

Abstract
MT4-MMP (MMP17) belongs to a unique subset of membrane type-matrix metalloproteinases that are anchored to the cell surface via a glycosylphosphatidylinositol moiety. However, little is known about its biochemical properties. Here, we report that MT4-MMP is displayed on the cell surface as a mixed population of monomeric, dimeric, and oligomeric forms. Sucrose gradient fractionation demonstrated that these forms of MT4-MMP are all present in lipid rafts. Mutational and computational analyses revealed that Cys564, which is present within the stem region, mediates MT4-MMP homodimerization by forming a disulfide bond. Substitution of Cys564 results in a more rapid MT4-MMP turnover, when compared with the wild-type enzyme, consistent with a role for dimerization in protein stability. Expression of MT4-MMP in Madin-Darby canine kidney cells enhanced cell migration and invasion of Matrigel, a process that requires catalytic activity. However, a serine substitution at Cys564 did not reduce MT4-MMP-stimulated cell invasion of Matrigel suggesting that homodimerization is not required for this process. Deglycosylation studies showed that MT4-MMP is modified by N-glycosylation. Moreover, inhibition of N-glycosylation by tunicamycin diminished the extent of MT4-MMP dimerization suggesting that N-glycans may confer stability to the dimeric form. Taken together, the data presented here provide a new insight into the characteristics of MT4-MMP and highlight the common and distinct properties of the glycosylphosphatidylinositol-anchored membrane type-matrix metalloproteinases.
Keywords: Cell Migration, Cell Surface Enzymes, Disulfide, Glycosylphosphatidylinositol Anchors, Matrix Metalloproteinase (MMP), Metalloprotease
Introduction
Matrix metalloproteinases (MMPs)2 are zinc-dependent endopeptidases responsible for the hydrolytic cleavage of multiple proteins and thus play key roles in regulation of diverse physiological processes. Because uncontrolled MMP activity can lead to tissue damage and can contribute to pathological conditions, the functions of the various members of the MMP family are strictly regulated, so that their proteolytic tasks are accomplished only within a suitable spatiotemporal window meant to sustain normal cell function. As in many other proteolytic systems, substrate specificity by MMPs is achieved, in part, by specific localization of the protease and its substrate(s) at precise cellular sites. To cover a wide range of cellular locations, the MMP family includes both soluble and membrane-anchored variants. The latter subclass of MMPs, known as membrane type-MMPs (MT-MMPs), is equipped with membrane-anchoring domains, which specifically target the proteases to plasma membranes. The MT-MMPs are classified as transmembrane or GPI-anchored MT-MMPs, a classification that is based on the presence of either a transmembrane domain or a GPI anchor (1). Although much information is available on the regulation and function of transmembrane MT-MMPs, there is a conspicuous paucity of knowledge on the GPI-anchored MT-MMPs. The GPI-MT-MMPs includes two enzymes, MT4-(MMP17) and MT6-MMP (MMP25), that share similar structural features and biochemical properties (2). The presence of a GPI anchor confers a unique subcellular distribution and regulation to MT4- and MT6-MMP, setting them apart from the other MT-MMPs. Moreover, contrary to the transmembrane MT-MMPs, which are insensitive to TIMP-1 inhibition, the GPI-anchored MT-MMPs are similarly inhibited by both TIMP-1 and TIMP-2 (2–5). However, like all MT-MMPs, MT4- and MT6-MMP are produced in a latent inactive form that is activated by cleavage of an RXXR motif located between the pro-domain and the catalytic domain by furin-like convertases (2). Previously, we reported some of the biochemical and biological properties of MT6-MMP (5, 6). The focus of this report is MT4-MMP.
MT4-MMP was originally cloned from a human breast carcinoma cDNA library (7–9). Analyses of MT4-MMP mRNA expression in human and mouse tissues showed readily detectable mRNA in brain, ovary, uterus, spleen, and leukocytes (eosinophils, lymphocytes, and monocytes but not neutrophils) (7, 10–14). The physiological role(s) of MT4-MMP is still unclear. However, several studies provided information on MT4-MMP function in physiological and pathological conditions. Mice lacking MT4-MMP show no evident pathologies, when compared with wild-type mice (14), suggesting that MT4-MMP is not required for normal development and survival. Deletion of MT4-MMP protects mice against interleukin-1β-induced loss of articular glycosaminoglycans implicating MT4-MMP in control of cartilage aggrecan integrity, possibly through regulation of aggrecanase activation (15, 16). A recent report described that MT4-MMP null mice exhibit abnormally diminished thirst, possibly due to disrupted thirst regulation in the brain (17). In pathological conditions, MT4-MMP protein expression was found to be elevated in human osteoarthritic cartilage (15), breast carcinomas (18), and in head and neck cancer (19). Functionally, ectopic expression of MT4-MMP in MDA-MB-231 breast cancer cells was shown to promote tumor growth and lung metastasis (18). Ectopic expression of MT4-MMP in a tongue squamous cell carcinoma cell line also increased lung colonization after tail vein inoculation (19), further demonstrating the ability of MT4-MMP to promote experimental metastasis. However, conflicting results were reported regarding the ability of MT4-MMP to promote in vitro cell invasion (18–21), and therefore the mechanism(s) by which MT4-MMP enhances metastasis remains unclear.
Structurally, MT4-MMP contains all the typical MMP domains, including the pro-domain, catalytic domain, hinge, and hemopexin-like domain (22). MT4-MMP, like MT6-MMP, also possesses a short stem region of 50 residues that links the hemopexin-like domain with the GPI anchor (2). The stem region of MT4- and MT6-MMP contains two and three cysteine residues, respectively, which in the case of MT6-MMP are involved in enzyme dimerization (2, 5, 6). Indeed, surface MT6-MMP is displayed as a disulfide-linked homodimer. At present, the profile of MT4-MMP forms and whether MT4-MMP is able to generate homodimers has not been reported. In this study, we examine the profile of MT4-MMP expressed by tissues and cells and the role of the cysteine residues in the stem region in enzyme dimerization and its implication for enzyme function. The data presented here shed new light on the biochemical characteristics of this unique and poorly studied MT-MMP.
EXPERIMENTAL PROCEDURES
Cell Culture
MDCK (CCL-34) cells were obtained from the American Type Culture Collection (ATCC) and cultured in minimum essential medium (MEM) supplemented with 10% fetal bovine serum (FBS), 1% antibiotics penicillin and streptomycin sulfate, 1 mm sodium pyruvate and were maintained at 37 °C in an atmosphere of 95% air and 5% CO2.
Recombinant Proteins and Antibodies
The rabbit monoclonal antibody (r-mAb) against the catalytic region (EP1270Y) of human MT4-MMP was obtained from Epitomic (Burlingame, CA) or Abcam Inc. (Cambridge, MA). A rabbit polyclonal antibody to human caveolin was purchased from BD Biosciences. The mAb against β-actin was obtained from Sigma. Human recombinant TIMP-2 was expressed and purified to homogeneity, as described previously (23).
cDNA Constructs and Transfections
The full-length human MT4-MMP cDNA was a generous gift from Dr. Motoharu Seiki (University of Tokyo, Japan). The MT4-MMP cDNA (GenBankTM accession number AB021225.2) was subcloned into the expression plasmid pcDNATM 3.1/myc-His (−) A (Invitrogen) to generate pcMT4 for stable transfection. Single Cys → Ser substitutions at Cys564 and Cys566 were generated by the QuikChangeTM site-directed mutagenesis kit (Stratagene, La Jolla, CA) using sequence-specific primers, and wild-type pcMT4 cDNA, as the template. The sequences of the inserts and junctions in the vectors were verified by DNA sequencing. MDCK cells were stably transfected with plasmids encoding the cDNA of either wild-type MT4-MMP (referred to as MDCK-MT4 cells), MT4-MMP cysteine mutants (referred to as MDCK-MT4C564S or MDCK-MT4C566S cells), or the cDNA of catalytically inactive MT4-MMP bearing an E252A substitution (referred to as MDCK-MT4E/A cells). Forty eight hours after transfection, the cells were selected for drug resistance, Geneticin® (G418 sulfate, Invitrogen). Pooled populations were selected using conventional approaches. MDCK-transfected cells with pcDNA3.1 without inserts (empty vector and thus referred to as EV) were also generated. The full-length mouse MT4 MMP cDNA was obtained from Open Biosystems Products (Huntsville, AL) and subcloned into the pcDNATM 3.1/myc-His(−) A vector for expression.
Preparation of Cell Lysates and Immunoblot Analyses
Cells were solubilized in cold lysis buffer (25 mm Tris-HCl (pH 8.0), 100 mm NaCl, 1% IGEPAL CA-630, and 60 mm octyl glucoside) supplemented with 20 mm N-ethylmaleimide (Sigma) and protease inhibitor (PI) tablet (Complete, Mini, EDTA-free, Roche Applied Science). Briefly, the cells were lysed with cold lysis buffer for 1 h on ice, and the cell debris was removed by centrifugation (13,000 rpm) for 15 min at 4 °C. Protein concentration was determined by the BCA kit (Pierce). An aliquot of each lysate was mixed with 4× Laemmli sample buffer with or without 2.5% β-mercaptoethanol (β-ME), boiled (95 °C, 5 min), and resolved by SDS-PAGE followed by immunoblot analyses using various antibodies.
Surface Biotinylation and PI-PLC Treatment
MDCK cells stably expressing wild-type or mutant MT4-MMP, in 6-well plates, were surface-biotinylated with the cell-impermeable EZ-link-sulfo-NHS-biotin (Pierce), as described previously (6). The cells were then lysed with 0.25 ml/well of cold lysis buffer on ice. After a brief centrifugation, equal protein amounts of the supernatants were incubated with NeutrAvidin beads (Pierce) for 2 h at room temperature. The beads were washed three times with harvest buffer (0.5% SDS, 60 mm Tris-HCl (pH 7.5), 2 mm EDTA) supplemented with 2.5% Triton X-100, and bound proteins were eluted with 2× Laemmli SDS sample buffer with or without 2.5% β-ME. Samples were boiled and resolved by SDS-PAGE followed by transfer to a nitrocellulose membrane. The proteins were detected with r-mAb EP1270Y. In some experiments, MDCK-MT4 cells were washed with cold phosphate-buffered saline (PBS) and treated with 0.3 IU/well of phosphatidylinositol-specific phospholipase C (PI-PLC, Molecular Probes) in serum-free medium for 30 min at 37 °C in a tissue culture incubator. The supernatant was clarified and concentrated with Microcon® centrifugal filter devices (Millipore) in the presence of EDTA-free protease inhibitor (PI) mixture. An aliquot of each concentrated supernatant was mixed with 4× Laemmli SDS sample buffer with or without β-ME, boiled, and resolved by SDS-PAGE followed by immunoblot analysis.
Deglycosylation of MT4-MMP
To inhibit O- or N-glycosylation, MDCK-MT4 cells were treated with either 2 mm benzyl-2-acetamido-2-deoxy-α-d-galactopyranoside (BGN, Sigma) or tunicamycin (1 or 5 μg/ml, Sigma), respectively, diluted in dimethyl sulfoxide (DMSO, final concentration 0.2%). Control cells received 0.2% of DMSO. In some experiments, MDCK-MT4 cells received tunicamycin (5 μg/ml) and the broad spectrum MMP inhibitor BB94 (10 μm). The cells were then incubated (20 h) at 37 °C in serum-free media (for BGN treatment) or complete media (for tunicamycin treatment) in a tissue culture incubator. Then the cells were lysed in 150–200 μl of ice-cold lysis buffer, and the lysates were resolved by reducing SDS-PAGE followed by immunoblot analyses, as described above. In vitro enzymatic deglycosylation of MT4-MMP was performed using the GlycoProTM enzymatic deglycosylation kit according to the manufacturer's instructions (ProZyme, San Leandro, CA). Briefly, 80 μg of MDCK-MT4 lysates, prepared as described above, were incubated with several glycosidases, including N-glycanase (PNGase F), sialidase ATM, and O-Glycanase® or combination of all three in 50 μl of reaction buffer (50 mm sodium phosphate (pH 7.0)) supplemented with 0.4% SDS, 200 mm β-mercaptoethanol, and 0.75% Nonidet P-40. After a 24-h incubation at 37 °C, the reaction was stopped by boiling the samples in reducing Laemmli sample buffer. An equal amount of each sample was resolved by 10% SDS-PAGE followed by immunoblot analyses.
Lipid Raft Isolation
The procedure was adopted from Kim et al. (24) and Zhuang et al. (25). Briefly, cells from three 100-mm Petri dishes were washed and detached with PBS containing 2 mm EDTA on ice. The cells were collected by centrifugation, and the cell pellet was resuspended in 0.5 ml of buffer A (25 mm MES, 150 mm NaCl (pH 6.5)) on ice. An equal volume of the same buffer containing 2% Triton X-100, 2 mm sodium orthovanadate, and PI mixture was added. The cell suspension was homogenized with Dounce homogenizer and incubated on ice. After 30 min, the homogenate was adjusted to 35% sucrose by adding an equal volume of 70% sucrose in buffer A. One ml of the homogenate was placed at the bottom of an ultracentrifuge tube, and two 4-ml layers of 30 and 5% sucrose in buffer A were overlaid above it. Centrifugation was performed at 100,000 × g for 24 h in a Beckman SW41 rotor (Beckman Coulter). A light scattering band was observed between the 5 and 30% sucrose interphase. Nine 1-ml fractions were collected from the top of the gradient and resolved by SDS-PAGE following by immunoblot analyses.
Preparation of Crude Plasma Membrane Fraction
Crude plasma membrane fractions were prepared as described previously (6). Briefly, MDCK-MT4 cells were washed with PBS and scraped into TBS (25 mm Tris-HCl (pH 7.4), and 50 mm NaCl) containing PI mixture. After centrifugation, the cell pellet was resuspended in TBS supplemented with 8.5% sucrose and homogenized in a Dounce homogenizer. After removal of cell debris at 19,000 × g for 30 min at 4 °C, the supernatant was subjected to ultracentrifugation at 100,000 × g for 75 min at 4 °C. The pellet containing crude plasma membrane was resuspended in TBS containing PI and stored at −80 °C. The protein concentration was determined by the BCA procedure (Pierce).
MT4-MMP Turnover
For evaluation of MT4-MMP turnover, serum-starved MDCK cells expressing wild-type or mutant (cysteine mutants) MT4-MMP cells were incubated with 50 μg/ml cycloheximide (CHX, Sigma) in serum-free MEM. At each designated time point (0–6 h), the cells were lysed with 0.2 ml of ice-cold lysis buffer. Protein concentration was determined by BCA kit (Pierce), and equal amounts of the lysates were resolved by reducing 10% SDS-PAGE followed by immunoblot analyses using r-mAb EP1270Y. Densitometric analyses of the bands in the blots were performed with ImageJ software (Version 1.42q, by Wayne Rasband, NIH, Bethesda) and are represented in graphs as percentage of control, 100%, time 0.
Cell Migration and Invasion Assays
For determination of cell migration, MDCK-EV, MDCK-MT4, and MDCK-MT4E/A cells (1 × 105/well) suspended in serum-free MEM were seeded into the upper chamber of 6.5-mm diameter (8.0-μm pore size) TranswellsTM (Costar). The lower chamber contained MEM supplemented with 5% FBS as chemoattractant. The migration of cells was allowed to proceed at 37 °C in a humidified atmosphere with 5% CO2 over a 6-h period. After incubation, the cells that migrated to the lower side of the filter were fixed and counted as described below. The experiment was repeated three times, and each treatment was done in quadruplicate. Invasion assays were performed in 6.5-mm diameter TranswellTM chambers (Costar). The upper side of the polycarbonate membrane (8.0-μm pore size) was coated with 80 μg of Matrigel basement membrane (BD Biosciences). MDCK-EV, MDCK-MT4, MDCK-MT4E/A, MDCK-MT4C564S, and MDCK-MT4C566S cells (1 × 105 cells/well) were seeded on the upper side of the filter in serum-free MEM. The lower chamber was filled with MEM containing 5% FBS. Some wells received MDCK-MT4 cells that were preincubated (30 min) with 200 nm purified human recombinant TIMP-2, and the medium in the lower chambers was also supplemented with 200 nm rTIMP-2. The chambers were placed in an incubator at 37 °C in a humidified atmosphere of 95% air and 5% CO2. After 40 h, the cells on the upper side of the filters were mechanically removed, and the cells that had migrated to the lower side were fixed and stained with Diff-Quick staining kit (Andwin Scientific). The filters were photographed under a light microscope, and the cells were counted in five representative fields. The experiment was repeated three times, and each treatment was done in triplicate.
Computational Method
The homology method by SWISS-model identified a template for the hemopexin-like domain (residues 333–526) from the existing crystal structures but not the stem domain. A more fundamental residue-by-residue analysis was carried out using San Diego Supercomputer Center Biology Workbench and the homology alignment analysis between the MT4-MMP sequence and the MT6-MMP sequence (6). A global alignment between the two MT-MMP stems was carried out using Prot and Gonnet scoring matrices (26). Opening and extension gap penalties were given values of −19 and 0, respectively, to achieve the maximal gap extension and the minimum of gap introduction. Similar to modeling performed for MT6-MMP, the secondary structure for the MT4-MMP stem sequence was predicted by Garnier-Osguthorbe-Robson (27) and Chou-Fasman (28) methods. The modeling started by building a strand structure within Sybyl (Tripos, 8.0) and the stem region of MT4-MMP, from Gly527 to Ala576 (including 621 atoms), was examined. The terminal residue Gly527 in the simulation was protonated, and the C-terminal Ala576 was deprotonated. Atom charges and types were assigned with the 2002 library of the amino acids in AMBER 9.0. Solvent molecules (a total of 52,871 TIP3P water molecules) were added to encompass the peptide. The entire system of the stem peptide and water complex was energy-minimized using the method described previously (29, 30).
Statistical Analysis
Statistical analyses were performed using t test using SigmaPlot 10.0 software. Values for all experiments are expressed as means ± S.E. of a minimum of three independent experiments.
RESULTS AND DISCUSSION
Profile of MT4-MMP Forms
We previously demonstrated that MT6-MMP, a close homologue of MT4-MMP, is found in a disulfide-linked homodimeric form (5, 6). Therefore, we wished to determine whether MT4-MMP also generates species consistent with dimerization. Previous studies demonstrated expression of MT4-MMP protein in cell lines and eosinophils (10, 19, 21, 31). However, these studies could not be used as a reference, because the full spectrum of MT4-MMP species at reducing and nonreducing conditions was not shown (10, 19) or protein expression was not displayed by immunoblotting (21, 31). Attempts to detect natural MT4-MMP in several cell lines were unsuccessful in our hands, possibly due to the low levels of expression. Mouse brain, lungs, and uterus have been shown to express high levels of MT4-MMP mRNA (14, 17). Therefore, we examined mouse brain and uterus tissues (Fig. 1A, lanes 1 and 2, respectively) for MT4-MMP protein using the r-mAb EP1270Y. Reactivity of this antibody with mouse MT4-MMP was confirmed using mouse MT4-MMP expressed in COS-1 cells (data not shown). The mouse tissues were homogenized under conditions that prevent oxidation, and the samples were resolved by reducing and nonreducing SDS-PAGE, followed by immunoblot analyses. Under nonreducing conditions (Fig. 1A, upper panel), mMT4-MMP was detected in both tissues as an ∼120- and ∼55–57-kDa form. After reduction, the ∼120-kDa species was converted to an ∼55-kDa form (Fig. 1A, lower panel). Based on the molecular mass and the known presence of two cysteine residues in the stem region of mouse MT4-MMP, we posited that the ∼120-kDa species represents enzyme dimers (referred to as MT4D in the figures), whereas the ∼55-kDa species represents the monomeric form (referred to as MT4M). To further characterize the protein forms of MT4-MMP, we generated pool populations of MDCK cells expressing human MT4-MMP (MDCK-MT4). The cells were analyzed for MT4-MMP species at the cell surface and in the cell lysates under nonreducing and reducing conditions. Under nonreducing conditions (Fig. 1B, lane 2, upper panel), surface MT4-MMP was detected as species of ∼240, ∼120, and ∼55–57 kDa, which were consistent with being oligomeric (MT4O), dimeric (MT4D), and monomeric (MT4M) MT4-MMP forms, respectively. It should be noted that although the monomeric and dimeric forms were readily detected at the cell surface, the detection of the oligomeric form was inconsistent. However, it should be kept in mind that differences in the relative levels of the MT4-MMP forms may be due in part to a differential affinity of this particular antibody for each enzyme species due to epitope reactivity under reducing and nonreducing conditions.
FIGURE 1.

Profile of MT4-MMP species. A, mouse brain (lane 1) and uterus (lane 2) tissues were homogenized in cold lysis buffer as described under “Experimental Procedures.” Homogenates (80 μg/lane) were resolved by 7.5% (−β-ME, upper panel) or 10% (+β-ME, lower panel) SDS-PAGE followed by transfer to a nitrocellulose membrane. Membranes were probed with r-mAb EP1270Y directed against the catalytic domain of MT4-MMP. B, MDCK-MT4 (lanes 2 and 4) and MDCK-EV (lanes 1 and 3) cells were surface-biotinylated as described under “Experimental Procedures.” The cells were lysed in 200 μl of cold lysis buffer, and protein concentrations were determined. One-third of the total lysate from each treatment was kept in the cold (total lysate); whereas the rest of each fraction was subjected to the pulldown assay of NeutrAvidin beads. The bound proteins were eluted with 2× Laemmli SDS sample buffer and divided in 2 aliquots; one received β-ME, and the other did not. The total lysates were also divided in two fractions; one was supplemented with β-ME and the other was not. The samples were resolved by 7.5% SDS-PAGE for reducing conditions (upper panel) and 10% SDS-PAGE for nonreducing conditions (lower panel) and then processed for immunoblot analysis using antibody EP1270Y. C, crude plasma membranes were prepared from MDCK-EV and MDCK-MT4 cells as described under “Experimental Procedures.” Twenty μg of each plasma membrane fraction were resolved by 7.5% (−β-ME, upper panel) or 10% (+β-ME, lower panel) SDS-PAGE followed by immunoblot analysis with antibody EP1270Y. D, MDCK-EV and MDCK-MT4 cells were treated with or without 0.3 IU/well of PI-PLC. After 30 min, the PI-PLC-released fraction was collected, clarified, and concentrated as described under “Experimental Procedures.” An aliquot of each concentrated supernatant was mixed with Laemmli SDS-sample buffer with or without β-ME, boiled, and resolved by 7.5% (−β-ME, upper panel) or 10% (+β-ME, lower panel) SDS-PAGE followed by immunoblot analysis with r-mAb EP1270Y. MT4O, MT4-MMP oligomeric form; MT4D, MT4-MMP dimeric form; MT4M, MT4-MMP monomeric form; pro-MT4, precursor/latent MT4-MMP.
Similarly to the cell surface fraction, total cell lysates resolved under nonreducing conditions showed species of MT4-MMP consistent with being oligomeric, dimeric, and monomeric forms (Fig. 1B, lane 4, upper panel). However, the lysates displayed an additional immunoreactive species of ∼72 kDa both under reducing and nonreducing conditions. At present, the precise nature of the 72-kDa species (Fig. 1B, lane 4) is unknown. However, based on its molecular mass, we surmise that this species represents the intracellular precursor form of MT4-MMP (before transamidase cleavage of the hydrophobic C-terminal region and incorporation of the GPI anchor) and/or the latent species of GPI-anchored MT4-MMP, and thus it is referred to as pro-MT4. Upon reduction, surface MT4-MMP (Fig. 1B, lane 2, lower panel) converted to a single species of ∼60 kDa. In the total cell lysates (Fig. 1B, lane 4, lower panel) reduced MT4-MMP was detected as two species of ∼72- and ∼57 kDa, which probably represent the precursor/latent form and active form of MT4-MMP, respectively. We also noticed a slight difference in electrophoretic mobility between surface and intracellular MT4M (Fig. 1B, lane 2 versus lane 4, respectively), which suggested the possibility of differential glycosylation between surface (mature) and intracellular forms (discussed later).
To further analyze the profile of MT4-MMP species, we examined crude plasma membranes isolated from MDCK-MT4 and MDCK-EV cells under reducing and nonreducing conditions. As shown in Fig. 1C, plasma membranes of MDCK-MT4 cells display the three forms of MT4-MMP (Fig. 1C, lane 2, upper panel), in agreement with the results of the surface biotinylation (Fig. 1B, lane 2). To further aid in distinguishing surface from intracellular protease forms, MDCK-MT4 cells were treated with PI-PLC, which is known to release GPI-anchored proteins from the cell surface. Immunoblot analysis of the PI-PLC-released fraction, under nonreducing conditions, revealed the ∼120- (MT4D) and ∼57 (MT4M)-kDa species, further indicating that these are GPI-anchored forms present on the cell surface (Fig. 1D, lane 4, upper panel). In contrast, neither the oligomeric species nor the precursor/pro-forms of MT4-MMP were detected in the PI-PLC-released fraction. The lack of detection of the oligomeric form in this fraction may be due to resistance to PI-PLC attack or levels of expression that are below the detection limit under the experimental conditions used. Reduced MT4-MMP released by PI-PLC was displayed as a single form of ∼57 kDa (Fig. 1D, lane 4, lower panel). Collectively, both the tissue and the cell data with natural and recombinant MT4-MMP, respectively, as indicated by the surface biotinylation and PI-PLC treatment, suggest that the physiological structural organization of MT4-MMP comprises monomeric and dimeric/oligomeric species.
GPI-anchored proteins are known to associate with glycosphingolipids and cholesterol-enriched membrane microdomains referred to as lipid rafts. To investigate the distribution of MT4-MMP forms in lipid rafts, we evaluated the buoyant density of MT4-MMP in discontinuous sucrose gradients after extraction of MDCK-MT4 cells with Triton X-100. After ultracentrifugation, the fractions were collected and resolved in reducing and nonreducing gels. These analyses showed that MT4-MMP was entirely recovered in the fraction containing the detergent-resistant lipid rafts (Fig. 2, fraction 5, upper and middle panels), as indicated by the presence of caveolin, a lipid raft marker. When resolved under nonreducing conditions, this fraction displayed all forms of MT4-MMP, namely the monomeric, dimeric, and oligomeric forms. Under reducing conditions, this fraction displays the 57-kDa monomeric form of MT4-MMP (Fig. 2, fraction 5, middle panel), as expected. These data indicate that, under the experimental conditions used here, all MT4-MMP forms, like MT6-MMP (5, 6), localize in lipid rafts, consistent with the affinity of the GPI anchor for these lipid microdomains.
FIGURE 2.

MT4-MMP is targeted to lipid rafts. MDCK-MT4 cells were homogenized in cold lysis buffer containing 1% Triton X-100 and protease inhibitors as described under “Experimental Procedures” and subjected to step sucrose gradient. After centrifugation at 100,000 × g for 24 h at 4 °C, nine fractions, 1 ml of each, were collected from the top of the gradient, and an equal volume of each fraction was mixed with 4× Laemmli SDS-sample buffer with or without β-ME. The samples were resolved by 7.5% (−β-ME, upper panel) or 10% (+β-ME, lower panel) SDS-PAGE followed by immunoblot analysis with r-mAb EP1270Y. The blot with reduced protein samples was re-probed with a polyclonal antibody to human caveolin. MT4O, MT4-MMP oligomeric form; MT4D, MT4-MMP dimeric form; MT4M, MT4-MMP monomeric form.
MT4-MMP Glycosylation
The differences in molecular mass between surface and intracellular MT4M (Fig. 1B, lane 2 versus lane 4, respectively) raised the question as to whether MT4-MMP is glycosylated. Previous analyses of putative glycosylation sites in members of the MT-MMP subfamily using the NetNGlyc (NetNGlyc 1.0) and NetOGlyc (NetOGlyc 2.0) programs predicted that the hinge region of MT4-MMP contains both O- and N-glycosylation sites at Ser302, Thr306, Ser323, Ser324, and Asn321, respectively (32). However, a newer version of the NetOGlyc program (NetOGlyc 3.1) (33) does not predict O-glycosylation sites in MT4-MMP. NetNGlyc 1.0 also predicts Asn140, in the catalytic domain of MT4-MMP, as a putative N-glycosylation site. However, Asn321 has a higher probability to be N-glycosylated than Asn140 according to the software.
To test whether MT4-MMP is glycosylated, we used BGN and tunicamycin, two established inhibitors of O- and N-glycosylation, respectively. BGN is a competitive inhibitor of galactosyltransferases, which blocks elongation of O-glycosyl chains. Tunicamycin inhibits eukaryotic N-acetylglucosamine transferases and blocks N-glycosylation. After treatment with the inhibitors, the MDCK-MT4 cell lysates were resolved by SDS-PAGE and subjected to immunoblot analyses. Changes in relative electrophoretic mobility of MT4-MMP from untreated and treated cells were considered to be indicative of glycosylation. As shown in Fig. 3A, BGN treatment of MDCK-MT4 cells did not alter the electrophoretic mobility of MT4-MMP when compared with untreated cells suggesting that MT4-MMP is not O-glycosylated (Fig. 3A), consistent with the prediction of the NetOGlyc 3.1 program. Interestingly, the program does predict that MT6-MMP is O-glycosylated. Consistently, we found that BGN treatment of cells (colon cancer HCT-116) expressing MT6-MMP caused a shift in the electrophoretic mobility of MT6-MMP3 suggesting that MT6-MMP, as opposed to MT4-MMP, is O-glycosylated.
FIGURE 3.

MT4-MMP is N-glycosylated. A–C, MDCK-MT4 cells were treated with either BGN (A) or tunicamycin (B and C), as described under “Experimental Procedures.” A, lysates of MDCK-MT4 cells treated with either DMSO (lane 1) or 2 mm BGN (lane 2). Ctrl, control. B, lysates of MDCK-MT4 cells treated with either DMSO (lane 1) or two concentrations of tunicamycin (Tunic.) (lane 2, 5 μg/ml; lane 3, 1 μg/ml). C, lysates of MDCK-MT4 cells treated with either DMSO (lane 1), 5 μg/ml tunicamycin (lane 2), or 5 μg/ml tunicamycin + 10 μm BB94 (lane 3). D, lysate from MDCK-MT4 cells was incubated with reaction buffer (lane 1) or with N-glycanase (N-Gly, lane 2), sialidase A (Sial.A, lane 3), O-glycanase (O-Gly, lane 4), or a combination of the three enzymes (lane 5), as described under “Experimental Procedures.” The reaction was stopped by adding reducing Laemmli SDS sample buffer (4×). The samples in A (30 μg/lane) were run under reducing conditions in 10% SDS-PAGE. The samples in B (lane 1, 5 μg/lane, and lanes 2 and 3, 30 μg/lane) were resolved by reducing conditions in 10% SDS-PAGE, which were run for a longer time to obtain better separation. Samples in C (lane 1, 7 μg/lane, and lanes 2 and 3, 25 μg/lane) were resolved by nonreducing 7.5% SDS-PAGE. Samples in D (lanes 1–5, 20 μg/lane) were resolved by reducing 10% SDS-PAGE. Detection of MT4-MMP (A–D) was performed by immunoblot analyses using r-mAb EP1270Y. MT4M, MT4-MMP monomeric form; MT4D, MT4-MMP dimeric form. Arrows indicate the change in MT4-MMP mobility. The band (∼72 kDa) above MT4M is likely the precursor/pro-form of MT4-MMP.
Treatment with tunicamycin caused a significant reduction in overall MT4-MMP expression levels. However, when higher levels of tunicamycin-treated cell lysates were loaded in the gel and the gel was run for a longer time, we clearly detected a downward shift in the electrophoretic mobility of the 57-kDa species (Fig. 3B, lanes 2 and 3) and to a lesser extent of the 72-kDa form of MT4-MMP, when compared with MT4-MMP displayed by lysates of control cells (Fig. 3B, lane 1), under reducing conditions. Under these conditions, the monomer in untreated cells was detected as a doublet, possibly representing a mixture of N-glycosylated species (Fig. 3B, lane 1).
When the samples were resolved under nonreducing conditions (Fig. 3C), the mobility shift in response to tunicamycin was less noticeable. However, we observed a significant reduction in MT4-MMP dimers in the tunicamycin-treated cells (Fig. 3C, lane 2). Because of the reduced levels of MT4-MMP expression, comparison of relative levels of monomer and dimers between untreated and treated cells was not feasible. However, the data could be analyzed in terms of changes in the monomer/dimer ratio within each condition. Semi-quantitative densitometry analyses showed that the monomer/dimer ratio was almost 1:1 in the untreated cells (Fig. 3C, lane 1). In contrast, the tunicamycin-treated cells displayed a monomer/dimer ratio of 5:1, suggesting that N-glycosylation is required for dimerization. To determine whether the decrease in dimerization was due in part to autocatalytic degradation, tunicamycin-treated cells were also incubated with the broad spectrum MMP inhibitor BB94. Although BB94 treatment caused a slight increase in the levels of both monomeric and dimeric forms when compared with cells treated with tunicamycin alone (Fig. 3C, lane 3 versus lane 2), it did not change the monomer/dimer ratio suggesting that the decrease in dimerization was not due to autocatalytic degradation.
To further examine MT4-MMP glycosylation, lysates of MDCK-MT4 cells were digested with various purified O- and N-glycanases and evaluated by SDS-PAGE. As shown in Fig. 3D, digestion with N-glycanase produced a clear downward shift in the molecular mass of the ∼57-kDa species of MT4-MMP (Fig. 3D, lane 2). However, N-glycanase digestion had no apparent effect on the mobility of the 72-kDa species. Possibly, this species escapes in vitro deglycosylation because of steric hindrance. Treatment with either O-glycanase or sialidase A had no effect on MT4-MMP migration profile (Fig. 3D, lanes 3 and 4). Because N-glycanase is known to remove virtually all N-linked oligosaccharides, this result further supports the assertion that MT4-MMP is N-glycosylated. Collectively, these data are consistent with MT4-MMP being modified by N-linked glycosylation and suggests that this modification might be required for protease dimerization. N-Linked glycosylation of MT4-MMP may contribute to the stability of the dimers by promoting noncovalent interactions. Alternatively, N-glycans may play a role during the process of MT4-MMP folding and generation of disulfide bonds (34). However, because of changes in overall cellular N-glycan production by tunicamycin, we cannot rule out the possibility that the effects observed on dimerization may be indirect. Thus, substitution at the putative N-glycosylation sites of MT4-MMP (Asn321 and Asn140) will help to address the role of N-glycosylation on protease organization and function in a more specific manner. Regardless, the experimental data presented here highlight a unique post-translational modification of MT4-MMP that may set it apart from other MT-MMPs, in general, and from its closely homologue MT6-MMP, in particular, and which may result in unique functional consequences.
Cys564 Mediates MT4-MMP Dimerization
We previously demonstrated that MT6-MMP generates disulfide-linked homodimers via cysteine residues within the stem region (6). Although MT6-MMP possesses three Cys residues at the stem region, MT4-MMP possesses two cysteine residues at positions 564 and 566 (ATG used as position 1), which are conserved across species. We hypothesized that these cysteine residues may form disulfide bridges and be responsible for MT4-MMP homodimerization. To test this hypothesis, we substituted Cys564 and Cys566 of the stem region for a serine residue (Fig. 4A). The resultant MT4-MMP mutants were stably expressed in MDCK cells and analyzed for the presence of high molecular weight species by reducing and nonreducing SDS-PAGE, followed by immunoblot analysis. As shown in Fig. 4B, cells expressing wild-type MT4-MMP display the 57- and 120-kDa forms on the cell surface (Fig. 4B, lane 6, upper panel, nonreducing conditions). Lysates of cells expressing wild-type MT4-MMP also displayed an additional high molecular weight form (MT4O) and an ∼70-kDa form (pro-MT4) (Fig. 4B, lane 2, upper panel). Cells expressing the Cys566 mutant of MT4-MMP displayed the monomeric form (MT4M) and low levels of the dimeric form (MT4D) on the cell surface (Fig. 4B, lane 8, upper panel), although their lysates contained the dimeric and oligomeric forms (Fig. 4B, lane 4, upper panel). In contrast, both the dimeric and oligomeric forms of MT4-MMP were absent in cells expressing the Cys564 mutant (Fig. 4B, lanes 3 and 7, upper panel), suggesting that Cys564 is essential for generation of these species. Higher levels of monomer (MT4M) were observed on the surface of MDCK cells expressing the Cys564 and Cys566 mutants, when compared with wild-type MT4-MMP (Fig. 4B, lanes 6–8, upper panel). Interestingly, a small shift in electrophoretic mobility was noted when the cysteine mutants were resolved under reducing conditions, which was more prominent with the Cys564 mutant (Fig. 4B, lower panel), possibly due to different folding of the mutants and/or differences in the extent of glycosylation as a consequence of alterations in dimerization.
FIGURE 4.

Role of cysteine residues in MT4-MMP dimerization. A, schematic diagram of MT4-MMP domains and cysteine residues located within the stem region. Cat., catalytic. B, effect of stem's cysteine substitutions on the profile of MT4-MMP forms. MDCK-EV, MDCK-MT4, and MDCK cells expressing the Cys564 or Cys566 mutants were surface-biotinylated as described under “Experimental Procedures.” The cells were lysed in 200 μl of cold lysis buffer (supplemented with protease inhibitors and 20 mm N-ethylmaleimide), and protein concentrations were determined. One-third of the lysate from each transfectant was kept in the cold (total lysate), whereas the rest of the fraction was subjected to NeutrAvidin bead pulldown assays. The bound proteins were eluted with 2× Laemmli SDS sample buffer and divided in 2 aliquots; one received β-ME, and the other did not. The total lysates were also divided in two fractions; one with β-ME, and the other without β-ME. The samples were resolved by 7.5% SDS-PAGE for nonreducing conditions (upper panel) and 10% SDS-PAGE for reducing conditions (lower panel) and then processed for immunoblot analysis using r-mAb EP1270Y. The blot for the total lysate samples (reducing conditions) was re-probed with a mAb against β-actin, to compare the overall levels of expression of the proteases (wild-type and Cys mutants) under steady-state conditions. MT4O, MT4-MMP oligomeric form; MT4D, MT4-MMP dimeric form; Pro-MT4, MT4-MMP precursor/latent form; MT4M, MT4-MMP monomeric form.
Analyses of MT4-MMP Stem Peptide by Computational Molecular Dynamics
Our mutational experiments indicated that MT4-MMP dimerization was completely prevented by mutations at Cys564 and diminished by mutations at Cys566. Interestingly, an alignment of the sequence of the stem region of MT4- and MT6-MMP shows that Cys564 of MT4-MMP corresponds to Cys530 of MT6-MMP and Cys566 to Cys532 (Fig. 5A). The analysis for the secondary structure of MT6-MMP stem was made recently (6). The results had revealed that the three cysteines in MT6-MMP stem region likely reside on a helical structure (Fig. 5A). Moreover, Cys530 and Cys534 are involved in an intramolecular disulfide bond, whereas Cys532 plays a role in MT6-MMP dimerization via disulfide bond formation (6). As indicated earlier, there are only two cysteines, Cys564 and Cys566, in the stem of MT4-MMP. If the similarity in structures of MT4-MMP and MT6-MMP were to extend to the motif for their respective dimerization, then a reasonable conclusion would be that Cys566 should be the cysteine involved in MT4-MMP dimerization, contrary to the mutagenesis results. The following computational analyses provide an explanation for the experimental data. The sequence analysis methods of Garnier-Osguthorbe-Robson (27) and Chou-Fasman (28) predicted conformations consistent with β-strand from Gly560 to Thr567 and for the two residues Gly575 to Ala576 and helix for Ser534 to Ala541 (Fig. 5A). The prediction of the secondary structure of the remaining residues was more difficult, but at least five residues, Gly533, Ala542, Asp559, Ser568, and Pro574, are involved to turns linked to either a β-strand or a helix. This analysis indicates that this 50-amino acid stretch in MT4-MMP is likely to contain flexible elements, which adds to the difficulty in structural elucidation. Based on the predicted structure for the stem region, we performed molecular dynamics simulations to gain further insight into the structural dynamics of cysteine residues within the stem region of MT4-MMP. This method allows for exploration of the conformational space that can be occupied by the given structural element. The side chain thiols of the two residues Cys564 and Cys566 did not approach each other significantly (C564-C566, Fig. 5B, panel I), which indicates that they are not able to form an intramolecular disulfide bond. Furthermore, we observed the stability of a structural segment starting from Tyr561 through Cys566 for a total of 3.5 ns of dynamics simulations (Y561-C566, Fig. 5B, panel II). The segment encompasses the predicted strand structure (…GYEVCSCT…, Fig. 5A). The distance between Tyr561 and Cys564 (Y561-C564, Fig. 5B, panel III) is in excess of 10 Å. As a result of analysis of the three distances, Fig. 5B, panels I–III, C564-C566, Y561-C566, and Y561-C564, a model is proposed for the structure of the strand portion in the stem peptide. The strand and its flanking regions form a motif (Fig. 6A) in which Cys564 is poised for the intermolecular disulfide bond formation by two MT4-MMP enzymes, as depicted in Fig. 6B. We noted that when Cys564 was mutated, the enzyme no longer formed the dimer, which is consistent with the proposed structure of Fig. 6B. An initially perplexing observation was that the Cys566 mutant displayed a poor but readily detectable level of residual dimerization. We attempted to understand this observation by in silico mutation at Cys566 (C566S). This mutant enzyme was submitted to dynamics simulation for the same duration of 3.5 ns. The stem region of the C566S mutant underwent a conformational change. The same distances that were measured in panels I–III of Fig. 5B are given for the mutant at Cys566 in panels IV, V, and VI of Fig. 5B. As is especially obvious from comparisons of Fig. 5B (panels I, IV, III, and VI), the mutant enzyme at position 566 would assume a subtly different conformation, as depicted in Fig. 6C (compared with Fig. 6A for the wild-type enzyme). The key feature of this conformational change for the C566S mutant is that Cys564 is now sheltered within the stem and is not readily available for intermolecular disulfide bond formation. This observation provides a plausible explanation for the reduced ability of the C566S mutant to generate homodimers despite the presence of the cysteine at position 564 (Fig. 4B, lane 8). Therefore, the computational and experimental data agree with a model of MT4-MMP dimerization mediated by disulfide bonds at Cys564 (Fig. 6D).
FIGURE 5.

Molecular dynamic simulations of MT4-MMP stem peptide. A, alignment of the stem regions of MT4-MMP and MT6-MMP. The segment of the MT4- and MT6-MMP stem region predicted to be a β-strand is in blue, and the helical segment is in red. B, explicit solvent molecular dynamics simulations of MT4-MMP stem peptide. Distances among residues C564-C566 (panel I), Y561-C566 (panel II), and Y561-C564 (panel III) in wild-type MT4-MMP stem region and in residues C564-S566 (panel IV), Y561-S566 (panel V), and Y561-S564 (panel VI) in the MT4-MMP Cys566 mutant were obtained from molecular dynamics simulations over a duration of 3.5 ns.
FIGURE 6.

Model of MT4-MMP dimerization interface. Stereo representation of four superimposed conformers of the stem peptide of MT4-MMP (A–C). A, stereo representations of superimposed conformers for the MT4-MMP stem of 3,500 conformations sampled from the 3.5-ns molecular dynamics simulation. B and C, model proposed for the dimerization of human MT4-MMP through the disulfide bond between two Cys564 thiols (B) and the Cys566 mutant (C). The strand portion is represented in the loop between Tyr561 and Cys566 (A) and between Tyr561 and Ser566 (C). Residues Tyr561, Cys564, and Cys566 (Ser566) are shown in capped stick. Loops are colored in green, and carbon atoms are colored in gray, oxygen in red, and sulfur in yellow. D, schematic depicting the location of disulfide bonds in MT4-MMP. The presence of the Cys335–Cys526 intramolecular disulfide bond in the hemopexin-like domain is based on the conserved structure of this domain in MMPs. The disulfide bonds of the Cys residues within the stem region are based on the data presented here.
Collectively, these observations indicate that the GPI-anchored MT-MMPs share a similar ability to generate disulfide-linked homodimers, a structural conformation that may regulate proteolysis and subcellular distribution (35). However, the emerging picture from multiple observations suggests that although MT6-MMP is mostly displayed as a dimer (6), surface MT4-MMP is found as a heterogeneous population of monomeric, dimeric, and oligomeric forms (Fig. 1B). This difference in supramolecular organization may be related to the number of cysteine residues within the stem region of MT6- and MT4-MMP and their ability to generate intra- or intermolecular disulfide-linked bonds. The stem region of MT6-MMP, as opposed to that of MT4-MMP, possesses three cysteine residues, two of which were proposed to generate an intramolecular disulfide bridge (6). Thus, only one cysteine is available for dimerization. In MT4-MMP, although Cys564 is involved in dimerization, Cys566 is free. This suggests the possibility that following dimerization MT4-MMP oligomerization proceeds via disulfide bond formation at Cys566. However, mutation of Cys566 did not dampen MT4-MMP oligomerization (Fig. 4B, lane 4, upper panel) suggesting that oligomerization may involve other domain interactions and/or macromolecular associations with other proteins that are stabilized by disulfide bridges and are SDS-resistant. At present, the factors regulating the dynamics and extent of MT4-MMP dimerization/oligomerization remain unknown. It will be interesting to determine whether monomeric and dimeric forms of MT4-MMP display a different substrate profile and whether fluctuations in the monomer/dimer ratio will alter the subcellular distribution (basal versus apical) of MT4-MMP, as reported with other GPI-anchored proteins (35–37).
Stability of MT4-MMP
Dimerization was shown to confer stability to MT6-MMP (6). To determine whether this is the case with MT4-MMP, we compared the turnover of MT4-MMP, wild-type, and cysteine mutants, after exposure of the cells to CHX to inhibit protein synthesis. At each time point (0–6 h) after CHX exposure, the cells were lysed and examined for MT4-MMP forms by immunoblotting (Fig. 7A), followed by quantification of protein levels by densitometry. Analyses of three independent experiments demonstrated that wild-type MT4-MMP was readily detectable even 6 h post-initiation of CHX treatment and displays a half-life of ∼4 h (Fig. 7B). In contrast, the Cys564 mutant exhibited a half-life of ∼1 h suggestive of faster turnover. The Cys566 mutant, however, exhibited a half-life of ∼2.5 h and thus was more stable than the Cys564 mutant, possibly because of to its ability to maintain partial dimerization. Degradation products of MT4-MMP could not be detected, because the antibody used is directed to the catalytic domain. Taken together, these data suggest that dimerization of MT4-MMP, as in the case of MT6-MMP (6), plays a role in regulation of protease turnover. Because the Cys564 MT4-MMP mutant showed undetectable dimerization (Fig. 4B), the CHX data also suggest that the monomeric form is more susceptible to degradation than the dimeric form. At present, the turnover rate of each MT4-MMP under steady-state conditions is unknown because of the unavailability of a reliable MT4-MMP precipitating antibody, which precluded us from conducting biosynthesis studies by pulse-chase analyses.
FIGURE 7.

Stability of wild-type and mutant MT4-MMP. A, serum-starved MDCK cells expressing wild-type MT4-MMP or cysteine mutants (Cys564 and Cys566) of MT4-MMP were incubated with 50 μg/ml CHX for 6 h in serum-free MEM. At each time point (0, 2, 4, and 6 h), the cells were lysed using the cold lysis buffer as described under “Experimental Procedures.” Equal amount of protein from each lysate was resolved by reducing 10% SDS-PAGE followed by immunoblotting analysis using r-mAb EP1270Y. The blots were re-probed with a mAb against β-actin, as a loading control. The experiment was repeated three times and representative blots are shown. B, blots from three independent experiments were subjected to densitometry analyses, as described under “Experimental Procedures.” Each value represents the mean ± S.D. of relative amount of wild-type or its cysteine mutants at each time point relative to time 0 (100%). MDCK-MT4 (●), MDCK-MT4C566S (■), and MDCK-MT4C564S (▿) cells. NS, not statistically significant. Asterisk, p < 0.001; Student's t test.
MT4-MMP Enhances in Vitro Cell Migration and Invasion
We wished to examine the effects of MT4-MMP expression and dimerization on enzyme activity. However, because a specific substrate of MT4-MMP could not be identified, analyses of catalytic competence were not possible. Moreover, MDCK-MT4 cells failed to activate pro-MMP-2 or pro-MMP-9, regardless of the presence of increasing concentration of TIMP-2 or TIMP-1 (data not shown), in agreement with English et al. (4). Thus, gelatinase activation could not be used to monitor MT4-MMP activity. In addition, MT4-MMP did not cleave the α1-proteinase inhibitor (data not shown), a known substrate of MT6-MMP (6, 38). Previous studies indicated a role for MT4-MMP in supporting in vitro cell invasion of Matrigel (19, 21, 31). Therefore, we examined whether MT4-MMP (wild type and mutants) supported the migration and invasion of MDCK cells using established in vitro assays. As shown in Fig. 8, expression of wild-type MT4-MMP enhanced both the migration (Fig. 8A) and invasion (Fig. 8B) of MDCK cells. These activities were dependent on catalytic activity, because expression of the E/A mutant of MT4-MMP in MDCK cells or addition of TIMP-2 had not effect on or diminished cell migration and invasion, respectively. Differences in invasion were not due to cell growth differences (on plastic or Matrigel) among the transfected cells (data not shown). Invasion of Matrigel by MDCK cells expressing the dimerization-deficient mutant C564S was similar to that of cells expressing wild-type MT4-MMP (Fig. 8B), suggesting that the ability of MT4-MMP to promote invasion of Matrigel is not dependent on protease homodimerization. Surprisingly, MDCK cells expressing the C566S mutant, which maintains a certain extent of dimerization, exhibited an invasive activity similar to that of MDCK-EV cells. The reason for this unexpected result with the C566S mutant on cell invasion is presently unknown, but it may be due to additional effects of this mutation on protease structure/function that are unrelated to dimerization but yet may still influence catalysis of Matrigel components. Regardless, these invasion results cannot be construed as a lack of importance of MT4-MMP oligomerization on enzymatic activity. On the contrary, we hypothesize that the catalytic competence of MT4-MMP depends on the nature of the substrate and its unique molecular interactions with various protease domains, and it is likely to be regulated by the protease structural conformation, including monomeric versus dimeric organization.
FIGURE 8.
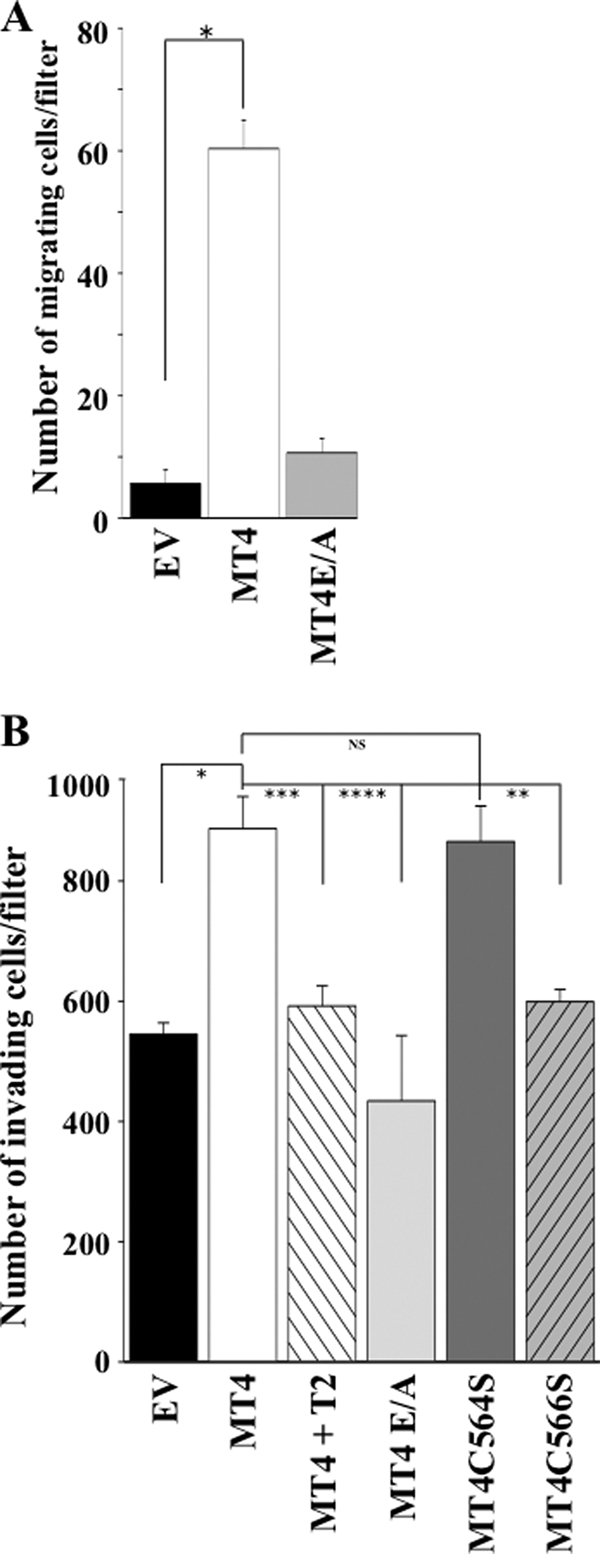
Effect of MT4-MMP on in vitro migration and invasion of MDCK cells. A, MDCK-EV, MDCK-MT4, and MDCK-MT4E/A cells in serum-free medium were seeded on top of 8-μm pore Transwell filters, and medium in the lower chamber was supplemented with 5% FBS, as described under “Experimental Procedures.” After 6 h of incubation in a tissue culture incubator, the number of cells that migrated to the lower side of the filter was counted in five representative fields. Each value represents the mean number of migrating cell/filter ± S.E. of three independent experiments. Asterisk, p < 0.001; Student's t test. B, MDCK-EV, MDCK-MT4, MDCK-MT4E/A, MDCK-MT4C564S, and MDCK-MT4C566S cells in serum-free medium were seeded on top of 8-μm pore Transwell filters coated with Matrigel, and medium in the lower chamber was supplemented with 5% FBS, as described under “Experimental Procedures.” After a 40-h incubation, the number of invading cells were fixed and stained. The cells were counted in five representative fields, and the number of invading cells per filter was plotted as the mean ± S.E. of three independent experiments. *, p < 0.004; **, p < 0.007; ***, p < 0.010; ****, p < 0.020; Student's t test. NS, statistically not significant.
Previous studies reported conflicting results in regards to the ability of MT4-MMP to stimulate in vitro invasion. Chabottaux et al. (18) and Hotary et al. (20) failed to demonstrate an invasion promoting activity of recombinant MT4-MMP expressed in human breast cancer MDA-MB-231 and monkey kidney COS cells, respectively. In contrast, Rizki et al. (21) and Hegedus et al. (31) showed that down-regulation of MT4-MMP by siRNA in human breast cancer T4-2 and MDA-MB-231 cells, respectively, inhibited Matrigel invasion. Likewise, Huang et al. (19) found that invasion through Matrigel, but not cell migration, of the tongue squamous cell carcinoma SAS cell line was enhanced by MT4-MMP expression. Conflicting invasion results may be due to a variety of factors, including cell type, levels of protease expression and activation, and/or the nature of the ECM barrier (39). Although the ECM degrading activity of natural MT4-MMP remains to be elucidated, previous studies using a bacterially expressed catalytic domain of mouse MT4-MMP showed that MT4-MMP exhibits a limited profile of ECM substrates when compared with MT1-MMP (3). Thus, it remains unclear whether the observed pro-metastatic activity of MT4-MMP found in vivo is directly related to its activity against ECM components and/or non-ECM proteins required for tumor cell invasion (40). Although more studies are needed to elucidate the proteolytic repertoire and function of MT4-MMP in physiological and pathological conditions, the data presented here provide new insight into the properties of MT4-MMP, a unique GPI-anchored MT-MMP.
This work was supported, in whole or in part, by National Institutes of Health Grants CA-61986-16 (to R. F.) and CA-122417 (to S. M.).
Q. Sun, A. Sohail, and R. Fridman, unpublished observations.
- MMP
- matrix metalloproteinase
- MT-MMP
- membrane type-MMP
- GPI
- glycosylphosphatidylinositol
- TIMP
- tissue inhibitor of metalloproteinase
- r-mAb
- rabbit monoclonal antibody
- MEM
- minimal Eagle's medium
- PI-PLC
- phosphatidylinositol-specific phospholipase C
- βME
- β-mercaptoethanol
- PI
- protease inhibitors
- CHX
- cycloheximide
- BGN
- benzyl 2-acetamido-2-deoxy-α-d-galactopyranoside
- MDCK
- Madin-Darby canine kidney cell
- ECM
- extracellular matrix.
REFERENCES
- 1. Zucker S., Pei D., Cao J., Lopez-Otin C. (2003) Curr. Top. Dev. Biol. 54, 1–74 [DOI] [PubMed] [Google Scholar]
- 2. Sohail A., Sun Q., Zhao H., Bernardo M. M., Cho J. A., Fridman R. (2008) Cancer Metastasis Rev. 27, 289–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. English W. R., Puente X. S., Freije J. M., Knauper V., Amour A., Merryweather A., Lopez-Otin C., Murphy G. (2000) J. Biol. Chem. 275, 14046–14055 [DOI] [PubMed] [Google Scholar]
- 4. English W. R., Velasco G., Stracke J. O., Knäuper V., Murphy G. (2001) FEBS Lett. 491, 137–142 [DOI] [PubMed] [Google Scholar]
- 5. Sun Q., Weber C. R., Sohail A., Bernardo M. M., Toth M., Zhao H., Turner J. R., Fridman R. (2007) J. Biol. Chem. 282, 21998–22010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhao H., Sohail A., Sun Q., Shi Q., Kim S., Mobashery S., Fridman R. (2008) J. Biol. Chem. 283, 35023–35032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Puente X. S., Pendás A. M., Llano E., Velasco G., López-Otín C. (1996) Cancer Res. 56, 944–949 [PubMed] [Google Scholar]
- 8. Kajita M., Kinoh H., Ito N., Takamura A., Itoh Y., Okada A., Sato H., Seiki M. (1999) FEBS Lett. 457, 353–356 [DOI] [PubMed] [Google Scholar]
- 9. Kinoh H., Hayashita H., Kajita M., Okada A., Seiki M. (1999) Cytogenet. Cell Genet. 87, 97–98 [DOI] [PubMed] [Google Scholar]
- 10. Gauthier M. C., Racine C., Ferland C., Flamand N., Chakir J., Tremblay G. M., Laviolette M. (2003) Int. J. Biochem. Cell Biol. 35, 1667–1673 [DOI] [PubMed] [Google Scholar]
- 11. Funatsu N., Inoue T., Nakamura S. (2004) Cereb. Cortex 14, 1031–1044 [DOI] [PubMed] [Google Scholar]
- 12. Nuttall R. K., Sampieri C. L., Pennington C. J., Gill S. E., Schultz G. A., Edwards D. R. (2004) FEBS Lett. 563, 129–134 [DOI] [PubMed] [Google Scholar]
- 13. Plaisier M., Koolwijk P., Hanemaaijer R., Verwey R. A., van der Weiden R. M., Risse E. K., Jungerius C., Helmerhorst F. M., van Hinsbergh V. W. (2006) Mol. Hum. Reprod. 12, 11–18 [DOI] [PubMed] [Google Scholar]
- 14. Rikimaru A., Komori K., Sakamoto T., Ichise H., Yoshida N., Yana I., Seiki M. (2007) Genes Cells 12, 1091–1100 [DOI] [PubMed] [Google Scholar]
- 15. Clements K. M., Flannelly J. K., Tart J., Brockbank S. M., Wardale J., Freeth J., Parker A. E., Newham P. (2011) Ann. Rheum Dis. 70, 683–689 [DOI] [PubMed] [Google Scholar]
- 16. Patwari P., Gao G., Lee J. H., Grodzinsky A. J., Sandy J. D. (2005) Osteoarthritis Cartilage 13, 269–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Srichai M. B., Colleta H., Gewin L., Matrisian L., Abel T. W., Koshikawa N., Seiki M., Pozzi A., Harris R. C., Zent R. (2011) PLoS One 6, e17099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chabottaux V., Sounni N. E., Pennington C. J., English W. R., van den Brûle F., Blacher S., Gilles C., Munaut C., Maquoi E., Lopez-Otin C., Murphy G., Edwards D. R., Foidart J. M., Noël A. (2006) Cancer Res. 66, 5165–5172 [DOI] [PubMed] [Google Scholar]
- 19. Huang C. H., Yang W. H., Chang S. Y., Tai S. K., Tzeng C. H., Kao J. Y., Wu K. J., Yang M. H. (2009) Neoplasia 11, 1371–1382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hotary K., Li X. Y., Allen E., Stevens S. L., Weiss S. J. (2006) Genes Dev. 20, 2673–2686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rizki A., Weaver V. M., Lee S. Y., Rozenberg G. I., Chin K., Myers C. A., Bascom J. L., Mott J. D., Semeiks J. R., Grate L. R., Mian I. S., Borowsky A. D., Jensen R. A., Idowu M. O., Chen F., Chen D. J., Petersen O. W., Gray J. W., Bissell M. J. (2008) Cancer Res. 68, 1378–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nagase H., Visse R., Murphy G. (2006) Cardiovasc. Res. 69, 562–573 [DOI] [PubMed] [Google Scholar]
- 23. Olson M. W., Gervasi D. C., Mobashery S., Fridman R. (1997) J. Biol. Chem. 272, 29975–29983 [DOI] [PubMed] [Google Scholar]
- 24. Kim J., Adam R. M., Solomon K. R., Freeman M. R. (2004) Endocrinology 145, 613–619 [DOI] [PubMed] [Google Scholar]
- 25. Zhuang L., Lin J., Lu M. L., Solomon K. R., Freeman M. R. (2002) Cancer Res. 62, 2227–2231 [PubMed] [Google Scholar]
- 26. Myers E. W., Miller W. (1988) Comput. Appl. Biosci. 4, 11–17 [DOI] [PubMed] [Google Scholar]
- 27. Garnier J., Osguthorpe D. J., Robson B. (1978) J. Mol. Biol. 120, 97–120 [DOI] [PubMed] [Google Scholar]
- 28. Chou P. Y., Fasman G. D. (1974) Biochemistry 13, 211–222 [DOI] [PubMed] [Google Scholar]
- 29. Onufriev A., Bashford D., Case D. A. (2000) J. Phys. Chem. B 104, 3712–3720 [Google Scholar]
- 30. Zhang W., Shi Q., Meroueh S. O., Vakulenko S. B., Mobashery S. (2007) Biochemistry 46, 10113–10121 [DOI] [PubMed] [Google Scholar]
- 31. Hegedüs L., Cho H., Xie X., Eliceiri G. L. (2008) J. Cell. Physiol. 216, 480–485 [DOI] [PubMed] [Google Scholar]
- 32. Wu Y. I., Munshi H. G., Sen R., Snipas S. J., Salvesen G. S., Fridman R., Stack M. S. (2004) J. Biol. Chem. 279, 8278–8289 [DOI] [PubMed] [Google Scholar]
- 33. Julenius K., Mølgaard A., Gupta R., Brunak S. (2005) Glycobiology 15, 153–164 [DOI] [PubMed] [Google Scholar]
- 34. Braakman I., Bulleid N. J. (2011) Annu. Rev. Biochem. 80, 71–99 [DOI] [PubMed] [Google Scholar]
- 35. Paladino S., Sarnataro D., Tivodar S., Zurzolo C. (2007) Traffic 8, 251–258 [DOI] [PubMed] [Google Scholar]
- 36. Caiolfa V. R., Zamai M., Malengo G., Andolfo A., Madsen C. D., Sutin J., Digman M. A., Gratton E., Blasi F., Sidenius N. (2007) J. Cell Biol. 179, 1067–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Cunningham O., Andolfo A., Santovito M. L., Iuzzolino L., Blasi F., Sidenius N. (2003) EMBO J. 22, 5994–6003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nie J., Pei D. (2004) Exp. Cell Res. 296, 145–150 [DOI] [PubMed] [Google Scholar]
- 39. Rowe R. G., Weiss S. J. (2009) Annu. Rev. Cell Dev. Biol. 25, 567–595 [DOI] [PubMed] [Google Scholar]
- 40. Chabottaux V., Ricaud S., Host L., Blacher S., Paye A., Thiry M., Garofalakis A., Pestourie C., Gombert K., Bruyere F., Lewandowsky D., Tavitian B., Foidart J. M., Duconge F., Noel A. (2009) J. Cell. Mol. Med. 13, 4002–4013 [DOI] [PMC free article] [PubMed] [Google Scholar]