

Abstract
Polo-like kinase-1 (Plk1) is essential for progression of mitosis and localizes to centrosomes, central spindles, midbody, and kinetochore. Ran, a small GTPase of the Ras superfamily, plays a role in microtubule dynamics and chromosome segregation during mitosis. Although Ran-binding protein-1 (RanBP1) has been reported as a regulator of RanGTPase for its mitotic functions, the action mechanism between Ran and RanBP1 during mitosis is still unknown. Here, we demonstrated in vitro and in vivo phosphorylation of RanBP1 by Plk1 as well as the importance of phosphorylation of RanBP1 in the interaction between Plk1 and Ran during early mitosis. Both phosphorylation-defective and N-terminal deletion mutant constructs of RanBP1 disrupted the interaction with Ran, and depletion of Plk1 also disrupted the formation of a complex between Ran and RanBP1. In addition, the results from both ectopic expression of phosphorylation-defective mutant construct and a functional complementation on RanBP1 deficiency with this mutant indicated that phosphorylation of RanBP1 by Plk1 might be crucial to microtubule nucleation and spindle assembly during mitosis.
Keywords: Cell Cycle, Microtubules, Mitosis, Protein Phosphorylation, Protein-Protein Interactions, Microtubule Nucleation, Phosphorylation, Polo-like Kinase 1, RanBP1
Introduction
Polo-like kinases are serine/threonine protein kinases that typically contain a catalytic domain in the N-terminal region and play numerous roles during M-phase progression (1–3). In mammalian cells, several types of Polo-like kinases have been identified based on the presence of polo-boxes (Plk1, Plk2 (or SNK), Plk3 (or Fnk/Prk), and Plk4 (or Sak)) (4). Plk1 is expressed from G2 phase onward and is degraded at the end of mitosis (5–7). Plk1 is required at several key steps in mitosis, including initiation of G2/M by phosphorylation of Cdc25C and mitotic cyclins (8–12), centrosome maturation (13, 14), and bipolar spindle formation (13–16). In addition, the polo-box domain of Plk1 can function as a dominant negative mutant and is essential for cytokinesis (17, 18). In several studies, Plk1 phosphorylation was found to induce polo-box targeting through a conformational change; however, catalytic activation did not appear to be a critical factor in the function of Plk1 (19, 20). These findings suggested that the polo-box of Plk1 plays an essential role in the function of Plk1, particularly in spatial distribution and physical interaction with substrates. In addition to posttranslational modification and structural function of Plk1 in mitotic cells, Plk1 has also been reported to be an important regulator of various target molecules involved in cell cycle-related events in many organisms. Ran is one of the Ras-related small GTP-binding proteins. Unlike Ras, Ran has no lipid modification site at the C-terminal end. Ran plays a role in various cellular processes, including nuclear transport, mitotic spindle assembly, nuclear envelope formation, and postmitotic nuclear assembly (21). Gradients of Ran-GTP are established through localization of its regulators, and the intrinsic rates of nucleotide exchange and hydrolysis of Ran are activated by RCC1, a guanine nucleotide exchange factor, and RanGAP1, a GTPase-activating protein-1 (22, 23). During interphase, RCC1 is localized on chromatin, whereas RanGAP1 is present in cytoplasm. Thus, RCC1 and RanGAP1 regulate the state of the guanine nucleotide, which binds to Ran. Conversely, during mitosis, RCC1 is phosphorylated by CDK1-cyclin B protein kinase, and its phosphorylation allows dynamic interaction with chromosomes (24). Similar to RCC1, phosphorylation of RanGAP1 is required at the onset of mitosis and remains associated with RanBP2 in the GTP-GDP state of the Ran cycle during progression through mitosis (25). Ran-binding protein-1 (RanBP1) is a cell cycle-dependent regulator of the Ran network in mammalian cells (26). Accumulation of RanBP1 occurs during mitosis, and its overexpression prolongs mitosis (27). Furthermore, disruption of bipolar spindle formation in Xenopus egg extracts was found to occur by the addition of RanBP1 (28). Consistent with these findings, RanBP1 is an inhibitor of RCC1 GEF activity, which suggests that an elevated concentration of RCC1 induces a reversal of the inhibitory effect of RanBP1 (29). Regulation of centrosome cohesion, chromosome segregation, and microtubule regulation during mitosis by mammalian RanBP1 through overexpression of RanBP1, antibody microinjection, and RNA interference experiments has been recently reported (30, 31). Ran-binding protein-3 (RanBP3), a member of the Ran-binding protein family, is phosphorylated by the Ras/ERK or PI3K/Akt signaling pathway. Phosphorylation of RanBP3 contributes to establishment of a Ran gradient and nucleocytoplasmic protein transport (32). Despite the dynamic functions of RanGTPase and its regulators, the clear mechanism of RanBP1 regulation in mammalian cells has not yet been elucidated. It will be of interest to determine whether RanBP1 is phosphorylated during mitosis and whether it is one of the targets of Plk1. In this study, we suggested that phosphorylation of RanBP1 by Plk1 plays a crucial role in the Ran network system, which is related to microtubule nucleation and function during mitosis.
EXPERIMENTAL PROCEDURES
Antibodies and Reagents
The following antibodies were used in this study: mouse monoclonal antibodies against FLAG (F1804, Sigma-Aldrich), α-tubulin (T5168, Sigma-Aldrich), γ-tubulin (T6557, Sigma-Aldrich), Plk1 (33-1700, Zymed Laboratories Inc.), and Ran (610340, Pharmingen); rabbit polyclonal antibodies against phospho-Plk1 (Thr-210) (KAP-CC107, Stressgene), RanBP1 (sc-28576, Santa Cruz Biotechnology, Inc., Santa Cruz, CA), RanBP1 (ab97659, Abcam), cyclin B1 (sc-752, Santa Cruz), and FLAG (F7425, Sigma-Aldrich); and goat polyclonal antibodies against Ran (sc-1156, Santa Cruz Biotechnology, Inc.) and actin (sc-1616, Santa Cruz Biotechnology, Inc.). Polyclonal antibody against phospho-RanBP1 was raised in rabbits against synthetic phosphopeptide CQEIKpTLEED. Secondary antibodies conjugated to HRP, FITC, and CY3 were purchased from GE Healthcare and Jackson ImmunoResearch Laboratories, respectively. Doxorubicin, mimosine, and nocodazole were obtained from Sigma-Aldrich.
Cell Culture, Transfection, and RNA Interference
HeLa cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (Hyclone) and a mixture of penicillin/streptomycin/fungizone (BioWhittaker) at 37 °C. To obtain cells arrested in the late G1 phase and early mitotic cells, cells were treated with mimosine (300 μm) for 16 h and nocodazole (100 ng/ml) for 10 h, respectively. The standard calcium chloride technique for transfection was conducted as previously described with modification (17, 19, 33). To deplete the endogenous Plk1 and RanBP1 in HeLa cells, a plasmid-based small interference RNA construct was purchased from SuperArray Bioscience Corp. (Frederick, MD).
Preparation of Cell Lysates and Kinase Assay
Cells were lysed in Nonidet P-40 lysis buffer (0.5% Nonidet P-40, 50 mm Hepes, pH 7.4, 150 mm NaCl, 1 mm DTT, 5 mm EGTA, 1 mm EDTA, 20 mm NaF, 20 mm β-glycerophosphate, 0.5 mm Na3VO4, 20 mm p-nitrophenyl phosphate) supplemented with protease inhibitors. Kinase reactions were conducted as described previously (34). Briefly, Plk1 protein was expressed in insect cells and purified using appropriate affinity column chromatography. The purified Plk1 protein was used in the kinase assays as a kinase source.
Preparation of DNA Constructs and Protein Expression
Plk1, Ran, and RanBP1 was amplified from human cDNA constructs, and point mutations at the indicated residues in the Plk or RanBP1 were generated by recombinant PCR. All PCR products were cloned into pGEX vector (Promega) and confirmed by DNA sequencing. The proteins were expressed in Escherichia coli by the induction of isopropyl-1-thio-β-galactopyranoside and confirmed by Western blot analysis using appropriate anti-RanBP1 antibody. To generate recombinant virus of Plk1, the pGEX constructs were transferred into pAcGHLT vector (Pharmingen), and the expressed proteins were purified for further experiments (17, 19). To express proteins in mammalian cells, all constructs were cloned into pCMV-Tag2 vector (Stratagene), which contains FLAG (DYKDDDDK) epitope in the N terminus of the gene, or pEGFP-N1 vector (Clontech) for detection of subcellular localization.
In Vitro Protein-binding Assay and Immunoprecipitation
To purify the GST-tagged proteins, the plasmids were transformed into E. coli strain DH5α, after which they were purified to homogeneity from cell extracts using glutathione-agarose 4B (Peptron). GST-conjugated proteins were incubated with mitotic HeLa extracts. After pulling down the beads, proteins bound to the tagged protein were detected using appropriate antibodies. For immunoprecipitation, HeLa cells were transfected with FLAG-tagged constructs. After 16 h of nocodazole treatment, mitotic cells collected by shake-off were resuspended in Nonidet P-40 lysis buffer. The total cell extracts were incubated with anti-FLAG/protein G-PLUS beads (Santa Cruz Biotechnology, Inc.) for 2 h at 4 °C. The beads were washed three times with Nonidet P-40 lysis buffer, and then finally, the bound proteins were analyzed by SDS-PAGE followed by Western blot analysis.
Flow Cytometry
Cells were harvested and fixed in ice-cold 70% ethanol for 16 h at 4 °C. They were incubated with 100 μg/ml RNase A for 2 h at room temperature and treated with 10 μg/ml propidium iodide for detection of DNA amounts. The bivariate levels of green fluorescent protein (GFP) and PI-DNA content (red) fluorescence were determined using a FACSCaliburTM system (BD Biosciences). A gate was set to select GFP-positive cells with a GFP signal that was at least 40 times stronger than that of the negative cells. 30,000 events were evaluated and analyzed using the BD CellQuest ProTM software (BD Biosciences) and Modfit LTTM 3.0 version (Verity Software House, Topsham, ME).
Fluorescence Microscopy
HeLa cells were cultured on coverslips and were transfected as previously described. After 40 h of post-transfection, the cells were fixed in 4% paraformaldehyde-PBS solution and then treated with ice-cold methanol. Cells were incubated with blocking buffer containing 10% FBS in PBS for 30 min and treated with specific antibodies in 5% FBS in PBS. Cells were then rinsed and incubated with secondary antibody conjugated to appropriate fluorochrome (Jackson ImmunoResearch) and DAPI (10 μg/ml) for detection of chromosomes. Finally, cells were analyzed by confocal microscopy with an LSM700 imaging system (Carl Zeiss).
RESULTS
Plk1, Ran, and RanBP1 Are Co-localized in Microtubule Regions during Early Mitosis
Ran is a small GTPase required for nucleo-cytoplasmic transport, spindle and nuclear assembly, and regulation of cell division (35–37). In its role as a nucleocytoplasmic transporter, Ran was found in the nuclear region, whereas GTPase-activating protein, RanGAP1, and its accessory protein, RanBP1, were localized in the cytoplasm during interphase. At the beginning of mitosis, when the nuclear envelope is broken down, nuclear Ran and cytoplasmic RanBP1 and RanGAP1 are recruited predominantly to the chromosomal region and kinetochore and regulate centrosome cohesion and mitotic spindle organization (27, 38–41). We previously reported on screening of Ran as a Plk1 target molecule in mitosis as well as the importance of the interaction between Ran and Plk1 in recruitment of Ran-binding protein (38). Based on these reports, we speculated that Plk1, Ran, and RanBP1 proteins might be co-localized into the chromosomal or microtubule nucleation region through breakdown of the nuclear envelope during early mitosis, and we evaluated subcellular localization of endogenous proteins by microscopic analysis of HeLa cells. Cells were arrested during early mitosis by treatment with nocodazole and were co-stained with anti-RanBP1 and anti-Plk1 or anti-Ran antibodies. As expected, during interphase, Ran and RanBP1 were localized in the nuclear region and in cytoplasm, respectively (Fig. 1A, a). However, in the early mitotic phase, both Ran and RanBP1 were concentrated in the vicinity of the chromosomes, particularly in the area of spindle formation (Fig. 1A, b). Plk1 and RanBP1 were co-localized at the spindle appearance site (Fig. 1B, b) but not in the centrosomal region during early mitosis (Fig. 1, B and C, panels b). Co-localization of Plk1 and RanBP1 was not clearly detected during the late mitotic stage (data not shown). Depletion of endogenous Plk1 using shRNA transfection resulted in a decrease of the interaction between Ran and RanBP1 (Fig. 2, top, lanes 2). Although the endogenous protein level of Plk1 was decreased in mitosis, cells were not defective and arrested in mitotic phase, which was confirmed by Western blots using anti-cyclin B1 (Fig. 2, bottom, α-cycB1).
FIGURE 1.

Ran, RanBP1, and Plk1 localize in the regions of microtubule nucleation in early mitosis. A, localization of endogenous Ran and RanBP1 in interphase (a) and in early mitotic cells (b). HeLa cells treated as described under “Experimental Procedures” were fixed and subjected to indirect immunofluorescence with anti-Ran (red) and anti-RanBP1 (green). DNA was detected by DAPI staining (blue). B, for the detection of endogenous Plk1 and RanBP1 in interphase (a) and early mitotic HeLa cells (b), proteins were stained with anti-Plk1 (green) and anti-RanBP1 (red) antibodies and evaluated by confocal microscopy. C, co-localization of endogenous RanBP1 and γ-tubulin in interphase (a) and in mitosis (b). Proteins were stained with anti-γ-tubulin (green) and anti-RanBP1 (red) antibody and then evaluated by confocal microscopy. Scale bars, 5 μm.
FIGURE 2.
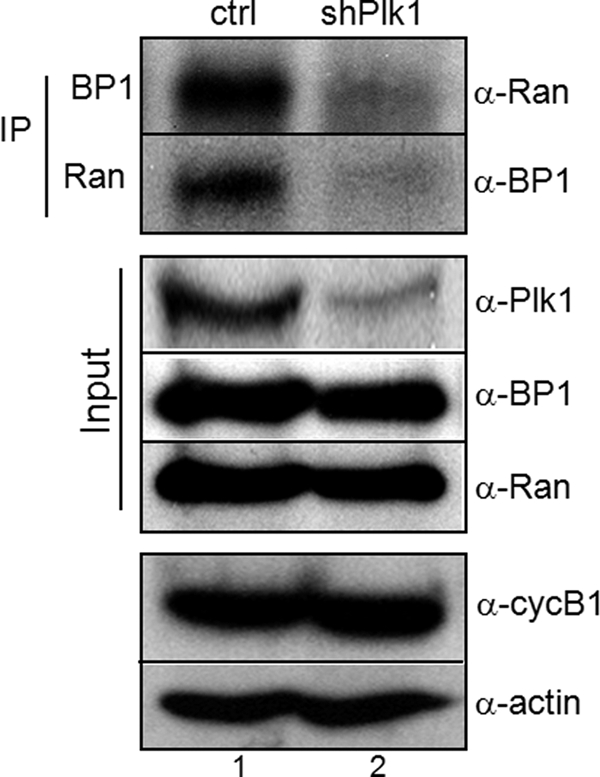
Interaction between Ran and RanBP1 was decreased in depletion of Plk1. In vivo interaction between Ran and RanBP1 was performed by coimmunoprecipitation. Cells were transfected with vector (lane 1, ctrl) or Plk1 shRNA (lane 2, shPlk1). After a 30-h incubation in fresh media, cells were treated with nocodazole for an additional 10 h to arrest in early mitosis. Endogenous Ran and RanBP1 were immunoprecipitated (IP), and bound proteins were detected by immunoblot analysis using appropriate antibodies (top panels, α-Ran or α-BP1). Protein amounts used in this experiment were estimated by actin expression (α-actin).
RanBP1 Is Phosphorylated by Plk1 as a Possible Target Protein
According to the Plk1-dependent manner in the interaction between RanBP1 and Ran, we attempted to determine whether or not RanBP1 can be phosphorylated by Plk1. For assessment of RanBP1 phosphorylation sites, an in vitro kinase assay was performed using His-tagged Plk1, which was purified from insect cells (19). Based on amino acid sequence analysis, there are two possible consensus sequences for phosphorylation by Plk1 in the N-terminal region of RanBP1, 38EIKTLEE and 58FASEND (Fig. 3A). GST-tagged RanBP1 was used as substrate in the kinase assay. Upon incubation with His-Plk1, GST-RanBP1 was strongly phosphorylated by Plk1 (Fig. 3B, lane 1). In addition, phosphoamino acid analysis revealed that threonine and serine residues of RanBP1 could be phosphorylated by Plk1 and that the intensity of phosphorylation on the serine residue was stronger than that on the threonine residue (Fig. 3C). In order to determine whether these residues of RanBP1 are phosphorylation sites or not, we constructed a deletion mutant (ΔN, deletion of residues 1–60) and point mutants, where threonine 40 and serine 60 were substituted with valine and alanine, respectively. In an in vitro kinase assay, phosphorylation signals of T40V and S60A mutants were decreased (Fig. 3D, top, lanes 2 and 3) in comparison with that of wild type RanBP1 (Fig. 3D, top, lane 1). The deletion constructs, ΔN and GST protein, were not phosphorylated (Fig. 3D, top, lanes 4 and 5). Taken together, these data demonstrated that RanBP1 can be phosphorylated by Plk1 on both threonine 40 and serine 60 and that these sites might be important for RanBP1 function in early mitosis.
FIGURE 3.

RanBP1 is phosphorylated by Plk1. A, a schematic structure of mammalian RanBP1. RBD, Ran-binding domain containing the nuclear localization signal (NLS); CRS, cytoplasmic retention signal; NES, nuclear export signal. Threonine 40 (T40) and serine 60 (S60) residues in the N-terminal region are conserved in various species as Plk1 substrate-consensus sequences. B, purified RanBP1 proteins were phosphorylated by Plk1 in vitro. An in vitro kinase assay was conducted as described under “Experimental Procedures.” GST-RanBP1 was phosphorylated by Plk1 ([32P] lane 1), but GST alone was not ([32P] lane 1). The total protein amounts were shown by Coomassie Brilliant Blue staining (CBB; bottom). C, phosphorylation site(s) of RanBP1 in B was analyzed by phosphoamino acid analysis. S, phosphoserine; T, phosphothreonine; Y, phosphotyrosine. D, phosphorylation signals of mutant constructs of RanBP1 ([32P] lanes 2–4) decreased more than that of wild type RanBP1 ([32P] lane 1). Protein amounts were shown by Coomassie Brilliant Blue staining (CBB; bottom).
RanBP1 Phosphorylation on Thr-40 by Plk1 Is Essential for Interaction with Ran
GTP hydrolysis requires the interaction between RanGTP and RanGTPase-activating protein (RanGAP1) and is stimulated by Ran-binding protein 1 or Ran-binding protein 2. RanGAP1 is phosphorylated by mitotic kinase cyclin B1-Cdk1, and this modification is important for interaction with Ran (25). Based on the regulatory mechanism of RanGAP, in order to determine whether RanBP1 phosphorylation by Plk1 was required for the occurrence of an interaction with Ran, we conducted a protein binding assay by immunoprecipitation. For detection of in vivo interaction in early mitotic cells, wild type and other mutant constructs of RanBP1 tagged with FLAG were expressed transiently in cells. Immunoprecipitation of RanBP1 proteins with anti-FLAG antibody resulted in coimmunoprecipitation of mitotic Ran protein with wild type and S60A point mutants of RanBP1 (Fig. 4A, top, lanes 2 and 4), whereas neither T40V, T40V/S60A, nor ΔN mutant construct interacted with endogenous Ran (Fig. 4A, lanes 3, 5, and 6). An in vitro protein pull-down assay with the GST-RanBP1 construct also showed a result similar to that observed with immunoprecipitation (Fig. 4B). These data indicated that the interaction between Ran and RanBP1 is dependent on Plk1 in mitotic cells and that RanBP1 phosphorylation by Plk1 might play a pivotal role in its interaction with Ran. Taken together, these data suggested that RanBP1 phosphorylation on Thr-40 is necessary for stable interaction between Ran and RanBP1. Although the Ser-60 residue was also phosphorylated by Plk1 in vitro, we had no interest with regard to its function in further study, because this phosphorylation had no effect on the interaction with Ran.
FIGURE 4.

Thr-40 phosphorylation of RanBP1 is essential for interaction with Ran. A, in vivo protein binding assay by immunoprecipitation. Various RanBP1 constructs containing FLAG tag were transfected for 24 h and then subjected to nocodazole treatment for 10 h to arrest in early mitosis. RanBP1s overexpressed were immunoprecipitated using anti-FLAG antibody (IP: FLAG). Bound Ran proteins were detected by Western blot (IB: α-Ran). The protein amounts are shown in the bottom panels (Input). B, in vitro protein binding assay by pull-down using fusion proteins. Various RanBP1 mutants were constructed as fusion proteins using maltose-binding protein (Mal) (lanes 1 and 2) or glutathione S-transferase (GST) (lanes 3–7) and were purified by pull-down of appropriate beads. Bound Ran proteins were detected by anti-Ran antibody (top, α-Ran). The protein amounts are shown at the bottom (Input).
RanBP1 Phosphorylation on Thr-40 by Plk1 Occurs in a Mitosis-specific Manner
To investigate the question of whether or not Thr-40 phosphorylation of RanBP1 occurs in a mitosis-specific manner in vivo, we constructed a Thr-40 phospho-specific antibody (α-pT40). Endogenous RanBP1 from late G1 cell extracts was weakly detected by the pT40 antibody (Fig. 5A, c, lane 1), whereas mitotic RanBP1 was strongly detected by the phosphoantibody (Fig. 5A, c, lane 2). Depletion of Plk1 in mitotic cells resulted in decreased Thr-40 phosphorylation of RanBP1 (Fig. 5A, c, lane 4) in comparison with that in control cells (Fig. 5A, c, lane 3). To determine whether RanBP1 phosphorylation on Thr-40 was regulated through mitotic progression, cells were synchronized in prometaphase by treatment with nocodazole and were then allowed to exit mitosis. Cell lysates were prepared at each time point, as indicated, and proteins were analyzed by Western blots. According to mitotic progression, the signal of phospho-RanBP1 decreased in a pattern similar to that observed with phospho-Plk1 (Fig. 5B, b and e). These data demonstrated that phosphorylation of Thr-40 occurs in a cell cycle-dependent manner, like Plk1 activity, and that RanBP1 can be a target of Plk1 during mitotic progression. Furthermore, Plk1 might regulate the interaction between Ran and RanBP1 through RanBP1 phosphorylation on the Thr-40 residue.
FIGURE 5.
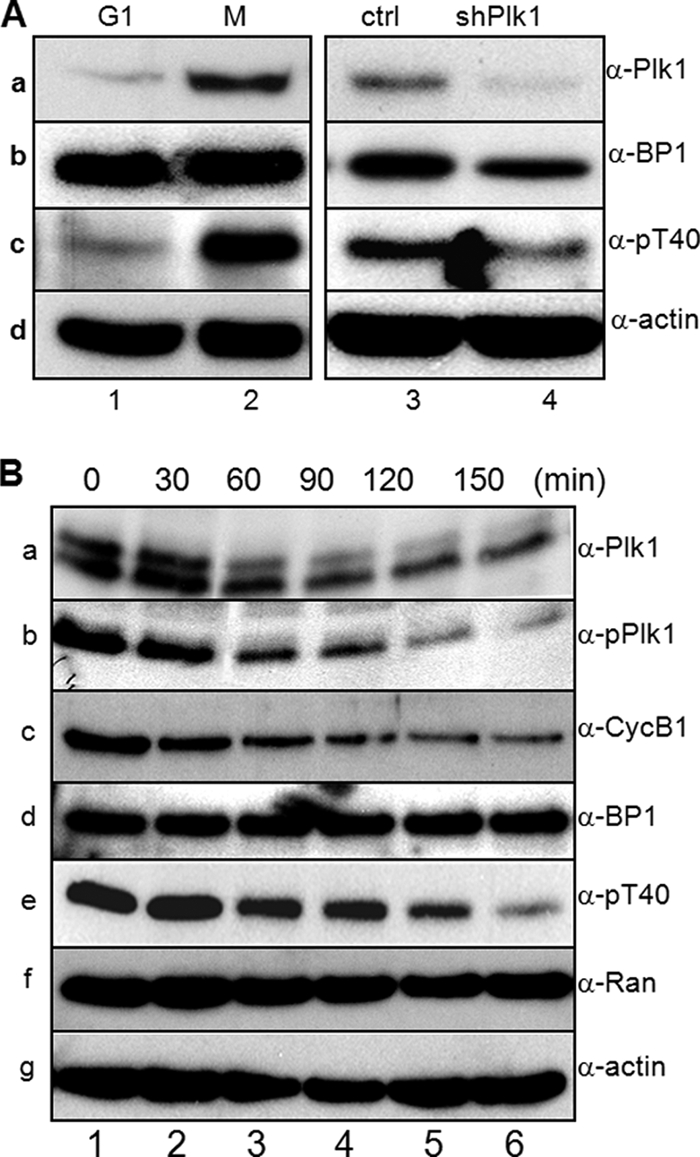
Plk1 phosphorylates Thr-40 of RanBP1 during mitotic progression. A, verification of anti-phospho-RanBP1 antibody (α-pT40). HeLa cells were synchronized in G1 (lane 1) and early mitosis (lane 2) and were transfected with vector (lane 3) or Plk1 shRNA for depletion of Plk1 (lane 4). Endogenous proteins were analyzed by Western blot using anti-Plk1 (a), anti-RanBP1 (b), anti-pT40 (c), and anti-actin (d) antibodies. B, Thr-40 phosphorylation of RanBP1 occurs in a cell cycle-dependent manner. After mitotic arrest by nocodazole treatment, cells were allowed to exit mitosis and harvested at the indicated time points (0–150 min). Endogenous proteins were analyzed by Western blot analysis. a, anti-Plk1 antibody (α-Plk1); b, anti-phospho-Plk1 antibody (α-pPlk1) on Thr-210; c, anti-cyclin B1 antibody (α-CycB1); d, anti-RanBP1 antibody (α-BP1); e, anti-phospho-RanBP1 antibody (α-pT40) on Thr-40; f, anti-Ran antibody (α-Ran); g, anti-actin antibody (α-actin).
RanBP1 Phosphorylation on Thr-40 Is Required for Microtubule Formation during Early Mitosis
In previous studies, RanBP1 or RCC1 regulated either dynamic features of the centrosome or appropriate chromosome segregation in human cells (30, 31, 42). Phosphorylation of RCC1 allows for dynamic interaction of RCC1 with chromosomes during mitosis (43). Similar to RCC1, RanGAP1 is phosphorylated by Cdk1, and phosphorylated RanGAP1 localizes into kinetochores and spindle microtubules during mitosis (44). These reports indicated that phosphorylation in these proteins might be important for mitotic progression related to Ran. In order to determine whether RanBP1 phosphorylation by Plk1 affects its appropriate localization during cell cycle progression, FLAG-RanBP1 constructs (FLAG-WT and -T40V) were expressed in mitotic HeLa cells. Formation of aster microtubules was observed in prophase in both control cells and RanBP1-expressing cells, (Fig. 6A, top and middle, α-tubulin); however, it was not formed in cells expressing T40V (Fig. 6A, bottom, α-tubulin). Moreover, overexpression of RanBP1-WT resulted in formation and attachment of microtubules to condensed chromosomes in metaphase in the same phenotype as control (Fig. 6B, top and middle, α-tubulin). However, overexpression of the T40V mutant resulted in disruption of bipolar spindle formation under chromosome condensation in metaphase (Fig. 6B, bottom, α-tubulin), indicating that T40V overexpression did not result in formation of microtubule asters or proper assembly of the spindle during early mitosis. As well as abnormal spindle formation, overexpression of the T40V construct resulted in an increase of undivided and multiple nuclei and hypertropic nucleus in comparison with those in RanBP1-WT-overexpressing cells (Fig. 6D, graph). To investigate the importance of phosphorylation on Thr-40 of RanBP1 on microtubule nucleation in early mitosis, we performed the functional complementation experiment. For this, RanBP1 phosphorylation-defective mutant (T40V) was overexpressed in cells depleted of RanBP1 by using an shRNA construct (Fig. 7A). In a previous study (31), cyclin B1 failed to associate with spindle microtubules in RanBP1-depleted mitosis, resulting in disruption of proper localization on spindle microtubule asters. As expected, when the RanBP1 was depleted, cyclin B1 did not associate with microtubule asters in early mitosis (31) (Fig. 7B, b, cyclinB1). When the wild type construct of RanBP1 was overexpressed in cells depleted of RanBP1, localization of cyclin B1 was recovered (Fig. 7B, c, cyclinB1). However, when phosphorylation-defective mutant (T40V) of RanBP1 was expressed, cyclin B1 did not localize on spindle microtubules (Fig. 7B, d). These data suggested that the phosphorylation-defective mutant (T40V) of RanBP1 does not overcome the defect induced by RanBP1 depletion and that Thr-40 phosphorylation might be important for appropriate microtubule nucleation during early mitosis; even the endogenous level of RanBP1 is important for progression of mitosis.
FIGURE 6.

Expression of phosphorylation-defective mutant of RanBP1 causes abnormal spindle formation and mitosis-defective phenotypes. RanBP1 wild type and T40V mutant constructs were transfected into HeLa cells for 24 h and then treated with nocodazole for 10 h for arrest in early mitosis. Cells on a coverslip were fixed with 4% paraformaldehyde/methanol as described under “Experimental Procedures.” The amount of expressed RanBP1s was estimated by the intensity of α-FLAG staining (FLAG) by confocal microscopic analysis. Endogenous α-tubulin pattern (α-tubulin) was examined by confocal microscopic analysis in cells in prophase (A) and in metaphase (B). Cells in specific mitotic phase were discriminated by chromosomal DNA conformation. C, frequency of mitotic abnormalities in cells overexpressing RanBP1 wild type (BP1-WT) and T40V mutant (BP1-TV) (a). The percentage of cells with undivided/multiple nuclei or hypertropic nucleus is indicated as a white bar or gray bar, respectively. Abnormal phenotypes in cells overexpressing RanBP1 T40V mutant are shown in b. Scale bars, 10 μm.
FIGURE 7.

Phosphorylation on Thr-40 of RanBP1 is involved in the association with microtubules and cyclin B1. A, cells were transfected with RanBP1-specific shRNA for depletion of RanBP1. Cells on a coverslip were fixed with 4% paraformaldehyde/methanol as described under “Experimental Procedures.” B, localization of endogenous cyclin B1 (cyclinB1) was detected by using anti-cyclin B1 antibody (red). RanBP1-specific shRNA transfected in cells was confirmed by intensity of GFP (green). a, early mitotic cell for control; b, cells transfected with RanBP1-specific shRNA construct; c, overexpression of RanBP1-WT in cells depleted of RanBP1; d, overexpression of RanBP1-T40V in cells depleted of RanBP1. Scale bar, 10 μm.
DISCUSSION
Ran is a small GTPase that belongs to the Ras superfamily. In addition to playing a role in nucleocytoplasmic transport during interphase, Ran and its effectors play key roles in centrosome duplication, mitotic spindle assembly, chromosome segregation, and kinetochore microtubule localization during mitosis (21, 22, 40, 43). Moreover, recent studies have reported on the importance of phosphorylation of these proteins by mitotic kinases in regulation of Ran function during mitosis. For example, RCC1 is a specific target of cyclin B1-Cdk1 during mitosis (24, 43, 45), and phosphorylated RCC1 plays a role in maintaining the balance between RanGTP and RanGDP. In addition to RCC1, RanGAP1 associated with RanBP2 is also phosphorylated by Cdks during mitotic progression (25). In addition, Ras/Akt signaling pathways modulate phosphorylation and function of RanBP3, which regulates the Ran gradient through control of RCC1 activity (32). RanBP1 is also regulated in order to ensure appropriate localization of mitotic regulatory factors on spindle microtubules and to perform chromosome segregation through modulation of RanGTP gradients during mitosis (31, 45). However, it is still unclear how RanBP1 activity is regulated in mammalian cells, especially during mitosis. In this report, we attempted to suggest that mitotic Plk1 and RanBP1 might be recruited to microtubule regions through interaction with Ran during early mitosis, following RanBP1 phosphorylation by Plk1. The increasing level of mitotic RanBP1 induced unusual spindles and delayed progression into late mitotic stage in mammalian cells (30), and overexpression of T40V mutant resulted in an increase of abnormal aspects, compared with those of cells containing RanBP1-WT (Fig. 6C). In addition to ectopic expression of a phosphorylation-defective mutant of RanBP1, functional complementation of RanBP1 depletion and Plk1 depletion experiments indicated that RanBP1 phosphorylation was dependent on Plk1 activity and that phosphorylation might be essential for appropriate microtubule aster formation (Figs. 5A and 7). Finally, we suggested that Plk1 might be one of the regulators of RanBP1 and that a direct interaction between RanBP1 and Ran might be regulated by phosphorylation of RanBP1. Because mitosis-specific phosphorylation of RanBP1 and its strong interaction with Ran could be important to the understanding of Ran-RanBP1 functions in mitosis, we speculated on the mechanism of their interactions. General models describing the mechanism of how the polo-box domain of Plk1 interacts with its specific substrates have been suggested by Lowery et al. (46). The “distributive phosphorylation” is one of the working mechanisms of Plk1. According to this model, a substrate or a target protein binds to the polo-box domain of Plk1, and the third protein can be recruited to this target protein as a docking protein and finally phosphorylated by Plk1. Both Thr-40 and Ser-60 of RanBP1 are consensus sites for Plk1 phosphorylation (47). These residues are located within the Ran-binding domain, which includes a nuclear localization signal, and are evolutionarily conserved among various species (Fig. 3A). Although Thr-40 phosphorylation was found to be weaker than that on the Ser-60 residue in the in vitro kinase assay (Fig. 3D), this site might be important for the interaction with Ran during mitosis (Fig. 4, A and B). Furthermore, Ran and RanBP1 did not interact in Plk1-depleted cells (Fig. 2), indicating that RanBP1 phosphorylation by Plk1 is essential for stronger interaction between RanBP1 and Ran, although these two proteins can interact temporally in cells. Several reports have shown that the timing and spatial coordination of the Ran network system during mitotic progression might be associated with RanGTP/GDP gradients and activation of mitotic kinases. Remarkably, RanBP1 activity is required for appropriate localization/function of Ran in regions of aster microtubules and chromosomes (31, 45). Thr-40 phosphorylation of RanBP1 through mitotic progression was a unique feature of mitotic RanBP1 and was discriminated from interphasic RanBP1 (Fig. 5A). Guarguaglini et al. (27) reported low levels of RanBP1 during the G1 phase, which increased during progression from the S phase to mitosis in NIH/3T3 cells. However, RanBP1 was constitutively expressed in many tumor cells in an E2F-dependent manner (26). Therefore, we evaluated the levels of RanBP1 expression in several tumor and primary cell lines. Constitutive and relatively high levels of RanBP1 were observed throughout the cell cycle in various tumor cells (HeLa, cervical carcinomas; KB, mouse epithelial carcinomas; MCF, human mammary tumor cells; SK-Hep1, hepatomas), whereas endogenous levels of RanBP1 in primary cultured cells were low and were found to be cell cycle-dependent (MC3T3, murine fibroblasts; PDL, periodontal ligaments; HGF, human gingival fibroblasts) (data not shown). In summary, we suggested that Plk1 phosphorylates RanBP1 during early mitosis just after disruption of the nuclear membrane and that Ran is then recruited to the mitotic microtubule region around chromosomes for establishment of normal microtubule aster formation, spindle formation, and chromosome segregation during mitosis. However, if phosphorylation on Thr-40 of RanBP1 by Plk1 did not occur, strong interaction between Ran and RanBP1 was disrupted or became unstable, followed by induction of mitotic defects in further mitotic progression (Fig. 8). Future studies will be conducted in order to determine whether RanBP1 is correlated with other mitotic kinases as well as Plk1 during mitotic progression and how they are involved in mitotic Ran function, taken together with other effectors, including RanGAP and other Ran-binding proteins.
FIGURE 8.

A diagram of interactions among Ran, RanBP1 and Plk1. In interphase (G2), Ran and RanBP1 are localized in nucleus and cytoplasm, respectively. According to entry into mitosis, in which Plk1 is phosphorylated and activated, Ran and RanBP1 interact weakly in the cytoplasm. When interaction between Ran and Plk1 occurs, RanBP1 can be phosphorylated by Plk1, and the Ran-RanBP1 complex will be stable. RanBP1 phosphorylation and stable interaction between Ran and RanBP1 are important for microtubule nucleation and spindle maturation.
Footnotes
This work was supported by National Research Foundation of Korea Grant R31-10069 (World Class University program) and Grant 2009-0093829 (Priority Research Centers Program) funded by the Ministry of Education, Science, and Technology.
REFERENCES
- 1. Donaldson M. M., Tavares A. A., Hagan I. M., Nigg E. A., Glover D. M. (2001) J. Cell Sci. 114, 2357–2358 [DOI] [PubMed] [Google Scholar]
- 2. Glover D. M., Hagan I. M., Tavares A. A. (1998) Genes Dev. 12, 3777–3787 [DOI] [PubMed] [Google Scholar]
- 3. Lane H. A., Nigg E. A. (1997) Trends Cell Biol. 7, 63–68 [DOI] [PubMed] [Google Scholar]
- 4. Barr F. A., Silljé H. H., Nigg E. A. (2004) Nat. Rev. Mol. Cell Biol. 5, 429–440 [DOI] [PubMed] [Google Scholar]
- 5. Fang G., Yu H., Kirschner M. W. (1998) Mol. Cell 2, 163–171 [DOI] [PubMed] [Google Scholar]
- 6. Lindon C., Pines J. (2004) J. Cell Biol. 164, 233–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Uchiumi T., Longo D. L., Ferris D. K. (1997) J. Biol. Chem. 272, 9166–9174 [DOI] [PubMed] [Google Scholar]
- 8. Abrieu A., Brassac T., Galas S., Fisher D., Labbé J. C., Dorée M. (1998) J. Cell Sci. 111, 1751–1757 [DOI] [PubMed] [Google Scholar]
- 9. Karaïskou A., Jessus C., Brassac T., Ozon R. (1999) J. Cell Sci. 112, 3747–3756 [DOI] [PubMed] [Google Scholar]
- 10. Kumagai A., Dunphy W. G. (1996) Science 273, 1377–1380 [DOI] [PubMed] [Google Scholar]
- 11. Ouyang B., Pan H., Lu L., Li J., Stambrook P., Li B., Dai W. (1997) J. Biol. Chem. 272, 28646–28651 [DOI] [PubMed] [Google Scholar]
- 12. Toyoshima-Morimoto F., Taniguchi E., Shinya N., Iwamatsu A., Nishida E. (2001) Nature 410, 215–220 [DOI] [PubMed] [Google Scholar]
- 13. Lane H. A., Nigg E. A. (1996) J. Cell Biol. 135, 1701–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sunkel C. E., Glover D. M. (1988) J. Cell Sci. 89, 25–38 [DOI] [PubMed] [Google Scholar]
- 15. Ohkura H., Hagan I. M., Glover D. M. (1995) Genes Dev. 9, 1059–1073 [DOI] [PubMed] [Google Scholar]
- 16. Qian Y. W., Erikson E., Li C., Maller J. L. (1998) Mol. Cell Biol. 18, 4262–4271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jang Y. J., Lin C. Y., Ma S., Erikson R. L. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 1984–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Seong Y. S., Kamijo K., Lee J. S., Fernandez E., Kuriyama R., Miki T., Lee K. S. (2002) J. Biol. Chem. 277, 32282–32293 [DOI] [PubMed] [Google Scholar]
- 19. Jang Y. J., Ma S., Terada Y., Erikson R. L. (2002) J. Biol. Chem. 277, 44115–44120 [DOI] [PubMed] [Google Scholar]
- 20. Ji J. H., Jang Y. J. (2006) J. Biochem. Mol. Biol. 39, 709–716 [DOI] [PubMed] [Google Scholar]
- 21. Clarke P. R., Zhang C. (2008) Nat. Rev. Mol. Cell Biol. 9, 464–477 [DOI] [PubMed] [Google Scholar]
- 22. Bischoff F. R., Ponstingl H. (1991) Nature 354, 80–82 [DOI] [PubMed] [Google Scholar]
- 23. Bischoff F. R., Krebber H., Kempf T., Hermes I., Ponstingl H. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 1749–1753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H. Y., Zheng Y. (2004) Genes Dev. 18, 512–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Swaminathan S., Kiendl F., Körner R., Lupetti R., Hengst L., Melchior F. (2004) J. Cell Biol. 164, 965–971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Di Matteo G., Fuschi P., Zerfass K., Moretti S., Ricordy R., Cenciarelli C., Tripodi M., Jansen-Durr P., Lavia P. (1995) Cell Growth Differ. 6, 1213–1224 [PubMed] [Google Scholar]
- 27. Guarguaglini G., Renzi L., D'Ottavio F., Di Fiore B., Casenghi M., Cundari E., Lavia P. (2000) Cell Growth Differ. 11, 455–465 [PubMed] [Google Scholar]
- 28. Kalab P., Pu R. T., Dasso M. (1999) Curr. Biol. 9, 481–484 [DOI] [PubMed] [Google Scholar]
- 29. Bischoff F. R., Krebber H., Smirnova E., Dong W., Ponstingl H. (1995) EMBO J. 14, 705–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Di Fiore B., Ciciarello M., Mangiacasale R., Palena A., Tassin A. M., Cundari E., Lavia P. (2003) J. Cell Sci. 116, 3399–3411 [DOI] [PubMed] [Google Scholar]
- 31. Tedeschi A., Ciciarello M., Mangiacasale R., Roscioli E., Rensen W. M., Lavia P. (2007) J. Cell Sci. 120, 3748–3761 [DOI] [PubMed] [Google Scholar]
- 32. Yoon S. O., Shin S., Liu Y., Ballif B. A., Woo M. S., Gygi S. P., Blenis J. (2008) Mol. Cell 29, 362–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen C., Okayama H. (1987) Mol. Cell Biol. 7, 2745–2752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lee K. S., Erikson R. L. (1997) Mol. Cell Biol. 17, 3408–3417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Arnaoutov A., Dasso M. (2003) Dev. Cell 5, 99–111 [DOI] [PubMed] [Google Scholar]
- 36. Dasso M. (2002) Curr. Biol. 12, R502–R508 [DOI] [PubMed] [Google Scholar]
- 37. Quimby B. B., Dasso M. (2003) Curr. Opin. Cell Biol. 15, 338–344 [DOI] [PubMed] [Google Scholar]
- 38. Jang Y. J., Ji J. H., Ahn J. H., Hoe K. L., Won M., Im D. S., Chae S. K., Song S., Yoo H. S. (2004) Biochem. Biophys. Res. Commun. 325, 257–264 [DOI] [PubMed] [Google Scholar]
- 39. Keryer G., Di Fiore B., Celati C., Lechtreck K. F., Mogensen M., Delouvee A., Lavia P., Bornens M., Tassin A. M. (2003) Mol. Biol. Cell 14, 4260–4271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Peloponese J. M., Jr., Haller K., Miyazato A., Jeang K. T. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 18974–18979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Joseph J., Tan S. H., Karpova T. S., McNally J. G., Dasso M. (2002) J. Cell Biol. 156, 595–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Moore W., Zhang C., Clarke P. R. (2002) Curr. Biol. 12, 1442–1447 [DOI] [PubMed] [Google Scholar]
- 43. Hutchins J. R., Moore W. J., Hood F. E., Wilson J. S., Andrews P. D., Swedlow J. R., Clarke P. R. (2004) Curr. Biol. 14, 1099–1104 [DOI] [PubMed] [Google Scholar]
- 44. Arnaoutov A., Dasso M. (2005) Cell Cycle 4, 1161–1165 [DOI] [PubMed] [Google Scholar]
- 45. Li H. Y., Ng W. P., Wong C. H., Iglesias P. A., Zheng Y. (2007) Cell Cycle 6, 1886–1895 [DOI] [PubMed] [Google Scholar]
- 46. Lowery D. M., Lim D., Yaffe M. B. (2005) Oncogene 24, 248–259 [DOI] [PubMed] [Google Scholar]
- 47. Nakajima H., Toyoshima-Morimoto F., Taniguchi E., Nishida E. (2003) J. Biol. Chem. 278, 25277–25280 [DOI] [PubMed] [Google Scholar]