

Summary
We have developed an integrated genetic, genomic and computational approach to identify and characterize genes involved in myoblast fusion in Drosophila. We first used fluorescence activated cell sorting to purify mesodermal cells both from wild-type embryos and from twelve variant genotypes in which muscle development is perturbed in known ways. Then, we obtained gene expression profiles for the purified cells by hybridizing isolated mesodermal RNA to Affymetrix GeneChip arrays. These data were subsequently compounded into a statistical meta-analysis that predicts myoblast subtype-specific gene expression signatures that were later validated by in situ hybridization experiments. Finally, we analyzed the myogenic functions of a subset of these myoblast genes using a double-stranded RNA interference assay in living embryos expressing green fluorescent protein under control of a muscle-specific promoter. This experimental strategy led to the identification of several previously uncharacterized genes required for myoblast fusion in Drosophila.
Keywords: cell-cell fusion, myoblast, mesoderm, myogenesis, muscle development, Drosophila, genomics, gene expression profiling
1. Introduction
Cell fusion is necessary for the development of different human tissues, including bone, placenta and muscle. During myogenesis, mononucleated myoblasts fuse with each other to form nascent, multinucleated functional myofibers. Thus, normal muscle growth and regeneration of injured tissue require the fusion of new myoblasts. Also, there are several myopathies related to defects in myoblast fusion (1–3). However, the molecular mechanisms underlying these pathologies are largely unknown.
Studies done in mammalian cells in vitro and in Drosophila embryos have demonstrated that myoblast fusion involves an ordered set of specific cellular events: first, myoblasts recognize and adhere, then, alignment occurs through the parallel apposition of the membranes of elongated myoblasts with myotubes or other myoblasts, and, finally, membrane union takes place between the aligned plasma membranes in small areas of cytoplasmic continuity, with vesiculation of the excess plasma membrane in the fusion area (2). Genetic analysis combined with light and electron microscopy of Drosophila fusing myoblasts have assigned the function of specific proteins to these particular cellular processes, providing an entry point for studying the molecular mechanisms underlying myoblast fusion (4). In spite of recent progress in this area, there are still many unanswered questions related to the molecular basis of muscle fusion, including the role of the cytoskeleton in cell shape changes that occur in fusing myoblasts, the identification and functional analysis of molecules responsible for the actual fusion of muscle cell membranes, and the mechanisms that govern the invariant size of each muscle, as determined by the number of fusion events that occur during formation of a particular myofiber.
Compared to vertebrate systems, Drosophila offers several advantages as an experimental organism for studying muscle development. First, the relatively short generation time of Drosophila allows myogenesis to be analyzed in vivo in a more rapid manner. Second, Drosophila offers outstanding genetic resources for studying muscle development. Third, this model organism has a smaller number of genes which has the advantage of circumventing potential functional redundancies inherent to mammalian genomes. And fourth, many of the key components responsible for myogenesis at the molecular level are well conserved between flies and humans, so that knowledge about fly muscle development is highly relevant to human biology and disease.
In an attempt to better understand the molecular mechanisms underlying myoblast fusion, we have undertaken an integrated genetic, genomic and computational strategy to determine which genes are expressed during and are essential for Drosophila myoblast fusion. We hyothesized that a functional genomic approach would reveal fusion genes that were not identified in previous forward genetic screens (1, 5, 6). Specifically, we used fluorescence-activated cell sorting (FACS) to purify embryonic myoblasts marked with green fluorescent protein (GFP). Included in the analysis were embryos derived from wild-type and from genetically modified strains in which mesoderm development was perturbed in predictable and informative ways (Fig. 1 and Fig. 2). The RNA from these cell populations was hybridized to Affymetrix GeneChip arrays, and pairwise comparisons were made to detect genes that are differentially expressed between the mesoderm and the rest of the embryo, as well as between the mutant mesodermal cells and their wild-type counterparts. Then, we combined the results from all the microarray data and subjected the resulting compendium of expression profiles to a statistical meta-analysis that was designed to reveal candidate genes expressed in each of the two fusing myobast populations, founder cells (FCs) and fusion-competent myoblasts (FCMs) (7). Finally, we validated hundreds of these predictions by gene-specific in situ hybridization, and assessed the functions of a selected group of myoblast genes using double-stranded RNA interference (RNAi) analysis of muscle development in whole living embryos (6). This integrated approach uncovered several previously uncharacterized genes that are involved in myoblast fusion (Fig. 3E,F and data not shown), with many more candidates remaining to be analyzed by RNAi.
Figure 1. Experimental strategy to obtain gene expression signatures of purified Drosophila embryonic mesodermal cells.
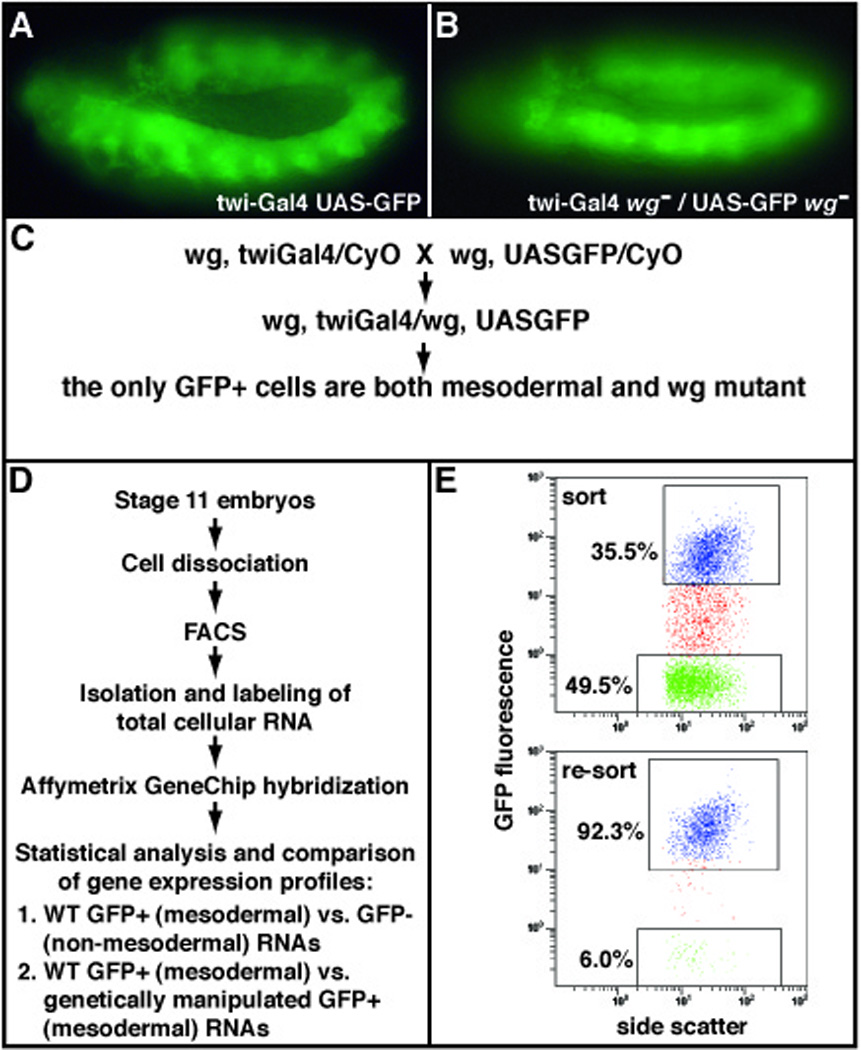
(A) Transgenic, stage 11, Drosophila embryo expressing Gal4 under the control of the twi promoter and GFP under the UAS regulatory sequence (21), resulting in GFP-positive mesodermal cells. (B) Transgenic, stage 11, wingless (wg) mutant embryo with GFP-positive mesodermal cells. (C) Genetic crossing scheme to obtain homozygous mutant GFP-positive mesodermal cells. Strains bearing independently generated recombinant chromosomes having the mutant gene of interest (for example, wg) and either the twiGal4 or UASGFP transgenes are crossed. (D) A representative fluorescence-activated cell sorting (FACS) experiment to obtain total RNA from mesodermally purified cells. (E) Representative FACS scatter plots before (top panel) and after (bottom panel) the separation of GFP-positive and -negative cell populations. Top panel, upper box: GFP-positive sort in blue; top pannel, lower box: GFP-negative sort in green. Bottom panel, upper box: in blue are shown the re-sorted GFP-positive cells to verify that purity obtained from the primary sort was greater than 90%.
Figure 2. Statistical meta-analysis of an expression profiling compendium to predict myoblast specific gene signatures.
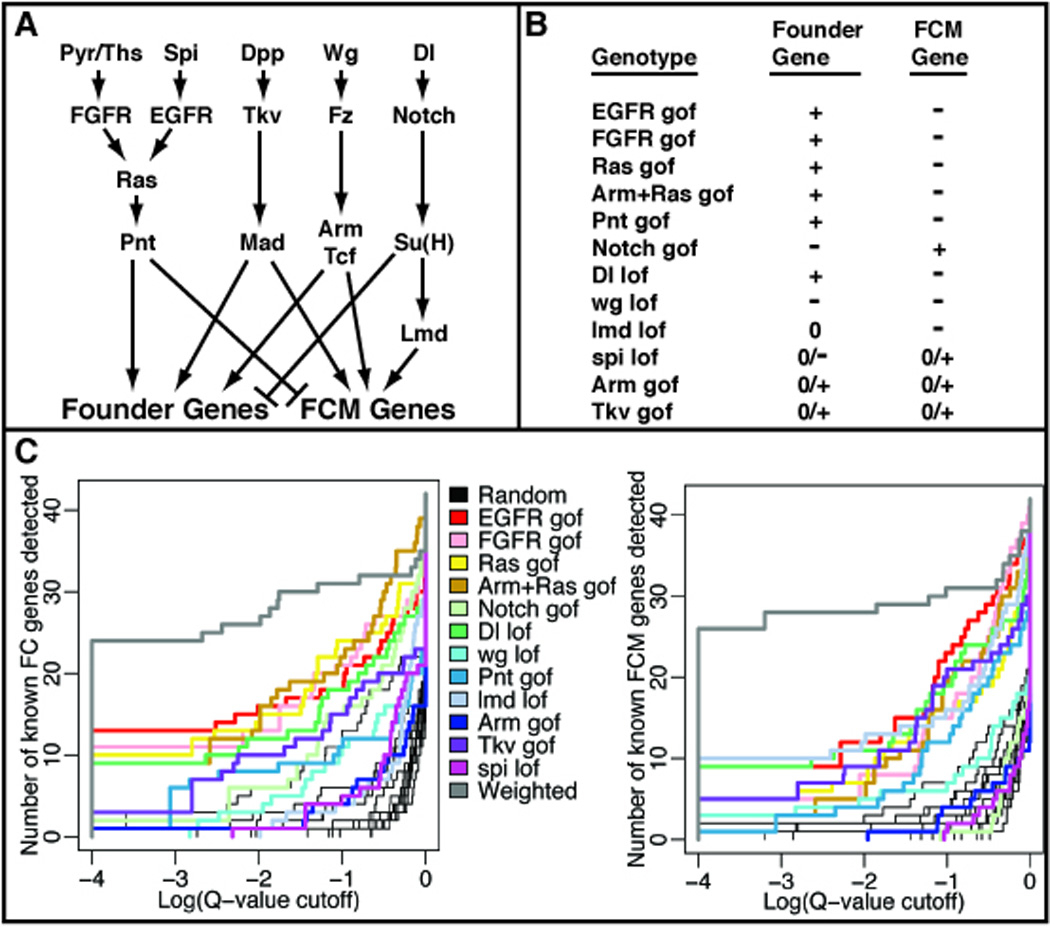
(A) Summary of the signaling pathways and transcription factors that positively and negatively regulate the expression of myoblast subpopulation-specific genes (founder genes and fusion-competent myoblast (FCM) genes). (B) Expected gene expression changes for founder or FCM genes in each of the 12 genetic perturbations used in our study (6). (C) Detection curves showing the number of genes from the training set (see below) detected, as a function of q-value (predicted measurement of false positive rate in Log scale), for FC genes (left) and FCM genes (right). In each panel, the predictive value of individual genotype/wild-type comparisons (various colors; see legend) are compared to randomly generated rankings (thin black lines) and to composite rankings derived from a combination of all datasets (gray). To avoid introducing biases for or against any genotype, the training sets were composed of known genes from the literature as well as the mesodermally enriched genes that had been verified by in situ hybridization in this study to be FC or FCM genes.
Figure 3. RNAi analysis of genes necessary for myoblast fusion.
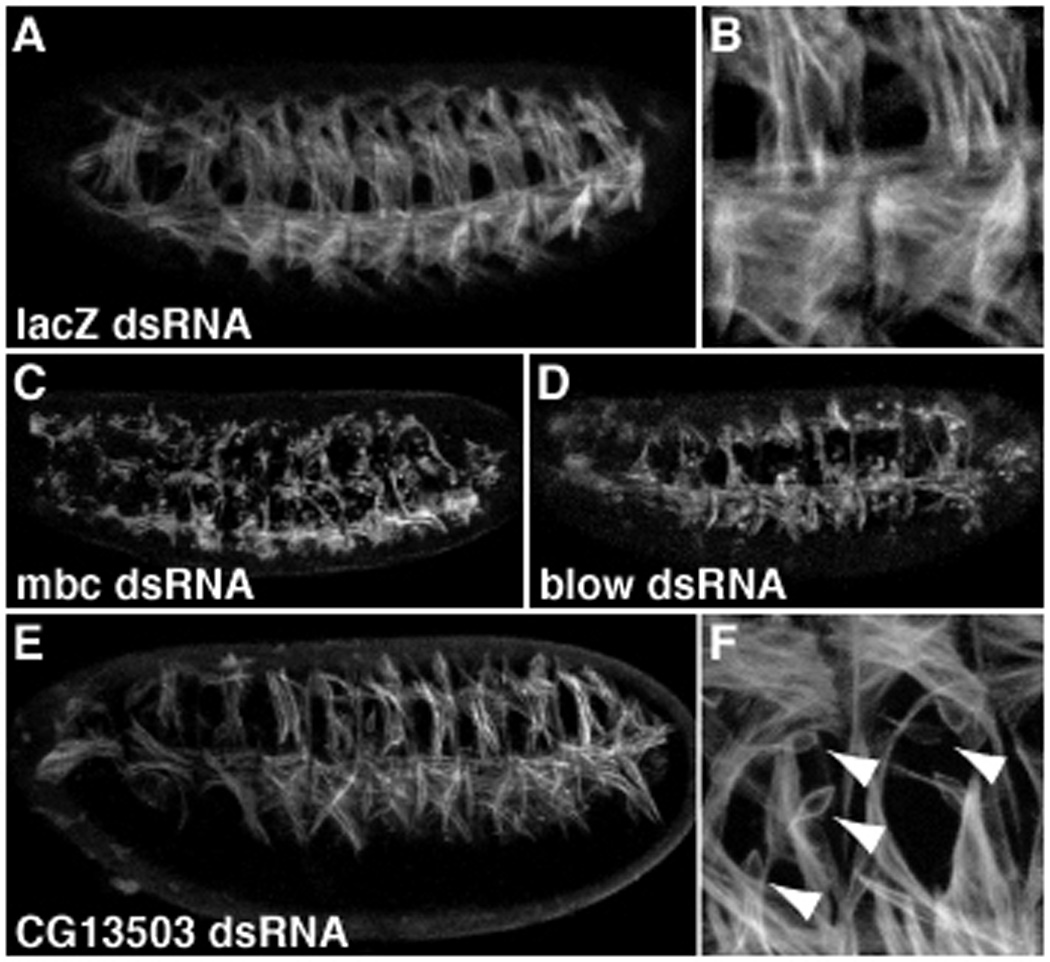
Live transgenic embryos expressing tau-GFP under the control of the myosin heavy chain promoter (20) were injected with negative control lacZ dsRNA (A and B), positive control myoblast city (mbc, C) or blown fuse (blow, D) dsRNA, and gene-specific test dsRNA (E and F). (E and F) RNAi for CG13503, solas, shows abundant unfused myoblasts (arrowheads) indicating that this gene is required for myoblast fusion. Since this gene is the homolog of the vertebrate WH2 actin-binding protein, and its expression is restricted to FCMs, we hypothesized that it is required for the unique cytoskeletal rearrangements occuring in FCMs during myoblast fusion (6).
2. Materials
2.1 Generating appropriate fly strains
The following Drosophila stocks were used to obtain both wild-type and genetically modified mesodermal cells expressing GFP: Twi-Gal4 UAS-2EGFP (8), Twi-Gal4 alone (9) see note 4.4), UAS-λTop (constitutively activated EGFR) (10), UAS-Dof UAS-λ-Htl (constitutively activated Heartless FGFR together with Downstream of FGFR/Heartbroken/Stumps) (11, 12), UAS-Ras1Act (activated Ras) (13), UAS-PntP2VP16 (activated Pointed) (14), UAS-TkvQD (activated Thick veins Decapentaplegic receptor) (15), UAS-ArmS10 (activated Armadillo) (16), UAS-ArmS10; UAS-Ras1Act, Twi-Gal4 wgCX4/CyO, wgIG22 UAS-2EGFP, UAS-Nintra (17), Twi-Gal4 lmd1/TM3 ftz-lacZ, UAS-2EGFP lmd2/TM3 ftz-lacZ, Twi-Gal4 DlX/TM3 ftz-lacZ, UAS-2EGFP Dl X/TM3 ftz-lacZ.
2.2 Collecting the embryos
Fly incubator, yeast paste, molasses plates (cook 378g of molasses with 65g of bacteriological agar in 2L of water for 45 min, when it cools down to 55–60° add 30 ml of tegosept: 80g of methyl 4-hydroxybenzoate and 750 ml of reagent alcohol), fly cages (400 ml Tripour plastic beakers perforated with a hot 25 gauge needle).
2.3 Preparation of embryonic single cell suspensions for flow cytometry
plastic squeeze bottles, camel hair brush, 70 micron strainer (Falcon), weighing balance, glass dounce homogenizers (VWR 62400-620, vendor: Wheaton) with loose-fitting pestle (clearance 0.0035-0.005 inch), desktop clinical centrifuge, hemocytometer (optional), 5 ml round bottom plastic tubes, 40 micron nylon mesh (www.smallparts.com), sterile plastic transfer pipets. Solutions: 50% bleach, 0.01% Triton X-100, 8% Fetal Bovine Serum (FBS from SIGMA) in Schneider’s medium (Gibco).
2.4 Sorting GFP positive and negative cells by flow cytometry
Fluorescent activated cell sorter (in our case was EPICS Altra with Hyper Sort Option from Beckman Coulter), 5 ml round bottom plastic tubes, carry-on cooler to transport cells on ice. Solutions: Seecof saline (6 mM Na2HPO4, 3.67 mM KH2PO4, 106 mM NaCl, 26.8 mM KCl, 6.4 mM MgCl2, 2.25 mM CaCl2, pH 6.8). Sterilize by filtering; do not autoclave. “RNAlater” from Ambion.
2.5 Obtaining total RNA from sorted cells
Beckman J6-M centrifuge with JS 5.2 rotor (or equivalent centrifuge), 2ml Eppendorf tubes. Bioanalyzer microfluidics platform (Agilent); alternatively, use a spectrophotometer and RNA electrophoresis gel to check the quantity and quality of the total RNA.
Solutions: Schneider’s Drosophila Medium (Gibco), Trizol Reagent (Life Technologies, Inc.). Isopropanol. RNAase free water (e.g. from Ambion).
2.6 Hybridization on Affymetrix chips
GeneChip Drosophila Genome Arrays. The hybridizations are done at a microarray core facility that will provide all the reagents, apparatus, and experimental expertise required for the procedure.
2.7 Data analysis
Data analyses involved the use of the statistical programming language R (http://www.r-project.org), the Bioconductor suite of bioinformatics R packages (http://www.bioconductor.org), and the goldenspike R package (http://www.elwood9.net/spike).
2.8 Validation of the predicted gene expressions
2.8.1
T7 and SP6 RNA polymerase (Roche). Digoxigenin-labeled nucleotide mix (Roche). RNAase inhibitor (Roche).
2.8.2
Drosophila Gene Collection (DGC1 and 2 http://www.fruitfly.org/DGC/index.html) and Drosophila embryonic primary cDNA.
2.8.3
Antibodies: Anti-Digoxigenin-AP (Roche), Rabbit anti-Lmd (from H. Nguyen, (18) was used at 1:1000. Rabbit anti-beta-galatosidase (Promega) was used at 1:500.
2.8.4
The following stocks were used to determine gene expression patterns in mutant backgrounds: Twi-Gal4, UAS-Ras1Act, DlX/TM3 ftz-lacz, lmd1/TM3 ftz-lacz. The enhancer trap line rp298lacz was used to test for localization of gene expression to founder cells (19).
2.9 Functional analysis of newly identified myoblast genes
RNA interference assay: MEGAscript RNAi kit (Ambion), Drosophila embryonic primary cDNA, DEPC-treated 1X injection buffer (5mM KCl, 0.1 mM PO4, pH 7.8), MHC-tau-GFP embryos (20). Microinjector, needle puller or commercially available microinjection needles, molasses plates, 50% bleach, halocarbon oil (series 700, from VWR, part number 700-1).
3. Methods
3.1 Generating appropriate fly strains
Specific gene promoter-driven GFP or Gal4/UAS (21) system-based strategies are some of the possibilities to express GFP (or other fluorescent protein) in the desired population of cells. Choose a strain of flies that expresses the highest levels of GFP (or other fluorescent protein). Engineering several copies of the reporter gene or using di-cistronic versions of GFP will increase the intensity (8). This strategy maximizes the fluorescence intensity and provides a higher recovery of cells with the greatest purity (see Note 2).
3.2 Collecting the embryos
3.2.1
Maintain flies in large plastic cages (400 ml tripour beakers) at 25º C, feeding them using molasses plates streaked with yeast paste. Put 700–1000 flies per cage. More flies prevent sufficient ventilation and results in the flies sticking to the sides of the cage.
3.2.2
Change molasses plates (w/ yeast) one time a day for 2–3 days.
3.2.3
Change plate, allow flies to pre-lay for 1 period of 2 hours on fresh food (prelaying for 2 periods of 2 hours improves the yield of the first collection if it is not initially adequate, also see Note 1), change plate every two hours and let the embryos age to the appropriate stage for cell dissociation. To obtain myoblasts that have already been specified but have not yet undergone fusion, use stage 11 embyros (~5.5–7.5 hours post egg laying at 25°C).
3.3 Preparation of embryonic single cell suspensions for flow cytometry
Single cell suspensions are obtained by homogenizing embryos with a Dounce tissue grinder (colloquially, “douncing”) as follows (protocol modified from (22):
3.3.1
To prepare the eggs for douncing, start approximately 20–30 minutes before the time assigned for the actual dissociation of the eggs.
3.3.2
Wash eggs and yeast from plates using dH2O from a squeeze bottle, loosen eggs with a camel hair brush and pour the eggs through a 70 micron strainer (the weight of the strainer should be measured beforehand), wash the materials in the strainer until the yeast is removed.
3.3.3
Blot the strainer with filter paper to remove excess liquid and weigh the strainer to obtain the wet-weight of the collected eggs.
3.3.4
Immerse the strainer in 50% (v/v) bleach to cover the eggs and decorionate them for 5 mins.
3.3.5
During dechorionation, fill the douncer with 7ml Schneider medium and keep on ice.
3.3.6
After dechorionation, wash the bleach completely from the eggs in the strainer with 0.01% (v/v) Triton X-100 from a squeeze bottle, rinse with dH2O and with a final rinse in Schneider medium, then brush the eggs into the douncer. Important: try not to put more than 0.03 g of embryos per douncer, as it reduces the yield of single cell suspension. Divide the total weight of embryos by 0.03 to estimate the number of douncers needed.
3.3.7
Use loose pestle to gently but firmly dounce to the bottom. Give 7 strokes. Push up and down, only, without rotating the pestle. From now on, keep the tubes on ice.
3.3.8
Transfer dounced materials from two douncers (7+7ml) into one conical centrifuge tube (15ml Falcon tube). Spin at 40g (418 rpm in a desktop clinical centrifuge) for 5 mins to pellet the tissue and cell debris, clumps and vitelline membranes. Single cells and yolk are in the supernatant.
3.3.9
Transfer the supernatant to a clean tube and spin at 380g for 10 min (1255rpm in a desktop clinical centrifuge) to pellet single cells, then discard the supernatant. Resuspend the cells with 8% FBS in Schneider’s Medium (1 ml per tube).
3.3.10
(Optional) Draw 40 µl of the cell suspension and count the cells using a hemocytometer.
3.3.11
Add another 1 to 2 ml of 8%FBS in Schneider’s Medium once the cells are resuspended.
3.3.12
Sieve the cells trough a 40 micron nylon mesh into 5 ml round bottom Falcon tube as follows. Cut the mesh in approximately 7×7cm pieces. Hold the tube and the mesh with one hand and hold the mesh with your thumb to create a funnel to which you will apply the cell suspension with a sterile plastic transfer pipet. Pipet 1 ml of cells into the funnel-shaped mesh, then sieve the cells through the mesh. Repeat the pipeting and sieving of the cells to sieve the rest of the volume of cells from that tube.
3.3.13
(Optional) Rinse the original tube with another 1 ml of 8% FBS in Schneider’s Medium and rinse the mesh with the same solution. Discard the mesh.
3.3.14
Repeat the entire process for each tube of cells.
3.3.15
Fill the tubes to 4-4.5 ml with 8% FBS in Schneider’s Medium. Maintain the tubes on ice and take to the sorting facility.
3.4 Sorting GFP-positive and -negative cells by flow cytometry
At the flow cytometry facility, specify the following (see Note 4):
3.4.1
Use Seecof Saline as the FACS running buffer (Drosophila cells have a higher osmolarity than mammalian cells (23).
3.4.2
Turn on the cooler so the cells that are being sorted are kept at 4°C.
3
Run the cell sorter between 5000-10,000 cells per second to maximize cell viability.
3.4.4
Calibrate the FACS for GFP-positive and GFP-negative windows using cells derived from a negative control strain (see Note 3).
3.4.5
Check that the purity of the GFP-positive cells is at least 90%. This can be achieved by re-sorting a small aliquot of both populations of sorted cells before continuing with the bulk of the cells (Fig. 1 E).
3.4.6
Prepare labeled 5 ml round bottom tubes with 1.5 ml of “RNAlater” solution to collect the cells while they are being sorted.
3.4.7
After one collecting tube is full, ask the FACS operator to invert that tube to mix the cells with the “RNAlater” solution. After this step, all tubes must be maintained on ice.
3.5 Isolating total RNA from sorted cells
3.5.1
When the FACS runs from the two consecutive two-hour egg collections are finished, dilute the cells in “RNAlater” with Schneider’s Medium (dilution is necessary because “RNAlater” is very viscous and would prevent later pelleting of the cells by centrifugation). Place ~30 mls of sorted cells (up to six tubes) in a 50 ml Falcon tube and fill it with Schneider’s Medium. Sorted cell suspensions are then diluted with Schneider medium so that RNAlater is no more than 20% of the total volume.
3.5.2
Centrifuge at 4400rpm (4900G) in a Beckman J6-M centrifuge with the JS 5.2 rotor (or equivalent centrifuge) for 15 min; decant the supernatant.
3.5.3
Resuspend the cells in 200–300 µl of Trizol and transfer to a 2 ml Eppendorf tube.
3.5.4
Precipitate the RNA in isopropanol (follow the Trizol manufacturer’s instructions for total RNA isolation). Keep RNA at −80°C until it is analyzed on a Bioanalyzer. Resuspend the RNA in 20–50 µl of RNAase-free water from Ambion. The RNA needs to be at least at 430 ng/µl to start the first strand synthesis of cDNA at the Affymetrix facility (assuming that 3µg of labeled RNA will be hybridized on each chip).
3.6 Hybridization of Affymetrix GeneChips
Total cellular RNA (2.5–3 µg) from each genotype is labeled in one round of linear amplification and used for hybridization to a single Affymetrix GeneChip using standard methods recommended by the manufacturer (http://www.affymetrix.com/support/technical/manual/expression_manual.affx). Each RNA sample is independently labeled and hybridized in triplicate (see Note 6).
3.7 Data analysis
3.7.1
In order to find genes with differential expression between two conditions or samples, we chose an analysis method that showed optimized detection of spiked-in control RNAs (24). This method involved the calculation of multiple expression summaries per probe set, using the Tukey-Biweight and median polish summary methods, and testing for differential expression using a regularized t-statistic metric as described in (25).
3.7.2
In order to identify genes that were specifically expressed in the two different population of myoblasts, FCs and FCMs, we pooled all the data from the expression profiling experiments for all 12 different genotypes and performed a statistical meta-analysis. This strategy allowed us to rank all the genes based on the similarity of their expression patterns with the canonical FC (or FCM) patterns of expression based on our prior observations (6). In order to do this meta-analysis, we devised a metric which is basically a sum of the t-statistics for each genotype-to-wild-type comparison, with each term multiplied by 1 if the expected response is increased expression in the genotype, or −1 if the expected response is down-regulation (Fig.2 B). Also, each term is multiplied by a weighting factor that was chosen to optimize the detection of a training set of known FC (or FCM) genes. The resulting summation gives us an overall metric for how well each gene follows the canonical FC (or FCM) expression pattern (Fig. 2 C).
3.7.3
The detailed description of the computational analysis is not provided here due to space limitations, but can be found in the main text and Supplementary Experimental Procedures, Method E of Estrada et al (2006).
3.8 Validation of the predicted gene expression patterns
3.8.1
Validations were done by embryonic in situ hybridizations both in wild type and the following mutant backgrounds: Constitutively activated Ras (Ras1Act) and Delta (DlX) mutant embryos produce an expansion of FCs at the expense of FCMs (13, 26–28). Thus, any gene the expression of which is expanded in these mutants compared to the wild-type expression is considered to be expressed in FCs. lameduck mutant embryos (lmd1) fail to develop FCMs; thus any gene the expression of which is reduced in these mutants compared to the wild type expression is considered to be expressed in FCMs. Gene-specific digoxigenin-labeled antisense RNA probes were synthesized using cDNA clones obtained from the Drosophila Gene Collection (DGC1 and 2, http://www.fruitfly.org/DGC/index.html). For genes without an available cDNA, gene-specific PCR primers were designed and 0–18 hour embryonic RNA was used for standard first-strand cDNA synthesis followed by PCR amplification. A microtiter plate method was used for parallel synthesis of multiple probes (http://www.fruitfly.org/about/methods/RNAinsitu.html).
3.8.2
Antibody stainings were carried out as described (13) on Dl or lmd mutant embryos where we used the beta-galactosidase staining from a lacZ-marked TM3 balancer chromosome to identify the homozygotes. Beta-galactosidase staining of the enhancer trap line rp298lacz was used to test for localization of gene expression to founder cells (19).
3.9 Functional analysis of newly identified genes
RNA interference assay: Gene segments for dsRNA synthesis were selected to be 300 to 700 bp in length and common to all predicted splice variants of the targeted gene. To avoid off-target RNAi effects (29, 30) regions were chosen that lack any consecutive 18 bp of identity to another predicted gene in the Drosophila melanogaster genome. These sequences were PCR-amplified from primary embryonic cDNA using primers which incorporated T7 promoters on both ends. Purified PCR product was transcribed in vitro and purified using the MEGAscript RNAi kit, precipitated, resuspended and diluted to 2 mg/mL in DEPC-treated 1X injection buffer (31). Dechorionated MHC-tau-GFP embryos (20) were injected mid-ventrally during the syncytial blastoderm stage, then allowed to develop to stage 16–17 before assessment. Each gene was initially injected and scored blindly, with negative control (lacZ dsRNA) and positive control (mbc or blow dsRNA) injections performed in parallel (Fig. 3). Only embryos that developed robust GFP expression and that lacked obvious major morphological defects (typically 60–80% of those injected) were included in the analysis.
Acknowledgments
We would like to thank Stephen S. Gisselbrecht and Sung E. Choe for comments on the manuscript. This work was funded by the Howard Hughes Medical Institute and the National Institues of Health.
Footnotes
To maximize the number of embryos collected, aim to collect at dawn or dusk when female flies are most active at laying eggs. You can also change their circadian clock by appropriately controlling the experimental light cycle to suit your own convenience.
One of the limiting steps in this genomic approach is gathering sufficient RNA to hybridize to the Affymetrix GeneChips, especially for mutants where only one fourth of the collected embryos has the desired genotype and expresses the fluorescent protein marker for cell sorting (Fig.1 B and C). It is important to do a pilot FACS run at the sorting facility (Fig.1 E) in order to establish optimum conditions for achieving adequate separation of the GFP-positive and -negative cell populations such that sufficient quantities of high-quality total RNA will be obtained.
To calibrate the FACS and optimize the separation of GFP-positive and GFP-negative cells, use a strain of flies that is related to the study strain except that it should lack GFP expression. We used Twi-Gal4 alone as our negative control. Yolk autofluorescence of Drosophila embryos needs to be differentiated from GFP expression.
Talk to the people who run the FACS facility about sorting Drosophila cells. In many cases, this will be the first time that they will be dealing with Drosophila, so they need to follow your instructions about the running buffer and the sorting speed.
A typical experiment involves the collection of 350–1500 mg (wet weight) of embryos from 7000–12,000 flies, the sorting of 1–3 × 107 total cells from the initial suspension, and the isolation of 1.6–8 × 106 GFP-positive cells. At least six independent cell collections were pooled for gene expression profiling of a given genotype. Approximately 1 microgram of total RNA was obtained from 1 × 106 cells.
We used three technical replicates of pooled embryo collections to optimize statistical significance. Although biological replicates—that is, independent embryo collections for RNA isolation and for each labeling reaction and hybridization—are ideal for statistical power, this approach was neither practical nor cost-effective (in terms of required FACS time) for obtaining sufficient precisely staged RNA for each chip hybridization. Thus, we pooled multiple RNA collections from different cell sorting runs to minimize the variation inherent among collections and to obtain adequate RNA from each genotype.
References
- 1.Chen EH, Olson EN. Unveiling the mechanisms of cell-cell fusion. Science. 2005;308(5720):369–373. doi: 10.1126/science.1104799. [DOI] [PubMed] [Google Scholar]
- 2.Horsley V, Pavlath GK. Forming a multinucleated cell: molecules that regulate myoblast fusion. Cells Tissues Organs. 2004;176(1–3):67–78. doi: 10.1159/000075028. [DOI] [PubMed] [Google Scholar]
- 3.Pomerantz J, Blau HM. Nuclear reprogramming: a key to stem cell function in regenerative medicine. Nat Cell Biol. 2004;6(9):810–816. doi: 10.1038/ncb0904-810. [DOI] [PubMed] [Google Scholar]
- 4.Chen EH, Olson EN. Towards a molecular pathway for myoblast fusion in Drosophila. Trends Cell Biol. 2004;14(8):452–460. doi: 10.1016/j.tcb.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 5.Dworak HA, Sink H. Myoblast fusion in Drosophila. Bioessays. 2002;24:591–601. doi: 10.1002/bies.10115. [DOI] [PubMed] [Google Scholar]
- 6.Estrada B, Choe SE, Gisselbrecht SS, et al. An Integrated Strategy for Analyzing the Unique Developmental Programs of Different Myoblast Subtypes. PLoS Genet. 2006;2(2):e16. doi: 10.1371/journal.pgen.0020016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baylies MK, Michelson AM. Invertebrate myogenesis: looking back to the future of muscle development. Curr Opin Genet Dev. 2001;11:431–439. doi: 10.1016/s0959-437x(00)00214-8. [DOI] [PubMed] [Google Scholar]
- 8.Halfon MS, Gisselbrecht S, Lu J, Estrada B, Keshishian H, Michelson AM. New fluorescent protein reporters for use with the Drosophila Gal4 expression system and for vital detection of balancer chromosomes. Genesis. 2002;34:135–138. doi: 10.1002/gene.10136. [DOI] [PubMed] [Google Scholar]
- 9.Greig S, Akam M. Homeotic genes autonomously specify one aspect of pattern in the Drosophila mesoderm. Nature. 1993;362:630–632. doi: 10.1038/362630a0. [DOI] [PubMed] [Google Scholar]
- 10.Queenan AM, Ghabrial A, Schüpbach T. Ectopic activation of torpedo/Egfr, a Drosophila receptor tyrosine kinase, dorsalizes both the eggshell and the embryo. Development. 1997;124:3871–3880. doi: 10.1242/dev.124.19.3871. [DOI] [PubMed] [Google Scholar]
- 11.Michelson AM, Gisselbrecht S, Buff E, Skeath JB. Heartbroken is a specific downstream mediator of FGF receptor signalling in Drosophila. Development. 1998;125:4379–4389. doi: 10.1242/dev.125.22.4379. [DOI] [PubMed] [Google Scholar]
- 12.Vincent S, Wilson R, Coelho C, Affolter M, Leptin M. The Drosophila protein Dof is specifically required for FGF signaling. Mol Cell. 1998;2:515–525. doi: 10.1016/s1097-2765(00)80151-3. [DOI] [PubMed] [Google Scholar]
- 13.Carmena A, Gisselbrecht S, Harrison J, Jiménez F, Michelson AM. Combinatorial signaling codes for the progressive determination of cell fates in the Drosophila embryonic mesoderm. Genes Dev. 1998;12:3910–3922. doi: 10.1101/gad.12.24.3910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Halfon MS, Carmena A, Gisselbrecht S, et al. Ras pathway specificity is determined by the integration of multiple signal-activated and tissue-restricted transcription factors. Cell. 2000;103(1):63–74. doi: 10.1016/s0092-8674(00)00105-7. [DOI] [PubMed] [Google Scholar]
- 15.Nellen D, Burke R, Struhl G, Basler K. Direct and long-range action of a DPP morphogen gradient. Cell. 1996;85(3):357–368. doi: 10.1016/s0092-8674(00)81114-9. [DOI] [PubMed] [Google Scholar]
- 16.Pai L-M, Orsulic S, Bejsovec A, Peifer M. Negative regulation of Armadillo, a Wingless effector in Drosophila. Development. 1997;124:2255–2266. doi: 10.1242/dev.124.11.2255. [DOI] [PubMed] [Google Scholar]
- 17.Lieber T, Kidd S, Alcamo E, Corbin V, Young MW. Antineurogenic phenotypes induced by truncated Notch proteins indicate a role in signal transduction and may point to a novel function for Notch in nuclei. Genes and Development. 1993;7:1949–1965. doi: 10.1101/gad.7.10.1949. [DOI] [PubMed] [Google Scholar]
- 18.Duan H, Skeath JB, Nguyen HT. Drosophila Lame duck, a novel member of the Gli superfamily, acts as a key regulator of myogenesis by controlling fusion-competent myoblast development. Development. 2001;128:4489–4500. doi: 10.1242/dev.128.22.4489. [DOI] [PubMed] [Google Scholar]
- 19.Nose A, Isshiki T, Takeichi M. Regional specification of muscle progenitors in Drosophila: the role of the msh homeobox gene. Development. 1998;125:215–223. doi: 10.1242/dev.125.2.215. [DOI] [PubMed] [Google Scholar]
- 20.Chen EH, Olson EN. Antisocial, an intracellular adaptor protein, is required for myoblast fusion in Drosophila. Dev Cell. 2001;1:705–715. doi: 10.1016/s1534-5807(01)00084-3. [DOI] [PubMed] [Google Scholar]
- 21.Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 22.Donady JJ, Fyrberg EA. Mass culturing of Drosophila embryonic cells in vitro. Tissue Culture Association Manual. 1977;3:685–687. [Google Scholar]
- 23.Singleton K, Woodruff RI. The osmolarity of adult Drosophila hemolymph and its effect on oocyte-nurse cell electrical polarity. Dev Biol. 1994;161(1):154–167. doi: 10.1006/dbio.1994.1017. [DOI] [PubMed] [Google Scholar]
- 24.Choe SE, Boutros M, Michelson AM, Church GM, Halfon MS. Preferred analysis methods for Affymetric GeneChips revealed by a wholly-defined control dataset. Genome Biol. 2005;6:R16. doi: 10.1186/gb-2005-6-2-r16. 1−. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Baldi P, Long AD. A Bayesian framework for the analysis of microarray expression data: regularized t -test and statistical inferences of gene changes. Bioinformatics. 2001;17(6):509–519. doi: 10.1093/bioinformatics/17.6.509. [DOI] [PubMed] [Google Scholar]
- 26.Ruiz-Gomez M, Coutts N, Suster ML, Landgraf M, Bate M. myoblasts incompetent encodes a zinc finger transcription factor required to specify fusion-competent myoblasts in Drosophila. Development. 2002;129(1):133–141. doi: 10.1242/dev.129.1.133. [DOI] [PubMed] [Google Scholar]
- 27.Carmena A, Buff E, Halfon MS, et al. Reciprocal regulatory interactions between the Notch and Ras signaling pathways in the Drosophila embryonic mesoderm. Dev Biol. 2002;244:226–242. doi: 10.1006/dbio.2002.0606. [DOI] [PubMed] [Google Scholar]
- 28.Bour BA, Chakravarti M, West JM, Abmayr SM. Drosophila SNS, a member of the immunoglobulin superfamily that is essential for myoblast fusion. Genes & Dev. 2000;14:1498–1511. [PMC free article] [PubMed] [Google Scholar]
- 29.Ma Y, Creanga A, Lum L, Beachy PA. Prevalence of off-target effects in Drosophila RNA interference screens. Nature. 2006;443(7109):359–363. doi: 10.1038/nature05179. [DOI] [PubMed] [Google Scholar]
- 30.Kulkarni MM, Booker M, Silver SJ, et al. Evidence of off-target effects associated with long dsRNAs in Drosophila melanogaster cell-based assays. Nat Methods. 2006;3(10):833–838. doi: 10.1038/nmeth935. [DOI] [PubMed] [Google Scholar]
- 31.Kennerdell JR, Carthew RW. Use of dsRNA-mediated genetic interference to demonstrate that frizzled and frizzled 2 act in the wingless pathway. Cell. 1998;95:1017–1026. doi: 10.1016/s0092-8674(00)81725-0. [DOI] [PubMed] [Google Scholar]