

Abstract
Shewanella oneidensis strain MR-1 utilizes soluble and insoluble ferric ions as terminal electron acceptors during anaerobic respiration. The components of respiratory metabolism are localized in the membrane fractions which include the outer membrane and cytoplasmic membrane. Many of the biological components that interact with the various iron forms are proposed to be localized in these membrane fractions. To identify the iron-binding proteins acting either as an iron transporter or as a terminal iron reductase, we used metal-catalyzed oxidation reactions. This system catalyzed the oxidation of amino acids in close proximity to the iron binding site. The carbonyl groups formed from this oxidation can then be labeled with fluoresceinamine (FLNH2). The peptide harboring the FLNH2 can then be proteolytically digested, purified by HPLC and then identified by MALDI-TOF tandem MS. A predominant peptide was identified to be part of SO2907 that encodes a putative TonB-dependent receptor. Compared with wild type (wt), the so2907 gene deletion (ΔSO2907) mutant has impaired ability to reduce soluble Fe(III), but retains the same ability to respire oxygen or fumarate as the wt. The ΔSO2907 mutant was also impacted in reduction of insoluble iron. Iron binding assays using isothermal titration calorimetry and fluorescence tryptophan quenching demonstrated that a truncated form of heterologous-expressed SO2907 that contains the Fe(III) binding site, is capable of binding soluble Fe(III) forms with Kd of approximate 50 μm. To the best of our knowledge, this is the first report of the physiological role of SO2907 in Fe(III) reduction by MR-1.
Keywords: Iron Metabolism, Oxidation-Reduction, Oxygen Radicals, Reactive Oxygen Species (ROS), Receptors, Protein Oxidation, Iron Chelator, Metal-catalyzed Oxidation
Introduction
Iron is the fourth most-abundant element in the Earth's crust. The redox transition between the Fe2+ and Fe3+ is an important biogeochemical process and is believed to play an important role in environmental biogeochemistry (1). Microorganisms from the Archaea and Bacteria domains which persist in water, soils and sediments are capable of metabolically exploiting the redox potential between Fe2+/Fe3+, either using Fe2+ as an electron donor or utilizing Fe3+ as a terminal electron acceptor (TEA)2 (2). The use of iron as the TEA is referred to as dissimilatory iron reduction (DIR). It has been suggested that microbial DIR predates the aerobic respiration (3). Interest in DIR has intensified upon discovery of the ability of DIR microbes in bioremediation of contaminated environments polluted with heavy and radioactive metals such as U6+ and Tc7+ (4, 5).
A model organism extensively studied for DIR is Shewanella, which is a diverse genus of facultative anaerobic γ-proteobacteria (6). Shewanella is capable of respiring a multitude of organic, inorganic, soluble, and insoluble TEAs (7, 8). For the insoluble TEAs, Shewanella has evolved mechanisms for the cytoplasmic membrane (CM)-localized electrons of respiration to traverse the periplasm and the outer membrane (OM) (9). Two studies (10, 11) have proposed that flavins serve as electron shuttling agents between Shewanella whole cells and the distal metal oxides. Flavins, however, do not seem to play as important of a role in the reduction of soluble metallic TEAs (11).
Whereas data suggest that most environmental metallic TEAs are insoluble, there are studies that suggest that Shewanella has also evolved a system for utilizing soluble metallic TEAs. Kinetic studies by Ruebush et al. (12) show that reduction of soluble TEAs occurs preferentially at the CM/periplasm. Work from Lies et al. (13) also suggested that reduction of soluble TEAs occurs mainly in the periplasm and CM. The recent results from Gescher et al. (14) are consistent with the importance of CM/periplasm-localized reduction of soluble iron forms. These workers found that E. coli transformed and expressing a CM-localized protein involved in DIR in Shewanella, cymA, is able to utilize nitrilotriacetate-chelated ferric ion (NTA-Fe3+) as a TEA. Furthermore, Shewanella has been suggested to produce endogenous Fe3+ chelators when grown with insoluble iron (15). Also, soluble organic-Fe3+ complexes have been detected using voltammetric techniques (15). Jones et al. (16) recently identified two mutants of S. oneidensis that are not able to produce soluble organic-Fe3+ complexes. These mutants were found to be deficient in anaerobic growth in both soluble and insoluble Fe3+. They suggested that the solubilization of iron oxide by endogenous iron chelators plays an important role in DIR of insoluble metal oxides.
If indeed soluble iron reduction occurs predominantly in the periplasm, the question remains on the route by which soluble metal complex is able to gain access to the periplasm. To identify the components involved in iron transport for the DIR process, we have used metal-catalyzed oxidation (MCO) of proteins. MCO has been used as a powerful tool to identify the amino acids involved in metal binding to proteins (17–20). The oxidants generated by MCO are highly reactive, having short half lives (21). As such, they are not able to diffuse far and therefore only the amino acids within close proximity to the metal binding site are oxidized (21, 22). The most well-documented type of oxidative modification is the formation of carbonyl groups from MCO (22). The detection of protein carbonyl groups therefore can be used to probe the location of the metal binding sites. The carbonyls can be detected by formation of Schiff bases with chromophores or fluorophores (19).
In this work we used fluoresceinamine to label carbonyl groups which are introduced into proteins as a result of MCO of total membrane (TM) fractions of S. oneidensis MR-1. The fluoresceinamine-labeled peptide was identified through tandem MS/MS. We found that Tyr-800 of a putative TonB-dependent receptor protein, which was encoded by SO2097, was oxidized. Compared with wild type (wt), the so2097 gene deletion mutant exhibited compromised growth rate in media containing soluble iron as the TEA. However, it maintained similar growth rates to that of wt when fumarate or oxygen was used as the TEA. We also expressed a truncated form of the TonB receptor and quantified its iron-binding capacity. Collectively, all these results suggest for the first time that so2097 is involved in reduction of soluble Fe3+ by MR-1, most likely via transporting soluble Fe3+ into the periplasm.
EXPERIMENTAL PROCEDURES
Materials
Ascorbate was purchased from Sigma. Fluoresceinamine was from Fluka, Sigma Aldrich. Sodium cyanoborohydride was from Acros Organics (Morris Plains, NJ). Trypsin Gold, mass spectrometry grade, was purchased from Promega (Madison, WI).
Bacteria Growth
S. oneidensis MR-1 (ATCC 700550) was grown by the method of Ruebush (12). Iron-grown cultures were harvested anaerobically when the aqueous Fe2+ concentration reached 30 mm as measured with ferrozine (23).
Iron-catalyzed Oxidation of Proteins
TM fractions were isolated according to the method of Ruebush (12) except all steps were performed anaerobically with all solutions purged with argon. Purified TM fractions (1 mg of protein) were incubated in the oxygen-saturated-oxidation buffer (100 mm HEPES, pH 7.5, and 1 mm ascorbate) in the absence or presence of 100 μm Fe3+ in a final volume of 100 μl for 1 h at 37 °C. Reactions were terminated by addition of 1 mm EDTA. The oxidized TM proteins were subsequently treated with 1% SDS (w/v) and heated to 100 °C for 1 min. Then 6 μl of 0.25 m fluoresceinamine (FLNH2, dissolved in 0.52 m NaOH) and 5 μl of freshly-prepared 0.4 m NaCNBH3 in 100 mm HEPES, pH 7.5, were added, and the volume was adjusted to 150 μl by the addition of 100 mm HEPES buffer, pH 7.5. After incubation at 37 °C for 1 h, the protein was precipitated with 1 ml of 30% (w/v) trichloroacetic acid (TCA), and was allowed to sit for 1 h in -20 °C and then centrifuged at 14,000 × g for 10 min. The supernatant was discarded, and the residual reagents were removed by washing the pellet three times with 1 ml of 10 mm HCl in ethanol: ethyl acetate (1:1). The precipitate was dissolved in 100 μl 0.1 m NaOH by incubating for 15 min at 37 °C. Protein content was determined by the method of Bradford (24).
Protein Carbonyl Detection
Aliquots (10 μl) from the TM oxidation reaction mixture (∼100 μg of protein) were incubated with 5 μl of 10% SDS and 40 μl of 10 mm 2,4-dinitrophenyl hydrazine (DNPH) (prepared in 2 N HCl). After incubation for 30 min at room temperature, 50 μl of 100% TCA was added to precipitate the protein. The mixture was centrifuged at 15,000 × g for 20 min. The pellet was washed three times with ice-cold 10 mm HCl in ethanol: ethyl acetate (1:1). The pellet was redissolved in the rehydration buffer and then analyzed by Western blotting.
Proteins were separated by SDS-PAGE (12%) and transferred to a nitrocellulose membrane (Whatman, Protran BA 83). After blocking with 5% BSA (w/v) in 10 mm Tris-Cl, 0.15 m NaCl (Tris-buffered saline) containing 0.05% (w/v) Tween 20 (TBST) for 30 min at room temperature, the membrane was incubated overnight at 4 °C with mouse anti-dinitrophenyl (DNP) monoclonal-antibodies (Sigma-Aldrich, 1:10,000). After washing three times with TBST, antigen-antibodies complexes were visualized with alkaline-conjugated anti-mouse IgG and AP substrate (ImmobilonTM Western, Millipore).
Proteolytic Digestion
The pH of the protein solution (in 0.1 m NaOH) was adjusted to 8.0 by adding aliquots of 1.5 m Tris-Cl, pH 8.0, and was vacuum dried in a SpeedVac concentrator (Salvent) under low heat. The pellet was re-dissolved in the 6 m urea, 100 mm Tris-Cl, pH 8.0 to yield a final protein concentration of 10 μg/μl. The protein mixture was first reduced with 0.25 mm dithiothreitol for 1 h at room temperature. Then 10 mm iodoacetamide was added and the mixture was incubated for 1 h at room temperature. Another 0.25 mm dithiothreitol was added to consume any unreacted iodoacetamide. The concentration of urea was reduced to ∼0.6 m by diluting the reaction mixture with 775 μl of water. To this solution was added 20 μg of trypsin and the digestion was carried out overnight at 37 °C. The reaction was stopped by adding concentrated acetic acid to adjust the pH below 6. The sample was dried twice after addition of 100 μl of de-ionized H2O to the dried pellet.
HPLC Separation of Digested Peptides
Peptides from the digested protein were separated on a C-18 reverse phase column (particle size 3.5 μm, 4.6 × 150 mm) from Waters (Waters Corp.) using a Shimazu HPLC. The linear gradient was 0.5% B (v/v) to 100% B in 60 min at a flow rate of 0.5 ml/min (A: 0.3% (v/v) formic acid in distilled deionized water, B: 0.3% formic acid in 100% acetonitrile). The HPLC was monitored at 220 nm and 440 nm using a UV diode-array detector. Peak fractions were collected using the auto sample collector, pooled as indicated, and dried in a speed vac concentrator. Dried samples were redissolved in 6 m guanidine-HCl, 20 mm phosphate buffer, pH 6.5. The peptide was further purified by reverse phase HPLC using a gradient of 10 to 80% acetonitrile (in water) over 60 min at 0.5 ml/min. Fractions with absorption at both 220 nm and 440 nm were collected, pooled, and dried as earlier described. The sample was dried in a speed vac concentrator and stored at −80 °C.
MALDI-TOF MS/MS Analysis
Purified peptide was directly deposited onto MALDI plate and overlaid with 0.5 μl of 5 mg/ml α-cyano-4-hydroxycinnamic acid matrix solution in 1:1 acetonitrile:H2O with 0.1% (v/v) trifluoroacetic acid. Peptide mass spectra were obtained with a MALDI-TOF/TOF mass spectrometer (5800 Proteomic Analyzer, Applied Biosystems, Europe) in the positive ion reflector mode. Spectra were obtained in the mass range between 800 and 4,000 Da with ABScier laser set at 3280. Spectra were processed and analyzed by the Global Protein Server Work station (Applied Biosystems), which uses internal Mascot (Matrix Science) software for searching peptide mass fingerprints and MS/MS data.
Mutant Generation
The procedure for making a SO2907 deletion mutant (ΔSO2907) was similar to that described previously (25). Briefly, upstream and downstream regions flanking SO2907 were PCR amplified with primers: SO2907-5O: 5′-TATATTCCCAAAGAACATGTACC; SO2907-5I: 5′-CGTTAGCTGCAGACCTAGGAACATATCATTCTCCCTTAACACTTG; SO2907-3O; 5′-TTCCTAGGTCTGCAGCTAACGTTTCCATCAGGGAAGATAGATAAC; SO2907-3I; 5′-GGTTCGCCCACTGAGCCAAGTG.
The resulting PCR products were fused by overlap extension PCR. The fused PCR product was cloned into a suicide vector. The resulting construct (pLS197) was introduced into MR-1 cells through conjugation. Following selections, the ΔSO2907 mutant (LS528) was validated by PCR with primers: SO2907-FO: 5′-ACCAAAAAGCTTTCTGAGTAACTTTC; SO2907-RO: 5′-CTATACTCTTAGTGGGTTGTGCC.
Growth Curve Determination
Both ΔSO2907 and wt of S. oneidensis MR-1 were maintained on a Luria-Broth (LB) agar plate. A single colony was used to inoculate LB liquid medium, which was then grown at room temperature aerobically overnight. The culture was centrifuged at 10,000 × g for 2 min. The pellet was washed twice with HEPES-buffered saline (100 mm HEPES, pH 7.0 with 0.7% (w/v) sodium chloride) and was resuspended in 1/10 volume of the same solution. The absorbance at 600 nm was determined, and then the culture was used as an inoculant where 1 ml of a solution that measured 0.1 absorbance at 600 nm was added to 100 ml of defined argon-purged M1 minimum medium as described by Myers and Nealson (6). The TEAs used to determine the growth curve included 50 mm ferric-citrate, 25 mm fumarate, 10 mm goethite, or 10 mm ferrihydrite.
Acridine Orange Staining
For cell staining and counting, 0.2 ml culture was filtered through a 0.2 mm Whatman Nuclephore Track-Etch membrane filter. Dilution of sample in 10 mm HEPES buffer, pH 7.0 was made as needed. The sample was then stained with 1 ml of a 0.1% (w/v) aqueous solution of filtered acridine orange in 50 mm bicarbonate buffer, pH 9.2, and was filtered (0.2 mm Whatman Nuclephore Track-Etch membrane filter) after 1 min. The membrane was then mounted on a drop of immersion oil on a microscope slide. Another drop of immersion oil was placed on top of the membrane before sealing with a coverslip. The slide was viewed immediately under 100× oil objective lens using the Zeiss Axioimager fluorescent microscopy. The numbers of bacteria per milliliter were estimated from a count of at least three random chosen microscope field circumscribed by an eyepiece micrometer. A total of 50–100 bacteria per field were counted.
Cloning and Expressing TonB-A and TonB-B Protein
We first attempted to express the whole SO2907 protein but were not able to obtain properly folded protein. We then expressed a truncated form of the protein. DAS software (27) was used to predict the transmembrane domains. Two portions predicted to be localized in the extracellular region were cloned using primers summarized in Table 1. TonB-A does not contain the peptide identified earlier while TonB-B does contains the putative iron-binding peptide. The protein sequence TonB-A and TonB-B are shown in supplemental Fig. S1. TonB-A and TonB-B genes were cloned and ligated into the BamHI and XhoI sites of pET21a (Novagen, USA). Both TonB-B and TonB-A protein were expressed in Escherichia coli BL21(DE3). Expression was induced by adding 0.1 mm isopropyl-1-thio-β-d-galactopyranoside when the optical density reaches ∼0.6 at 600 nm. Purification of both proteins followed the procedure as previously described (28) except that 0.5% N-lauroylsarcosine was added to all solutions. Protein was dialyzed against 20 mm Tris-Cl, pH 8.0 with the buffer changed every 12 h for 2 days.
TABLE 1.
Primers used for PCR
| Forward primer (5′-3′) | Reverse primer (5′-3′) | |
|---|---|---|
| TonB-A | GCATCCGGATCCGCATTCGACGCAAAATTTGGT | ATCTTACTCGAGATAAGAGTCAGTTTCAATGTT |
| TonB-B | GCATCCGGATCCGGTGACTTAGGTTATGCTGTT | ATCTTACTCGAGGAATTTCACTTTGTATCCTGC |
| TonB-B (Y800F) | TTGATTGCTAAATTCATTGATTCCTAT | ATAGGAATCAATGAATTTAGCAATCAA |
| TonB-B (D803A) | GCTAAATACATTGCCTCCTATGATGGT | ACCATCATAGGAGGCAATGTATTTAGC |
Isothermal Titration Calorimetry and Data Analysis
Isothermal titration calorimetry (ITC) was used to measure iron binding affinity constants. The fundamental principle of ITC has been described elsewhere (29). The experiment was performed as previously described (28). Briefly, 20 μm TonB-A or TonB-B protein was titrated with 0.1 mm NTA -Fe3+ or ferric citrate which was prepared by the method of Wang et al. (45). Prior to all measurements, all solutions were degassed under vacuum with stirring. A series of at least 15 titrations was carried out before the heat release reached saturation.
A single set of independent binding sites model was employed to analyze the calorimetric data (30). We assume that the TonB-B protein (M) contains one type of binding sites for the ligand (L) with binding affinities (Ka) and stoichiometry (n). To fit the experimental data, the concentration of all species in equilibrium, free and bound, after each injection i, must be calculated from the total concentration of TonB-B protein ([MT]) and NTA-Fe3+ ([LT]) in the system. Based on the mass conservation, the free ligand concentration [L] can be described in Equation 1,
where p is the fractional saturation. The binding affinity Ka is given by Equation 2.
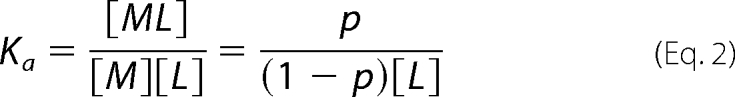 |
Combining Equations 1 and 2 gives the quadratic Equation 3.
Solving Equation 3 gives Equation 4.
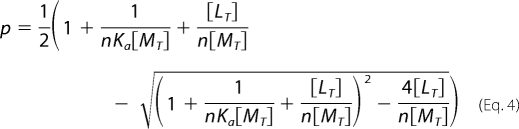 |
The integral heat of binding (Q) after the ith injection is given by Equation 5,
where V0 is the cell volume, ΔH is the enthalpy change of the binding. A nonlinear fit based on Equation 5 to the hyperbolic saturation curve in the integral mode (Q versus [LT]) yields the parameters Ka, and n from the experiment.
Quantification of Iron Binding by Fluorometry
Tryptophan fluorescences of TonB-A and TonB-B were measured in 1 ml of quartz cuvette at 25 °C with a SLM Aminco Fluorometer (SLM Instruments, Inc.). The excitation and emission wavelengths were 291 and 351 nm, respectively. Control experiments, consisting of a similar sample solution but containing only the buffer as the titrant, were subtracted from experimental data obtained from the TonB-A and TonB-B titrations. For ferric ion binding, spectra were obtained on a 20 μm TonB-A or TonB-B and ferric ion was titrated from a stock solution of NTA-Fe3+ over the concentration specified in the figure legends. The iron binding constant of each protein was calculated according to Winzor and Sawyer (31).
Site-directed Mutagenesis
Plasmid pET21a-tonB-B was used as the template for the QuikChange site-directed mutagenesis (Stratagene) with the procedures following manufacturer's instructions. Two mutations were made: Y800F and D803A. Forward and reverse primers to obtain desired point mutations are shown in Table 1. The sequence shown in bold represent bases introduced to obtain the desired mutation, which was subsequently confirmed by DNA sequencing.
RESULTS
Iron-catalyzed Oxidation of TM Proteins
We first utilized MCO to oxidize membrane-associated proteins isolated from anaerobically grown S. oneidensis MR-1 with iron as the TEA. TM fractions were incubated with the protein oxidation system of ascorbate and Fe3+ (1 mm and 0.1 mm, respectively). MCO-derived carbonyls were then reacted with either DNPH or FLNH2. The oxidized proteins from the TM were separated on SDS-PAGE and then visualized by Western blot analysis using an antibody to DNPH. As shown in Fig. 1, DNPH is detected in the MCO-treated samples, and none were detected from control TM samples (without oxidation treatment) (Fig. 1).
FIGURE 1.

Western blot of DNPH-labeled oxidized membrane proteins. Proteins were oxidized by the MCO and the protein carbonyls were labeled with DNPH. After 12% SDS-PAGE, the DNPH-derivatized protein carbonyls of the oxidized proteins (10 μg) were visualized by Western blotting. DNPH was visualized by using mouse monoclonal antibodies to DNP. The control sample was not treated with the MCO system.
In vitro oxidation of TM proteins was further examined by FLNH2-labeling of the carbonyl groups (Fig. 2). After MCO, TM proteins were labeled with FLNH2 and then precipitated with 30% TCA. The protein pellet was then washed with ethanol/ethyl acetate to remove the un-reacted FLNH2. Because FLNH2 absorbs light at 440 nm, the ratio 440 nm to 280 nm absorbance is an indicator of the amount of FLNH2 incorporated into the carbonyl groups. The 440 nm/280 nm ratio increased at 30 min, and reached maximum after 2 h. We choose 2 h incubation time for all subsequent work.
FIGURE 2.

FLNH2 labeling of iron-catalyzed oxidized membrane proteins. The TM (1 mg of protein) was incubated with 0.1 mm Fe3+, and 1 mm ascorbate in 100 mm HEPES, pH 7.5 aerobically. Samples were removed at specified times and reacted with FLNH2 as described under “Experimental Procedures.” The control sample is the TM fraction without treatment from the MCO system. The A440/A280 ratio reflects the extent of carbonyl formation. Experiments were performed in triplicates. Data were represented as mean ± S.D.
HPLC Separation of Peptides
After MCO and reactions with FLNH2, the TM proteins were digested with trypsin and then analyzed by reverse phase HPLC. Both 220 nm (peptide absorbance) and 440 nm absorbance (FLNH2) were monitored. Comparison of HPLC profile monitoring 220 nm absorption (Fig. 3) revealed the control and oxidized samples yielded very similar if not identical tryptic digestion profiles. When 440 nm absorbance was monitored, five peaks were detected from the oxidized TM fraction (Fig. 3). Peaks 2, 3, 4 were observed in all TM MCO reactions whereas peaks 1 and 5 sometimes varied in relative area and were not even present in some experiments (data not shown). Therefore, we choose peaks 2, 3 and 4 for further analysis by MS/MS.
FIGURE 3.

HPLC chromatogram of tryptic peptides of FLNH2-derivatized TM proteins. Peptides from oxidized proteins are in red and the control non-oxidized proteins are black. Left panel: elution profile monitored at 220 nm associated with peptide absorbance. Right panel, elution profile monitored at 440 nm associated with FLNH2 absorbance.
MALDI-MS/MS Identification of Peptides
Peak 2 was then further purified manually by HPLC. The eluted sample was dried and then analyzed by MALDI-TOF MS. The results indicated that the sample contained two peptides, one of m/z 2,172 and the other m/z 2,533. The product ion of 2,172 was searched against the S. oneidensis database using Mascot software and was identified to be a tryptic digest of SO2907 (YIDSYDGETPQQQEVGTNK) which encodes a putative TonB-dependent receptor protein. The peptide sequence was confirmed through b and y ions in the MALDI-MS/MS spectrum (data not shown). The other product ion with m/z at 2,533 did not hit anything in the data base. Further analysis helped identify the peptide. The results are consistent with the HPLC not resolving the unmodified peptide (as identified above) with that of the MCO product of the peptide. Our logic is as follows. The 361 Da difference between the m/z of these two peaks can be accounted for by (i) oxidation of a Tyr (800) to topaquinone (plus 30 Da), (ii) the addition of the FLNH2 (plus 347 Da), (iii) dehydration of one water (minus 18), and (iv) by reduction of the Schiff base (plus 2) (scheme 1). Topaquinone is a known MCO product of Tyr (32). The MALDI-MS/MS spectrum confirmed the modification of tyrosine and the peptide sequence (Table 2 and supplemental Fig. S2). The data are consistent with FLNH2 adding to the carbonyls formed on the Tyr in the N terminus of the peptide.
SCHEME 1.

The proposed mechanism of tyrosine modification via iron-catalyzed protein oxidation.
TABLE 2.
Peak assignments of the fragmented peptide with m/z of 2533
| B/Y ions | Fragment composition | m/z |
|---|---|---|
| Y4 | KNTG | 419.14 |
| Y7 | KNTGVEQ | 775.29 |
| Y8 | KNTGVEQQ | 903.34 |
| Y10 | KNTGVEQQQP | 1128.43 |
| Y11 | KNTGVEQQQPT | 1229.49 |
| Y13 | KNTGVEQQQPTEG | 1415.55 |
| Y16 | KNTGVEQQQPTEGDYS | 1780.63 |
| B8 + 361 | Y*IDSYDGE | 1304.31 |
| B9 + 361 | Y*IDSYDGET | 1405.31 |
| B11 + 361 | Y*IDSYDGETP | 1630.43 |
| B15 + 361 | Y*IDSYDGETPQQQEV | 2115.64 |
SO2097 Gene Deletion Mutant
The identification of SO2907 as an iron-binding protein (and having a role in DIR) was further confirmed by mutagenesis studies (and also by ITC studies described below). To assess the role of SO2907 in Fe3+ reduction, the so2907 gene deletion (Δso2097) mutant was generated. We then examined whether ΔSO2907 mutant had any phenotype in Fe3+ reduction. We measured the rate and extent of aerobic growth of both wild type and ΔSO2907 cultures using LB media (Fig. 4A). ΔSO2907 exhibited the same growth rate and attained the same maximal optical density as the wild type. Likewise, cultures were inoculated in M1 media with lactate as the electron donor and fumarate as the electron acceptor. Both ΔSO2907 and wild type share similar growth rate as well as the maximal optical density. These results are consistent with SO2907 not being involved in iron assimilation (where iron is taken up into the cell probably through siderophore participating pathway) (33).
FIGURE 4.

Growth and iron reduction characterization of wild type and ΔSO2907. Panel A, growth curve of wild type (filled symbols) and ΔSO2907 (open symbols) under aerobic conditions with LB (triangles, left axis) or anaerobically with fumarate as the TEA (circles, right axis). The fumarate growth medium contained 30 mm lactate and 50 mm fumarate. Panel B, growth curve (circles) and Fe3+ reduction (triangles) of wt (filled symbols) and ΔSO2907 (open symbols) growing anaerobically with citrate-Fe3+ as the TEA. The growth medium contained 30 mm lactate and 50 mm citrate-Fe3+. Panel C, growth curve (circles) and Fe3+ reduction (triangles) of wt (filled symbols) and ΔSO2907 (open symbols) growing anaerobically with 10 mm goethite as the TEA. Experiments (except for the cell counts of panel C) were performed in triplicates. Data were represented as mean ± S.D.
ΔSO2907 mutant showed a much slower growth rate and iron reduction rate relative to wild type when citrate-Fe3+ was used as the TEA (Fig. 4B). The doubling time for ΔSO2907 was determined to be 3.3 h compared with 1.6 h for the wild type. To avoid potential complications from the citrate in the experiments (citrate can be used as an addition carbon source), we also utilized NTA-Fe3+ and identical results were observed (data not shown). ΔSO2907 was also tested for its capacity to utilize the insoluble iron oxides goethite and ferrihydrite as the TEA. Growth curves showed that ΔSO2907 grows just slightly slower than wild type on ferrihydrite and even a little more slower on goethite as the TEA (Fig. 4C, data not shown for ferrihydrite). These results are consistent with TonB being involved in reduction of soluble Fe3+ and playing a lesser role in reduction of insoluble Fe3+ oxides by S. oneidensis MR-1.
Iron Binding Properties of SO2907
Truncated constructs of SO2907 were heterologously expressed in E. coli. The full-length protein formed inclusion bodies and attempts to refold and solubilize the protein failed. DAS software (27) was used to predict the transmembrane domain of SO2907. Two parts of the protein, TonB-A and TonB-B, were expressed that do not contain a transmembrane domain. Both TonB-A and TonB-B are predicted to be extracellularly exposed with TonB-B containing the peptide identified above (MS/MS) that harbored the iron-binding sequence. In contrast, TonB-A does not. The two His-tagged proteins were heterologously expressed in E. coli and purified by Ni-agarose (supplemental Fig. S3). The ability of these two purified proteins to bind iron was assayed with NTA-Fe3+ and citrate-Fe3+. ITC measurements show that the Kd for NTA-Fe3+ binding to TonB-B is 50 ± 7 μm (Fig. 5A) while no binding could be detected for TonB-A binding to NTA-Fe3+. The binding of TonB-B to citrate-Fe3+ was also investigated and similar results were observed (data not shown).
FIGURE 5.

Iron binding of truncated SO2907 protein. Panel A, ITC measurement of NTA-Fe3+ binding to TonB-A (circles), TonB-B (inverted triangles), and TonB-B (Y800F) (squares) at pH 7.5 (50 mm HEPES, 100 mm NaCl) and 25 °C. Titration plots were derived from the integrated heats of binding, corrected for heats of dilution. The black line represents the best non-linear fit to the data assuming a single-set binding model. Panel B, the inset shows fluorescence spectra of TonB-B and the quenching by additions of NTA-Fe3+. Spectra were obtained on a 20 μm solution of TonB-B. Both proteins and ligand samples were prepared in 50 mm HEPES, pH 7.4 and 100 mm NaCl. The bind curve was generated from analysis of the fluorescence quenching measurements following the method of Winzor (31). Apparent binding affinity is summarized in the text.
Because TonB-B contains two Trp residues, we examined whether the binding of chelated iron could be studied through fluorescence quenching experiments. We found that the fluorescence is strongly quenched by NTA-Fe3+ (Fig. 5B) while no apparent fluorescence quenching was observed by addition of concentrated NTA (supplemental Fig. S4). The experimentally-determined binding affinities (58 ± 9 μm) were found to be similar to those determined by ITC (50 ± 7 μm). Again, no evidence was found for iron binding to TonB-A. Therefore, both fluorescence binding experiments and ITC support the conclusion that TonB-B, which contains the postulated iron binding peptides, binds soluble Fe3+.
Identification of Tyr-800 in Iron Binding
To determine the residues involved in iron binding by SO2907, we utilized site-directed mutagenesis on the gene encoding the TonB-B protein. Tyr-800, the residue identified through protein oxidation studies to be involved in iron binding was mutated along with Asp-803. Asp-803 is in close proximity to Tyr-800. Wild type (wt) and mutants (Y800F and D803A) were expressed in E. coli BL21(DE3) (pET21a-tonB-B) and purified (data not shown). The iron binding constants were obtained by ITC as previously performed for wt TonB-B. Compared with the wt, D803A exhibits similar if not the same binding isothermal trace when titrated with NTA-Fe3+ (data not shown). In contrast, the Y800F mutant yielded a change in the Kd (Fig. 5A). The calculated Kd for Y800F is 92 ± 9 μm. This is ∼80% greater than that of wt which is consistent with Tyr-800 being involved in iron binding by TonB-B.
DISCUSSION
There are currently three proposed mechanism for how Shewanella is able to utilize insoluble metal oxides as TEAs in respiratory metabolism (9): (i) electron shuttles (e.g. flavins) (10, 11), which deliver the reducing equivalents from the CM to the extracellular matrix, (ii) direct contact (either through hemeproteins and/or with nanowires (25, 34)), and (iii) metal chelators (13, 15), which serve to solubilize the metal oxides such that they can contact with membrane-associated electron-transport components. Experimental evidence exists for all three mechanisms. Also, the three mechanisms are not mutually exclusive. Furthermore, different mechanism may be operative when the TEAs are soluble metals (e.g. U6+ or chelated ferric ion). As such, it would seem plausible that reduction of low molecular weight ferric species would also occur in the periplasm where it can gain contact with the CM. While iron exists primarily as insoluble, solid phase minerals at pH at or above circumneutral, ferric ion can be soluble when chelated and indeed soluble biological chelated iron forms have been detected in the environment (15, 35, 36). Evidence also shows that the utilization of soluble ferric ion is of physiological relevance. Voltammetric techniques have provided evidence for the production of iron chelators by Shewanella (16). The importance of soluble iron reduction is also supported by the recent work of Gescher et al. (14) showing that the expression of S. oneidensis CymA in E. coli confers it the ability to use soluble ferric ion as a TEA (as measured by growth). Work from our laboratory (12) has also shown that the reduction of soluble iron forms is all catalyzed by the CM. The reduction of insoluble iron forms is also totally dependent upon the CM, however, OM components facilitate the process (12). In addition to soluble ferric iron, a number of other soluble TEAs are shown to be reduced by the CM which include fumarate (37), nitrate, nitrite, DMSO (38) and U(VI) (39).
The present study is actually not the first to suggest a role for the Ton-B-dependent receptor (SO2907) in DIR. Using two-dimensional gels, we previously showed that SO2907 is up-regulated in cultures grown with ferric citrate as the TEA (12). These growth conditions contained 50 mm ferric citrate and at such high concentrations, would indicate that SO2907 is not involved in the classic, iron-limiting siderophore-mediated iron acquisition process (33). Using the MCO system to identify the iron binding site of proteins, this putative TonB-dependent receptor (locus name SO2907) was the major protein to be labeled.
It is widely known that for Gram-negative bacteria such as E. coli, the iron uptake system relies on a TonB-dependent transportation system (see reviews (40, 41)). Basically, Gram-negative bacteria use iron-binding specific transporters in the OM to deliver iron complexes to the periplasmic space. This transport is proposed to be driven by the CM-dependent proton motive force involving a complex of proteins (TonB, ExbB, ExbD) (41). The TonB-dependent receptors differ from porins in several fundamental ways, including their high iron binding affinities and specificity for substrate binding and low transport rate.
The SO2907 protein was examined using SWISS-MODEL workplace (42). The results from the sequence alignments indicated that SO2907 belongs to the OM-channel protein family. It contains two conserved domains: TonB-dependent receptor β-barrel domain (residues 554–884) and TonB-dependent receptor plug domain (residues 52–167). Both conserved domains are typically found in other OM TonB-dependent receptors involved in iron transport. One example is FepA from E. coli serving as ligand-gated transporter for ferric chelates and vitamin B12 (43).
The importance of SO2907 in DIR is supported by the phenotype of the knock-out mutant, ΔSO2907. ΔSO2907 exhibited the same growth rate and final yield when grown with either molecular oxygen or fumarate as the TEA. Only with iron as the TEA did ΔSO2907 have a slower growth rate and slower rate of iron reduction. The greatest decrease was observed with soluble iron forms citrate-Fe3+ and NTA-Fe3+. A decrease was also observed with goethite or ferrihydrite but the extent of this decrease was not as much as that with soluble Fe3+. This finding is consistent with the transport of chelated iron by TonB as the rate-limiting step when soluble iron forms are used as TEA. However, when using iron oxides, either other steps are more rate-limiting than ferric ion transport or other mechanisms are involved (e.g. electron shuttle agents (11)).
Using a truncated form of SO2907, the portion identified by MCO, we determined the binding constant to chelated iron by both ITC and fluorometry. Both methods yielded a binding constant of ∼50 μm. Through site-directed mutagenesis, we have identified Tyr-800 as one of iron binding residues on SO2907. This is consistent with the result of oxidation studies where the Tyr-800 is modified. The iron binding affinity of SO2907 is in contrast to traditional TonB receptors which typically show iron-siderophore binding affinities in the range of 0.1 and 100 nm (44). For an example, the dissociation constant of FepA binding to ferric-enterobactin is lower than 0.1 nm (26). The relative weaker binding of SO2907 to iron citrate or iron-NTA is consistent with this protein not being involved in iron assimilation. This is confirmed by its identical growth curve to wild type under fumarate and oxygen respiratory conditions. As such, our results with two-dimensional gels (12) show that SO2907 is up-regulated under iron-rich conditions. Therefore, we propose that SO2907 may function as a low-affinity iron transporter inside the CM/periplasm for DIR. The weak absorption of iron citrate can mediate rapid diffusion of soluble iron inside the periplasm/CM for DIR.
Our results here provide a mechanism for transport of chelated ferric species across the OM into the periplasm where it can contact the electrons of respiratory metabolism within the CM. We speculate that the relative contribution of this process is dependent upon the nature of the TEA. With soluble ferric species, SO2907 appears to play a significant role whereas with insoluble iron oxides, it apparently plays a minor role (or is not the rate-limiting step) in the overall respiratory pathway. Nevertheless, our results show the high plasticity of Shewanella's respiratory system possessing a variety of mechanisms to utilize a wide range of TEAs.
Supplementary Material
Acknowledgments
We thank the Mass Spectrometry Facility in the Penn State University Hershey Medical Center for MS data analysis. We thank Dr. Donald Bryant and Dr. Jeffrey Catchmark (Penn State University) for the use of fluorometer, ITC, and fluorescent microscopy.
This work was supported by the NSF Environmental Molecular Sciences Institute program (CHE-0431328) through the Penn State Center for environmental Kinetics Analysis and by DOE Grant ER64399 0013153.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S4.
- TEA
- terminal electron acceptor
- FLNH2
- fluoresceinamine
- DNPH
- 2,4-dinitrophenylhydrazine
- NTA
- nitrilotriacetic acid
- MCO
- metal-catalyzed oxidation
- TM
- total membranes
- CM
- cytoplasmic membrane
- TCA
- trichloroacetic acid
- DIR
- dissimilatory iron reduction
- ITC
- isothermal titration calorimetry.
REFERENCES
- 1. Nealson K. H., Banfield J. F. (1997) in Geomicrobiology: Interactions Between Microbes and Minerals in Rev. Miner. (America M. S. ed.) Washington, D.C [Google Scholar]
- 2. Weber K. A., Achenbach L. A., Coates J. D. (2006) Nat. Rev. Microbiol. 4, 752–764 [DOI] [PubMed] [Google Scholar]
- 3. Perry A. J., Rowcliff D. J. (1973) J. Mater. Sci. 8, 904–907 [Google Scholar]
- 4. Fredrickson J. K., Zachara J. M., Kennedy D. W., Duff M. C., Gorby Y. A., Li S. M. W., Krupka K. M. (2000) Geochim. Cosmochim. Acta 64, 3085–3098 [Google Scholar]
- 5. Fredrickson J. K., Zachara J. M., Kennedy D. W., Kukkadapu R. K., McKinley J. P., Heald S. M., Liu C. X., Plymale A. E. (2004) Geochim. Cosmochim. Acta 68, 3171–3187 [Google Scholar]
- 6. Myers C. R., Nealson K. H. (1988) Science 240, 1319–1321 [DOI] [PubMed] [Google Scholar]
- 7. Nealson K. H., Saffarini D. (1994) Annu. Rev. Microbiol. 48, 311–343 [DOI] [PubMed] [Google Scholar]
- 8. Tiedje J. M. (2002) Nat. Biotechnol. 20, 1093–1094 [DOI] [PubMed] [Google Scholar]
- 9. Gralnick J. A., Newman D. K. (2007) Mol. Microbiol. 65, 1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Marsili E., Baron D. B., Shikhare I. D., Coursolle D., Gralnick J. A., Bond D. R. (2008) Proc. Natl. Acad. Sc.i U.S.A. 105, 3968–3973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. von Canstein H., Ogawa J., Shimizu S., Lloyd J. R. (2008) Appl. Environ. Microbiol. 74, 615–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ruebush S. S., Brantley S. L., Tien M. (2006) Appl. Environ. Microbiol. 72, 2925–2935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lies D. P., Hernandez M. E., Kappler A., Mielke R. E., Gralnick J. A., Newman D. K. (2005) Appl. Environ. Microbiol. 71, 4414–4426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gescher J. S., Cordova C. D., Spormann A. M. (2008) Mol. Microbiol. 68, 706–719 [DOI] [PubMed] [Google Scholar]
- 15. Taillefert M., Beckler J. S., Carey E., Burns J. L., Fennessey C. M., DiChristina T. J. (2007) J. Inorg. Biochem. 101, 1760–1767 [DOI] [PubMed] [Google Scholar]
- 16. Jones M. E., Fennessey C. M., DiChristina T. J., Taillefert M. (2010) Environ. Microbiol. 12, 938–950 [DOI] [PubMed] [Google Scholar]
- 17. Temple A., Yen T. Y., Gronert S. (2006) J. Am. Soc. Mass. Spectrom. 17, 1172–1180 [DOI] [PubMed] [Google Scholar]
- 18. Amici A., Levine R. L., Tsai L., Stadtman E. R. (1989) J. Biol. Chem. 264, 3341–3346 [PubMed] [Google Scholar]
- 19. Climent I., Tsai L., Levine R. L. (1989) Anal. Biochem. 182, 226–232 [DOI] [PubMed] [Google Scholar]
- 20. Stadtman E. R., Oliver C. N. (1991) J. Biol. Chem. 266, 2005–2008 [PubMed] [Google Scholar]
- 21. Stadtman E. R. (1990) Free Radic. Biol. Med. 9, 315–325 [DOI] [PubMed] [Google Scholar]
- 22. Stadtman E. R. (1993) Annu. Rev. Biochem. 62, 797–821 [DOI] [PubMed] [Google Scholar]
- 23. Stookey L. L. (1970) Anal. Chem. 42, 779–781 [Google Scholar]
- 24. Bradford M. M. (1976) Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 25. Gorby Y. A., Yanina S., McLean J. S., Rosso K. M., Moyles D., Dohnalkova A., Beveridge T. J., Chang I. S., Kim B. H., Kim K. S., Culley D. E., Reed S. B., Romine M. F., Saffarini D. A., Hill E. A., Shi L., Elias D. A., Kennedy D. W., Pinchuk G., Watanabe K., Ishii S., Logan B., Nealson K. H., Fredrickson J. K. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 11358–11363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Newton S. M., Igo J. D., Scott D. C., Klebba P. E. (1999) Mol. Microbiol. 32, 1153–1165 [DOI] [PubMed] [Google Scholar]
- 27. Cserzö M., Wallin E., Simon I., vonHeijne G., Elofsson A. (1997) Protein Eng. 10, 673–676 [DOI] [PubMed] [Google Scholar]
- 28. Qian Y., Paquete C. M., Louro R. O., Ross D. E., LaBelle E., Bond D. R., Tien M. (2011) Biochemistry 50, 6217–6224 [DOI] [PubMed] [Google Scholar]
- 29. Wiseman T., Williston S., Brandts J. F., Lin L. N. (1989) Anal. Biochem. 179, 131–137 [DOI] [PubMed] [Google Scholar]
- 30. Perozzo R., Folkers G., Scapozza L. (2004) J. Recept. Sig. Transd. 24, 1–52 [DOI] [PubMed] [Google Scholar]
- 31. Winzor D. J., Sawyer W. H. (1995) Quantitative Characterization of Ligand Binding, Wiley-Liss, New York [Google Scholar]
- 32. Ali F. E., Barnham K. J., Barrow C. J., Separovic F. (2004) J. Inorg. Biochem. 98, 173–184 [DOI] [PubMed] [Google Scholar]
- 33. Madigan M. T., Martinko J. M., Parker J. (2000) Brock Biology of Microorganisms., Prentice Hall [Google Scholar]
- 34. Lower B. H., Shi L., Yongsunthon R., Droubay T. C., McCready D. E., Lower S. K. (2007) J. Bacteriol. 189, 4944–4952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Campbell J. A., Stromatt R. W., Smith M. R., Koppenaal D. W., Bean R. M., Jones T. E., Strachan D. M., Babad H. (1994) Anal. Chem. 66, A1208–A1215 [Google Scholar]
- 36. Nancharaiah Y. V., Schwarzenbeck N., Mohan T. V., Narasimhan S. V., Wilderer P. A., Venugopalan V. P. (2006) Water Res. 40, 1539–1546 [DOI] [PubMed] [Google Scholar]
- 37. Pealing S. L., Cheesman M. R., Reid G. A., Thomson A. J., Ward F. B., Chapman S. K. (1995) Biochemistry 34, 6153–6158 [DOI] [PubMed] [Google Scholar]
- 38. Schwalb C., Chapman S. K., Reid G. A. (2003) Biochemistry 42, 9491–9497 [DOI] [PubMed] [Google Scholar]
- 39. Wade R., DiChristina T. J. (2000) FEMS Microbiol. Lett. 184, 143–148 [DOI] [PubMed] [Google Scholar]
- 40. Postle K., Kadner R. J. (2003) Mol. Microbiol. 49, 869–882 [DOI] [PubMed] [Google Scholar]
- 41. Postle K., Larsen R. A. (2007) Biometals 20, 453–465 [DOI] [PubMed] [Google Scholar]
- 42. Arnold K., Bordoli L., Kopp J., Schwede T. (2006) Bioinformatics 22, 195–201 [DOI] [PubMed] [Google Scholar]
- 43. Buchanan S. K., Smith B. S., Venkatramani L., Xia D., Esser L., Palnitkar M., Chakraborty R., van der Helm D., Deisenhofer J. (1999) Nat. Struct. Biol. 6, 56–63 [DOI] [PubMed] [Google Scholar]
- 44. Postle K. (1999) Nat. Struct. Biol. 6, 3–6 [DOI] [PubMed] [Google Scholar]
- 45. Wang Z. M., Liu C. X., Wang X. L., Marshall M. J., Zachara J. M., Rosso K. M., Dupuis M., Fredrickson J. K., Heald S., Shi L. (2008) Appl. Environ. Microbiol. 74, 6746–6755 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.