

Abstract
The asparaginyl hydroxylase, factor-inhibiting hypoxia-inducible factor (HIF), is central to the oxygen-sensing pathway that controls the activity of HIF. Factor-inhibiting HIF (FIH) also catalyzes the hydroxylation of a large set of proteins that share a structural motif termed the ankyrin repeat domain (ARD). In vitro studies have defined kinetic properties of FIH with respect to different substrates and have suggested FIH binds more tightly to certain ARD proteins than HIF and that ARD hydroxylation may have a lower Km value for oxygen than HIF hydroxylation. However, regulation of asparaginyl hydroxylation on ARD substrates has not been systematically studied in cells. To address these questions, we employed isotopic labeling and mass spectrometry to monitor the accrual, inhibition, and decay of hydroxylation under defined conditions. Under the conditions examined, hydroxylation was not reversed but increased as the protein aged. The extent of hydroxylation on ARD proteins was increased by addition of ascorbate, whereas iron and 2-oxoglutarate supplementation had no significant effect. Despite preferential binding of FIH to ARD substrates in vitro, when expressed as fusion proteins in cells, hydroxylation was found to be more complete on HIF polypeptides compared with sites within the ARD. Furthermore, comparative studies of hydroxylation in graded hypoxia revealed ARD hydroxylation was suppressed in a site-specific manner and was as sensitive as HIF to hypoxic inhibition. These findings suggest that asparaginyl hydroxylation of HIF-1 and ARD proteins is regulated by oxygen over a similar range, potentially tuning the HIF transcriptional response through competition between the two types of substrate.
Keywords: Enzyme Catalysis, Hydroxylase, Hypoxia, Mass Spectrometry (MS), Post-translational Modification, 2-Oxoglutarate-dependent Dioxygenase, Ankyrin Repeat Domain, Hypoxia-inducible Factor, SILAC
Introduction
Factor-inhibiting HIF5 (FIH) is an iron(II)- and 2-oxoglutarate-dependent dioxygenase that was first identified as a negative regulator of the HIF transcriptional response (1–3). In the HIF pathway, FIH catalyzes the post-translational hydroxylation (PTH) of a conserved Asn residue in the C-terminal transactivation domain of HIF-α (HIF1-CAD) that renders the CAD unable to bind the essential transcriptional co-activator p300/CBP. FIH couples PTH of prime substrate to the decarboxylation of 2-oxoglutarate (2-OG) in a reaction that is absolutely dependent on the availability of molecular oxygen as a co-substrate. Because the Km value of FIH for oxygen is above the physiological range, FIH can function as a cellular oxygen sensor (4, 5). Thus, hypoxic inhibition of FIH promotes CAD activity and a robust HIF transcriptional response.
More recently, members of an alternative class of substrates have been identified as targets for FIH-mediated hydroxylation. A range of functionally diverse proteins sharing a common protein-protein interaction motif known as the ankyrin repeat domain (ARD) have been shown to be substrates of FIH, including the intracellular domain of Notch-1 (6, 7), p105, IkBα (8), suppression of cytokine signaling box protein 4 (ASB4) (9), MYPT1 (10), Tankyrase-2, Rabankyrin-5, RNase L (11, 12), and ankyrinR (13). ARDs have been well characterized structurally and, irrespective of function, share the same basic architecture consisting of a variable number of 33-residue repeats that individually fold into paired antiparallel α-helices followed by a β-hairpin loop. In each case the target Asn is positioned at a distinct site within the hairpin loop that links individual repeats (14). Interestingly, the target Asn residue is semi-conserved and forms part of an “ankyrin repeat consensus” that, together with the degenerate hydroxylation motif derived from the relatively small subset (n = 12) of ARD substrates defined to date, suggests that FIH-dependent PTH could extend to many of the ∼300 ARD-containing proteins in the human proteome (15).
Despite the apparent ubiquity of this modification on ARD proteins, and in contrast to the well defined signaling role in the HIF pathway, the biological consequence(s) of FIH-catalyzed ARD PTH are not clear. Mass spectrometric methods have been successful in proving the existence of this modification across a range of ARD proteins and cellular backgrounds. Collectively, these studies show that ARD-containing substrates are often multiply hydroxylated and that the level of hydroxylation can vary between sites in the same ARD. Hydroxylation is incomplete at most sites that have been examined. It is unclear whether this represents a steady state common to all protein molecules, whether it is a reflection of the progressive accumulation of hydroxylation at target sites, or whether it is evidence for the operation of a reversal process. Resolution of these possibilities is of considerable interest. On proteins that are not intrinsically labile, dynamically regulated signaling modifications such as phosphorylation and ubiquitination are often enzymatically reversed as part of the regulatory process. Because many ARD proteins are not intrinsically labile, a dynamic signaling role would likely be predicated on the existence of a reversal process. Conversely, progressive accumulation of an irreversible modification would have the potential to encode time-dependent functions such as protein half-life.
The notion that FIH interacts with multiple ARD substrates also has implications for the oxygen-sensing role of FIH on the HIF pathway, and it has been postulated that ARD proteins serve to “fine-tune” the HIF transcriptional response by binding and sequestering FIH (15). In support of this model, in vitro studies of the Notch family of substrates indicate that ARDs bind to FIH with a 50-fold higher affinity compared with HIF-1α (6), and certain ARD proteins can compete with the HIF-1α transactivation domain for hydroxylation when co-expressed in transfected cells (7). Given that the affinity of FIH for its substrate decreases upon hydroxylation (6), it has been proposed that hypoxic inhibition of hydroxylation of the ARD “pool” could modulate the signal/response curve of HIF-1α activity (16). For this to occur, the hydroxylation status of the ARD pool would have to be equally sensitive or more sensitive to decreasing oxygen levels than HIF-1α. It is not clear whether this is the case. In vitro kinetic studies of FIH have revealed Km values for oxygen that are an order of magnitude lower with Notch-1 than with HIF1-CAD as substrate, suggesting that Notch-1 hydroxylation might not be regulated at oxygen levels that inhibit HIF1-CAD hydroxylation (17). However, given the large number of potential ARD substrates and the possibility that different ARDs substrates are differentially affected by hypoxia, it is difficult to predict oxygen-regulated characteristics in cells from these data. The sensitivity of ARD hydroxylation to oxygen levels and FIH cofactor availability has not been systematically studied in cells.
To address these issues, we embarked upon a detailed proteomic study focusing on the regulation of hydroxylation of a number of prototypical ARD-containing FIH substrates, as well as the HIF1-CAD. We utilized an isotope label-based mass spectrometric approach termed SILAC (stable isotope labeling by amino acids in cell culture) to monitor the dynamics of hydroxylation on protein that was synthesized within a defined time interval under defined conditions of oxygen and FIH-cofactor availability. Our findings provide no evidence of reversal of hydroxylation on the substrates examined. Hydroxylation at sites in ARD proteins accrued over time but generally remained incomplete, even in normoxic cultures fully supplemented with FIH cofactors. Studies of oxygen and cofactor dependence revealed marked site-specific regulation of ARD hydroxylation under conditions similar to those affecting hydroxylation of HIF1-CAD proteins, consistent with the proposal that variable hydroxylation of the ARD protein pool has the potential to generate flexible interactions with HIF-signaling pathways.
EXPERIMENTAL PROCEDURES
Cell Culture
The human embryonic kidney cell line HEK293 were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (v/v) fetal calf serum, 2 mm l-glutamine, 50 IU·ml−1 penicillin, and 50 μg·ml−1 streptomycin. Stable transfectants were generated in the HEK293 background and maintained in G418 (500 μg/ml geneticin, Invitrogen). Where indicated, cells were treated with 1 mm dimethyloxalylglycine (DMOG; Frontier Scientific, Lancashire, UK), 100 μg/ml cycloheximide (Sigma), 25 μm MG132 (Enzo Life Sciences, Exeter, UK), 50 μm l-ascorbate (Sigma), or 40 μm FeCl2 (Sigma). 100× l-ascorbate and iron (FeCl2) stock solutions were made immediately prior to addition onto the cell cultures. Hypoxic incubations of cells were performed in an in vivo2 400 hypoxic workstation (Ruskinn Technologies, Bridgend, UK), and media and plastic culture dishes were pre-equilibrated under hypoxic conditions (0.2 or 1.0% O2) for 24 h prior to the start of the experiment. Atmospheric oxygen concentrations in hypoxic work stations were confirmed at the start and end of every experiment using an independent oxygen probe (Z210 Oxygen Analyzer, HiTech Instruments, Bedfordshire, UK).
Plasmids and Transfections
cDNAs encoding full-length human Rabankyrin-5, mouse Notch1 ARD (mN1ARD, residues 1899–2105), and the C-terminal transactivation domain (CAD) of HIF-1α(652–826) were amplified by PCR and ligated into p3×FLAG-CMV-10 (Sigma). The integrity of all constructs was verified by DNA sequence determination. Stable transfectants were generated from HEK293 cells by FuGENE 6 transfection according to the manufacturer's instructions (FuGENE6TM transfection reagent; Roche Applied Science) with p3×FLAG CMV-10 constructs encoding Rabankyrin-5, Rabankyrin-5/HIF1-CAD fusion, and mN1ARD/HIF1-CAD fusion, followed by selection in G418. Clones were picked as individual colonies and maintained in G418 with the highest expressing clone of each transfectant, as determined by FLAG immunoblotting, selected for analyses.
Knockdown of FIH Expression Using siRNA (Small Interfering RNA)
siRNA sequences targeting human FIH (target sequence 91) and control lamin duplexes have been described previously (19) and were synthesized by Dharmacon (Lafayette, CO). Cells were transfected twice at 24-h intervals using a 20 nm dose of duplex and Dharmafect reagent (Dharmacon), according to the manufacturer's instructions.
Immunoblotting
For immunoblotting of FIH and FLAG constructs, cells were lysed in Jie's buffer (10). For immunoblotting of HIF-1α and HIF1α-N803OH, cells were lysed directly into 3× SDS sample buffer and sonicated briefly. Lysates were resolved by SDS-PAGE, electroblotted onto PVDF membranes (Millipore, Watford, UK), and probed using primary antibodies to HIF-1α (BD Biosciences), hydroxy-HIF-1α (Asn803 (20)), FIH (FIH antibody NB100-428, Novus Biologicals, Cambridge, UK), monoclonal anti-FLAG M2-peroxidase (HRP) Clone M2 (Sigma), or β-actin-HRP (Abcam, Cambridge, UK). Horseradish peroxidase-conjugated anti-mouse secondary antibodies (Dako, Cambridge, UK) were used with either SuperSignal West Dura or SuperSignal West Femto (Thermo Fisher Scientific, Leicestershire, UK) to visualize immunoreactive species.
SILAC Protocol
HEK293 cells expressing FLAG-tagged proteins were labeled by serial passage in arginine- and lysine-deficient DMEM (Thermo Fisher Scientific) containing 10% (v/v) dialyzed fetal bovine serum (Invitrogen) supplemented with either normal (“light”) isotopic abundance (0.68 μm) l-lysine and (0.54 μm) l-arginine or with “heavy” isotopic forms of l-lysine (U-13C6; Lys6) and l-arginine (U-13C6; Arg6) at identical concentrations (light amino acids, Sigma; heavy amino acids, Cambridge Isotope Laboratories, Hook Hampshire, UK). To prevent conversion of isotope-coded arginine to proline in cells (21), all media were supplemented with 200 mg/liter l-proline (Sigma). Cells were lysed in Jie's buffer (10), and FLAG-tagged proteins were immunopurified from heavy and light lysates by FLAG affinity gel (Sigma). Samples were eluted in ammonium hydroxide and either resolved by SDS-PAGE or desalted by methanol/chloroform precipitation prior to digestion in bicarbonate buffer using sequencing-grade trypsin (Sigma) as described previously (22).
Mass Spectrometry
Liquid chromatography-mass spectrometry (LC-MS) was performed on Waters and Agilent quadrupole time-of-flight (Q-TOF) platforms. The Waters platform consisted of a nanoAcquity UPLC coupled to a Waters Q-TOF premier mass spectrometer (Waters). Analyte was loaded onto a 250-mm × 75-μm inner diameter C18 column (1.7-μm particle size; Waters) and eluted using a 90-min gradient of 2–45% (v/v) solvent B (solvent A, 0.1% formic acid in H2O; solvent B, 99.9% acetonitrile, 0.1% formic acid in H2O). Data-independent MSE acquisition was used, with scans of 1.5 s alternating between low collision energy (4 eV) and high collision energy (ramping from 15 to 40 eV) as described previously (22). The Agilent platform consisted of an Agilent 1200 series HPLC-Chip system coupled to an Agilent 6520 Q-TOF mass spectrometer. Analyte was loaded onto an Agilent large capacity chip (II), containing a 150-mm 300-Å C18 column. A gradient from 0 to 40% (v/v) solvent B over 47 min was used to elute the peptides (solvent A, 2% acetonitrile, 0.1% formic acid in H2O; solvent B, 95% acetonitrile, 0.1% formic acid in H2O). Data-dependent acquisition was performed on the mass spectrometer, with MS scans of 0.5 s followed by up to 6 MS/MS scans of 0.5 s. Fragmented masses were placed on an exclusion list for 90 s. For samples containing Rabankyrin-5 and the Rabankyrin-5/HIF1-CAD construct, an inclusion list containing m/z values of the hydroxylated peptides and their unhydroxylated partners was specified in order that these peptides were preferentially chosen for fragmentation.
Identification of Peptides
During the quantitation process, peptides were identified using their accurate mass and retention time (AMT) (23). This approach was facilitated by the high mass accuracy (<10 ppm) of the Q-TOF platforms and reproducible chromatography of the UPLC and HPLC-Chip systems. To establish AMT signatures for the peptides of interest, identification was performed on datasets from both platforms using database search engines. On the Waters platform, identification of the Rabankyrin-5-hydroxylated peptides has been described previously (12). Identification of the HIF1-CAD peptide was performed as described previously (12), with annotated spectra and chromatograms given in supplemental Figs. S1–S3. On the Agilent platform, peptide identifications were performed using the in-house Central Proteomics Facilities Pipeline (24). Briefly, raw data were converted into MzXML format using Trapper version 4.3.0 (25) and uploaded to the pipeline. Database searches were performed against a concatenated target/decoy version of IPI_Human 3.65 (86379 target, 86379 decoy protein sequences) using Mascot version 2.3.01 (Matrix Science, London, UK), X!Tandem version 2008.12.01.1 (26), and OMSSA version 2.1.9 (27). Mass tolerances were set at 50 ppm for precursor and 0.1 Da for fragment ions. Fully tryptic specificity with a single missed cleavage was permitted, and carbamidomethylation on cysteine was specified as a fixed modification. Oxidation of methionine and asparagine was specified as variable modifications, with labeled (13C6) arginine and lysine as additional variable modifications for SILAC datasets. Peptide identifications from each search engine were subsequently validated using PeptideProphet (28) and combined using iProphet (29). Protein identifications were inferred from peptides using ProteinProphet (30). Peptide identifications were filtered to a 1% false discovery rate using the target-decoy method (31) prior to manual inspection of MS/MS spectra. Confident identifications, containing b- and y-ion ladders spanning modification sites, were required for both hydroxylated and unhydroxylated species of peptides to establish their AMT signatures. Where an unlabeled or heavily labeled form was not identified, the AMT signature was calculated by addition or subtraction of the monoisotopic mass of the SILAC label, assuming identical retention time. The supplemental Figs. S4–S24 provide annotated spectra and chromatograms for each peptide on the Agilent platform.
Quantitation of Peptides
All quantitation was performed using the intensity of peptide ions in MS scans. In each case, peptides to be quantitated were identified using their accurate mass and retention time, obtained as explained previously. Extracted ion chromatograms were generated for each peptide mass, using an m/z window of ± 0.1 Da. Retention times of peaks were matched to the AMT signature with a tolerance of ±2 min to allow for variation due to column replacement. MS spectra across the elution peaks of the peptides were inspected to ensure that the match was to the monoisotopic peak of a peptide ion and that co-eluting peptides did not interfere with quantitation. Data obtained on the Waters platform was quantitated manually using MassLynx version 4.1 SCN639. Abundance values were obtained by summing the intensity of the monoisotopic peak of a peptide in all low energy MS scans across its elution peak. Quantitation of samples analyzed on the Agilent platform was performed using MassHunter Qualitative Analysis version B.03.01. Abundance measurements were obtained by integration of the area under the elution peak for a peptide, using default parameters. Where the software's integration function failed to correctly identify the boundaries of the peak, manual integration was performed. No smoothing or other processing of the chromatograms was used.
Statistical Analysis
Unless otherwise stated, quantitative data are presented as the means ± S.E. for three independent experiments. Statistical significance was determined using a one-way analysis of variance (ANOVA) with Dunnett's post hoc test using SPSS statistics 17.0; differences were considered significant if p values were <0.05.
RESULTS
Dynamics of Asparagine Hydroxylation in ARD Proteins
To understand factors contributing to the regulation of steady-state levels of hydroxylation on ARD proteins, we began by testing for evidence of reversal of these hydroxylations in cells. As a first step we analyzed the decay of hydroxylation within the prototypic ARD protein Rabankyrin-5 following inhibition of protein synthesis by cycloheximide and inhibition of “forward” FIH-catalyzed hydroxylation by DMOG. Stable transfectants expressing FLAG-tagged Rabankyrin-5 were treated with cycloheximide for 6 h, in the presence of DMOG. Asparaginyl hydroxylation at sites within Rabankyrin-5 was monitored by UPLC-MS following anti-FLAG immunopurification and tryptic digestion. Analysis of hydroxylation at Asn752 (Fig. 1A) revealed no substantial change in the extent of hydroxylation at this site following treatment with cycloheximide (Chx) and DMOG (76% hydroxylation versus 77% in untreated cells). Similar results were observed for Asn485, where overall levels of hydroxylation were lower (22% hydroxylation in cells exposed to cycloheximide and DMOG versus 21% in untreated cells (data not shown)). Thus, under these conditions, hydroxylated and total protein species decay in parallel, indicating that hydroxylation at these sites in Rabankyrin-5 is not significantly reversed over this period of time.
FIGURE 1.

Assessment of asparagine hydroxylation reversal on FIH targets by cycloheximide and DMOG treatment. A, extracted ion chromatograms (m/z 772.71 and 778.02) corresponding to unhydroxylated (dashed line) and hydroxylated (solid line) forms of the tryptic Rabankyrin-5 peptide containing the target Asn752 residue (N752, SGCDVNSPRQPGANGEGEEEAR [M + 3H]3+) bearing a single missed cleavage. Nano-UPLC-MSE chromatography analysis illustrating no significant change in the proportion of hydroxylated to unhydroxylated peptide signals upon inhibition of FIH-mediated catalysis and new protein synthesis: control (upper panel, 77%); +CHX/+DMOG (lower panel, 76%). B, extracted ion chromatograms (m/z 1061.16) corresponding to the mass of the hydroxylated (solid line) form of the tryptic HIF-1α peptide containing the target Asn803 residue (N803, LLGQSMDESGLPQLTSYDCEVNAPIQGSR [M + 3H]3+). Background traces (BKGD, dashed line). Nano-UPLC-MSE analysis demonstrates no appreciable reduction in the extent of hydroxylation upon inhibition of FIH-mediated catalysis and new protein synthesis in the context of the Rabankyrin-5/HIF1-CAD fusion protein: control (upper panel, >98%), +CHX/+MG132 (middle panel, >98%), +CHX/+MG132/DMOG (lower panel, >98%). The signal intensity of reference peaks is indicated in parentheses on the chromatograms.
To extend the analysis to a different class of FIH substrates, we fused the HIF-1α C-terminal activation domain (HIF1-CAD) to the C terminus of Rabankyrin-5 and prepared stable transfectants expressing the FLAG-tagged Rabankyrin-5/HIF1-CAD fusion protein. Addition of the HIF1-CAD sequence to the C terminus of Rabankyrin-5 markedly destabilized Rabankyrin-5, resulting in reduced protein expression during the cycloheximide chase. Thus, we added a proteasomal inhibitor (MG132) to the cycloheximide-treated cells in the presence or absence of DMOG to retrieve sufficient material for mass spectrometric analysis (UPLC-MS). Addition of proteasomal inhibitors during the cycloheximide chase also precluded the possibility that differential proteasomal proteolysis of the hydroxylated and unhydroxylated species could mask a reversal activity. In the absence of cycloheximide, UPLC-MS analysis revealed the HIF1-CAD to be essentially fully (>98%) hydroxylated on Asn803 (Fig. 1B). Following concurrent exposure of cells to cycloheximide and DMOG, there was no appearance of unhydroxylated HIF1-CAD. Rather, levels of hydroxylation remained at >98% after 6 h of cycloheximide indicating parallel decay of hydroxylated and total protein.
Because some ARD proteins are long lived, we considered the possibility that reversal of hydroxylation might occur over a longer period of time. However, cycloheximide-associated toxicity precludes its use over an extended period. Therefore, a SILAC chase protocol was developed to enable measurements of the decay of asparaginyl hydroxylation over a longer time course (Fig. 2A). Cells expressing FLAG-tagged Rabankyrin-5 were grown in SILAC media supplemented with heavy isotopes of lysine and arginine (heavy medium) for 5 days to allow for near-complete incorporation of the mass label into cells. Heavy medium was removed, and cells were overlaid with normal light medium supplemented with DMOG to inhibit FIH. Cells were harvested at time points up to 36 h, and peptides corresponding to sites of asparaginyl hydroxylation within Rabankyrin-5 were analyzed by UPLC-MS. Data for Rabankyrin-5 Asn485 are summarized in Fig. 2B. Chromatograms of light and heavy labeled Rabankyrin-5 Asn485 peptides are shown for all chase time points in supplemental Fig. S25. Following the switch to light medium supplemented with DMOG, hydroxylated light peptide decreased to zero, confirming efficient inhibition of FIH by DMOG (data not shown). As expected, the percentage of heavy label incorporation (overall heavy label incorporation) decreased with time as new protein was synthesized (Fig. 2B). In heavy labeled material, quantification of hydroxylated and unhydroxylated peptides encompassing Asn485 demonstrated no substantial decrease in the extent of hydroxylation (Fig. 2B, HEAVY Ox%) over the 36-h time course of the experiment, indicating parallel decay of Asn485-hydroxylated and total protein during the chase. These results indicate that no substantial reversal of hydroxylation occurs at this site even over an extended period of time. Finally, we assayed the hydroxylation status of the endogenous HIF-1α protein in von Hippel-Lindau (VHL)-defective cells using a pre-validated monoclonal antibody that specifically recognizes an epitope containing the hydroxylated form of Asn803 (20, 32). The increased sensitivity of this reagent over our mass spectrometric assays allowed us to assess the kinetics of hydroxylation on the endogenous protein. In VHL-competent cells, HIF-1α protein levels are tightly regulated by the VHL gene product (pVHL), which is a critical component of a ubiquitin ligase complex that targets HIF-α for oxygen-dependent proteolysis. Under normal oxygen levels, HIF-1α is rapidly degraded with a half-life of <5 min by the ubiquitin-proteasome pathway (33). However, in VHL-defective cells, this pathway is inactive, and HIF-1α has a half-life of over 60 min, permitting assay for processes capable of reversing the hydroxylation over similar periods of time. To this end, we compared the decay of total HIF-1α versus that of hydroxylated HIF-1α in VHL-defective RCC4 cells that were treated with DMOG 20 min prior to blockade of protein synthesis with cycloheximide (Fig. 3). No difference in the rate of decay of hydroxylated versus total HIF-1α signal was discerned. Thus, under a range of experimental conditions, we were unable to find evidence for an activity reversing hydroxylation of asparaginyl residues, either in HIF-1α or in a prototypical ARD protein.
FIGURE 2.

No evidence for reversal of ARD hydroxylation over an extended period as determined by SILAC-chase methodology. A, synopsis of SILAC-chase methodology. HEK293 cells stably transfected with Rabankyrin-5 were grown in media containing heavy isotopes (Heavy media; Lys6/Arg6) for 5 days to achieve >90% incorporation of the mass label. Cells were subsequently “chased” in normal media (Light media; Lys0/Arg0), supplemented with 1 mm DMOG, to inhibit FIH-mediated hydroxylation. Following inhibition of FIH, the extent of hydroxylation on the pre-existing heavy labeled material was monitored over 36 h by LC-MS to determine the proportion of material that was hydroxylated over time. B, quantitation of heavy hydroxylation at Asn485 in Rabankyrin-5 over 36 h following addition of light media and DMOG. Data points were derived from extracted ion chromatograms of m/z 560.53 and 564.53 corresponding to heavy labeled forms of the unhydroxylated and hydroxylated tryptic Asn485 Rabankyrin-5 peptide, AAGAGNEAAALFLATNGAHVNHR ([M + 4H]4+) and expressed as percentage hydroxylation (Heavy Ox %). The overall percentage of heavy label incorporation, calculated from the sum of unhydroxylated and hydroxylated Asn485 heavy peptides over light forms of the peptide are depicted (black triangle), showing the expected removal of the heavy label over time. Raw data (extracted ion chromatograms for Asn485 peptides) are presented in supplemental Fig. S25 along with control data confirming the efficacy of DMOG treatment on the newly synthesized light material (supplemental Fig. S26).
FIGURE 3.

Assessment of hydroxylation reversal on endogenous HIF-1α by immunoblotting with an antibody specific to hydroxy CAD (Asn803). i, representative immunoblot of RCC4 cell lysates probed with antibodies to HIF-1α and hydroxylated HIF-1α (HIF1α N803-OH) following treatment with 1 mm DMOG and cycloheximide. Results are representative of four independent experiments. ii, densitometric analysis of relative band intensities (mean ± S.E., n = 4) of hydroxylated and total HIF-1α protein normalized to the β-actin loading control. Values were plotted with the starting level (t = 0) set to 1.
Kinetics of Asparaginyl Hydroxylation in Cells
The finding that FIH-catalyzed hydroxylation is not subject to a reversal process, at least under the conditions of these experiments, raises an important question as to how the level of hydroxylation relates to the age of a protein species. For instance, it is unclear whether incomplete hydroxylation, which is observed at many sites in ARD proteins, represents ongoing hydroxylation during the lifetime of the protein species or reaches the observed level rapidly after synthesis. To analyze this, we treated cells with cycloheximide, without inhibiting FIH activity, and monitored the hydroxylation status of specific sites within Rabankyrin-5 by UPLC-MS for up to 10 h. At all Asn-hydroxylation sites that resolved chromatographically, the prevalence of the hydroxylated species increased gradually during the experiment, indicating that the extent of hydroxylation on a particular protein species does indeed progress over time. Data for Asn752 is shown in Fig. 4A and reveals that, at this site, hydroxylation increases essentially to completion over a 10-h period.
FIGURE 4.

Accrual of asparagine hydroxylation over time. A, extracted ion chromatograms of m/z 772.71 and 778.02 corresponding to unhydroxylated (dashed line) and hydroxylated (solid line) forms of the tryptic Rabankyrin-5 peptide encompassing Asn752; SGCDVNSPRQPGANGEGEEEAR ([M + 3H]3+). Nano-UPLC-MSE chromatography analysis demonstrating increase in the proportion of material that was hydroxylated over time in cells treated with cycloheximide (Chx) for 4 h (middle panel) and 10 h (lower panel). The signal intensity of reference peaks is indicated in parentheses on the chromatograms. B, panels i and ii, pulsed SILAC experiment demonstrating increase in the proportion of material that was hydroxylated at Asn485 and Asn797 in Rabankyrin-5 over an extended 48-h chase period in light media (Lys0/Arg0) following 24-h pulse labeling in media containing heavy isotopes (heavy media; Lys6/Arg6) (hydroxylation percentages shown for chase period only); nano-UPLC-MSE analysis. Two independent biological replicates are shown, and data presented are from one representative experiment.
To monitor changes in Asn hydroxylation over a longer period of time, at sites manifesting lower levels of hydroxylation, a SILAC pulse-chase methodology was employed. Stable transfectants expressing FLAG-tagged Rabankyrin-5 were pulsed with heavy SILAC medium for 24 h before switching to light medium for 48 h. The relative abundance of hydroxylated and unhydroxylated peptides in the heavy labeled protein was monitored and used to calculate changes in the extent of hydroxylation over the time course of the chase. Data for two sites at which hydroxylation was readily quantified (Asn485 and Asn797) are summarized in Fig. 4B. This revealed that time-dependent accrual of hydroxylation continued over the course of the experiment but did not always run to completion, reaching 40 and 65% at Asn485 and Asn797, respectively, after 48 h. Interestingly, the rate at which hydroxylation accrued at both sites was not linear over the course of the experiment and plateaued over time.
Effect of Asparagine Hydroxylation on Protein Half-life
The accrual of ARD hydroxylation over time suggests that it could potentially regulate age-dependent properties such as degradation. To address this, we measured the effects of FIH-dependent hydroxylation on the half-life of Rabankyrin-5. In the first instance, we monitored the degradation of endogenous Rabankyrin-5 following siRNA-mediated suppression by cycloheximide chase. Monitoring Rabankyrin-5 degradation during an 8-h cycloheximide chase did not reveal a major effect of hydroxylation on the stability of this ARD substrate, although the half-life of endogenous Rabankyrin-5 was clearly greater than the 8-h cycloheximide treatment (data not shown). Given the relatively long half-life of Rabankyrin-5, we sought a more sensitive assay that was not confounded by cycloheximide-associated toxicity. To test for effects of hydroxylation on ARD protein degradation over a longer period of time, stable transfectants expressing FLAG-tagged Rabankyrin-5 were transfected with FIH-directed siRNA or control siRNA at 24-h intervals. 24 h after the second siRNA transfection, cells were pulsed with heavy SILAC media for 18 h. Cells were subsequently chased in light media, and material was analyzed by HPLC-MS at 12-h intervals for 48 h. Decay of heavy labeled Rabankyrin-5 during the chase period was calculated from measurements of the sum of the abundance of all Rabankyrin-5-derived peptides that do not contain sites of hydroxylation (Fig. 5A). Knockdown of FIH was confirmed by immunoblotting (Fig. 5C) and demonstrated by MS to result in near complete suppression of hydroxylation of Rabankyrin-5 (Fig. 5B). Closely similar half-life values for Rabankyrin-5, in cells treated with FIH-directed and control siRNA, indicates that FIH-mediated hydroxylation has no discernible effect on the stability of Rabankyrin-5 under these conditions. Annotated spectra of heavy labeled hydroxylated and unhydroxylated peptides encompassing Rabankyrin-5 Asn485, along with chromatograms showing retention times on the Agilent platform, are shown in supplemental Figs. S7–S9.
FIGURE 5.

Hydroxylation has minimal effect on the stability of overexpressed Rabankyrin-5. A, Rabankyrin-5 half-life was similar between cells treated with control siRNA (black triangle) or siRNA specific to FIH (gray circle) as demonstrated by a similar loss of heavy label signal during 48-h chase period in light media (Lys0/Arg0). B, confirmation that siRNA-mediated knockdown of FIH suppressed the hydroxylation of heavy labeled Rabankyrin-5 at Asn485 at the start (t = 0) and end (t = 48 h) of the chase period. Note, the apparent increase in heavy hydroxylation observed 48 h after label washout was noted previously (Fig. 4B); it most likely reflects the accrual of hydroxylation as the average age of the heavy labeled material increases. Raw data (extracted ion chromatograms for Asn485 peptides) are presented in supplemental Fig. S27. C, immunoblot demonstrating siRNA-mediated knockdown of FIH at the protein level through the duration of the chase period.
Regulation of ARD Hydroxylation
FIH is a member of the 2-oxoglutarate dependent dioxygenase family of enzymes, whose activity is dependent on iron, ascorbate and 2-OG as co-factors and co-substrates, as well as on availability of molecular oxygen. Although in vitro studies have confirmed these co-factor/co-substrate requirements for FIH, and have defined the kinetic properties of recombinant FIH with respect to these molecules (34, 35), it is difficult to predict responses in cells from these data. Given that we were unable to detect any reversal of asparaginyl hydroxylation under the conditions of these experiments, we argued that an analysis of changes in the level of hydroxylation at sites of FIH-catalyzed modification should directly reflect modulation of FIH activity in cells.
To determine whether availability of iron and/or ascorbate were limiting for FIH-catalyzed hydroxylation in cells cultured under standard conditions, we wished to define the effect of co-factor supplementation on sites of hydroxylation within Rabankyrin-5 that were incompletely hydroxylated. To avoid confounding effects from signals arising from pre-existing protein, which was present in cells prior to the experiment, we used SILAC methodology to confine the MS analysis to protein species whose expression was concurrent with that of the test condition. Accordingly, cells stably expressing FLAG-tagged Rabankyrin-5 were cultured in “heavy” SILAC medium supplemented with varying concentrations of l-ascorbate (50 to 1000 μm), iron (20 to 320 μm), or a combination of both (20 μm iron + 100 μm l-ascorbate) for 24 h. Following anti-FLAG immunoprecipitation and trypsinolysis, “heavy” labeled peptides bearing sites of FIH-catalyzed asparaginyl hydroxylation were analyzed by UPLC-MS. Results shown in Fig. 6 reveal that addition of 50 μm l-ascorbate substantially increased hydroxylation. No further increase in hydroxylation was observed at higher concentrations of ascorbate (data not shown). In contrast, no consistent effect of iron supplementation was observed, either alone or in combination with l-ascorbate (data not shown). Similarly, addition of exogenous cell-permeable 2-OG had a negligible effect on the hydroxylation of Rabankyrin-5, indicating that intracellular 2-OG levels are not limiting for FIH under standard culture conditions (data not shown). Representative data for the effect of 50 μm l-ascorbate or 40 μm iron for 24 h are presented in Fig. 6. Hydroxylation at all sites in the ARD of Rabankyrin-5 that could be robustly quantified by MS increased in response to ascorbate supplementation (Asn316, Asn485, Asn649, and Asn797), with the sites manifesting the lowest levels of hydroxylation showing the largest proportional increase in hydroxylation. Annotated spectra of heavy labeled hydroxylated and unhydroxylated Rabankyrin-5 Asn316, Asn485, Asn649, and Asn797 peptides, along with chromatograms showing retention times on the Agilent platform are shown in supplemental Fig. S4–S15.
FIGURE 6.
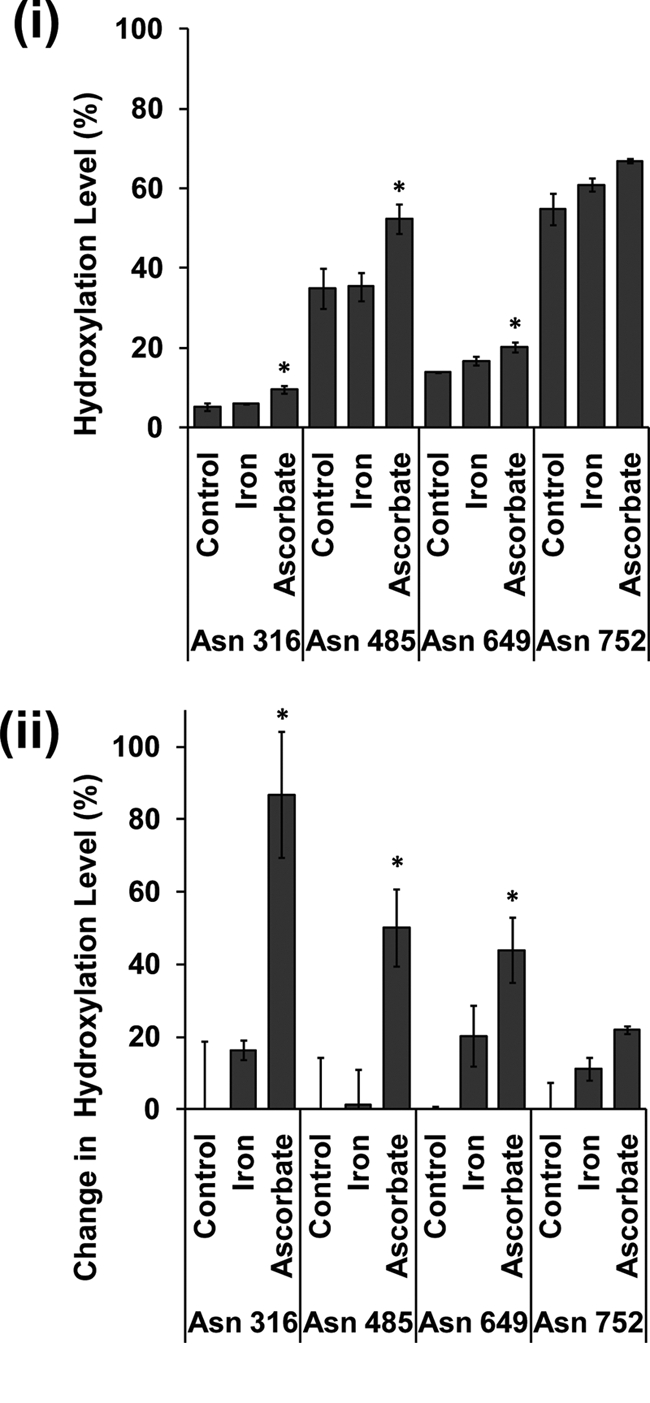
Effect of ferrous iron and l-ascorbate supplementation on FIH-dependent hydroxylation of Rabankyrin-5. Heavy SILAC media were added to cells stably expressing FLAG-tagged Rabankyrin-5 at the time of iron (40 μm) and ascorbate (50 μm) supplementation. Heavy labeled peptides encompassing known hydroxylation sites were quantified by LC-MS/MS. Data presented are from three independent biological replicates (mean ± S.E.); peptide loss was observed for Asn316, Asn649, Asn752 (n = 2). i shows the level of hydroxylation; ii, illustrates the change in the level of hydroxylation at named sites following co-factor addition. This value is derived from ((level in test condition − level in control condition)/level in control condition) × 100. *, p < 0.05 compared with control by one-way ANOVA with post hoc Dunnett's test (n = ≥2).
In vitro experiments have revealed kinetic differences between FIH-catalyzed hydroxylation of certain ARD proteins and the HIF1-CAD. In particular, the Km values for ARD polypeptide substrates have been reported to be much lower than those reported for the HIF1-CAD and a much lower apparent Km value for oxygen has been reported for FIH-catalyzed hydroxylation of Notch1 ARD as opposed to FIH-catalyzed hydroxylation of the HIF1-CAD (17). To determine whether these sites differ in their sensitivity to hypoxia in cells, we constructed further stable transfectants expressing mouse Notch1 (mN1) ARD fused to the same CAD polypeptide as the Rabankyrin-5/HIF1-CAD fusion protein. We then compared the effects of hypoxia on different sites of hydroxylation in the two ARD proteins with the effects of hypoxia on asparagine hydroxylation of the HIF1-CAD in each context. As before, SILAC methodology was used to confine the analysis of protein hydroxylation to specific conditions of oxygenation, with heavy SILAC medium being added to cells at the start of the 24-h hypoxic exposure.
Surprisingly, given the in vitro data suggesting that ARD proteins may be better substrates for FIH than the HIF1-CAD, in material derived from cells expressing the Rabankyrin-5/HIF1-CAD fusion protein, the HIF1-CAD was more strongly hydroxylated than sites within the Rabankyrin-5 ARD. Interestingly, analysis in hypoxia revealed a significant difference in the response of the ARD and HIF1-CAD sites to reduced oxygen. While hydroxylation of each of the three sites in Rabankyrin-5 was clearly inhibited at 1% oxygen, hydroxylation of the HIF1-CAD was largely unaffected and only shows a 25% reduction in more severe (0.2%) hypoxia (Fig. 7A). To provide a more direct comparison with substrates that have been compared in vitro by ourselves and by others (6, 7), we next analyzed material from transfectants expressing the mNotch1/HIF1-CAD fusion protein. As before, heavy SILAC medium was added to the cells at the start of the 24-h hypoxic exposure, enabling comparative analysis of the effects of hypoxia on sites of hydroxylation within the fusion protein. In keeping with previous data indicating that mNotch-1 Asn1945 is a very good substrate for FIH-catalyzed hydroxylation, under oxygenated conditions this site was essentially fully (>98%) hydroxylated, like that in the HIF1-CAD, permitting comparison of the effect of hypoxia on two sites that were similarly hydroxylated in normoxic cells (Fig. 7B). Again, hydroxylation of the HIF1-CAD was barely suppressed by 1% oxygen but more severely suppressed by 0.2% oxygen. In contrast with Rabankyrin-5, the response of the ARD in mNotch1 was broadly similar to that of the HIF1-CAD, although a somewhat greater reduction in hydroxylation was observed at 0.2% oxygen than with the HIF1-CAD. Taken together, these results indicate that, in cells, ARD hydroxylation is not intrinsically less sensitive to hypoxia than hydroxylation of the HIF1-CAD. Rather, sensitivity to reduction of hydroxylation in hypoxia appears to be related to the level of hydroxylation under basal conditions, with less completely hydroxylated sites being more sensitive.
FIGURE 7.

ARD and CAD hydroxylation are inhibited by hypoxia. Heavy SILAC media were added to cells stably expressing FLAG-tagged Rabankyrin-5/HIF1-CAD (A) and FLAG-tagged NOTCH (mN1ARD)/HIF1-CAD (B) fusion proteins for 24 h at the indicated hypoxic exposure. Heavy labeled peptides encompassing known hydroxylation sites were quantified by LC-MS/MS. Data presented are from three independent biological replicates (mean ± S.E.); peptide loss was observed for Asn316 and Asn649 (n = 2). i shows the level of hydroxylation; ii, illustrates the change in the level of hydroxylation at named sites following hypoxic exposure. This value is derived from ((level in test condition − level in control condition)/level in control condition) × 100. *, p < 0.05 compared with normoxic control by one-way ANOVA with post hoc Dunnett's test (n = ≥2). **, p < 0.01 compared with normoxic control by one-way ANOVA with post hoc Dunnett's test (n = ≥2).
DISCUSSION
The catalytic properties of FIH have been extensively studied in vitro using purified recombinant enzyme and polypeptide substrates representing the sequences of the HIF1-CAD and ARD-containing proteins (4–6, 17). Although these studies have suggested large differences in the kinetics of hydroxylation on these two major classes of FIH substrate, comparative studies have not been performed in vivo, in part because of difficulties in monitoring levels of protein hydroxylation within cells. To address this, we have applied SILAC technology to monitor hydroxylation at specific sites in a series of transfectants stably expressing a range of HIF1-CAD and ARD polypeptides at levels sufficient for analysis by MS. Importantly, the pulsed SILAC approach enabled the study of both accrual and decay of hydroxylation under a defined set of experimental conditions.
Measurements of the decay of hydroxylated species following the catalytic inhibition of FIH demonstrated near identical decay of hydroxylation and hydroxylated protein species for both HIF1-CAD and ARD proteins, revealing that hydroxylation of these proteins is not significantly reversed, at least under the experimental conditions examined. Together with evidence from this and other studies that the catalysis of asparaginyl hydroxylation at these sites is entirely dependent on FIH (i.e. the enzyme is nonredundant) (8, 34), this indicates that the levels of hydroxylation on proteins examined under specific conditions should be an accurate reflection of the activity of FIH.
Using SILAC to focus the analysis of hydroxylation on proteins that were synthesized under particular conditions of oxygen availability, we demonstrated that hydroxylation at sites within ARD proteins was inhibited by hypoxia in a site-dependent manner. Rather than ARD protein hydroxylation being universally more resistant to hypoxia than the HIF1-CAD, as might be predicted from in vitro studies demonstrating much higher affinity of ARD proteins for FIH and higher rates of FIH-catalyzed hydroxylation (6, 17), we observed that particular sites of ARD protein hydroxylation were more sensitive to hypoxia than others and that most ARD protein sites were more sensitive to hypoxia than the HIF1-CAD. Interestingly, we also found that when HIF1-CAD polypeptide sequences were fused to ARD polypeptides to present both hydroxylation sites at identical concentration in an identical context, the HIF1-CAD was preferentially hydroxylated. Thus, it appears that within cells, factors other than those derived from in vitro kinetic studies using recombinant proteins and peptides contribute to the regulation of asparaginyl hydroxylation at different sites.
Further SILAC-based analyses conducted under conditions of varying co-factor availability revealed that FIH-catalyzed hydroxylation was enhanced by supplementation of the tissue culture medium with ascorbate but not 2-OG or iron. The studies therefore confirm in vitro data indicating that, like many 2-OG-dependent dioxygenases, FIH is an ascorbate-requiring enzyme (35). However, consistent with the observation that asparaginyl hydroxylation within the HIF1-CAD is relatively insensitive to inhibition by iron chelators (32), they suggest that FIH activity is relatively resistant to variation in cellular iron availability.
Consistent with the absence of a reversal process for asparaginyl hydroxylation, when SILAC labeling was used to monitor the hydroxylation of previously synthesized proteins, we found that hydroxylation accrued over a considerable period of time. Nevertheless, even in fully oxygenated cell cultures supplemented with FIH co-factors, hydroxylation did not proceed to completion and appeared to plateau at certain sites during the course of the 48-h SILAC chase. The reason for this unexpected observation is unclear. We have previously reported that hydroxylation can increase the thermodynamic stability of an ARD (37, 38). Because structural studies indicate that the ARD must unfold to allow interaction of the target asparagine with the FIH catalytic site (6), it is possible that hydroxylation at one site could constrain hydroxylation of another that is in close proximity. However, on at least some ARD proteins, the effect of hydroxylation on thermodynamic stability appears to be minimal suggesting there could be other explanations (37). For instance, because the ARD functions as a protein interaction domain (14), an alternative possibility is that other ARD-protein interactions restrict hydroxylation.
Given that hydroxylation of the prototypical ARD-protein Rabankyrin-5 accrued over time, potentially encoding age-dependent properties, we tested for FIH-dependent effects on the half-life of the protein. However, we did not observe any differences between half-life measured either by cycloheximide chase or SILAC. This result is consistent with a recent report on IκBα, another ARD protein with an intrinsically shorter half-life that was unaltered by hydroxylation (39). Whether asparaginyl hydroxylation could have effects on protein half-life on much longer lived proteins is unclear. Interestingly, deamidation of certain asparaginyl residues to aspartate residues has been proposed to function as a biological clock over periods of days to weeks (40). Hydroxylation of asparaginyl residues may be predicted to affect either this process or the activity of the repair enzyme, protein-l-isoaspartate(d-aspartate) O-methyltransferase. However, deamidation rates are strongly dependent on the amino acid sequence proximal (+1) to the deamidation site, and residues predisposing to rapid deamidation are less common in ARD proteins (14, 40).
To date, the function of ARD protein hydroxylation is unclear, although hydroxylation of the HIF1-CAD has a clear role in the regulation of the HIF transcriptional cascade. The existence of nonfunctional low stoichiometry unregulated post-translational modifications are predicted from proteome-wide studies, such as the recently described phosphoproteome (36). However, our studies reveal that ARD hydroxylation is strongly regulated by oxygen levels in a manner that is similar to HIF-1α and occurs at high stoichiometry. We and others have previously proposed that competition between ARD and HIF hydroxylation could result in effective sequestration of FIH by the unhydroxylated ARD protein pool and thus modulate the oxygen-sensing function of FIH (6, 15, 16). Several predictions can be made as to the biological impact of ARD cross-competition on the HIF transcriptional response. Mathematical models predict that substrate competition by the ARD pool serves to focus HIF-CAD hydroxylation into a narrower range of oxygen tensions and to sharpen the signal/response curve at the oxygen threshold (16). In support of this, in a separate study we observed that the sensitivity of FIH to graded hypoxia in cells was sharper than for the related prolyl hydroxylase enzymes that also modify HIF (32). The hydroxylation status of the ARD pool also has the potential to encode the strength and duration of a hypoxic insult, which would be predicted to impinge upon the HIF signaling cascade. In this scenario, the long lived ARD-containing proteins would accumulate in an unhydroxylated state and would be predicted to act as a sink for FIH. This could manifest as a delay in CAD hydroxylation upon re-oxygenation that is related to the severity and duration of the preceding hypoxic episode (15). The current data revealing that ARD protein hydroxylation shows variable, site-specific oxygen dependence within the same range as the HIF1-CAD is compatible with the operation of these processes.
More complete and more quantitative analysis of post-translational modifications using SILAC and other MS methodologies should reveal whether this aspect of signaling through asparaginyl hydroxylation is unusual or reflects the common existence of large pools of competitive substrates for enzymes catalyzing post-translational modifications.
Supplementary Material
Acknowledgments
We acknowledge the Computational Biology Research Group, Medical Sciences Division, Oxford, United Kingdom, for use of their services in this project. We are grateful to David Mole and Rebecca Konietzny (University of Oxford) for helpful discussions and technical support.
This work was supported in part by the Biotechnology and Biological Sciences Research Council, the European Union (7th Framework), and the Wellcome Trust (to P. J. R.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S27.
- HIF
- hypoxia-inducible factor
- AMT
- accurate-mass and retention time
- ARD
- ankyrin repeat domain
- DMOG
- dimethyloxalylglycine
- FIH
- factor inhibiting HIF
- CAD
- C-terminal transactivation domain
- p300/CBP
- p300 and cAMP response element-binding protein
- PTH
- post-translational hydroxylation
- SILAC
- stable isotope labeling by amino acids in cell culture
- UPLC-MS
- ultraperformance liquid chromatography-mass spectrometry
- VHL
- von Hippel-Lindau
- ANOVA
- analysis of variance
- 2-OG
- 2-oxoglutarate.
REFERENCES
- 1. Hewitson K. S., McNeill L. A., Riordan M. V., Tian Y. M., Bullock A. N., Welford R. W., Elkins J. M., Oldham N. J., Bhattacharya S., Gleadle J. M., Ratcliffe P. J., Pugh C. W., Schofield C. J. (2002) J. Biol. Chem. 277, 26351–26355 [DOI] [PubMed] [Google Scholar]
- 2. Lando D., Peet D. J., Gorman J. J., Whelan D. A., Whitelaw M. L., Bruick R. K. (2002) Genes Dev. 16, 1466–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Mahon P. C., Hirota K., Semenza G. L. (2001) Genes Dev. 15, 2675–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ehrismann D., Flashman E., Genn D. N., Mathioudakis N., Hewitson K. S., Ratcliffe P. J., Schofield C. J. (2007) Biochem. J. 401, 227–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Koivunen P., Hirsilä M., Günzler V., Kivirikko K. I., Myllyharju J. (2004) J. Biol. Chem. 279, 9899–9904 [DOI] [PubMed] [Google Scholar]
- 6. Coleman M. L., McDonough M. A., Hewitson K. S., Coles C., Mecinovic J., Edelmann M., Cook K. M., Cockman M. E., Lancaster D. E., Kessler B. M., Oldham N. J., Ratcliffe P. J., Schofield C. J. (2007) J. Biol. Chem. 282, 24027–24038 [DOI] [PubMed] [Google Scholar]
- 7. Zheng X., Linke S., Dias J. M., Zheng X., Gradin K., Wallis T. P., Hamilton B. R., Gustafsson M., Ruas J. L., Wilkins S., Bilton R. L., Brismar K., Whitelaw M. L., Pereira T., Gorman J. J., Ericson J., Peet D. J., Lendahl U., Poellinger L. (2008) Proc. Natl. Acad. Sci. U.S.A. 105, 3368–3373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cockman M. E., Lancaster D. E., Stolze I. P., Hewitson K. S., McDonough M. A., Coleman M. L., Coles C. H., Yu X., Hay R. T., Ley S. C., Pugh C. W., Oldham N. J., Masson N., Schofield C. J., Ratcliffe P. J. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 14767–14772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ferguson J. E., 3rd, Wu Y., Smith K., Charles P., Powers K., Wang H., Patterson C. (2007) Mol. Cell. Biol. 27, 6407–6419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Webb J. D., Murányi A., Pugh C. W., Ratcliffe P. J., Coleman M. L. (2009) Biochem. J. 420, 327–333 [DOI] [PubMed] [Google Scholar]
- 11. Yang M., Chowdhury R., Ge W., Hamed R. B., McDonough M. A., Claridge T. D., Kessler B. M., Cockman M. E., Ratcliffe P. J., Schofield C. J. (2011) FEBS J. 278, 1086–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Cockman M. E., Webb J. D., Kramer H. B., Kessler B. M., Ratcliffe P. J. (2009) Mol. Cell. Proteomics 8, 535–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang M., Ge W., Chowdhury R., Claridge T. D., Kramer H. B., Schmierer B., McDonough M. A., Gong L., Kessler B. M., Ratcliffe P. J., Coleman M. L., Schofield C. J. (2011) J. Biol. Chem. 286, 7648–7660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Li J., Mahajan A., Tsai M. D. (2006) Biochemistry 45, 15168–15178 [DOI] [PubMed] [Google Scholar]
- 15. Cockman M. E., Webb J. D., Ratcliffe P. J. (2009) Ann. N.Y. Acad. Sci. 1177, 9–18 [DOI] [PubMed] [Google Scholar]
- 16. Schmierer B., Novák B., Schofield C. J. (2010) BMC Syst. Biol. 4, 139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wilkins S. E., Hyvärinen J., Chicher J., Gorman J. J., Peet D. J., Bilton R. L., Koivunen P. (2009) Int. J. Biochem. Cell Biol. 41, 1563–1571 [DOI] [PubMed] [Google Scholar]
- 18. Deleted in proof.
- 19. Stolze I. P., Tian Y. M., Appelhoff R. J., Turley H., Wykoff C. C., Gleadle J. M., Ratcliffe P. J. (2004) J. Biol. Chem. 279, 42719–42725 [DOI] [PubMed] [Google Scholar]
- 20. Lee S. H., Jeong Hee, Moon, Eun Ah, Cho, Ryu S. E., Myung Kyu Lee. (2008) J. Biomol. Screen. 13, 494–503 [DOI] [PubMed] [Google Scholar]
- 21. Bendall S. C., Hughes C., Stewart M. H., Doble B., Bhatia M., Lajoie G. A. (2008) Mol. Cell. Proteomics 7, 1587–1597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu D., Suenaga N., Edelmann M. J., Fridman R., Muschel R. J., Kessler B. M. (2008) Mol. Cell. Proteomics 7, 2215–2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pasa-Toliæ L., Masselon C., Barry R. C., Shen Y., Smith R. D. (2004) BioTechniques 37, 621–624 [DOI] [PubMed] [Google Scholar]
- 24. Trudgian D. C., Thomas B., McGowan S. J., Kessler B. M., Salek M., Acuto O. (2010) Bioinformatics 26, 1131–1132 [DOI] [PubMed] [Google Scholar]
- 25. Keller A., Eng J., Zhang N., Li X. J., Aebersold R. (2005) Mol. Syst. Biol. 1, 2005.0017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. MacLean B., Eng J. K., Beavis R. C., McIntosh M. (2006) Bioinformatics 22, 2830–2832 [DOI] [PubMed] [Google Scholar]
- 27. Geer L. Y., Markey S. P., Kowalak J. A., Wagner L., Xu M., Maynard D. M., Yang X., Shi W., Bryant S. H. (2004) J. Proteome Res. 3, 958–964 [DOI] [PubMed] [Google Scholar]
- 28. Keller A., Nesvizhskii A. I., Kolker E., Aebersold R. (2002) Anal. Chem. 74, 5383–5392 [DOI] [PubMed] [Google Scholar]
- 29. Deutsch E. W., Shteynberg D., Lam H., Sun Z., Eng J. K., Carapito C., von Haller P. D., Tasman N., Mendoza L., Farrah T., Aebersold R. (2010) Proteomics 10, 1190–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nesvizhskii A. I., Keller A., Kolker E., Aebersold R. (2003) Anal. Chem. 75, 4646–4658 [DOI] [PubMed] [Google Scholar]
- 31. Elias J. E., Gygi S. P. (2007) Nat. Methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 32. Tian Y. M., Yeoh K. K., Lee M. K., Eriksson T., Kessler B. M., Kramer H. B., Edelmann M. J., Willam C., Pugh C. W., Schofield C. J., Ratcliffe P. J. (2011) J. Biol. Chem. 286, 13041–13051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maxwell P. H., Wiesener M. S., Chang G. W., Clifford S. C., Vaux E. C., Cockman M. E., Wykoff C. C., Pugh C. W., Maher E. R., Ratcliffe P. J. (1999) Nature 399, 271–275 [DOI] [PubMed] [Google Scholar]
- 34. Zhang N., Fu Z., Linke S., Chicher J., Gorman J. J., Visk D., Haddad G. G., Poellinger L., Peet D. J., Powell F., Johnson R. S. (2010) Cell Metab. 11, 364–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Flashman E., Davies S. L., Yeoh K. K., Schofield C. J. (2010) Biochem. J. 427, 135–142 [DOI] [PubMed] [Google Scholar]
- 36. Olsen J. V., Blagoev B., Gnad F., Macek B., Kumar C., Mortensen P., Mann M. (2006) Cell 127, 635–648 [DOI] [PubMed] [Google Scholar]
- 37. Hardy A. P., Prokes I., Kelly L., Campbell I. D., Schofield C. J. (2009) J. Mol. Biol. 392, 994–1006 [DOI] [PubMed] [Google Scholar]
- 38. Kelly L., McDonough M. A., Coleman M. L., Ratcliffe P. J., Schofield C. J. (2009) Mol. Biosyst. 5, 52–58 [DOI] [PubMed] [Google Scholar]
- 39. Devries I. L., Hampton-Smith R. J., Mulvihill M. M., Alverdi V., Peet D. J., Komives E. A. (2010) FEBS Lett. 584, 4725–4730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Robinson N. E., Robinson A. B. (2004) Molecular Clocks: Deamidation of Asparaginyl and Glutaminyl Residues in Peptides and Proteins, pp. 1–419, Althouse Press, Cave Junction, OR [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.