

Background: CRAG expression in the Purkinje cells of mice expressing polyQ resulted in clearance of the polyQ and rescue from ataxia.
Results: CRAG induces transcriptional activation of c-Fos-dependent AP-1 via SRF.
Conclusion: CRAG enhances the cell survival signal against the cytotoxicity of polyQ partially via c-Fos-dependent AP-1 activation.
Significance: Our findings extend the possible use of targeted delivery of CRAG as a gene therapy for polyglutamine diseases.
Keywords: AP-1 Transcription Factor, Fos, Gene Therapy, Neurodegeneration, Polyglutamine Disease
Abstract
We previously demonstrated that CRAM (CRMP5)-associated GTPase (CRAG), a short splicing variant of centaurin-γ3/AGAP3, facilitated degradation of expanded polyglutamine protein (polyQ) via the nuclear ubiquitin-proteasome pathway. Taking advantage of this feature, we also showed that lentivirus-mediated CRAG expression in the Purkinje cells of mice expressing polyQ resulted in clearance of the polyQ aggregates and rescue from ataxia. However, the molecular basis of the function of CRAG in cell survival against polyQ remains unclear. Here we report that CRAG, but not centaurin-γ3, induces transcriptional activation of c-Fos-dependent activator protein-1 (AP-1) via serum response factor (SRF). Mutation analysis indicated that the nuclear localization signal and both the N- and C-terminal regions of CRAG are critical for SRF-dependent c-Fos activation. CRAG knockdown by siRNA or expression of a dominant negative mutant of CRAG significantly attenuated the c-Fos activation triggered by either polyQ or the proteasome inhibitor MG132. Importantly, c-Fos expression partially rescued the enhanced cytotoxicity of CRAG knockdown in polyQ-expressing or MG132-treated cells. Finally, we suggest the possible involvement of CRAG in the sulfiredoxin-mediated antioxidant pathway via AP-1. Taken together, these results demonstrated that CRAG enhances the cell survival signal against the accumulation of unfolded proteins, including polyQ, through not only proteasome activation, but also the activation of c-Fos-dependent AP-1.
Introduction
We previously identified a novel neuronal GTPase, which was named CRAG2 for CRAM (CRAMP5)-associated GTPase, because CRAG co-immunoprecipitated from neonatal rat brain with anti-CRMP5 antibody (1). CRAG is an alternative splicing variant of centaurin-γ3/AGAP3, the function of which has not yet been elucidated. CRMPs (semaphorin response mediator proteins) are believed to be required for repulsive factor semaphorin-mediated axon guidance (2–4), suggesting that CRAG is involved in semaphorin signaling. Interestingly, nuclear translocation of CRAG was observed in response to semaphorin-3A and ultraviolet (UV) irradiation in a reactive oxygen species (ROS)-dependent manner (1). Therefore, CRAG may play an important role in oxidative stress-mediated signaling during neuronal development.
Upon semaphorin stimulation or UV irradiation, CRAG formed unique nuclear inclusions that co-localized with an enlarged ring-like structure of promyelocytic leukemia protein body, which is often seen in brains of polyglutamine disease (PD) patients (1). This result prompted us to examine the role of CRAG in PD, which are inherited neurodegenerative diseases caused by the accumulation of expanded polyglutamine protein (polyQ) (5, 6). In our previous experiments, CRAG was found to interact with polyQ and facilitate degradation of polyQ via the nuclear ubiquitin-proteasome pathway, thereby attenuating the cytotoxicity triggered by the accumulation of polyQ in cultured cells. In contrast, CRAG knockdown by small interfering RNA blocked the nuclear translocation of polyQ and enhanced polyQ-mediated cell death (1). These results suggest that CRAG protects neuronal cells against polyQ-mediated cytotoxicity.
CRAG expression is very high in the developing brain and decreases thereafter in the adult brain. This developmentally regulated expression of CRAG may be closely related to the onset of polyglutamine disease. Therefore, targeted expression of CRAG is a potential gene therapy for polyglutamine disease. Indeed, lentivector-mediated expression of CRAG in the Purkinje cells of mice extensively cleared polyQ aggregates and re-activated dendritic differentiation, resulting in a striking rescue from ataxia (7). Our in vivo data suggest the usefulness of targeted delivery of CRAG as a gene therapy for PD.
In this study, we report that CRAG protects neuronal cells against accumulation of unfolded proteins, including polyQ, by switching the AP-1 content from c-Jun homodimers to c-Fos/c-Jun heterodimers, which mediates the cell survival pathway. Our findings further extend the possible use of targeted delivery of CRAG as a gene therapy for PD. Finally, the implication of CRAG in an antioxidant pathway is discussed.
EXPERIMENTAL PROCEDURES
Cell Culture, Transfection, Viability Assay, and Luciferase Assay
Neuro2A cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin at 37 °C, in 5% CO2, in a humidified chamber. Neuro2a cells were transfected with Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. ATP reduction assays were performed using the CellTiter-Glo Luminescent Cell Viability Assay kit (Promega). Luciferase assay were performed using dual-luciferase reporter assay system (Promega).
Antibodies
Anti-CRAG rabbit polyclonal antibody was described previously (1). Anti-α-tubulin and anti-FLAG antibodies were from Sigma. Anti-HA antibody was from BabCO. Anti-peroxiredoxin-SO3 and anti-peroxiredoxin 2 antibodies were from AbFrontier. Anti-c-Jun and anti-caspase-3 antibodies were from Cell Signaling. Anti-c-Fos antibody was from Santa Cruz Biotechnology. Anti-Myc antibody was from Roche Applied Science.
Immunofluorescence Microscopy
Cells were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 10 min at room temperature, then washed twice with 0.2% Tween 20 in PBS, permeabilized with 0.2% Triton X-100 in PBS for 10 min, washed four times with PBS, and blocked with 3% bovine serum albumin in PBS, all at room temperature. For double staining, the cells were incubated with appropriate primary antibodies for 1 h at room temperature, washed three times with PBS, and then incubated with appropriate secondary antibodies for 30 min. The samples were washed as before, mounted using Fluorescent Mounting Medium (Dako), and analyzed using an Olympus IX81 confocal fluorescence microscope.
Co-immunoprecipitation and Western Blotting
Cells were lysed in lysis buffer (20 mm Tris-HCl, pH 7.4, 5 mm EDTA, 1% Triton X-100, 150 mm NaCl). The lysate was clarified by centrifugation at 15,000 × g for 10 min and immunoprecipitated with the appropriate antibody. Immunoprecipitates were washed three times with lysis buffer. Cell lysates were separated by SDS-PAGE and transferred to the PVDF membrane (Millipore). The blots were probed with the indicated antibodies, and protein bands on the blot were visualized by the enhanced chemiluminescence reagent (Millipore).
Expression Constructs
CRAG WT, GTPase mutant, NLS mutant, and GFP-70Q were described previously (1). HA-70Q-Myc-His was described previously (8). Centaurin-γ2-short, MAL mutant (C471), and c-fos cDNA were obtained from mouse brain by RT-PCR. Centaurin-γ2-short form with N-terminal HA epitope tag was created by PCR using the primers 5′-CCAGATCTCTATGAACTACCAGCAGCAGC-3′ and 5′-CAGCCCGCATTGTGCTGGGATCCGG-3′ and subcloned into pCMV5. MAL mutant (C471) form with N-terminal FLAG epitope tag was created by PCR using the primers 5′-CCGATATCATGACTCTGCTGGAGCCTGAG-3′ and 5′-CCTCTAGACTCATCACCCGTGCTGAGCAG-3′ and subcloned into pCMV5. c-fos with N-terminal FLAG epitope tag was created by PCR using the primers 5′-CCGAATTCGATGATGTTCTCGGGTTTCAAC-3′ and 5′-CACGCTGCTGGCCCTGTGAAAAGGATCCAC-3′ and subcloned into pCMV5. CRAG ΔN 60–395 with N-terminal HA epitope tag was created by PCR using the primers 5′-CCGGATCCCTATGTTCGCGCTCTCCAACT-3′ and 5′-AGTGCCTCTCCTGGCCCTGG-3′ and subcloned into pCMV5. Two SRF mutants (Δ413 and Δ338) were described previously (9). pAP-1-Luc plasmid and pSRF-Luc plasmid were from Stratagene. pRL-CMV was for use as an internal control reporter from Promega. Srxn-1-Luc was obtained from total genome of mouse brain by PCR using the primers 5′-CCACGCGTCTGGAGTGGACCTACTTTG-3′ and 5′-GGCTCGAGTCTCTTTATCCCTCG-3′ and subcloned into pGL3-basic vector. Srxn-1-Luc mutant1, Srxn-1-Luc mutant2, and Srxn-1-Luc mutant3 were created as described previously (10). For RNAi assay, sense and antisense oligonucleotides corresponding to the following target sequences were designed: 5′-CCATCCGAAAGCAGTCCAATT-3′ (siCRAG). Qiagen's thoroughly tested and validated AllStars Negative Control siRNA was used as a negative control.
RESULTS
CRAG Induces c-Fos-mediated AP-1 Activity
Our previous study suggested that CRAG is a modulator of promyelocytic leukemia protein function and dynamics (1). Recent studies indicate a role for promyelocytic leukemia protein intranuclear structures in the regulation of transcription, including AP-1, in response to various stresses or the induction of cell differentiation (11, 12). Therefore, we examined the relationship between AP-1 and CRAG. For this purpose, the effect of the overexpression CRAG and CRAG-related family members on AP-1 activity in Neuro2A cells was compared. A structural comparison of CRAG and CRAG-related family members is shown in Fig. 1A. CRAG is an alternative splicing variant of centaurin-γ3/AGAP3, and the regions common to CRAG and centaurin-γ3 are the GTPase domain and the nuclear localization signal (NLS). In addition, centaurin-γ3 contains PH, Arf GAP, and ankyrin repeat domains, which are absent in CRAG. Centaurin-γ2/AGAP1, which bears a close resemblance to centaurin-γ3, also possesses a short splicing variant similar to CRAG (13, 14).
FIGURE 1.

CRAG induces AP-1 activity. A, structural comparison of CRAG with centaurin-γ3/AGAP3 (G3) and centaurin-γ2/AGAP1 short form (G2-s). NLS, nuclear localization signal; ANK, ankyrin repeat; PH, pleckstrin homology domain. B, CRAG activates AP-1. Neuro2A cells were transfected with both pAP-1-Luc and pRL-CMV together with either empty expression vector or indicated vector. Luciferase activities were assessed 48 h after the transfection. Error bars indicate ±S.D. (n = 3). *, p < 0.05 (Student's t test). C, CRAG induces c-Fos activation. Lysates of Neuro2A cells as described above were immunoblotted with the indicated antibodies. Arrowheads indicate the positions of phosphorylated c-Fos and c-Jun. The protein levels of c-Fos and c-Jun normalized with tubulin are shown in the right panel when the control value was arbitrarily set 1.0. Error bars indicate ±S.D. (n = 4). *, p < 0.05; **, p < 0.01. D, subcellular localizations of CRAG, G3, and G2-s. Neuro2A cells transfected with either the empty vector, HA-CRAG, HA-G3, or HA-G2-s were immunostained with anti-HA (green) and Hoechst 33258 (blue). Scale bar, 5 μm.
A luciferase assay was performed on Neuro2A cells transfected with HA-tagged CRAG (HA-CRAG) or indicated plasmids and a reporter gene containing an AP-1 site (pAP-1-Luc). At 48 h after transfection, the luciferase activity in each cell lysate was measured as described under “Experimental Procedures.” Importantly, CRAG was found to induce AP-1 activity (Fig. 1B); however, no significant activation of AP-1 was observed in cells expressing centaurin-γ3 (G3) or centaurin-γ2 (G2, not shown). Slight activation of AP-1 was observed in cells expressing the short splicing variant of Centaurin-γ2 (G2-s). These results indicate that, among the Centaurin-γ/AGAP family, only CRAG is a potent activator of AP-1.
AP-1 is mainly composed of a c-Jun/c-Jun homodimer or a c-Fos/c-Jun heterodimer (15). To identify the molecular content of CRAG-mediated AP-1 activity, we performed immunoblot analysis with anti-c-Fos and anti-c-Jun antibodies. A mobility-shifted c-Fos band, which is indicative of the active form of c-Fos by phosphorylation (16–18), clearly appeared in cells overexpressing CRAG but not in cells overexpressing G3 or G2-s, although c-Jun activation was detected in cells expressing G2-s (Fig. 1C, left panel). Quantitative analysis of the relative protein expression further confirmed the specific activation of c-Fos by CRAG among the centaurin-γ/AGAP family members (Fig. 1C, right panel). These results demonstrated that CRAG induced AP-1 activity that was composed of a c-Fos/c-Jun heterodimer complex, which has been shown to promote neuronal cell survival. Therefore, CRAG-mediated neuronal cell survival may be partially due to c-Fos-dependent AP-1 activity.
To understand the relationship between AP-1 activity and nuclear localization, we compared the subcellular distribution of CRAG and other related members. Immunofluorescent analysis indicated that CRAG was localized in the nucleus, whereas other members were diffusely distributed in the cytoplasm or plasma membrane without nuclear localization (Fig. 1D). Therefore, the lack of significant AP-1 activation by other family members may be explained by a defect in nuclear translocation. Because the other members also contain NLS sequences, it is possible that they translocate to the nucleus and activate AP-1 after some modification(s) such as proteolysis or phosphorylation in response to extracellular stimulations.
Identification of CRAG Domains Required for AP-1 Activity
To determine which region(s) or domain(s) in CRAG are necessary for the transcriptional activation of AP-1, the effects of CRAG mutations and deletions on AP-1 activity were examined in a luciferase reporter assay. For this purpose, four mutants were generated as illustrated in Fig. 2A: a C-terminal deletion mutant (ΔC, 1–374) lacking the CRAG-specific region that is absent from the centaurin-γ3; an NLS mutant (K386E,R369E) that exhibits no nuclear localization; an N-terminal deletion mutant (ΔN, 60–395) lacking the glycine-rich domain, which is considered a protein-binding region (19–22); and a GTPase activity-defective mutant (S140N), which is constitutively GTP-bound. As shown in Fig. 2B, the C-terminal deletion mutant of CRAG (ΔC) could not activate AP-1 at all, indicating that the CRAG-specific C terminus is absolutely required for AP-1 activation. Similarly, AP-1 activation was significantly attenuated in cells expressing the NLS mutation, the N-terminal deletion, and the GTPase activity-defective mutant, although these mutants induced slight activation of AP-1. Immunoblot analysis indicated that c-Fos activation by these mutants was very low compared with wild-type CRAG. In contrast, c-Jun was activated in these mutants (Fig. 2B). Quantitative analysis of the relative protein expression confirmed the specific activation of c-Fos by CRAG compared with the mutants (Fig. 2C, right panel). To determine whether CRAG induced formation of c-Fos/c-Jun heterodimer, a co-immunoprecipitation assay was performed on cells expressing wild-type CRAG or CRAGΔC (Fig. 2D). The formation of c-Fos/c-Jun heterodimer was induced by wild-type CRAG, but not by CRAGΔC, indicating that CRAG enhanced the formation of c-Fos/c-Jun heterodimer. Taken together, these results demonstrated that the C terminus, NLS, N terminus, and GTPase activity of CRAG are required for c-Fos-mediated AP-1 activation via the formation of the c-Fos/c-Jun heterodimer.
FIGURE 2.

Identification of CRAG domains required for AP-1 activation. A, structural comparison of CRAG wild-type (WT) with various CRAG mutants. WT, CRAG WT; ΔC, CRAGΔC 1–374 mutant; NLSm, CRAG NLS mutant K386E R369E; ΔN, CRAGΔN 60–375 mutant; GTPm, CRAG GTPase mutant. B, AP-1 activities in cells expressing CRAG WT and mutants. Neuro2A cells were transfected with both pAP-1-Luc and pRL-CMV together with either empty expression vector or each indicated vector. Luciferase activities were assessed 48 h after the transfection. Error bars indicate ±S.D. (n = 3). *, p < 0.05; ***, p < 0.005 (Student's t test). C, effects of CRAG mutants on activations of c-Fos and c-Jun. Lysates of Neuro2A cells as described above were immunoblotted with the indicated antibodies. Arrowheads indicate the positions of phosphorylated c-Fos and c-Jun. The protein levels of c-Fos and c-Jun normalized with tubulin are shown in the right panel when the control value was arbitrarily set 1.0. Error bars indicate ±S.D. (n = 4). *, p < 0.05; **, p < 0.01. D, CRAG induces the formation of c-Fos/c-Jun heterodimer. Co-immunoprecipitation assay was performed on cells expressing vector, CRAG WT, or CRAGΔC. Cell lysates were immunoprecipitated with anti-c-Jun antibody, and the immunoprecipitates were immunoblotted with the indicated antibodies. E, subcellular localizations of CRAG WT and mutants. Neuro2A cells as described above were immunostained with anti-HA (green) antibody and Hoechst 33258 (blue). Scale bar, 5 μm.
To understand the relationship between AP-1 activation and the intracellular localization of CRAG mutants, their subcellular distribution in Neuro2A cells were compared (Fig. 2E). Immunofluorescent analysis indicated that, in contrast to the cytoplasmic and nuclear localization pattern of wild-type CRAG, the CRAG ΔC and NLS mutants exhibited a diffuse cytoplasmic pattern without nuclear localization. This result suggested that not only the NLS but also the C-terminal region of CRAG are necessary for nuclear translocation. Conversely, the GTPase mutant and CRAGΔN showed a nuclear localization pattern similar to wild-type CRAG. Therefore, although both the GTPase and N terminus are not required for nuclear translocation, they may be involved in the regulation of AP-1 transcriptional activation in the nucleus.
To gain insight into the molecular mechanism of CRAG-induced c-Fos activation, we focused on SRF activity, because SRF is a known upstream molecule of c-Fos (23, 24). To examine whether CRAG activates SRF, a luciferase assay was performed on Neuro2A cells transfected with constructs containing CRAG or CRAG-related family members and reporter genes containing an SRF-binding site (pSRF-Luc). As shown in Fig. 3A, CRAG was found to induce SRF activity; however, no significant activation of SRF was observed in cells expressing centaurin-γ3 (G3) or the centaurin-γ2 short variant (G2-s). To determine which region(s) or domain(s) of CRAG are necessary for the transcriptional activation of SRF, the effect of various CRAG mutants on SRF activation was examined in the luciferase reporter assay. As shown in Fig. 3B, the C-terminal deletion mutant of CRAG (ΔC), the NLS mutant (K386E,R369E), and the N-terminal deletion mutant (ΔN, 60–395) could not activate SRF. In contrast, the GTPase activity-defective CRAG mutant (S140N) induced SRF activity, although to a lower level than wild-type CRAG. These results demonstrated that the C terminus, the N terminus, and the NLS of CRAG are required for SRF activation. The pattern of SRF activation by the CRAG mutants is closely correlated with that of c-Fos-dependent AP-1 activation by the CRAG mutants, suggesting that CRAG induces c-fos transcription via SRF.
FIGURE 3.

CRAG mediates c-Fos-dependent AP-1 activation via SRF. A, CRAG activates SRF. G3, centaurin-γ3; G2-s, centaurin-γ2 short form. B, identification of CRAG domains required for SRF activation. WT, CRAG WT; ΔC, CRAGΔC 1–374 mutant; NLSm, CRAG NLS mutant K386E R369E; ΔN, CRAGΔN 60–375 mutant; GTPm, CRAG GTPase mutant. C, inhibitory effects of two SRF mutants and SRF cofactor MAL mutant on CRAG-induced SRF activation. D, effects of SRF inhibitions on CRAG-induced AP1 activities. (C–E) SRF cofactor MAL mutant (C471) and two SRF mutants (Δ413 and Δ338) are described under “Experimental Procedures.” A–D, Neuro2A cells were transfected with both pSRF-Luc and pRL-CMV together with either empty vector or the indicated vector. Luciferase activities were assessed 48 h after the transfection. Error bars indicate ±S.D. (n = 3). *, p < 0.05; **, p < 0.01; ***, p < 0.005 (Student's t test). E, effects of SRF inhibitions on CRAG-induced c-Fos activations. Lysates of Neuro2A cells as described above were immunoblotted with the indicated antibodies.
To test the possibility of SRF-dependent c-Fos activation by CRAG, we examined the effects of SRF inhibition on CRAG-induced activation of c-Fos and AP-1. For this purpose, we used two SRF mutants (Δ413 and Δ338) and a mutant of the SRF cofactor MAL (C471), which were all previously demonstrated to inhibit SRF activity (9, 25). We confirmed that these mutants blocked CRAG-induced SRF activation (Fig. 3C). As shown in Fig. 3D, these mutants also blocked CRAG-induced AP-1 activation. Similarly, immunoblot analysis indicated that these mutants inhibited CRAG-induced c-Fos activation (Fig. 3E). We further confirmed that CRAG induces the transcription of c-fos mRNA via SRF (not shown). Taken together, we concluded that CRAG activates c-Fos-dependent AP-1 via SRF.
CRAG Mediates c-Fos-mediated AP-1 Activation Triggered by Accumulation of polyQ and Misfolded Proteins
Next we examined the effect of CRAG knockdown on AP-1 activation triggered by the accumulation of expanded polyQ (70Q). Because it has been reported that polyQ activates AP-1 (26–28), we determined whether polyQ activated AP-1 in Neuro2A cells and obtained a similar result (Fig. 4A). Importantly, CRAG knockdown by a specific siRNA reduced polyQ-induced AP-1 activation by approximately one-half (Fig. 4A). To analyze the content of AP-1, we performed immunoblot analysis to detect the activation of c-Fos and c-Jun. Similarly, polyQ-induced activation of c-Fos and c-Jun was attenuated by CRAG knockdown (Fig. 4B). Therefore, these results suggested that CRAG is involved in polyQ-induced activation of AP-1 containing the c-Fos/c-Jun heterodimer.
FIGURE 4.

CRAG knockdown and CRAG dominant negative mutant reduce polyQ- and MG132-induced c-Fos and AP-1 activities. A and B, effect of CRAG knockdown on polyQ-induced AP-1 (A) and c-Fos/c-Jun activations (B). C and D, effect of CRAG knockdown on MG132-induced AP-1 (C) and c-Fos/c-Jun activations (D). E and F, effect of expression of CRAGΔC (ΔC) on polyQ-induced AP-1 (E) and c-Fos/c-Jun activations (F). G and H, effect of expression of CRAGΔC on MG132-induced AP-1 (G) and c-Fos/c-Jun activations (H). Luciferase assay was performed with Neuro2A cells transfected with both pAP-1-Luc and pRL-CMV with indicated vector and/or siRNA (sc: scramble siRNA, siCRAG: CRAG-specific siRNA). For MG132 treatment, Neuro2A cells were treated with either DMSO or 10 μm MG132 for 24 h. Lysates of Neuro2A cells transfected with the indicated vector and/or siRNA were immunoblotted with indicated antibodies. The protein levels of c-Fos and c-Jun normalized with tubulin are shown in the right panel when the control value was arbitrarily set at 1.0. Error bars indicate ±S.D. (n = 4). *, p < 0.05; **, p < 0.01; ***, p < 0.005 (Student's t test).
To determine whether c-Fos activation by CRAG is a polyQ-specific phenomenon or a general reaction in response to the accumulation of misfolded proteins, cells were treated with the proteasome inhibitor MG132. Interestingly, treatment with MG132 resulted in an ∼6-fold increase in AP-1 activation (Fig. 4C). Furthermore, immunoblot analysis indicated strong activation of both c-Fos and c-Jun by MG132 (Fig. 4D), suggesting that MG132 caused a large accumulation of misfolded proteins and intensive cytotoxicity compared with polyQ expression and induced cell survival signaling via c-Fos activation in Neuro2A cells. Most importantly, siRNA knockdown of CRAG drastically reduced MG132-induced AP-1 and c-Fos activation (Fig. 4, C and D). In contrast, no obvious change in MG132-induced c-Jun activation was observed following CRAG knockdown. These results demonstrated that CRAG plays a pivotal role in c-Fos activation in response to not only polyQ, but also the general accumulation of misfolded proteins.
CRAG has a specific C terminus that is not included in centaurin-γ3, and the C-terminal deletion mutant (CRAGΔC) failed to localize to the nucleus despite maintaining the NLS. Therefore, we predicted that CRAGΔC functions as a dominant negative mutant. To verify this possibility, we examined the effect of CRAGΔC on AP-1 and c-Fos activation triggered by polyQ and MG132, respectively. As expected, CRAGΔC suppressed AP-1 activation triggered by both polyQ and MG132 (Fig. 4, E and G). In particular, polyQ-mediated AP-1 activity was completely abolished by co-expression of CRAGΔC (Fig. 4E). Wild-type CRAG induced c-Fos activation in the presence of polyQ or MG132, whereas CRAGΔC did not induce c-Fos activation (Fig. 4, F and H). Most strikingly, MG132-mediated c-Fos activation was strongly inhibited by co-expression of CRAGΔC (Fig. 4H). In contrast, the inhibitory effect of CRAGΔC on c-Jun induction was only partial (Fig. 4H), suggesting that the c-Jun/c-Jun homodimer still formed. Taken together, these results demonstrated that CRAGΔC acts as a potent dominant negative mutant and the C terminus of CRAG is critical for CRAG to function and promote activation of c-Fos and AP-1.
A Protective Role for CRAG-induced c-Fos Activation in Both polyQ- and MG132-induced Cytotoxicity
It is highly possible that CRAG enhances the cell survival signal in response to polyQ-induced cell toxicity via not only proteasomal degradation of polyQ but also c-Fos activation. To evaluate this possibility, we investigated whether c-Fos expression could rescue Neuro2A cells from the polyQ-induced cell toxicity enhanced by CRAG knockdown. We assessed apoptosis by measuring the production of cleaved caspase-3, a marker of cell apoptosis, using immunoblot analysis with anti-cleaved caspase-3 antibody. CRAG siRNA knockdown significantly increased the production of polyQ-mediated cleaved caspase-3, consistent with our previous observation (Fig. 5A). Importantly, c-Fos expression significantly reduced the polyQ-mediated cleaved caspase-3 production enhanced by CRAG knockdown. Quantitative analysis of the relative amount of cleaved caspase-3 protein confirmed c-Fos-dependent rescue from polyQ-induced cytotoxicity (Fig. 5A, lower panel). Similarly, the ATP reduction assay indicated that c-Fos partially decreased the polyQ-mediated ATP reduction ratio enhanced by CRAG knockdown (Fig. 5B).
FIGURE 5.
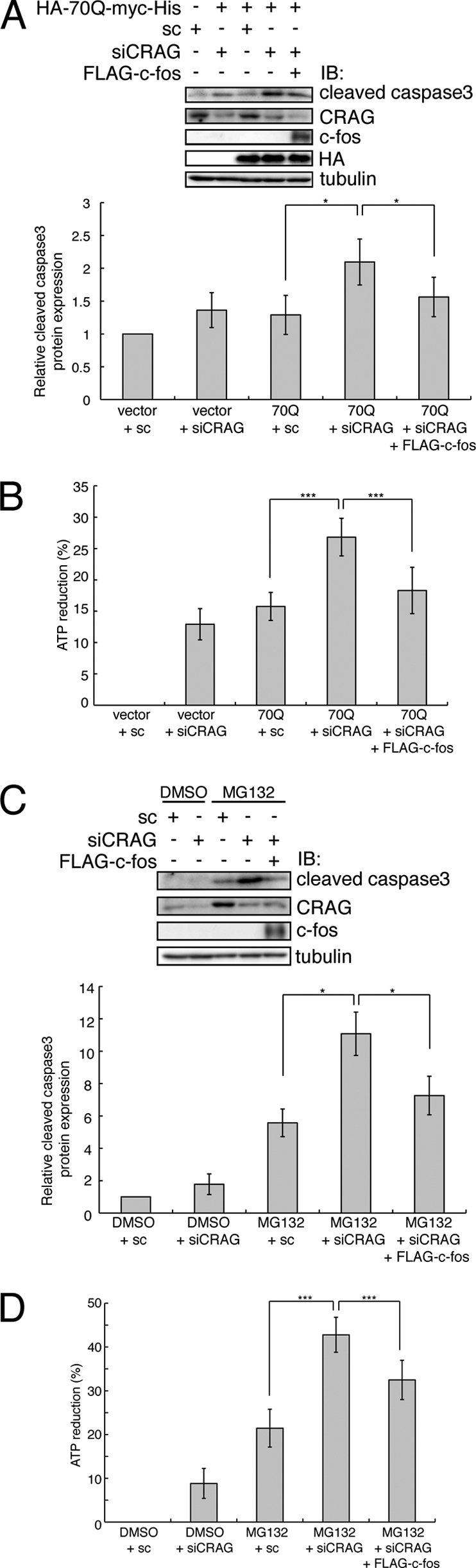
Expression of c-Fos partially rescued from polyQ- and MG132-induced cytotoxicity enhanced by CRAG knockdown. Effect of c-Fos on cytotoxicity enhanced by CRAG knockdown in cells expressing polyQ (A and B) and with MG132 treatment (C and D). Lysates of Neuro2A cells transfected with the indicated vector(s) were immunoblotted with the indicated antibodies. The protein levels of cleaved caspase3 normalized are shown in the upper panel when the control value was arbitrarily set at 1.0 (A and C). For MG132 treatment, Neuro2A cells were treated with either DMSO or 10 μm MG132 for 24 h. An ATP reduction assay was performed on Neuro2A cells as described above (B and D). Error bars indicate ±S.D. (n = 5). *, p < 0.05; ***, p < 0.005 (Student's t test).
We then examined whether c-Fos expression could rescue cells from MG132-induced cell toxicity enhanced by CRAG knockdown. Expression of c-Fos attenuated the MG132-mediated cleaved caspase-3 production enhanced by CRAG knockdown (Fig. 5C). Furthermore, the ATP reduction assay also revealed that c-Fos expression partially decreased the MG132-mediated ATP reduction ratio enhanced by CRAG knockdown (Fig. 5D). Taken together, these results demonstrate that CRAG-mediated c-Fos activation plays a protective role in both polyQ and MG132-induced cell toxicity.
AP-1 complexes consisting of c-Fos/c-Jun heterodimers regulate transcription by binding to the AP-1 sequence found in antioxidant enzyme genes. Intrinsic antioxidant defenses are important for neuronal survival against oxidative stress induced by unfolded protein accumulations, including polyQ (29, 30). One of the major antioxidant defenses is the thioredoxin-peroxiredoxin (Prx) system (31). Prxs are a family of peroxidases that reduce hydroperoxides. During catalysis, the cysteine residue in the active site of Prx enzymes undergoes reversible oxidation to sulfinic acid, and sulfiredoxin-1 (Srxn-1) is responsible for reversal of the resulting enzyme inactivation (32, 33). Because Srxn-1 was reported to be up-regulated by synaptic activity via AP-1 (10), we examined whether CRAG activates Srxn-1 via AP-1 (Fig. 6A). A luciferase assay demonstrated that CRAG activated Srxn-1 and that activation was dependent on the NLS (Fig. 6A). Mutational analysis indicated that both the AP-1 binding sites in the promoter region of Srxn-1 are critical for CRAG-mediated Srxn-1 activation (Fig. 6B). Indeed, quantitative RT-PCR analysis indicated that Srxn-1 mRNA expression in Neuro2A cells was induced ∼1.3-fold by CRAG expression (Fig. 6C). Furthermore, we found that CRAG significantly suppressed hydrogen peroxide-induced hyperoxidation of Prx (Fig. 6D). In contrast, siRNA knockdown of CRAG significantly increased hydrogen peroxide-induced hyperoxidation of Prx (Fig. 6E). Taken together, these results suggested that CRAG induces Srxn-1 activation, at least in part, via AP-1. A schematic model for the CRAG-mediated c-Fos and AP-1 signaling pathway is illustrated in Fig. 6F.
FIGURE 6.

CRAG activates antioxidative pathway via AP-1-mediated Srxn-1 activation. A, CRAG activates Srxn-1. Neuro2A cells were transfected with both Srxn-1-Luc and pRL-CMV together with either empty vector or indicated vector. Luciferase activities were assessed 48 h after the transfection. Error bars indicate ±S.D. (n = 3). *, p < 0.05 (Student's t test). B, both AP-1 sites in the Srxn-1 promoter are required for CRAG-dependent Srxn-1 activation. There are two AP-1 sites in the Srxn-1 promoter, and three different mutants were generated as indicated in the figure. The effect of CRAG expression on luciferase activity of Srxn-1 promoter mutants was assessed 48 h after the transfection. Error bars indicate ±S.D. (n = 4). *, p < 0.05; **, p < 0.01 (Student's t test). C, induction of Srxn-1 mRNA by CRAG. Neruro2A cells were transfected with either control vector or CRAG. At 48 h after transfection, quantitative RT-PCR was performed to quantify Srxn-1 mRNA in control and CRAG-transfected cells. D, CRAG reduces hydrogen peroxide-induced Prx-SO2/3H. Neruro2A cells were transfected with either control vector or CRAG. At 48 h after transfection, cells were treated with 200 μm hydrogen peroxide for 1 h, following incubation in the fresh medium. Cells were harvested at the indicated time. Lysates of Neuro2A cells were immunoblotted with anti-Prx-SO2/3H antibody. E, CRAG knockdown enhanced hydrogen peroxide-induced Prx-SO2/3H accumulation. Neruro2A cells were transfected with either control or CRAG-specific siRNA. At 48 h after transfection, cells were treated with 100 μm hydrogen peroxide for 1 h, following incubation in the fresh medium. Cells were harvested at the indicated time. Lysates of Neuro2A cells were immunoblotted with anti-Prx-SO2/3H antibody. F, a schematic model for the CRAG-mediated AP-1 signaling pathway.
DISCUSSION
PD are a group of nine neurodegenerative disorders characterized by the presence of a toxic polyglutamine expansion in specific target proteins. Using cells and mouse models, we have shown that CRAG is a useful gene therapy target for PD through the degradation of polyQ (1, 7). However, the molecular basis of CRAG function in cell survival remains unknown. In this study, we demonstrated that CRAG enhances cell survival against the accumulation of unfolded proteins, including polyQ, through not only proteasome activation but also transcriptional activation of c-Fos-dependent AP-1 via SRF.
In neuronal cells, AP-1 has opposing roles in apoptosis and survival (15). Several lines of evidence have demonstrated that polyQ leads to activation of c-Jun-mediated AP-1 via the stress kinase JNK, which is implicated in neuronal death (26–28). Consistent with this, overexpression of a dominant negative mutant of c-Jun, as well as pharmacological inhibition of JNK, strongly protected neuronal cells against apoptosis induced by polyQ (28). Therefore, c-Jun activation is an early event in the pathogenesis of polyglutamine diseases. In contrast, the immediate early gene c-fos encodes a transcription factor that forms heterodimers with c-Jun family proteins, and the resulting AP-1 complexes regulate transcription by binding to the AP-1 sequence found in many cellular genes. Emerging evidence indicates that c-Fos is essential for the regulation of neuronal cell survival versus death, suggesting that c-Fos is a key regulator of the apoptosis/survival decision (34, 35). A previous study demonstrated that c-fos mutant mice have more severe kainic acid-induced seizures, increased neuronal excitability, and neuronal cell death than control mice and that c-Fos regulates the expression of brain-derived neurotrophic factor (35), whose reduction has been implicated in the pathogenesis of polyglutamine diseases. Indeed, increasing brain-derived neurotrophic factor levels ameliorated PD phenotypes (36, 37). Therefore, CRAG-mediated c-Fos activation may explain the potent protective effect of CRAG against the cytotoxicity of unfolded protein accumulation, including polyQ. Because CRAG facilitates the degradation of polyQ through the ubiquitin-proteasome pathway, we examined whether c-Fos activation is involved in CRAG-mediated polyQ degradation. We found that c-Fos did not promote the degradation of polyQ (supplemental Fig. S1). Therefore, the c-Fos-dependent rescue from polyQ-induced cytotoxicity in CRAG-knockdown cells may not be due to proteasomal activation and degradation of polyQ. On the other hand, we found that CRAG activates SRF, an upstream transcriptional factor of c-Fos. Because SRF has many target genes promoting cell growth, survival, and differentiation, it is possible that SRF is involved in the proteasomal activation and degradation of polyQ. Further analysis is needed to clarify the roles of SRF in the CRAG-mediated polyQ degradation and c-Fos-independent neuronal cell survival pathway via SRF.
CRAG may be involved in semaphorin-mediated signaling due to its association with CRMP5, although the relationship between CRAG and CRMP5 is quite obscure. Semaphorin-3A has been previously reported to induce redox signaling by generating ROS through activation of the oxidoreductase MICAL, which directly interacts with the semaphorin-3A receptor plexin-A1 (38). Interestingly, in cultured rat hippocampal neurons, CRAG was found to translocate from the cytosol to the nucleus following stimulation of semaphorin-3A in an ROS-dependent manner,3 suggesting the possible involvement of CRAG in semaphorin-mediated redox signaling. Similarly, rapid nuclear translocation of CRAG was observed following UV irradiation in an ROS-dependent manner. Because CRAG knockdown revealed a vulnerability to oxidant stress, CRAG may play an important role in antioxidant signaling during brain development. In this study, we found that CRAG activated the antioxidant protein Srxn-1 via AP-1 and reduced hyperoxidation of Prx (Fig. 6). Thus, CRAG conveys an antioxidant signal, at least in part, via AP-1-mediated Srxn-1 activation, thereby contributing to neuronal cell survival and development. In addition, unfolded protein accumulations, including polyQ, have been shown to cause oxidative stress by generating ROS (29, 30). It is therefore considered that CRAG protects neuronal cell against cytotoxicity of unfolded protein accumulations, including polyQ, partially via the antioxidant pathway mediated by AP-1-dependent Srxn-1 activation.
A schematic model for the CRAG-mediated AP-1 signaling pathway is illustrated in Fig. 6F. In summary, CRAG promotes cell survival against the stress of unfolded proteins, including polyQ, by switching the AP-1 content from c-Jun homodimers, which induce the apoptotic pathway, to c-Fos/c-Jun heterodimers, which induce the cell survival pathway. Our findings extend the possible use of targeted delivery of CRAG as a gene therapy for PD.
Supplementary Material
This work was supported in part by Grants-in-Aid for scientific research from the Ministry of Education, Culture, Sports, Science and Technology and the Japan Society for the Promotion of Science (to T. F., R. I., and S. Y.), the Naito Foundation, the Takeda Science Foundation, the Mochida Memorial Foundation for Medical and Pharmaceutical Research, and the Uehara Memorial Foundation.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Fig. S1.
S. Nagashima, T. Fukuda, Y. Kubota, A. Sugiura, M. Nakao, R. Inatome, and S. Yanagi, unpublished data.
- CRAG
- CRAM (CRMP5)-associated GTPase
- CRMP
- semaphorin response mediator protein
- polyQ
- expanded polyglutamine proteins
- AP-1
- activation protein-1
- SRF
- serum response factor
- UV
- ultraviolet
- PML
- promyelocytic leukemia protein
- ROS
- reactive oxygen species
- NLS
- nuclear localization signal
- Prx
- peroxiredoxin
- Srxn-1
- sulfiredoxin-1
- PD
- polyglutamine disease.
REFERENCES
- 1. Qin Q., Inatome R., Hotta A., Kojima M., Yamamura H., Hirai H., Yoshizawa T., Tanaka H., Fukami K., Yanagi S. (2006) J. Cell Biol. 172, 497–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Inatome R., Tsujimura T., Hitomi T., Mitsui N., Hermann P., Kuroda S., Yamamura H., Yanagi S. (2000) J. Biol. Chem. 275, 27291–27302 [DOI] [PubMed] [Google Scholar]
- 3. Samaj J., Ovecka. M., Hlavacka. A., Lecourieux F., Meskiene I., Lichtscheidl I., Lenart P., Salaj J., Volkmann D., Bögre L., Baluska F., Hirt H. (2002) EMBO J. 21, 3296–3306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hotta A., Inatome R., Yuasa-Kawada J., Qin Q., Yamamura H., Yanagi S. (2005) Mol. Biol. Cell 16, 32–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Orr H. T. (2001) Genes Dev. 15, 925–932 [DOI] [PubMed] [Google Scholar]
- 6. Ross C. A. (1997) Neuron 19, 1147–1150 [DOI] [PubMed] [Google Scholar]
- 7. Torashima T., Koyama C., Iizuka A., Mitsumura K., Takayama K., Yanagi S., Oue M., Yamaguchi H., Hirai H. (2008) EMBO Rep. 9, 393–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sugiura A., Yonashiro R., Fukuda T., Matsushita N., Nagashima S., Inatome R., Yanagi S. (2011) Mitochondrion 11, 139–146 [DOI] [PubMed] [Google Scholar]
- 9. Matsuzaki K., Minami T., Tojo M., Honda Y., Saitoh N., Nagahiro S., Saya H., Nakao M. (2003) Genes Cells 8, 275–286 [DOI] [PubMed] [Google Scholar]
- 10. Papadia S., Soriano F. X., Léveillé F., Martel M. A., Dakin K. A., Hansen H. H., Kaindl A., Sifringer M., Fowler J., Stefovska V., McKenzie G., Craigon M., Corriveau R., Ghazal P., Horsburgh K., Yankner B. A., Wyllie D. J., Ikonomidou C., Hardingham G. E. (2008) Nat. Neurosci. 11, 476–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bernardi R., Papa A., Pandolfi P. P. (2008) Oncogene 27, 6299–6312 [DOI] [PubMed] [Google Scholar]
- 12. Salomoni P., Bernardi R., Bergmann S., Changou A., Tuttle S., Pandolfi P. P. (2005) Blood 105, 3686–3690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Meurer S., Pioch S., Wagner K., Müller-Esterl W., Gross S. (2004) J. Biol. Chem. 279, 49346–49354 [DOI] [PubMed] [Google Scholar]
- 14. Kahn R. A., Bruford E., Inoue H., Logsdon J. M., Jr., Nie Z., Premont R. T., Randazzo P. A., Satake M., Theibert A. B., Zapp M. L., Cassel D. (2008) J. Cell Biol. 182, 1039–1044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hess J., Angel P., Schorpp-Kistner M. (2004) J. Cell Sci. 117, 5965–5973 [DOI] [PubMed] [Google Scholar]
- 16. Murphy L. O., Smith S., Chen R. H., Fingar D. C., Blenis J. (2002) Nat. Cell Biol. 4, 556–564 [DOI] [PubMed] [Google Scholar]
- 17. Barber J. R., Verma I. M. (1987) Mol. Cell. Biol. 7, 2201–2211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Iwatsuki M., Inageda K., Matsuoka M. (2011) Toxicol. Appl. Pharmacol. 251, 209–216 [DOI] [PubMed] [Google Scholar]
- 19. Cartegni L., Maconi M., Morandi E., Cobianchi F., Riva S., Biamonti G. (1996) J. Mol. Biol. 259, 337–348 [DOI] [PubMed] [Google Scholar]
- 20. D'Orso I., Frasch A. C. (2002) J. Biol. Chem. 277, 50520–50528 [DOI] [PubMed] [Google Scholar]
- 21. Izzo A., Regnard C., Morales V., Kremmer E., Becker P. B. (2008) Nucleic Acids Res. 36, 950–962 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Li F., Parry D. A., Scott M. J. (2005) Mol. Cell. Biol. 25, 8913–8924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Treisman R. (1987) EMBO J. 6, 2711–2717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Knöll B., Nordheim A. (2009) Trends Neurosci. 32, 432–442 [DOI] [PubMed] [Google Scholar]
- 25. Miralles F., Posern G., Zaromytidou A. I., Treisman R. (2003) Cell 113, 329–342 [DOI] [PubMed] [Google Scholar]
- 26. Nishitoh H., Matsuzawa A., Tobiume K., Saegusa K., Takeda K., Inoue K., Hori S., Kakizuka A., Ichijo H. (2002) Genes Dev. 16, 1345–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Merienne K., Helmlinger D., Perkin G. R., Devys D., Trottier Y. (2003) J. Biol. Chem. 278, 16957–16967 [DOI] [PubMed] [Google Scholar]
- 28. Garcia M., Charvin D., Caboche J. (2004) Neuroscience 127, 859–870 [DOI] [PubMed] [Google Scholar]
- 29. Rubinsztein D. C., Carmichael J. (2003) Exp. Rev. Mol. Med. 5, 1–21 [DOI] [PubMed] [Google Scholar]
- 30. Wyttenbach A., Sauvageot O., Carmichael J., Diaz-Latoud C., Arrigo A. P., Rubinsztein D. C. (2002) Hum. Mol. Genet. 11, 1137–1151 [DOI] [PubMed] [Google Scholar]
- 31. Masutani H., Bai J., Kim Y. C., Yodoi J. (2004) Mol. Neurobiol. 29, 229–242 [DOI] [PubMed] [Google Scholar]
- 32. Biteau B., Labarre J., Toledano M. B. (2003) Nature 425, 980–984 [DOI] [PubMed] [Google Scholar]
- 33. Chang T. S., Jeong W., Woo H. A., Lee S. M., Park S., Rhee S. G. (2004) J. Biol. Chem. 279, 50994–51001 [DOI] [PubMed] [Google Scholar]
- 34. Yuan Z., Gong S., Luo J., Zheng Z., Song B., Ma S., Guo J., Hu C., Thiel G., Vinson C., Hu C. D., Wang Y., Li M. (2009) Mol. Cell. Biol. 29, 2431–2442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhang J., Zhang D., McQuade J. S., Behbehani M., Tsien J. Z., Xu M. (2002) Nat. Genet. 30, 416–420 [DOI] [PubMed] [Google Scholar]
- 36. Xie Y., Hayden M. R., Xu B. (2010) J. Neurosci. 30, 14708–14718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Spires T. L., Grote H. E., Varshney N. K., Cordery P. M., van Dellen A., Blakemore C., Hannan A. J. (2004) J. Neurosci. 24, 2270–2276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Terman J. R., Mao T., Pasterkamp R. J., Yu H. H., Kolodkin A. L. (2002) Cell 109, 887–900 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.