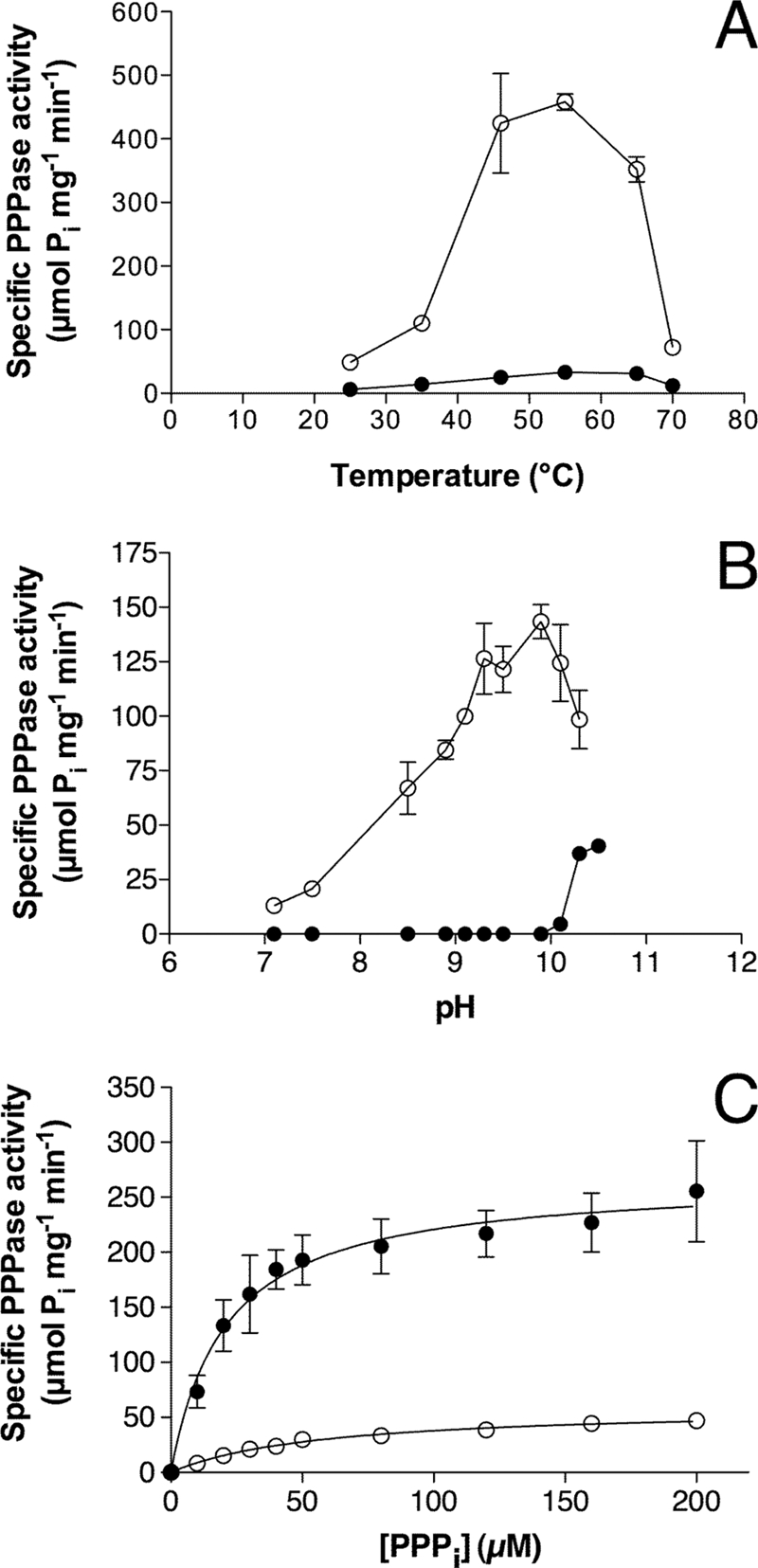
Kinetic properties of NeuTTM PPPase activity. The PPPi concentration was 0.5 mm. A, dependence on temperature in the presence of 5 mm Mg2+ at pH 9.7 (○) or 0.8 mm Mn2+ at pH 10.4 (●) is shown. B, pH dependence in the presence of 10 mm Mg2+ (○) or 1 mm Mn2+ (●) at 37 °C is shown. C, dependence on substrate concentration at pH 7.1 (○) and 9.7 (●) at 37 °C is shown. The curves were drawn by non-linear regression of the Michaelis-Menten equation (n = 3). The buffers used for different pH values are given under “Experimental Procedures.” Results are the mean ± S.D.
As Mn2+ is a good substituent for Mg2+ in several TTM members (7, 12, 29–31), we tested the possible activating effects of Mn2+ ions on NeuTTM PPPase activity. We found that it was a very poor substituent for Mg2+ at all temperatures tested and in the pH range 7–10 (Fig. 3, A and B). However, there was a significant Mn2+-dependent activity at very alkaline pH (10.0–10.5) (Fig. 3B).
Kinetic parameters (Km, kcat, and catalytic efficiency) were determined with different enzyme preparations and under different experimental conditions (Table 2). Using a freshly prepared His-tagged purified preparation, Michaelis-Menten kinetics were obtained (Fig. 3C). At pH 9.7 and 37 °C, Km was 21 ± 3 μm, and Vmax was 240 ± 30 μmol min−1 mg−1 in the presence of 5 mm Mg2+. Under such conditions, the substrate is essentially under the form of a Mg2+-PPPi complex, as Kd for the dissociation of the complex is low (∼1.6 10−6 m). The calculated kcat was 76 s−1 under these conditions (assuming a molecular mass of 19 kDa for the monomeric enzyme with one catalytic site). The catalytic efficiency of NeuTTM triphosphatase activity is thus very high, with kcat/Km = 3.6 106 m−1s−1. At neutral pH, kcat/Km is about 10 times lower (3.3 105 m−1s−1), but this is still high enough to consider that NeuTTM may be a genuine specific inorganic triphosphatase (PPPase). This is all the more plausible as kcat was even higher for the untagged enzyme (up to 7900 s−1 at 50 °C and pH 9.7, see Table 2). This is 2 orders of magnitude higher than kcat values obtained for CthTTM with its best substrates, PPPi and Gp4 (12, 13). This raises the possibility that in N. europaea the physiological function of NeuTTM might be the degradation of PPPi. It should be emphasized, however, that any firm conclusion concerning the biological role of this enzyme awaits the demonstration of the presence of inorganic triphosphate in Nitrosomonas or any other cell types.
Effects of Different Activators and Inhibitors on Recombinant NeuTTM
As their name suggests, enzymes from the TTM family are highly dependent on divalent metal cations. In common with other CYTH enzymes, NeuTTM PPPase had an absolute requirement for divalent metal ions, the highest activity being found with Mg2+ ions in the concentration range 2–10 mm (Fig. 4A). As already mentioned, Mn2+ was less effective as an activator (Fig. 4A), but half-maximum activation was obtained at 0.4 mm Mn2+, which corresponds to a very low concentration of free Mn2+.
FIGURE 4.
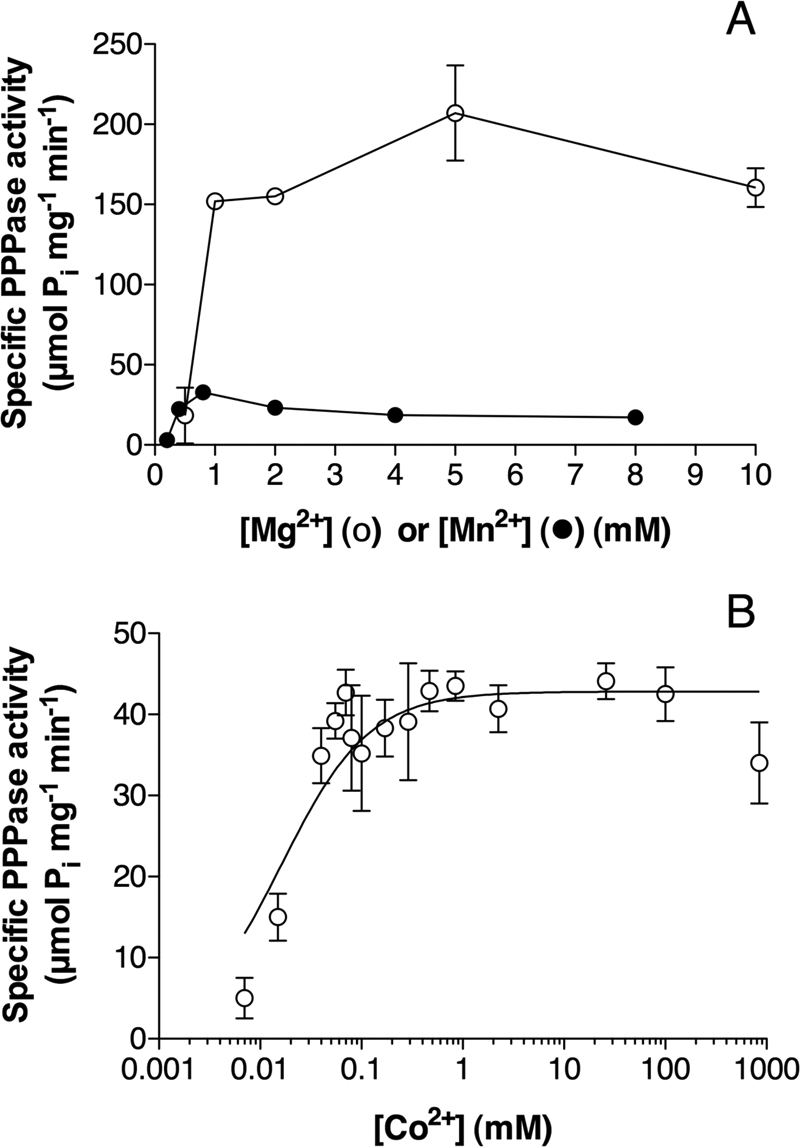
Activation of NeuTTM PPPase by divalent cations. A, shown is dependence of PPPase activity on Mg2+ (○) and Mn2+ (●) total concentration. The incubation was carried out in the presence of 50 mm Na-CHES (Mg2+, pH 9.7) or Na-CAPS (Mn2+, pH 10.4) at 37 °C. B, shown is the effect of increasing concentrations of Co2+ (calculated free Co2+ concentration assuming Kd = 8 nm for the Co-PPPi complex) on the PPPase activity. Conditions of experiment: PPPi, 0.5 mm; Na-CHES, 50 mm, pH 9.5, 45 °C. Results are the mean ± S.D, n = 3.
We also tested the activating effect of Co2+, which like Mn2+ is a good activator for several TTM enzymes (30). At optimal pH (9.5–9.7), Co2+ was a better activator than Mn2+, although the maximum activity was less than 10% that measured in the presence of 5 mm Mg2+ (Fig. 4B). The concentration of free Co2+ corresponding to half-maximum activation was extremely low (about 30–50 nm), indicating that the affinity of the binding site for Co2+ is even higher than for Mn2+.
As already reported for other CYTH enzymes such as Cet1 (30) and 25-kDa ThTPase (28), Ca2+ had no activating effect, but it was a potent inhibitor in the presence of Mg2+ (Fig. 5A); IC50 was 50–100 μm in the presence of 5 mm Mg2+. Mn2+ was also a potent inhibitor in the presence of Mg2+, at least at pH < 10 (IC50 ∼ 40 μm). As Mn2+ is also a weak activator, the inhibition by Mn2+ in the presence of Mg2+ was not complete. The curves showing the activating effect of increasing concentrations of Mg2+ were shifted to the right in the presence of Ca2+ (Fig. 5B), suggesting that the latter ion exerts its inhibitory effect at least partially by competition with Mg2+. We finally tested the effect of Zn2+, which has been shown to be a potent inhibitor of mammalian 25-kDa ThTPase (28). Zn2+ inhibited NeuTTM PPPase in the lower micromolar range (IC50 ∼ 1.5 μm) (Fig. 5C). In contrast to Ca2+, the inhibitory effect of Zn2+ was only marginally affected by the concentration of the activator Mg2+ (data not shown), suggesting that the two cations do not compete for the same binding site. This was also the case for 25-kDa ThTPase (28). Those data are in agreement with the assumption that Mg2+, Mn2+, Ca2+, and Co2+ may share a common binding site, whereas Zn2+ may bind to a peripheral “allosteric” site. In addition to Zn2+, Cd2+ and Cu2+ ions were also inhibitory but with higher IC50 values, 10 and 20 μm, respectively.
FIGURE 5.
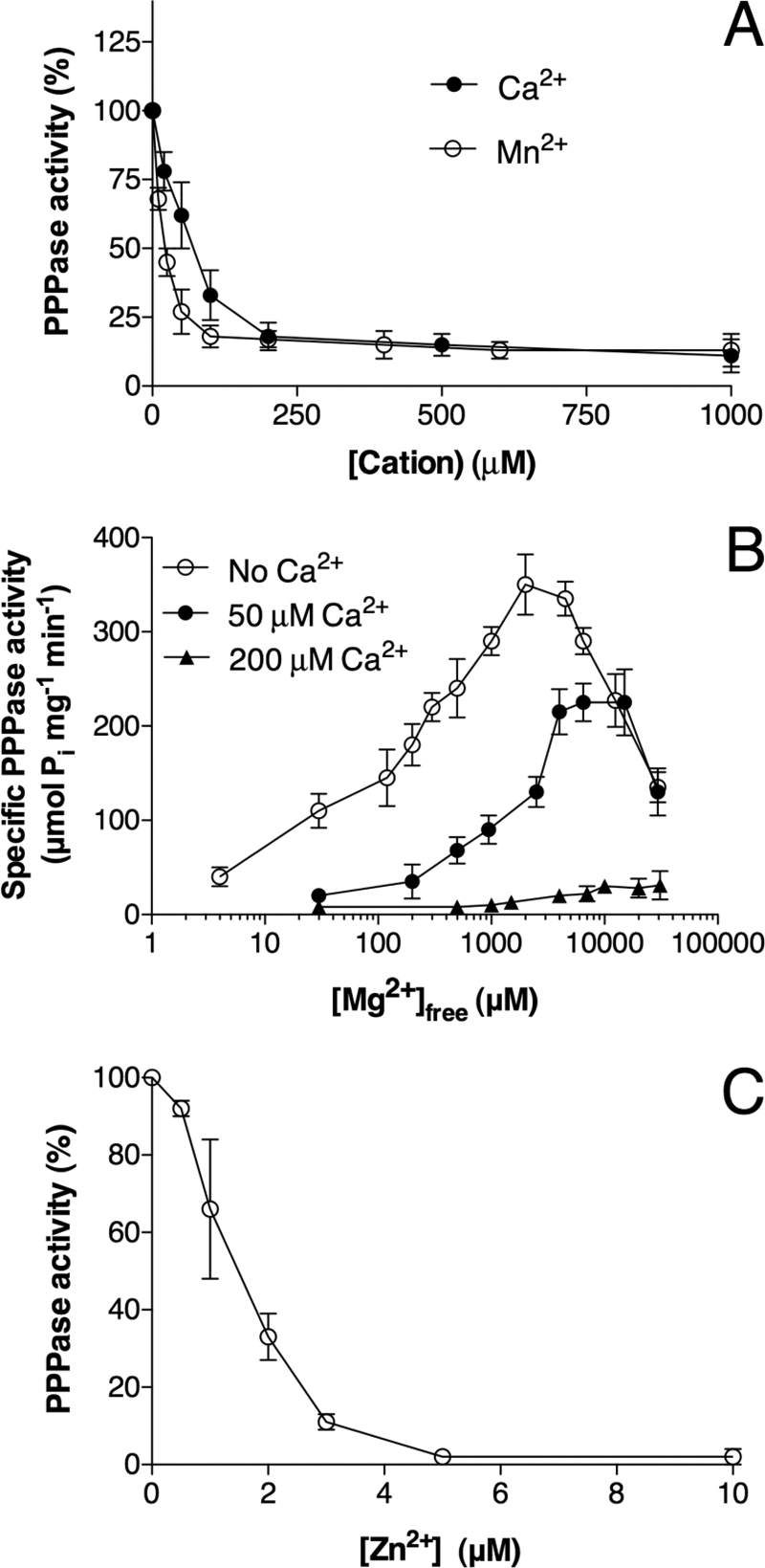
Inhibition of NeuTTM PPPase by divalent cations. A, inhibition by Ca2+ and Mn2+ in the presence of Mg2+ is shown. The incubation medium contained 0.5 mm PPPi, 5 mm MgCl2, 50 mm Na-CHES, pH 9.7, at 37 °C. B, shown is the effect of increasing concentrations of Mg2+ (calculated free Mg2+) on NeuTTM PPPase activity in the presence or absence of Ca2+ ions at 50 °C. The incubation medium contained 0.5 mm PPPi, 50 mm Na-CHES, pH 9.7. C, shown is the effect of increasing concentrations of ZnSO4 on the PPPase activity of NeuTTM. The incubation medium contained 0.5 mm PPPi, 5 mm MgCl2, 50 mm Na-CHES, pH 9.7, at 37 °C. Results are the mean ± S.D., n = 3.
Structural and Binding Properties of the NeuTTM Protein Analyzed by Native Mass Spectrometry
We first investigated the ability of the 19-kDa recombinant NeuTTM to dimerize in solution. The x-ray diffraction data already suggested that the protein crystallized as a dimer (Protein Data Bank code 2FBL). We carried out electrospray mass spectrometry of the concentrated His-tagged protein (total monomer concentration 10 μm) in 100 mm ammonium acetate, pH 6.5 (Fig. 6), and found that the protein was a mixture of monomer (noted M) and dimer (noted M2) forms, the dimer being more abundant. The dimer dissociated easily in the gas phase when the RF lens 1 voltage was increased from 50 to 150 V. This suggests that salt bridge interactions do not prevail at the dimerization interface. Indeed, the diffraction data show that the interface is mainly composed of hydrophobic amino acids, among which are Leu-29, -38, -40, -51, and -74 and Ile-36, -64, and -66 (supplemental Fig. S2), and this explains the lability of the dimers in electrospray.
FIGURE 6.

Mass spectra of His-tagged NeuTTM in the absence of added substrates and divalent cations. M, monomer (theoretical mass = 19,987.4 Da); M2, dimer (theoretical mass = 39,974.8 Da). A, shown is a spectrum recorded in harsher source conditions (RF lens 1 voltage = 150 V); the dimer was partially destroyed, but the mass accuracy was higher. B, shown is a spectrum recorded in soft source conditions (RF lens 1 voltage = 50 V), showing that the dimer is the most abundant species in solution. The protein concentration was 10 μm in 100 mm ammonium acetate, pH 6.5.
We then studied the complexes of NeuTTM, with substrates alone (PPPi, ATP, and ThTP) in the presence of Mg2+. In all cases the substrates remained tightly bound to NeuTTM at all voltages, in line with ionic interactions in these complexes. Fig. 7 shows the mass spectra of NeuTTM alone (Fig. 7A) and of the binary mixture with PPPi (Fig. 7B), ATP (Fig. 7C), and ThTP (Fig. 7D). With PPPi, at the concentrations used (10 μm NeuTTM and 25 μm PPPi), the peak of the free protein is not distinguishable from the noise. It can, therefore, be estimated that the concentration of free protein is less than 8.5% that of the total protein concentration. This means that the dissociation constant for the monomer-substrate complex under the conditions stated above must be lower than 1.5 μm. Moreover, the protein dimers contain exclusively two PPPi molecules, as seen from the uneven charge states 15+ and 13+. This strongly contrasts with ATP and ThTP, where the free protein is predominant, and both the monomer and dimer bind one molecule of substrate on average. From the peak intensities, the Kd of the monomer-substrate complex is estimated to be at least 90 μm for ATP and ThTP. This is in agreement with the high Km value (800 ± 120 μm) measured for ATPase activity of NeuTTM (Table 2).
FIGURE 7.

Mass spectra showing high specificity of NeuTTM for PPPi compared with ATP and ThTP. A, shown is a spectrum of 10 μm protein alone in 100 mm NH4OAc, pH 6.5. B, conditions were as in A but with 25 μm PPPi. C, conditions were as in A but with 20 μm protein and 50 μm ATP. D, conditions were as in A with 25 μm ThTP instead of PPPi. M, monomer; M2, dimer.
Fig. 8 shows the mass spectra of charge state 8+ of the protein monomer in different mixtures with Mg2+ and/or PPPi. The protein alone shows a peak at m/z = 2499.4 (Fig. 8A), that remains predominant when the protein is mixed with a 10-fold excess of Mg2+ (Fig. 8B). We found that both monomers and dimers bind Mg2+ with moderate affinity. We also tested binary mixtures of the protein with Mn2+ and Zn2+, and the results suggest a higher affinity for Zn2+ (with multiple binding sites) than for Mg2+ and Mn2+ (data not shown). This is in agreement with the finding that the PPPase activity of NeuTTM is inhibited by micromolar concentrations of Zn2+ (Fig. 5C).
FIGURE 8.

Detailed identification of complexes of NeuTTM (10 μm) in binary and ternary mixtures with MgCl2 (0. 1 mm) and/or PPPi (25 μm). The spectra were all recorded in 100 mm ammonium acetate, pH 6.5, at an RF lens 1 voltage = 150 V for a more accurate mass determination of each complex. A, NeuTTM alone at charge state 8+ is shown. The main peak corresponds to the uptake of eight protons, and few sodium or potassium adducts are detected. B, NeuTTM binary mixture with Mg2+ is shown. The intensity of unresolved adducts peaks increases, but the fully protonated protein remains the most intense peak. C, shown is a NeuTTM binary mixture with PPPi. The fully protonated complex is minor, and the major complexes take up two monovalent cations. D, shown is a NeuTTM ternary mixture with PPPi and Mg2+. The enzyme-substrate complex with the uptake of one Mg2+ is clearly detected. M, monomer; M2, dimer.
A more detailed analysis of the masses of the NeuTTM-PPPi complexes in the absence of divalent cations revealed that they specifically take up two monovalent cations (the peak intensities of the complexes with zero and one monovalent cations being insignificant). Data from Fig. 8C show the presence of a complex with two Na+ ions. The latter presumably originates from the tripolyphosphate preparation, which is in the form of a pentasodium salt. A NeuTTM-PPPi-Na+-K+ complex was also detected, with K+ possibly originating from the protein preparation.
Finally, we investigated the properties of the ternary complexes (supposed to be catalytically active) of NeuTTM, with the substrate PPPi and the activator Mg2+. To avoid appreciable hydrolysis of PPPi, the spectra were recorded immediately after mixing the protein with Mg2+ and PPPi. Despite the high enzyme concentration, PPPase activity is low at room temperature and pH 6.5 (see Fig. 3), and complete hydrolysis of PPPi under the present conditions would probably take more than a few seconds. With MgCl2 (0.1 mm) in addition to PPPi, the enzyme-PPPi complex took up one Mg2+ ion rather than two monovalent cations (Fig. 8D). No free enzyme was observed. We can, therefore, assume that the Mg-PPPi complex (which is the true substrate) binds to the active site with high affinity, Kd being of the same order of magnitude than for free PPPi (<1.5 μm).
On the other hand, it is clear that the requirement for an activating Mg2+ ion is not simply due to the fact that it binds to PPPi. Although the true substrate is likely to be the Mg2+-PPPi complex, the divalent metal activator must also bind to a specific site on the protein, as full enzyme activity requires the presence of millimolar free Mg2+ in the incubation medium (Figs. 4A and 5B). This was also shown to be the case for RNA triphosphatase from Trypanosoma (29), and it is probably a general feature of CYTH proteins (3). In the case of NeuTTM, the metal ion presumably interacts with the carboxyl groups of Glu-4, Glu-6, Glu-114, and Glu-116 (see Fig. 1).
There is no evidence from the present data that the protein might bind more than one Mg2+ ion with appreciable affinity. The same conclusion was reached by Song et al. (9) for the 25-kDa mouse ThTPase. Although Iyer and Aravind (3) suggested that CYTH proteins may bind two divalent metal activators, this is probably not a general rule for this superfamily of proteins.
In-gel Activity of NeuTTM under Non-denaturing Conditions
In agreement with mass spectrometric data, two peaks of ∼20 and 40 kDa were obtained after separation on a size exclusion column (supplemental Fig. S3). The specific activities of the monomer and the dimer were, respectively, 245 and 169 μmol·min−1·mg−1 with 0.5 mm PPPi at 50 °C, suggesting that the catalytic activity was not much changed by dimerization. This was also confirmed by in-gel activity determination after polyacrylamide gel electrophoresis under non-denaturing conditions and incubation under optimal conditions (supplemental Fig. S4). Furthermore, when the monomer and the dimer were separated on the size exclusion column and then reinjected, both appeared pure, suggesting that no rapid conversion between the two forms occurs. These results suggest that dimerization may simply be an artifact associated with enzyme purification and without physiological significance.
Site-directed Mutagenesis
Alanine mutation of Lys-8 and Lys-85 were chosen because homologous residues were reported to be important for catalytic activity and substrate specificity of the closely related CthTTM (12). Indeed, the primary structures of CthTTM and NeuTTM have 73 positions of side-chain identity (12).
We found that in contrast to the K8A mutant of CthTTM, the K8A mutated NeuTTM remained highly active with PPPi as substrate and Mg2+ as activator, with a catalytic efficiency close to that of the recombinant wild-type enzyme, as both Km and kcat were increased (Table 2). This is an important difference with CthTTM. Indeed, Keppetipola et al. (12) found that the corresponding K8A mutation in CthTTM nearly completely abolished the PPPase activity of the enzyme while strongly stimulating its ATPase activity. According to Keppetipola et al. (12), Lys-8 would form a hydrogen bond with a backbone carbonyl at the break of the C-terminal helices α4-α5, stabilizing the open structure of the eight-stranded β-barrel where the substrate binds. Substituting Lys-8 by alanine would prevent this specific interaction between the β1 strand and α4-α5 helices, making the conformation less rigid. Our own data do not suggest that such a H-bond is required to maintain a rigid structure. Furthermore, with the mutated K8A enzyme, ATP remains a poor substrate (Table 2), even with Mn2+ as activator, the catalytic efficiency being 3 orders of magnitude lower than with PPPi. The present results suggest that either a very rigid conformation is not essential for a high and specific PPPase activity of NeuTTM or that Lys-8 is not involved in the stabilization of the enzyme conformation. Note that Keppetipola et al. (12) made their assumption on the basis of sequence similarities with NeuTTM and that the three-dimensional structure of CthTTM is not known.
Lys-85 is located at the bottom of the cleft that is supposed to contain the catalytic site, close to the arginine residues (Arg-39, -41, and -87) that are thought to bind the substrate phosphoryl groups electrostatically. In the case of the very similar CthTTM protein, the Lys-87 residue, which is homologous to Lys-85 in NeuTTM, has been found to be essential for catalysis, but the authors did not determine Km and Vmax (12).
The K85A mutant of NeuTTM was about 10 times less active than the non-mutated enzyme, but the optimal conditions for activity were essentially the same, i.e. pH 9.7, temperature ∼50 °C. The mutant was also much less active with ATP than with PPPi as substrate. The extrapolated Vmax value yielded a kcat around 21 s1 (at 50 °C) in the presence of 5 mm Mg2+ (Table 2), which is 14 times lower than kcat measured under the same optimal conditions for the non-mutated enzyme. On the other hand, the Km for PPPi was 1 or 2 orders of magnitude higher than for the non-mutated NeuTTM. Note that due to the high Km, it was not possible to use saturating substrate concentrations, increasing the uncertainty on both Km and Vmax. Although the catalytic efficiency of the enzyme appears to be rather strongly decreased by the K85A mutation, it cannot be concluded that Lys-85 is really essential for catalysis, as kcat remains relatively high.
Another important consequence of the K85A mutation is that the effects of divalent cations are profoundly altered; in contrast to the non-mutated enzyme, the K85A mutant is more strongly activated by Mn2+ than by Mg2+, and the inhibitory effects of Ca2+ and Zn2+ are less pronounced (supplemental Table S2). Actually, Zn2+ can even replace Mn2+ as activator, although it is less effective. It thus appears that deleting the positive charge of the side chain at position 85 alters the protein conformation in such a way that cations larger than Mg2+ become better activators. This suggests that Lys-85 may act as a “discriminator” residue, ensuring that the smaller and more physiological Mg2+ ion is the specific activator. Taken together the data suggest that Lys-85 is not directly involved in catalysis and is more likely to play an important (although not essential) role to properly orient the PPPi molecule in the enzyme-substrate complex. It is also important for the specificity of Mg2+ binding.
As neither Lys-8 nor -85 appear to be essential residues for catalysis, we suspected that Lys-52, which is highly conserved in CYTH proteins (3, 9), might play a more essential role. In CthTTM, mutation of Lys-52 resulted in the loss of 90–99% of the activity, depending on the substrate or activator used (12). In NeuTTM, the conservative K52R mutation resulted in a decreased Vmax by about 2 orders of magnitude (Table 2), and as the apparent Km is strongly increased (supplemental Table S5), the catalytic efficiency is at least 1000 times lower than that of the non-mutated enzyme. Mn2+ did not induce a significant activation of this mutant. Thus, in contrast to K85A, the specificity for activating ions does not appear to be changed. The fact that this conservative mutation strongly decreases the catalytic efficiency of the enzyme suggests that this highly conserved lysine residue plays an important role in the catalytic mechanism.
A puzzling property of NeuTTM as well as other enzymes of the CYTH protein family is their strong activation at alkaline pH (Fig. 3B). To check that this pH profile was not due to impaired binding of the substrate at lower pH, we plotted Vmax/Km as a function of pH (Fig. 9A). Our results show that the Km does not change much with pH, and the profile obtained is quite similar to the activity versus pH profile (Fig. 3B). We used the Dixon-Webb plot Log Vmax versus pH to estimate the pKa value for the main residue responsible for activity decrease between 9.7 and 7.0. As shown in Fig. 9B, the estimated pKa was close to 9.3 for the non-mutated enzyme. The same value was found for the K52R mutant. These data are in agreement with the proposal that the catalytic activity depends on a single deprotonation step with pKa ∼ 9.3. This may correspond to a general base that removes a proton from water; the resulting OH− would attack a phosphorus atom of the PPPi substrate.
FIGURE 9.

pH dependence of non-mutated NeuTTM and of the Y28F and K52R mutants. The reaction was carried out in the presence of 5 mm Mg2+ at 37 °C. The control values obtained in the absence of enzyme were subtracted. A, shown is Vmax/Km as a function of pH for the non-mutated enzyme. B, shown is pH dependence of Vmax of non-mutated (●) and K52R-mutated (▴) NeuTTM. C, activities of Y28F (○) and K52R (▴) mutants are expressed as the percentage of the respective value obtained at pH 9.7 (100%). The PPPi concentration was 0.1 mm. D, shown is pH dependence of Y29F-mutated NeuTTM, PPPi = 0.1 mm. Results are the mean ± S.D, n = 3–6.
As the pH-rate profile remains largely unaffected by K52R mutation, the decrease of activity between 9.7 and 7.0 cannot be ascribed to protonation of the Lys-52 amino group. This suggests the involvement of another residue able to interact with Lys-52 and acting as a general base in the hydrolytic mechanism. According to the crystal structure (Fig. 1B), only three residues are in a position to interact easily with Lys-52: Tyr-28, Glu-37, and Glu-61 (see also supplemental Fig. S6). Obviously, the carboxyl groups of glutamate residues could hardly exhibit such an alkaline pKa, and we suspected that the residue acting as the general base was Tyr-28 (pKa for the hydroxyl group of free tyrosine is ∼10.0) Indeed, the conservative mutation Y28F results in a strong loss of enzyme activity (∼2.6 μmol min−1 mg−1 at 100 μm PPPi, compared with ∼200 μmol min−1 mg−1 for the non-mutated enzyme, Fig. 3C). Surprisingly, however, the activity continuously increased when PPPi was raised to 1 mm (the highest testable concentration with our method) with no tendency to saturation (supplemental Fig. S5). This lack of saturation was observed at all pH values tested from 7.0 to 9.0 (data not shown) Thus, the kinetic parameters Km and Vmax could not be estimated with this mutant enzyme. The pH optimum of the Y28F mutant is shifted to lower values compared with the non-mutated (Fig. 3B) and the K52R mutant (Fig. 9C). We plotted the log of the specific activity versus pH at 37 °C for PPPi = 100 μm (Fig. 9D). It is obvious that there is an important shift in pH-rate profile toward lower pH values, and the Dixon-Webb plot yielded a pKa value ∼7.4. This apparent pKa was not strongly affected by changes in PPPi concentration (not shown).
This value might possibly reflect the pKa of the carboxyl group of a neighboring glutamate (Glu-37 or -61) that might also act as a general base, albeit less efficiently than Tyr-28. This would explain that the activity of the Y28F mutant still reaches relatively high values when PPPi is in large excess (supplemental Fig. S5). Taken together, those data suggest that Tyr-28 plays an important role in NeuTTM catalysis (possibly forming a catalytic dyad with Lys-52) and is at least partially responsible for the unusual high pH optimum of the enzyme. The relatively slight decrease in activity at pH ≥ 10 could result from a number of possibilities, e.g. charge alterations involving unidentified residues in or near the active site or destabilization of the protein structure.
In the model shown in Fig. 10, we hypothesize that a catalytic dyad is formed by the Tyr-28/Lys-52 pair, the nucleophilic nitrogen of Lys-52 being hydrogen-bonded with the phenolate oxygen of Tyr-28. The latter would act as the general base that removes a proton from water, and the resulting OH− attacks a phosphorus atom of the substrate PPPi (Fig. 10). Alternatively, Tyr-28 might act as a nucleophile to directly attack the γ-phosphorus atom of PPPi (Fig. 10). In the latter case, we would expect a covalent phosphotyrosyl intermediate to be formed that would be unusual for a hydrolase. When the catalytic mechanism involves a phosphoenzyme intermediate, the activity is generally inhibited by sodium orthovanadate acting as a transition state inhibitor. We tested the effect of sodium orthovanadate on the non-mutated NeuTTM PPPase activity, but we found no inhibition at concentrations up to 2 mm. However, the activity was very sensitive to fluoride with an IC50 as low as 30 ± 10 μm for NaF (results not shown). Fluoride is a well known inhibitor of acid phosphatases, but millimolar concentrations are required. However, inorganic pyrophosphatases are sensitive to fluoride at concentrations lower than 1 mm. In family I pyrophosphatases, where fluoride inhibition involves rapid and slow phases (32), there is much evidence that catalysis proceeds by direct phosphoryl transfer to water rather than via a phosphoryl enzyme intermediate (33, 34). The structure of the F−-inhibited complex has shown a fluoride ion replacing the nucleophilic water molecule (35). In family II pyrophosphatases, which belong to the DHH phosphoesterase superfamily (as defined by (5)), the IC50 for fluoride inhibition was even lower than for NeuTTM (∼12 μm) (36). In any case fluoride is believed to act as an analog of hydroxide (37). The detailed mechanism of PPPi hydrolysis by NeuTTM requires further investigation, but the lack of effect of vanadate and the strong inhibitory effect of fluoride argue against the existence of a covalent phosphoenzyme intermediate. Therefore, the model proposed in Fig. 10 seems to be the simplest and the most plausible.
FIGURE 10.
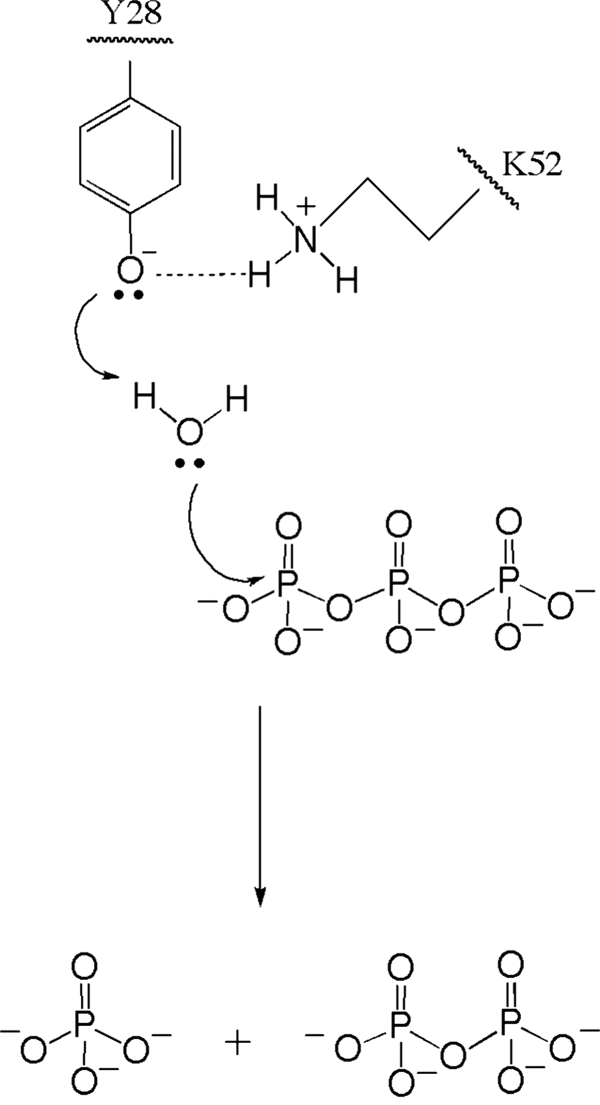
Proposed catalytic mechanism for PPPi hydrolysis by NeuTTM. The Lys-52/Tyr-28 pair is considered to form a catalytic dyad. The most simple and plausible mechanism is a general acid-general base catalysis with no covalent acyl-enzyme intermediate.
DISCUSSION
The present data demonstrate that the hypothetical protein NE1496, present in N. europaea (NeuTTM), is a functional phosphohydrolase with surprisingly high specificity, affinity, and catalytic efficiency for PPPi. This is also the first molecular characterization of a specific tripolyphosphatase. Several enzymes hydrolyzing PPPi have been characterized previously, but they are mainly exopolyphosphatases that, in addition to PPPi, hydrolyze other substrates such as, for instance, long chain polyphosphates or Gp4 (38–40). So far, only one enzyme was reported to be strictly specific for PPPi hydrolysis; it was purified from Neurospora crassa and found to be a dimer of 40-kDa subunits (41). Its sequence remains unknown, and unlike NeuTTM, it does not appear to have a high affinity for PPPi.
NeuTTM belongs to the superfamily of CYTH proteins (3) and was first referred to as a hypothetical protein with putative phosphatase or CyaB-like adenylyl cyclase activity by analogy with the A. hydrophila CYTH protein (4). However, it has become clear that the functional properties of CYTH proteins cannot be predicted from primary or even three-dimensional structure. In fact, the CYTH domain is a functionally versatile protein fold, characterized mainly by its ability to bind triphosphate compounds and catalyze their hydrolysis or other chemical transformations in the presence of divalent metal ion activators. Members of the CYTH superfamily exist in archaea, but none has been functionally characterized so far. In bacteria, two CYTH orthologs were shown to have an adenylyl cyclase activity (4, 11, 27), but under physiological conditions this activity is low, and its biological significance is not established. CthTTM, the ortholog from C. thermocellum hydrolyzes PPPi with a relatively good turnover (kcat = 3 s−1), but it also hydrolyzes other substrates such as Gp4 (kcat = 96 s−1) and, to a lesser extent, nucleoside triphosphates and long chain polyphosphates (12, 13). Thus, its possible physiological function remains undefined. In contrast, a clear biological role has been demonstrated for the CYTH orthologs of fungi and protozoa, which catalyze the first step of RNA capping in these organisms (7). In pluricellular eukaryotes, only the mammalian 25-kDa ThTPase has been characterized (1, 9, 28). Like NeuTTM, the latter enzyme is characterized by a high specificity and a high catalytic efficiency (kcat/Km = 6 106 m−1 s−1 for the bovine ThTPase (1)). On the basis that all known CYTH proteins hydrolyze triphosphates and that PPPi is the simplest triphosphate compound conceivable, PPPase activity might well be the primitive enzymatic activity in the CYTH protein family, which later evolved to hydrolyze more complex organic substrates such as RNA or ThTP in eukaryotes. It should be pointed out that a high affinity for PPPi was already demonstrated in the case of yeast RNA triphosphatases, where PPPi is a potent competitive inhibitor, albeit a poor substrate (29, 42, 43).
The present data show that despite low sequence similarity and profound structural differences, NeuTTM and 25-kDa ThTPase have several kinetic properties in common, i.e. requirement for Mg2+, inhibition by Ca2+ by competition with Mg2+, allosteric inhibition by Zn2+, alkaline pH optimum, and an optimal temperature around 50–60 °C. A. hydrophila and Y. pestis CyaB-like adenylyl cyclases are also Mg2+-dependent and have high pH and temperature optima (4, 11, 27). Although more detailed investigations are required, most of these features may possibly be functional signature properties of CYTH enzymes.
Our results shed some light on the particular conformation of the NeuTTM protein and the relationship between structural and kinetic properties of the enzyme. The open cleft structure (incomplete β-barrel) is in contrast to the closed tunnel conformation of other TTM proteins (7). It appears to be rather rigid and to be stabilized by hydrophobic interactions between the antiparallel β strands and the broken C-terminal α4-α5 helix (Fig. 1C). Hence, in contrast to mammalian 25-kDa ThTPase (9), NeuTTM is probably unable to form a tunnel-like structure even when a substrate is bound. This is in line with the very different substrate specificities of the two enzymes.
We propose a catalytic mechanism (Fig. 10) in which the hydroxyl group of Tyr-28 hydrogen-bonded to the nucleophilic nitrogen of Lys-52 acts as a nucleophile, attacking a water molecule to form OH−. Lys-52 was also considered essential in the enzymatic activity of CthTTM (12). To our knowledge, this is the first lysine-tyrosine dyad proposed for a hydrolase. Such a mechanism strongly contrasts with the classical serine-histidine dyad observed in many phosphatases.
Catalytic dyads based on tyrosyl and lysyl residues seem to be rare (44); nevertheless such a catalytic dyad has been suggested for the enol-keto tautomerization step of the NADP+-dependent malic enzyme (45). In that enzyme the Lys→Arg and Tyr → Phe mutations resulted in a decrease in kcat by 2 orders of magnitude as we observed here.
In the crystal structure described here, the distance between Lys-52 and Tyr-28 is 7.4 Å (supplemental Fig. S6), too much for a hydrogen bond between these two residues. However, as shown for ThTPase, the binding of the substrate induces an important conformational change (9). Such a conformational change in NeuTTM might bring the two residues close enough for an interaction.
Four glutamate residues, Glu-4, -6, -114, and -116, protrude from the bottom into the catalytic cleft. They are part of the CYTH consensus sequence and are thought to be involved in Mg2+ binding (3, 12). Our data show that the enzyme substrate complex binds only one Mg2+ ion (Fig. 8B). We may thus consider the possibility that this Mg2+ as well as probably Lys-85 stabilize and orient the substrate PPPi toward the catalytic dyad projecting from the ceiling of the cleft (Fig. 1B).
Concerning the quaternary structure, it is worth pointing out that NeuTTM crystallizes as a dimer (Protein Data Bank code 2FBL) and can also form dimers in solution. However, our results suggest that dimerization is probably not of physiological significance for NeuTTM. This is in contrast to yeast RNA triphosphatases Cet1 from S. cerevisiae and Pet1 from Schizosaccharomyces pombe, where dimerization is important for thermal stability and in vivo function (46).
The high affinity and specificity of NeuTTM for PPPi raises the question of the possible biological roles of both PPPi and the enzymes able to synthesize it. For PPPi, there are so far very few data concerning its enzymatic synthesis. In E. coli, it was shown long ago that PPPi can be produced by enzymatic cleavage of deoxyguanosine triphosphate (47). On the other hand, it is well known that PPPi is formed as an intermediate in the enzymatic synthesis of S-adenosylmethionine, but in this case it is not released in the cytosol (48). It was also shown that PPPi is an intermediate in naturally occurring pterins in Drosophila melanogaster (49). Like ThTP (50), PPPi was shown to be an alternative phosphate donor for protein phosphorylation in vitro (51), and therefore, like cAMP and possibly ThTP (15), PPPi might act as an intracellular signal.
However, in contrast to inorganic polyphosphate of higher molecular weight (>10 phosphoryl residues), PPPi and other very short chain polyphosphates have never been reported to exist in any organism (except in acidocalcisomes, specific organelles rich in calcium and polyphosphates in some protozoans (52)). In all likelihood this is simply due to the present lack of sensitive and specific detection methods. For polyphosphates of longer chain (15–750 residues), the problem was overcome thanks to sensitive assay methods (53). In E. coli, polyphosphates play an important role in the response to various forms of environmental stress (14, 54, 55). Polyphosphates may also act as energy stores or divalent cation chelators (14). The demonstration of similar physiological roles for PPPi awaits the development of an adequate method to measure its intracellular concentration.
Hence, the present results provide the first detailed characterization of a specific PPPase with high affinity for PPPi and high catalytic efficiency. Although it is not proven that the physiological function of the protein is to degrade PPPi in vivo, our study is a first step toward the understanding of the possible roles of short chain polyphosphates, which might turn out to be just as important as their long chain counterparts in cell biology.
Supplementary Material
Acknowledgments
Nucleic acid sequencing was performed by Véronique Dhennin, Genotranscriptomics Platform, GIGA, University of Liège. We thank Dr. Eric Oldfield for the gift of tetrapolyphosphate and Dr. Ilca Margineanu for help with the manuscript and some experiments. We are grateful to Dr. Alexei Savchenko for the gift of the pET15b vector for the expression of NeuTTM and helpful discussion. The structure of NeuTTM was determined by the Midwest Centre for Structural Genomics as part of NIH Protein Structure Initiative Grant GM074942.
This work was supported, in whole or in part, by National Institutes of Health Protein Structure Initiative Grant P50-GM62413-02 (to Alexei Savchenko (Banting and Best Department of Medical Research, University of Toronto, Toronto, Ontario, M5G 1L6, Canada). This work was also supported by Grants 2.4558.04 and 2.4508.10 from the Fonds de la Recherche Fondamentale Collective of the Fonds de la Recherche Scientifique (to L. B., E. D. P., and B. L.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and S2 and Figs. S1–S6.
- ThTP
- thiamine triphosphate
- ThTPase
- thiamine triphosphatase
- AC
- adenylyl cyclase
- Gp4
- guanosine 5′-tetraphosphate
- P4
- inorganic tetraphosphate
- PPPase
- inorganic triphosphatase
- PPPi
- inorganic triphosphate
- TTM
- triphosphate tunnel metalloenzyme
- NeuTTM
- N. europaea TTM
- CthTTM
- C. thermocellum TTM
- TAPS
- 3-{[2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino}-1-propanesulfonic acid
- CHES
- 2-(cyclohexylamino)ethanesulfonic acid.
REFERENCES
- 1. Lakaye B., Makarchikov A. F., Antunes A. F., Zorzi W., Coumans B., De Pauw E., Wins P., Grisar T., Bettendorff L. (2002) J. Biol. Chem. 277, 13771–13777 [DOI] [PubMed] [Google Scholar]
- 2. Gangolf M., Czerniecki J., Radermecker M., Detry O., Nisolle M., Jouan C., Martin D., Chantraine F., Lakaye B., Wins P., Grisar T., Bettendorff L. (2010) PLoS One 5, e13616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Iyer L. M., Aravind L. (2002) BMC Genomics 3, 33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sismeiro O., Trotot P., Biville F., Vivares C., Danchin A. (1998) J. Bacteriol. 180, 3339–3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aravind L., Koonin E. V. (1998) Trends Biochem. Sci. 23, 17–19 [DOI] [PubMed] [Google Scholar]
- 6. Tesmer J. J., Sunahara R. K., Johnson R. A., Gosselin G., Gilman A. G., Sprang S. R. (1999) Science 285, 756–760 [DOI] [PubMed] [Google Scholar]
- 7. Gong C., Smith P., Shuman S. (2006) RNA 12, 1468–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lima C. D., Wang L. K., Shuman S. (1999) Cell 99, 533–543 [DOI] [PubMed] [Google Scholar]
- 9. Song J., Bettendorff L., Tonelli M., Markley J. L. (2008) J. Biol. Chem. 283, 10939–10948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gallagher D. T., Smith N. N., Kim S. K., Heroux A., Robinson H., Reddy P. T. (2006) J. Mol. Biol. 362, 114–122 [DOI] [PubMed] [Google Scholar]
- 11. Gallagher D. T., Kim S. K., Robinson H., Reddy P. T. (2011) J. Mol. Biol. 405, 787–803 [DOI] [PubMed] [Google Scholar]
- 12. Keppetipola N., Jain R., Shuman S. (2007) J. Biol. Chem. 282, 11941–11949 [DOI] [PubMed] [Google Scholar]
- 13. Jain R., Shuman S. (2008) J. Biol. Chem. 283, 31047–31057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rao N. N., Gómez-García M. R., Kornberg A. (2009) Annu. Rev. Biochem. 78, 605–647 [DOI] [PubMed] [Google Scholar]
- 15. Lakaye B., Wirtzfeld B., Wins P., Grisar T., Bettendorff L. (2004) J. Biol. Chem. 279, 17142–17147 [DOI] [PubMed] [Google Scholar]
- 16. Bettendorff L., Nghiêm H. O., Wins P., Lakaye B. (2003) Anal. Biochem. 322, 190–197 [DOI] [PubMed] [Google Scholar]
- 17. Otwinowski Z., Minor W. (1997) in Methods in Enzymology (Carter C. W., Jr., Sweet R. M. eds.) pp. 307–326, Academic Press, New York: [DOI] [PubMed] [Google Scholar]
- 18. Weeks C. M., Shah N., Green M. L., Miller R., Furey W. (2005) Acta Crystallogr. Sect. A 61, C152 [Google Scholar]
- 19. Cohen S. X., Morris R. J., Fernandez F. J., Ben Jelloul M., Kakaris M., Parthasarathy V., Lamzin V. S., Kleywegt G. J., Perrakis A. (2004) Acta Crystallogr. D. Biol. Crystallogr. 60, 2222–2229 [DOI] [PubMed] [Google Scholar]
- 20. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 21. Emsley P., Cowtan K. (2004) Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 22. Lovell S. C., Davis I. W., Arendall W. B., 3rd, de Bakker P. I., Word J. M., Prisant M. G., Richardson J. S., Richardson D. C. (2003) Proteins 50, 437–450 [DOI] [PubMed] [Google Scholar]
- 23. Engh R. A., Huber R. (1991) Acta Crystallogr. Sect. A 47, 392–400 [Google Scholar]
- 24. Lanzetta P. A., Alvarez L. J., Reinach P. S., Candia O. A. (1979) Anal. Biochem. 100, 95–97 [DOI] [PubMed] [Google Scholar]
- 25. Simonović A. D., Gaddameedhi S., Anderson M. D. (2004) Anal. Biochem. 334, 312–317 [DOI] [PubMed] [Google Scholar]
- 26. Hill M., Dupaix A., Volfin P., Kurkdjian A., Arrio B. (1987) Methods Enzymol. 148, 132–141 [Google Scholar]
- 27. Smith N., Kim S. K., Reddy P. T., Gallagher D. T. (2006) Acta Crystallogr. Sect. F. Struct. Biol. Cryst. Commun. 62, 200–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lakaye B., Makarchikov A. F., Wins P., Margineanu I., Roland S., Lins L., Aichour R., Lebeau L., El Moualij B., Zorzi W., Coumans B., Grisar T., Bettendorff L. (2004) Int. J. Biochem. Cell Biol. 36, 1348–1364 [DOI] [PubMed] [Google Scholar]
- 29. Gong C., Martins A., Shuman S. (2003) J. Biol. Chem. 278, 50843–50852 [DOI] [PubMed] [Google Scholar]
- 30. Ho C. K., Pei Y., Shuman S. (1998) J. Biol. Chem. 273, 34151–34156 [DOI] [PubMed] [Google Scholar]
- 31. Bisaillon M., Shuman S. (2001) J. Biol. Chem. 276, 17261–17266 [DOI] [PubMed] [Google Scholar]
- 32. Baykov A. A., Fabrichniy I. P., Pohjanjoki P., Zyryanov A. B., Lahti R. (2000) Biochemistry 39, 11939–11947 [DOI] [PubMed] [Google Scholar]
- 33. Gonzalez M. A., Webb M. R., Welsh K. M., Cooperman B. S. (1984) Biochemistry 23, 797–801 [DOI] [PubMed] [Google Scholar]
- 34. Zyryanov A. B., Pohjanjoki P., Kasho V. N., Shestakov A. S., Goldman A., Lahti R., Baykov A. A. (2001) J. Biol. Chem. 276, 17629–17634 [DOI] [PubMed] [Google Scholar]
- 35. Heikinheimo P., Tuominen V., Ahonen A. K., Teplyakov A., Cooperman B. S., Baykov A. A., Lahti R., Goldman A. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 3121–3126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Fabrichniy I. P., Lehtiö L., Tammenkoski M., Zyryanov A. B., Oksanen E., Baykov A. A., Lahti R., Goldman A. (2007) J. Biol. Chem. 282, 1422–1431 [DOI] [PubMed] [Google Scholar]
- 37. Briley P. A., Eisenthal R., Harrison R. (1975) Biochem. J. 145, 501–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wurst H., Kornberg A. (1994) J. Biol. Chem. 269, 10996–11001 [PubMed] [Google Scholar]
- 39. Fang J., Ruiz F. A., Docampo M., Luo S., Rodrigues J. C., Motta L. S., Rohloff P., Docampo R. (2007) J. Biol. Chem. 282, 32501–32510 [DOI] [PubMed] [Google Scholar]
- 40. Tammenkoski M., Koivula K., Cusanelli E., Zollo M., Steegborn C., Baykov A. A., Lahti R. (2008) Biochemistry 47, 9707–9713 [DOI] [PubMed] [Google Scholar]
- 41. Egorov S. N., Kulaev I. S. (1976) Biokhimiia 41, 1958–1967 [PubMed] [Google Scholar]
- 42. Gong C., Shuman S. (2002) J. Biol. Chem. 277, 15317–15324 [DOI] [PubMed] [Google Scholar]
- 43. Issur M., Despins S., Bougie I., Bisaillon M. (2009) Nucleic Acids Res. 37, 3714–3722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gutteridge A., Thornton J. M. (2005) Trends Biochem. Sci. 30, 622–629 [DOI] [PubMed] [Google Scholar]
- 45. Kuo C. C., Lin K. Y., Hsu Y. J., Lin S. Y., Lin Y. T., Chang G. G., Chou W. Y. (2008) Biochem. J. 411, 467–473 [DOI] [PubMed] [Google Scholar]
- 46. Hausmann S., Pei Y., Shuman S. (2003) J. Biol. Chem. 278, 30487–30496 [DOI] [PubMed] [Google Scholar]
- 47. Kornberg S. R., Lehman I. R., Bessman M. J., Simms E. S., Kornberg A. (1958) J. Biol. Chem. 233, 159–162 [PubMed] [Google Scholar]
- 48. Pérez Mato I., Sanchez del Pino M. M., Chamberlin M. E., Mudd S. H., Mato J. M., Corrales F. J. (2001) J. Biol. Chem. 276, 13803–13809 [DOI] [PubMed] [Google Scholar]
- 49. Switchenko A. C., Primus J. P., Brown G. M. (1984) Biochem. Biophys. Res. Commun. 120, 754–760 [DOI] [PubMed] [Google Scholar]
- 50. Nghiêm H. O., Bettendorff L., Changeux J. P. (2000) FASEB J. 14, 543–554 [DOI] [PubMed] [Google Scholar]
- 51. Tsutsui K. (1986) J. Biol. Chem. 261, 2645–2653 [PubMed] [Google Scholar]
- 52. Moreno B., Urbina J. A., Oldfield E., Bailey B. N., Rodrigues C. O., Docampo R. (2000) J. Biol. Chem. 275, 28356–28362 [DOI] [PubMed] [Google Scholar]
- 53. Ault-Riché D., Fraley C. D., Tzeng C. M., Kornberg A. (1998) J. Bacteriol. 180, 1841–1847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kuroda A., Nomura K., Ohtomo R., Kato J., Ikeda T., Takiguchi N., Ohtake H., Kornberg A. (2001) Science 293, 705–708 [DOI] [PubMed] [Google Scholar]
- 55. Brown M. R., Kornberg A. (2008) Trends Biochem. Sci. 33, 284–290 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.