

Abstract
Studies in animal models have indicated that dietary isothiocyanates (ITCs) exhibit cancer preventive activities through carcinogen detoxification-dependent and -independent mechanisms. The carcinogen detoxification-independent mechanism of cancer prevention by ITCs has been attributed at least in part to their ability to induce apoptosis of transformed (initiated) cells (e.g. through suppression of IκB kinase and nuclear factor κB as well as other proposed mechanisms). In the current studies we show that ITC-induced apoptosis of oncogene-transformed cells involves thiol modification of DNA topoisomerase II (Top2) based on the following observations. 1) siRNA-mediated knockdown of Top2α in both SV40-transformed MEFs and Ras-transformed human mammary epithelial MCF-10A cells resulted in reduced ITC sensitivity. 2) ITCs, like some anticancer drugs and cancer-preventive dietary components, were shown to induce reversible Top2α cleavage complexes in vitro. 3) ITC-induced Top2α cleavage complexes were abolished by co-incubation with excess glutathione. In addition, proteomic analysis revealed that several cysteine residues on human Top2α were covalently modified by benzyl-ITC, suggesting that ITC-induced Top2α cleavage complexes may involve cysteine modification. Interestingly, consistent with the thiol modification mechanism for Top2α cleavage complex induction, the thiol-reactive selenocysteine, but not the non-thiol-reactive selenomethionine, was shown to induce Top2α cleavage complexes. In the aggregate, our results suggest that thiol modification of Top2α may contribute to apoptosis induction in transformed cells by ITCs.
Keywords: Apoptosis, Cancer Therapy, Chromosomes, DNA Damage, DNA Topoisomerase
Introduction
Many dietary components have been shown to possess cancer preventive activities (1). Among them, the isothiocyanates (ITCs)2 from cruciferous vegetables and other sources have been most extensively studied (2). ITCs such as benzyl ITC (BITC), phenethyl-ITC (PEITC), and sulforaphane (4-methylsulfinylbutyl-ITC (SFN), are converted from their glucosinolate precursors by the action of myrasinase (2).
ITCs are known to induce phase II enzymes, thereby detoxifying carcinogens and preventing initiation of carcinogenesis in animal models (3). In this carcinogen-detoxifying mechanism of cancer prevention, ITCs activate the nuclear factor E2-related factor (Nrf2) through a mechanism that involves their covalent modifications of the critical cysteine residues on Kelch-like ECH-associated protein 1 (Keap1), resulting in the release of active Nrf2 from the repressive Nrf2-Keap1 complex. Activated Nrf2 then stimulates the transcription of phase II carcinogen-detoxifying enzymes (4). However, it has also become increasingly evident that the ability of ITCs (as well as other dietary components such as selenite and epigallocatechin gallate) to detoxify carcinogens through phase II enzyme induction is not the only mechanism for their cancer preventive activity. First, ITCs are effective in cancer prevention in animals even when administered weeks after carcinogen application, which argues against the carcinogen-detoxifying mechanism as the sole mechanism for cancer prevention (5). Second, ITCs have been shown to prevent tumor formation in spontaneous carcinogenesis animal models where no carcinogen is involved (6). In addition, ITCs can suppress tumor growth in mice carrying human tumor xenografts (7). These studies argue for the existence of a carcinogen detoxification-independent mechanism for the cancer preventive activity of ITCs.
ITCs readily inhibit cell proliferation and induce apoptosis in various cancer cells (8). Indeed, ITC-induced apoptosis has been proposed to be responsible for the cancer-preventive activity in the post-initiation phase (8). It has been suggested that ITCs may selectively induce apoptosis of transformed cells during the initial stage of tumorigenesis (9). Consistent with this notion, transformed cells and cancer cells are much more sensitive to ITCs than their normal untransformed counterparts (10). The ability of ITCs to selectively induce apoptosis of transformed cells has been studied extensively. For example, ITCs are known to activate caspases, up-regulate pro-apoptotic proteins, and down-regulate anti-apoptotic proteins (8). The apoptosis-inducing activity of ITCs has also been linked to reactive oxygen species production (11), activation of the JNK pathway (12), suppression of IκB kinase and NF-κB (13), down-regulation of STAT3 (10), inhibition of tubulin polymerization (14), and proteasome inhibition (15). However, the exact molecular basis for the ITC ability to induce apoptosis of tumor cells remains unclear.
DNA topoisomerase IIα (Top2α), a tumor marker (16), is highly up-regulated in transformed and cancer cells due to activation of oncogenic Ras (17), inactivation of p53 tumor suppressor (18), and in some tumors, co-amplification with human epidermal growth factor receptor 2 due to the close proximity of their genes located on chromosome 17q21 (19, 20). Top2-targeting drugs, such as VP-16 (etoposide), doxorubicin, and mitoxantrone, are known to trap lethal Top2α-DNA covalent adducts, termed Top2α cleavage complexes, which are responsible for the antitumor activity of Top2-targeting drugs (21). Studies of thiol-reactive compounds such as β-lapachone, menadione, disulfiram (DSF), epigallocatechin gallate, and selenite have suggested a thiol modification mechanism for the induction of lethal Top2α-DNA covalent adducts in vitro (22–25).
In view of the importance of the thiol reactivity of ITCs in their cancer preventive activity (26), we have carried out the current studies to examine a possible role of thiol modification of Top2α in tumor cell killing by ITCs. Our current studies have demonstrated that the ITC sensitivity of transformed cells is in part determined by the Top2α protein level. In addition, ITCs can stimulate the formation of Top2α-DNA covalent adducts in vitro through a thiol modification mechanism. These results suggest that thiol modification of Top2α and, hence, the formation of Top2α-DNA covalent adducts may contribute to apoptosis induction in transformed cells by ITCs.
EXPERIMENTAL PROCEDURES
Chemicals and Reagents
BITC, PEITC, SFN, glutathione (GSH), selenomethionine, selenocysteine, VP-16 (etoposide), DSF, sodium selenite, and thiazolyl blue tetrazolium bromide were purchased from Sigma. Anti-α-tubulin antibodies were purchased from the Developmental Studies Hybridoma Bank at the University of Iowa. Anti-phospho-H2AX (Ser-139) clone JBW301 mouse antibodies were purchased from Millipore Co. Anti-β-actin (13E5) rabbit antibodies was purchased from Cell Signaling Technology. The anti-Top2β antibodies were purchased from Santa Cruz Biotechnology. The anti-Top2α antibodies were gift from Dr. Jaulang Hwang (IMB, Acdemia Sinica, Taiwan). The annexin V-FITC apoptosis detection kit was purchased from BD Pharmingen. Tissue culture media, DMEM, and RPMI 1640, were purchased from Invitrogen. All other tissue culture media and reagents were purchased from Invitrogen.
In Vitro Top2α-mediated DNA Cleavage Assay
Top2-mediated DNA cleavage assay was carried out as described previously (27). Briefly, the reaction mixture (20 μl total volume) containing 50 mm Tris-HCl, pH 8.0, 100 mm KCl, 10 mm MgCl2, 0.5 mm EDTA, 30 μg/ml bovine serum albumin, 1 mm ATP, 10 ng of 3′-end 32P-labeled YEpG DNA, 10 ng of purified human Top2α, and the compound of interest were incubated at 37 °C for 30 min. The reactions were terminated by the addition of 5 μl of 5% SDS and proteinase K to a final concentration of 200 μg/ml followed by incubation for an additional 120 min at 42 °C. DNA samples were separated in 1% agarose gel, 0.5× Tris-Phosphate-EDTA buffer (45 mm Tris-Phosphate, pH 8.0, 1 mm EDTA), 2.5 V/cm. Gels were dried onto Whatman No. 3MM chromatographic paper and autoradiographed at −80 °C using Eastman Kodak Co. XAR-5 films.
Apoptosis Assay
Annexin V Staining
To determine ITC-induced apoptosis, SV40-transformed MEFs were treated with different compounds. At the end of treatment cells were trypsinized using 0.5% trypsin-EDTA and washed twice with ice-cold 1× PBS. After washing, cells were subjected to annexin V staining by following the manufacturer's protocol. Briefly, cells were resuspended in 100 μl of 1× annexin V binding buffer followed by a 15-min staining in the dark with annexin V-FITC for phosphatidylserine translocation and propidium iodine for membrane integrity. An additional 400 μl of 1× annexin V binding buffer was added to each staining reaction. The samples were then subjected to flow cytometry analysis.
Nucleosomal DNA Fragmentation
For measuring ITC-induced chromosomal DNA fragmentation, 2.5 × 106 HL60 or HL60/MX2 cells were treated with different compounds for 4 h at 37 °C. Cells were then pelleted and lysed in 0.4 ml of lysis buffer (10 mm Tris-HCl, pH 8.0, 20 mm EDTA, and 0.2% Triton-X100) followed by incubation on ice for 20 min. After centrifugation in a microcentrifuge (12,000 × g) for 20 min, fragmented DNA in the supernatant was extracted with phenol/chloroform and precipitated with a volume of 3 m sodium acetate, pH 5.2, and 2 volumes of ethanol. DNA was pelleted by centrifugation, rinsed with 70% ethanol, and then resuspended in Tris-EDTA buffer containing 100 μg/ml RNase A. After 2 h of incubation at 37 °C, DNA samples were electrophoresed in 1.5% agarose gel, stained with ethidium bromide, and visualized under UV light.
Cell Culture and Cytotoxicity Assay
Top2β+/+ and top2β−/− primary MEFs were isolated from E13.5 mouse embryos as described previously (28). SV40-transformed MEFs were obtained by transfecting primary MEFs with the SV40 large T antigen-expressing plasmid, pAN2 (28). The human AML cell line HL-60 and its mitoxantrone-resistant variant HL-60/MX2 were obtained from ATCC. Oncogenic Ras-transformed MCF10A cells were generated by transfecting K-Ras-expressing plasmid into immortalized human breast MCF10A cells (63). MEFs and transformed MEFs were cultured in DMEM media containing 10% FetalPlex animal serum complex (Gemini Bio-Products), l-glutamine (2 mm), penicillin (100 units/ml), and streptomycin (0.1 mg/ml) in a humidified incubator with 5% CO2 at 37 °C. HL-60 and HL-60/MX2 cells were cultured under the same conditions except that RPMI 1640 was used instead of DMEM. MCF10A and K-Ras-transformed MCF-10A cells were cultured as described (30). For IC50 determination, cells were exposed continuously to various ITCs. Thiazolyl blue tetrazolium bromide assays were performed at the end of the fourth day. All assays were performed at least twice in triplicate. Serum deprivation of primary MEFs were performed using 0.2% instead of 10% FetalPlex on confluent cultures (2.5 × 105 cells/well of 6-well plate) for 48 h.
Gene Silencing by siRNAs
For silencing Top2α, subconfluent MEFs (SV40 T-transformed) and MCF10A (K-Ras-transformed) cells were cultured in Opti-MEM (Invitrogen) and transfected with 150 ng of mouse (sense, 5′-GUAUUAGAGUCACAAUUGA-3′; antisense, 5′-UCAAUUGUGACUCUAAUCA-3′) and human (sense, 5′-CAAGAAGUGUUCAGCUGUA-3′; antisense, 5′-UACAGCUGAACACUUCUUG-3′) Top2α-specific siRNAs (purchased from Sigma) using Oligofectamine (Invitrogen), respectively. Scrambled siRNAs were transfected as controls. 6 h post-transfection, cells were treated with specific ITCs for a period as indicated and immediately lysed in 6× SDS-PAGE sample loading buffer. To determine the efficiency of gene silencing, the Top2α protein level was determined by immunoblotting using anti-Top2α antibodies.
Detection of Covalent Adduction of BITC to Human Top2α in Vitro
We engineered a recombinant human TopIIα with sequences modified at both the N and C termini. The first 28 amino acids of the protein were replaced by the first 5 amino acids of yeast Top2 followed by sequences of a heart muscle kinase phosphorylation site and a hexahistidine tag. 125 amino acids were also truncated at C terminus (1405–1530). A GAL+/protease-deficient yeast strain, BCY123, was used as a host for expressing recombinant protein. Recombinant hTop2α was purified as described previously (31) except that POROS® HS column chromatography (Applied Biosystems) was used as a further purification step after Ni2+-nitrilotriacetic acid column. For detecting Top2α fragments with adducted BITC, 30 μg of recombinant hTop2α were preincubated in 100 μl of buffer (50 mm NH4HCO3, pH 7.5, 50 mm NaCl, 10 mm MgCl2, 0.1 mm EDTA, and 0.5 mm tris(2-carboxyethyl)phosphine) at 37 °C for 5 min. The reaction mixture was then incubated at 37 °C in the presence of BITC as indicated (time and dose). For kinetic analysis, samples were reacted with an excess amount of iodoacetamide (50 mm) for 30 min. Most of the modified enzyme formed insoluble aggregates and was collected by centrifuging at 10,000 × g for 10 min. To protect buried and unmodified cysteines with iodoacetamide, precipitant was resuspended with 100 μl of buffer (6 m urea, 50 mm iodoacetamide, 50 mm NH4HCO3, pH 7.5, 50 mm NaCl, 10 mm MgCl2, 0.1 mm EDTA and 0.5 mm tris(2-carboxyethyl)phosphine) for 30 min at 37 °C. After the reaction, enzyme aggregates were collected again by diluting urea 5-fold and centrifuging at 10,000 × g for 10 min. Tryptic peptide fragments were generated by digesting the aggregated enzyme in 50 mm NH4HCO3, pH 7.5, with trypsin (1:20 (w/w)) for 2 h at 37 °C and processed for mass spectrometry analysis described in the following. The digested fragments were first separated over an 18-min linear gradient from 0 to 60% (v/v) acetonitrile at 200 μl/min performed on a Zorbax C8 column (SB-C8, 2.1 × 50 mm (5 μm), Agilent Technology) coupled to a quadrupole-time of flight mass spectrometer (QSTAR XL, Applied Biosystems) equipped with a nano-electrospray ionization source. The mass spectra were acquired under a positive ionization mode in the range of 200 to 2000 m/z (atomic mass units).
Homology Modeling of the Human Top2α AB Prime Region
The homology model of the human Top2α was built from residues 410 to 1213 of the known sequence (CAA09762). The x-ray crystal structure of yeast topoisomerase II (PDB code 3L4K) was used as the template for model construction (32). All template searching and sequence alignment work was done using the pGenThreader algorithm (33). The template homodimer was prepared from the monomeric crystal structure using the symmetry routine in PyMOL (Schrödinger). The initial models were built from the pGenThreader alignment while maintaining the positions of the zinc ions and the nucleic acid from the template via the single-template approach using the Modeler program (9v8) (34–37). The lowest discrete optimized protein energy (DOPE) score model was selected for additional refinement. The double-stranded DNA was made whole in order to build a non-covalent Top2-DNA complex. The model was further refined via energy minimization using the Amber 11 suite of programs (38). The Amber99SB-ildn force field (39) was used with a 12 Å cutoff, and solvent was accounted-for using the generalized Born implicit solvent model (40). Illustrations were prepared using PyMOL.
RESULTS
The ITC Sensitivity Correlates with the Top2α Protein Level
ITCs have been shown to preferentially inhibit proliferation and induce apoptosis of tumor cells as compared with normal cells (9, 10). To determine whether oncogene activation may be responsible for the preferential sensitivity of tumor cells, the ITC sensitivity of MCF-10A cells and K-Ras-transformed MCF-10 cells was determined. As shown in Table 1, Ras-transformed MCF-10A cells were 2–5-fold more sensitive to different ITCs (i.e. BITC, PEITC, and SFN) than non-transformed MCF-10A cells. Ras-transformed MCF-10A cells also showed a 2-fold greater sensitivity toward VP-16 (a Top2-specific inhibitor) (21) than non-transformed MCF-10A cells. By contrast, both cells exhibited similar sensitivity toward camptothecin (CPT), a DNA topoisomerase 1-specific inhibitor (21). It was also observed that PEITC, like VP-16, induced at least 2-fold more DNA damage signal, γ-H2AX, in Ras-transformed MCF-10A cells than in non-transformed MCF-10A cells (Fig. 1A), whereas CPT induced the same amount of the γ-H2AX signal in both cells. It is interesting to note that the expression level of Top2α was about 2-fold higher in Ras-transformed MCF-10A cells than in non-transformed MCF-10A cells.
TABLE 1.
ITCs selectively inhibit the growth of Ras-transformed MCF-10A cells
MCF10A and K-Ras-transformed MCF-10A cells were exposed to various compounds as indicated. MTT assay was performed at the end of the fourth day as described under “Experimental Procedures.” Each experiment was performed in triplicate and repeated more than twice. The IC50 is calculated by regression curve-fitting.
| Cells | BITC | PEITC | SFN | VP-16 | CPT |
|---|---|---|---|---|---|
| μm | μm | μm | μm | nm | |
| MCF-10A | 15 ± 0.5 | 12 ± 0.8 | 23 ± 2.8 | 4.3 ± 0.2 | 70 ± 1.8 |
| K-Ras | 3.2 ± 0.7 | 3.4 ± 0.5 | 10 ± 1.3 | 2.0 ± 0.1 | 69 ± 3.2 |
FIGURE 1.
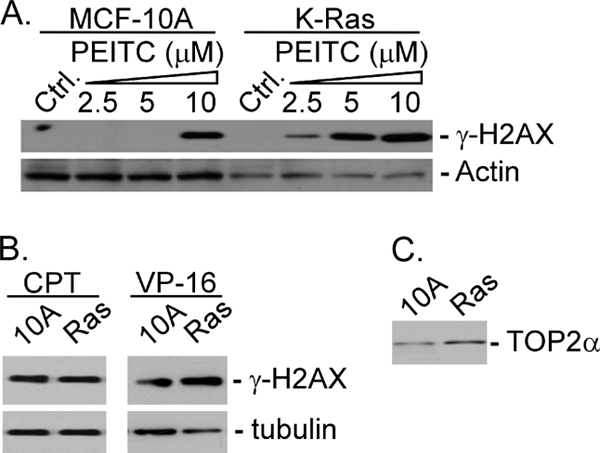
PEITC induces γ-H2AX more effectively in K-Ras-transformed cells. MCF-10A and K-Ras-transformed MCF-10A cells were treated with PEITC (see the indicated concentrations) (A) or CPT (10 μm) or VP-16 (10 μm) (B) for 6 h followed by immunoblotting using antibodies against γ-H2AX or β-actin. Top2α protein levels in MCF-10A and K-Ras-transformed MCF-10A cells were determined by immunoblotting using anti-human Top2α antibodies.
To test the possibility that the Top2α protein level may determine ITC sensitivity, the ITC sensitivity of HL-60 (Top2α-proficient) and HL-60/MX2 (Top2α-deficient) cells was determined using a nucleosomal DNA fragmentation assay for apoptosis induction. As shown in Fig. 2A, PEITC selectively induced the release of nucleosomal DNA fragments in HL-60 cells as compared with HL-60/MX2 cells. As a positive control, VP-16 was also shown to preferentially induce nucleosomal DNA fragmentation in HL-60 cells as compared with HL-60/MX2 cells. By contrast, CPT was equally active in inducing nucleosomal DNA fragmentation in both HL-60 and HL-60/MX2 cells (Fig. 2A). Similarly, BITC treatment caused nucleosomal DNA fragmentation in HL-60 cells, but not in HL-60/MX2 cells (Fig. 2B). SFN, although less active, also induced nucleosomal DNA fragmentation in HL-60 cells but not in HL-60/MX2 cells (Fig. 2C). It is noted that the efficiency of ITCs to induce apoptosis seems to parallel their IC50 (in the order of PEITC = BITC > SFN). These results suggest that the Top2α protein level correlates with ITC sensitivity in terms of growth inhibition, DNA damage, and apoptosis.
FIGURE 2.
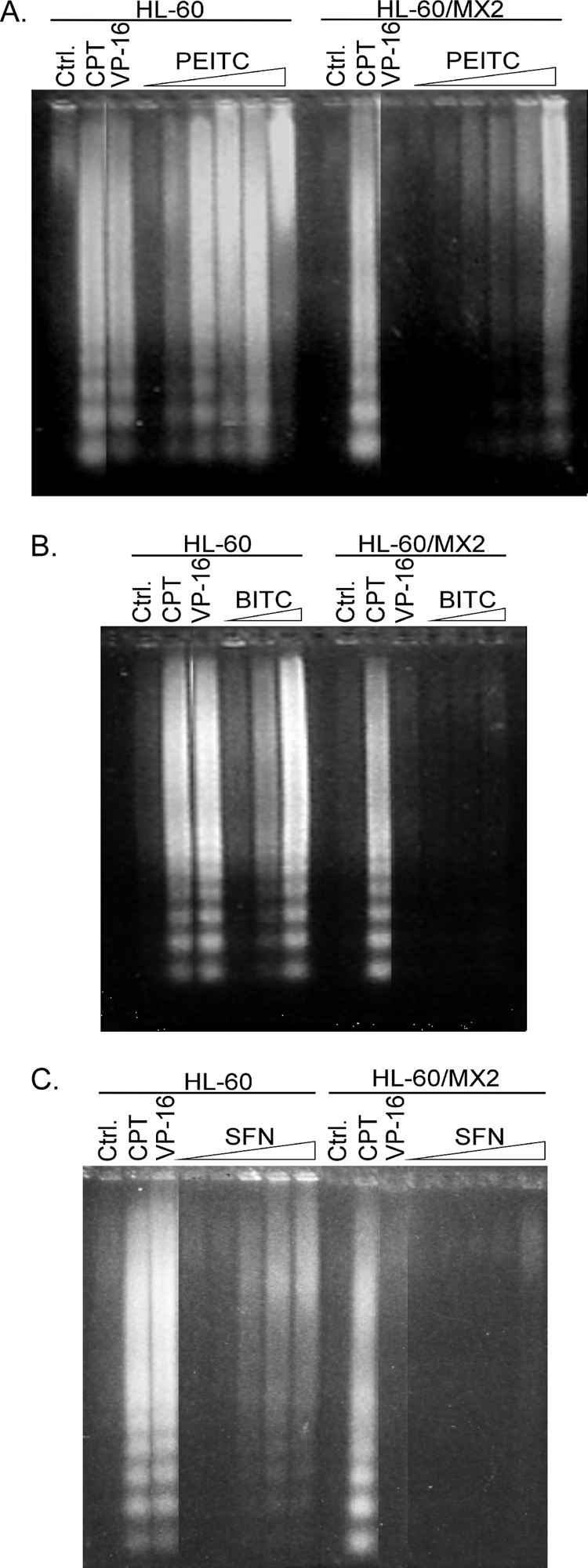
ITCs induce Top2α-dependent nucleosomal DNA fragmentation. HL60 and Top2-deficient HL60/MX2 cells were treated with PEITC (0.1/1/1.5/2/2.5/5 μm) (A), BITC (0.1/1/1.5 μm) (B), and SFN (1/2.5/5/10/20 μm) (C) for 4 h. As controls, cells were also treated with VP-16 (10 μm) or CPT (2 μm). Genomic DNA was isolated and subjected to agarose gel electrophoresis as described under “Experimental Procedures.”
siRNA-mediated Knockdown of Top2α Attenuates ITC-induced DNA Damage and Apoptosis
To test if the Top2α protein level determines ITC sensitivity, the Top2α protein level was knocked down using RNAi before ITC treatment. As shown in Fig. 3A, left panel, siRNA-mediated knockdown of Top2α in SV40-trasnformed MEFs (see the lower panel for Top2α knockdown efficiency) led to a significant decrease of the γ-H2AX signal induced by ITC (i.e. BITC, PEITC, and SFN) treatment. As positive and negative controls, respectively, Top2α silencing was also shown to reduce the VP-16-induced, but not CPT-induced, γ-H2AX signal (Fig. 3A, right panel). The effect of Top2α silencing (see Fig. 3B, lower panel) on ITC-mediated DNA damage was also studied in Ras-transformed MCF-10A cells. As shown in Fig. 3B, top panels, siRNA-mediated knockdown of Top2α reduced PEITC and VP-16 but not CPT-induced γ-H2AX signals. These results suggest that the Top2α protein level determines the DNA damage signal induced by ITCs.
FIGURE 3.

Top2α silencing attenuates ITC-induced DNA damage and apoptosis signals. A, SV40-transformed MEFs were transfected with Top2α or control siRNA for 6 h followed by treatment with VP-16 (10 μm), CPT (10 μm), BITC (5 μm), PEITC (10 μm), or SFN (20 μm) for 2 h. Immunoblotting was performed using anti-γ-H2AX antibodies. To determine the gene silencing efficiency, the Top2α protein level was measured by immunoblotting using anti-mouse Top2α antibodies. B, Ras-transformed MCF-10A cells were transfected with Top2α or control siRNA for 6 h followed by treatment with VP-16 (10 μm), CPT (2 μm), and PEITC (10 μm) for another 6 h. Immunoblotting was performed using anti-γ-H2AX antibodies. The Top2α gene silencing efficiency was determined as in A using anti-human Top2α antibodies. C, SV40 large T antigen-transformed MEFs were transfected with mouse Top2α-specific siRNA for 6 h followed by treatment with VP-16 (10 μm), hydrogen peroxide (150 μm), BITC (10 μm), PEITC (10 μm), or SFN (10 μm) for 12 h. The apoptotic population was determined by flow cytometry after staining with annexin V-FITC.
The effect of Top2α gene knockdown on ITC-induced apoptosis was also examined in transformed MEFs. As shown in Fig. 3C, Top2α knockdown decreased the annexin V-positive populations in ITC and VP-16 but not hydrogen peroxide-treated SV40-transformed MEFs. Again, it was noted that SFN was less effective in inducing apoptosis than BITC and PEITC. Together, our results suggest that the Top2α protein level is a determinant of ITC sensitivity in terms of both DNA damage and apoptosis induction in transformed cells.
ITC Sensitivity Is Reduced in Cells Depleted of Both Top2 Isozymes
There are two Top2 isozymes, Top2α and Top2β, in mammalian cells (41). Top2α is known to be a cell proliferation marker and is up-regulated in tumor cells due to multiple mechanisms (16–19). In non-transformed cells such as primary MEFs, Top2α levels peak at G2/M, and serum deprivation is known to drive MEFs into G1 phase, where the Top2α level is minimal (42). By contrast, Top2β is present at more or less constant levels in all cells (42). To deplete both Top2 isozymes in MEFs, primary MEFs (top2β−/−) were serum (0.2% FetalPlex)-deprived for 24 h to reduce the Top2α protein level (see Fig. 4A, inset). The cytotoxic effect of ITCs on serum-deprived MEFs (both top2β−/− and Top2β+/+) was then evaluated using the thiazolyl blue tetrazolium bromide assay. As shown in Table 2, the cytotoxic activity of ITCs was reduced 2–4-fold in top2β−/− MEFs as compared with Top2β+/+ MEFs, whereas those of H2O2 and bleomycin were approximately the same in both cells. These results suggest that the cytotoxic effect of ITCs is dependent on the protein level of the Top2β isozyme when Top2α expression was greatly reduced by serum deprivation, providing further support that ITC sensitivity is Top2-dependent. It is interesting to note that the cytotoxic activities of selenite and DSF were also Top2-dependent (about 4-fold) under the same conditions (Table 2). Both selenite and DSF have been previously shown to inhibit Top2 through the formation of Top2-DNA covalent adducts (24).
FIGURE 4.

ITC-induced γ-H2AX signal is Top2β-dependent in primary MEFs. Quiescent primary Top2β+/+ and top2β−/− MEFs were treated with PEITC (A), BITC (B), SFN (C), and selenite (D) for 2 h followed by immunoblotting analysis using anti-γ-H2AX and anti-α-tubulin antibodies. As controls, quiescent MEFs were also treated with VP-16 (100 μm) or hydrogen peroxide (150 μm) under the same conditions. E, quiescent primary Top2β+/+ MEFs were treated with dexrazozane (ICRF-187) for 24 h to induce Top2β down-regulation. Cells were then treated with BITC (5 μm), PEITC (10 μm), SFN (20 μm), VP-16 (100 μm), or H2O2 (250 μm) for 2 h. The levels of γ-H2AX and Top2β were determined by immunoblotting using anti-γ-H2AX and anti-mouse Top2β antibodies, respectively. c-PARP, cleaved PARP.
TABLE 2.
ITC cytotoxicity in quiescent primary MEFs is Top2β-dependent
Primary Top2β+/+ and top2β−/− MEFs were driven into quiescence in 0.2% serum for 24 h followed by treatment with various compounds. MTT assay was performed at the end of the fourth day as described under “Experimental Procedures.” Each experiment was performed in triplicate and repeated at least twice. The IC50 was calculated by regression curve-fitting.
| MEF | Selenium | BITC | PEITC | SFN | DSF | H2O2 | VP-16 | bleomycin |
|---|---|---|---|---|---|---|---|---|
| μm | μm | μm | μm | μm | μm | μm | μm | |
| Top2β+/+ | 18 ± 5.0 | 5.9 ± 0.3 | 7.3 ± 0.3 | 1.7 ± 0.4 | 1.7 ± 0.3 | 25 ± 3.2 | 4.1 ± 0.8 | 64 ± 1.8 |
| top2β −/− | 76 ± 10 | >20 | 20 ± 0.5 | 3.1 ± 0.4 | 5.7 ± 0.7 | 20 ± 5.0 | >200 | 69 ± 2.2 |
It was noted that the VP-16 sensitivity was reduced more than 50-fold in serum-deprived top2β−/− MEFs as compared with Top2β+/+ MEFs under the same serum deprivation conditions. The difference in Top2 dependence (2–4-fold versus >50-fold dependence) between VP-16 and ITCs on Top2 suggests that ITCs may exert its cytotoxic activity through both Top2-dependent and -independent mechanisms, whereas VP-16 cytotoxicity is exclusively through the Top2-dependent mechanism.
We also monitored the ITC-induced DNA damage signal (i.e. γ-H2AX) in serum-deprived (quiescent) Top2β+/+ and top2β−/− primary MEFs. As shown in Fig. 4, A–C, different ITCs induced a greater amount of the γ-H2AX signal in Top2β+/+ MEFs than in top2β−/− MEFs. As expected, VP-16 also induced less γ-H2AX signal in top2β−/− than in Top2β+/+ MEFs, whereas H2O2 induced an equal amount of the γ-H2AX signal in both cells. These results provide additional support for ITC-induced DNA damage being Top2-dependent. As an additional positive control, the selenite-induced γ-H2AX signal in primary MEFs was also monitored under the same conditions. As shown in Fig. 4D, selenite induced a higher level of γ-H2AX signal in Top2β+/+ MEFs than in top2β−/− MEFs, consistent with our previous conclusion that selenium compounds induce Top2-dependent DNA damage (24).
Previous studies have demonstrated that Top2β can be specifically and efficiently down-regulated by Top2 catalytic inhibitors (e.g. ICRF-193 and ICRF-187) through a proteasome pathway (43). Consequently, it is possible to deplete Top2β in MEFs using such inhibitors. As shown in Fig. 4E, inset, a 24-h treatment with ICRF-187 was able to significantly down-regulate Top2β in serum-deprived primary (Top2β+/+) MEFs, creating a condition where both Top2 isozymes were essentially null. Under such a condition, the ITC-induced, like VP-16-induced but not H2O2-induced γ-H2AX signal, was reduced by ICRF-187 (Fig. 4E), again suggesting that ITC-induced DNA damage is Top2-dependent.
In addition to the DNA damage signal γ-H2AX, we also monitored poly(ADP-ribose) polymerase (PARP) cleavage as an apoptosis end point in serum-deprived primary MEFs. As shown in Fig. 4, A–C, ITCs (i.e. PEITC, BITC, and SFN) induced less PARP cleavage (see the levels of cleaved PARP (c-PARP)) in top2β−/− than in Top2β+/+ MEFs. These results provide additional support for the notion that ITC-induced apoptosis is Top2-dependent. However, it is noted that under the same condition (see Fig. 4), VP-16 (100 μm), which induced more γ-H2AX signal than ITCs, induced less apoptosis (PARP cleavage) than ITCs in serum-deprived Top2β+/+ MEFs. This result could suggest that ITCs, unlike VP-16, which is highly Top2-specific, may also induce apoptosis through an additional Top2-independent mechanism.
ITCs Induce Top2α Cleavage Complexes in Vitro
The Top2 dependence in ITC sensitivity (i.e. growth inhibition, DNA damage, and apoptosis) could implicate the involvement of Top2-mediated DNA damage (i.e. the formation of Top2 cleavage complexes). To determine whether ITCs can induce Top2α-mediated DNA damage, purified hTop2α was used in an in vitro cleavage assay (see under “Experimental Procedures”). As shown in Fig. 5, A–C, PEITC, BITC, and SFN induced concentration-dependent DNA cleavages in the presence of hTop2α. These cleavages were reversed by a subsequent incubation with EDTA, indicating the reversibility of the cleavages, which is the hallmark of topoisomerase cleavage complexes (44). As positive controls, VP-16 and DSF were also demonstrated to induce reversible Top2α cleavage complexes (Fig. 5). Additionally, co-incubation with reduced GSH (0.5 mm) reduced the ability of BITC and DSF to induce DNA cleavage (Fig. 5D). By contrast, co-incubation of VP-16 with reduced GSH did not result in detectable changes in DNA cleavages (Fig. 5D). In the aggregate, these results suggest that ITCs can induce DNA Top2α cleavage complexes, and the thiol reactivity of ITCs is likely involved in their induction.
FIGURE 5.

ITCs induce reversible Top2 cleavage complexes in vitro. Left panels (A–C), ITCs induce DNA cleavage in the presence of purified hTop2α. DNA cleavage assay was performed as described in under “Experimental Procedures” using purified recombinant hTop2α. The concentrations of VM-26, VP-16, and disulfiram were 5, 20, and 100 μm, respectively. The concentrations of PEITC (A, left panel) and BITC (B, left panel) were from 0.01 μm to 10 mm with a 10-fold serial dilution, whereas concentrations of SFN (C, left panel) were from 1 μm to 10 mm. DNA cleavage was measured after a 30-min incubation. Right panels (A–C), post-reaction EDTA treatment reverses ITC-induced DNA cleavage. To test the reversibility of ITC-induced DNA cleavage, 50 mm EDTA (final concentration) was added to each cleavage reaction after a 30-min preincubation with PEITC (1 mm) (A), BITC (1 mm) (B), and SFN (10 mm) (C). D, ITC-induced Top2 cleavage complex is greatly reduced in the presence of glutathione (GSH). DNA cleavage was performed as described except that increasing concentrations of GSH (5 μm to 0.5 mm with 10-fold increments) was co-incubated with VP-16 (20 μm), disulfiram (100 μm), or BITC (100 μm). DNA cleavage products of all reactions were analyzed by 1% agarose gel electrophoresis followed by autoradiography. Organo-selenium induce reversible Top2 cleavage complexes in vitro. Left panels (E and F), organo-selenium induce DNA cleavage in the presence of purified hTop2α. DNA cleavage assay was performed as described under “Experimental Procedures” using purified recombinant hTop2α. The concentrations of VM-26, VP-16, and disulfiram were 5, 20, and 100 μm, respectively. The concentrations of selenocysteine (E, left panel) were from 0.1 μm to 10 mm with a 10-fold serial dilution. The concentrations of selenomethionine (F, left panel) were from 1 μm to 10 mm with a 10-fold serial dilution. DNA cleavage was measured after a 30-min incubation. Right panels (E and F), post-reaction EDTA treatment reverses ITC-induced DNA cleavage. To test the reversibility of organo-selenium-induced DNA cleavage, 50 mm EDTA (final concentration) was added to each cleavage reaction after a 30-min preincubation with selenocysteine (E; 10 mm), selenomethionine (F, 10 mm). After another 30 min, DNA cleavage was then measured. As controls, VM-26 (5 μm) or disulfiram (100 μm) were also included.
In addition to ITCs, the essential micronutrient selenium, another dietary component, has also been shown to exhibit cancer preventive activity (45). Studies have suggested that the cancer preventive activity of selenium, which requires supranutritional doses, is unrelated to its micronutrient function (46). Like ITCs, selenium compounds induce phase II enzymes through their thiol reactivity and also exhibit cancer preventive activity in the post-initiation phase (47). Previous studies have demonstrated that inorganic selenite can induce Top2 cleavage complexes in vitro through a thiol modification mechanism (24). Interestingly, as shown in Fig. 5E, left panel, selenocysteine (Se-Cys) also readily induced DNA cleavage at 10 μm in the presence of hTop2α. In addition, selenocysteine-induced DNA cleavage can be fully reversed by post-incubation with EDTA, suggesting the formation of reversible Top2α cleavage complexes. By contrast, selenomethionine, which was used in the failed multicenter SELECT prevention trial for prostate cancer (48) and is expected to be much less reactive with thiols, did not induce any detectable Top2α-mediated DNA cleavage even at 1 mm (Fig. 5F). These results suggest that the cancer preventive activity of selenium compounds, like that of ITCs, could depend on their thiol modification activity and may involve Top2α cleavage complexes. The failure of the SELECT prevention trial could be explained by the lack of (or weak) thiol reactivity of selenomethionine.
Human Top2α Is Covalently Modified by BITC in Vitro
To determine the key cysteine residue(s) on Top2α that could be modified by ITCs, proteomic analysis was performed using purified hTop2α in the presence of BITC. The kinetic analysis with BITC (0.1 mm; 1 and 5 min) indicated that of the 13 cysteine residues on hTop2α, Cys-300 was the most reactive (Fig. 6A). At 1 mm BITC (30 min), more cysteine residues were modified, including highly reactive ones (Cys-104, -170, -300, -392, -455, and -733) and modestly reactive ones (Cys-427, -997/1008, and -1045) (Fig. 6A). Cys-216, -405, and -862 were not modified by BITC under this condition.
FIGURE 6.

Modification of cysteine residues on Top2α by BITC. A, the determination of BITC-modified cysteine residues on human Top2α by TOF-MS (see under “Experimental Procedures.”) is shown. +, −, and ± indicate positive, negative, and weak detection of the covalently modified cysteine, respectively. B, location of Cys-300 (blue) in the ATPase domain of hTop2α (residues 29–411, adapted from the x-ray crystal structure PDB code 1ZXM of the symmetric homodimer) (29) is shown. C, location of residue C455 (blue) and residue C733 (light blue) in the AB prime region of hTop2α (from the homology model of the symmetric homodimer, see under “Experimental Procedures”) is shown. The catalytic tyrosine, Tyr-805, is colored dark gray.
DISCUSSION
Our current studies have demonstrated that ITC-induced apoptosis (evidenced by PARP cleavage and annexin V staining) and DNA damage (evidenced by γ-H2AX) is not only correlated with the Top2 protein levels but also causally linked to Top2 expression. The Top2 dependence of ITC sensitivity is unusual as elevated Top2 protein levels are shown to confer increased ITC sensitivity rather than ITC resistance in this study, whereas elevated expression of most target enzymes are known to lead to drug resistance rather than sensitivity (49). This unusual dependence suggests that the inhibition of the Top2 catalytic activity is most likely not responsible for ITC sensitivity. This unusual property of Top2 dependence is reminiscent of a class of Top2 inhibitors known as Top2 poisons (e.g. VP-16 and doxorubicin). These Top2 inhibitors kill tumor cells by converting Top2 enzymes into DNA cleaving “poisons” (21). Consequently, elevated expression of Top2 confers drug sensitivity rather than resistance (50). Indeed, we have demonstrated that ITCs, like VP-16, can poison Top2α as evidenced by induction of reversible Top2α cleavage complexes in vitro.
Our studies have also suggested that a thiol modification mechanism may be responsible for ITC-induced formation of Top2α cleavage complexes. A thiol modification mechanism for Top2 poisoning was initially proposed based on studies of β-lapachone and menadione (22), both of which are naphthoquinones capable of undergoing the Michael addition reaction to react rather specifically with thiol nucleophiles (e.g. protein thiols). It has been proposed that modification of the sulfhydryl group(s) of cysteine(s) on Top2α by these Michael acceptors leads to trapping of reversible Top2α cleavage complexes (51). This thiol modification mechanism has also been suggested to be responsible for the Top2α poisoning activity of selenite and epigallocatechin gallate (24, 52). It is interesting to point out that, like ITCs, selenite, and epigallocatechin gallate, many cancer-preventive dietary components, including curcumin, organosulfides, and retinoids (e.g. retinoic acid and retinamide) are also chemically reactive toward sulfhydryl groups (53, 54). Moreover, all these agents are known to induce similar cellular responses, including phase II enzyme induction through thiol modification of Keap1 and inhibition/suppression of IκB kinase/NF-κB (53, 55–58). Consequently, thiol modification of key regulatory enzymes may be responsible for the multitude of activities of these dietary components. Modification of Top2 by ITC may represent only one mechanism for ITC-induced apoptosis. In this regard, it is interesting to point out that our studies have revealed the presence of a Top2-independent mechanism for ITC-induced apoptosis in serum-starved primary top2β−/− MEFs, where the expression level of both Top2 isozymes is nil or close to nil (Fig. 4). The nature of this Top2-independent mechanism is currently unknown. However, ITCs is known to up-regulate pro-apoptotic proteins and down-regulate anti-apoptotic proteins (8). In addition, ITCs are known to suppress NF-κB, which could influence apoptosis (8). The possible interplay among these mechanisms may contribute to the pro-apoptotic activity of ITCs, and the contribution of each mechanism may depend on the functional status of the relevant target proteins.
Top2α is a tumor marker, and poisoning of Top2α by small molecules (e.g. anticancer drugs) is expected to lead to tumor-specific killing. The Top2α poisoning activity of ITCs, as demonstrated in the current studies, could contribute to the post-initiation phase of cancer prevention by ITCs through elimination of transformed cells. However, as we have demonstrated in Fig. 4, the Top2β isozyme is similarly involved in ITC-induced apoptosis. Top2β is known to be present at more or less constant levels in all cells and functions in gene activation/repression (59). It has been suggested that Top2β poisoning, unlike Top2α poisoning, can lead to tissue damage (e.g. doxorubicin-induced cardiotoxicity) and DNA sequence rearrangements (i.e. etoposide-induced t-AML) (43, 60). Consequently, the ability of ITCs to poison the Top2β isozyme could present a potential problem for the safe use of ITCs. Indeed, there is evidence suggesting that ITCs may paradoxically induce tumors at higher doses (61). However, due to the much higher abundance of Top2α relative to Top2β in tumor cells, low concentrations of ITCs could be safely used to kill tumor cells through Top2α poisoning with a minimal effect on Top2β (43, 60).
It has been suggested that the rate of thiol modification depends on both the chemical structure of the thiol-reactive compounds (e.g. the size of the alkyl side chains of ITCs) and the structural microenvironment in the vicinity of the -SH group (e.g. the adjacent amino acids to the cysteine residue) (14, 24). There are 13 cysteine residues on human Top2α. Identification of the key cysteine residues on Top2α that are responsible for ITC-mediated thiol modification coupled with structural analysis of the local environments surrounding the key cysteine residues may help design novel ITCs that are Top2α-selective, thereby alleviating the potential Top2β isozyme-mediated side effects. Our proteomic analysis has identified Cys-300 on hTop2α to be the most reactive cysteine residue toward BITC-mediated covalent modification. Cys-300 is located in the ATPase domain of hTop2α (see Fig. 6B). Although Cys-300 is the most reactive cysteine residue on hTop2α toward BITC, it may not be the critical cysteine residue responsible for ITC-induced poisoning of hTop2 as it is located in the ATPase domain, which is distant from the active site tyrosine (Tyr-805). It is possible that another cysteine(s) (e.g. Cys-455 and -733) located in the breakage-reunion domain (shown in Fig. 6C; Cys-455 is located about 29.6 Å, and Cys-733 is 19.0 Å away from the active site Tyr-805 as measured from Cα to Cα in the same chain of the homodimer), albeit less reactive, may be the critical one(s) for ITC-mediated Top2 poisoning. It is interesting to point out that Cys-392 and -405 of human hTop2α (located in the ATPase domain) have been shown to form adducts with thiol-reactive quinones (62). In our current studies, Cys-392 is reactive with BITC at 30 min in the presence of 1 mm BITC, whereas Cys-405 is not reactive at all with BITC under the same condition. Because Cys-392 is reactive with both quinones and BITC, it could be the critical cysteine residue for poisoning of Top2α by these thiol-reactive compounds. However, it is also possible that multiple cysteine residues on hTop2α, when covalent-modified, could lead to stabilization of hTop2α cleavage complexes, and different thiol-reactive compounds may modify a different cysteine residue(s) on hTop2α to achieve similar poisoning activity. Clearly, further studies through a combination of structural, biochemical, and mutational approaches are necessary to identify the critical cysteine residues(s) on hTop2 isozymes that is responsible for ITC-mediated poisoning of hTop2. Such studies could be important for the future design of Top2 isozyme-specific thiol-modifying Top2 poisons that can be used for cancer prevention and/or therapy.
This work was supported, in whole or in part, by National Institutes of Health Grants CA39662 and CA102463.
- ITC
- isothiocyanate
- BITC
- benzyl ITC
- PEITC
- phenethyl ITC
- SFN
- sulforaphane (4-methylsulfinylbutyl-ITC)
- Top2
- DNA topoisomerase II
- hTop
- human Top
- Nrf2
- nuclear factor E2-related factor
- Keap1
- Kelch-like ECH-associated protein 1
- DSF
- disulfiram
- CPT
- camptothecin
- PARP
- poly(ADP-ribose) polymerase
- Ctrl
- control
- MEF
- mouse embryonic fibroblast.
REFERENCES
- 1. Stan S. D., Kar S., Stoner G. D., Singh S. V. (2008) J. Cell. Biochem. 104, 339–356 [DOI] [PubMed] [Google Scholar]
- 2. Herr I., Büchler M. W. (2010) Cancer Treat. Rev. 36, 377–383 [DOI] [PubMed] [Google Scholar]
- 3. Zhang Y., Talalay P., Cho C. G., Posner G. H. (1992) Proc. Natl. Acad. Sci. U.S.A. 89, 2399–2403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dinkova-Kostova A. T., Holtzclaw W. D., Cole R. N., Itoh K., Wakabayashi N., Katoh Y., Yamamoto M., Talalay P. (2002) Proc. Natl. Acad. Sci. U.S.A. 99, 11908–11913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chung F. L., Conaway C. C., Rao C. V., Reddy B. S. (2000) Carcinogenesis 21, 2287–2291 [DOI] [PubMed] [Google Scholar]
- 6. Warin R., Chambers W. H., Potter D. M., Singh S. V. (2009) Cancer Res. 69, 9473–9480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Singh A. V., Xiao D., Lew K. L., Dhir R., Singh S. V. (2004) Carcinogenesis 25, 83–90 [DOI] [PubMed] [Google Scholar]
- 8. Cheung K. L., Kong A. N. (2010) AAPS J. 12, 87–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Trachootham D., Zhou Y., Zhang H., Demizu Y., Chen Z., Pelicano H., Chiao P. J., Achanta G., Arlinghaus R. B., Liu J., Huang P. (2006) Cancer Cell 10, 241–252 [DOI] [PubMed] [Google Scholar]
- 10. Sahu R. P., Srivastava S. K. (2009) J. Natl. Cancer Inst. 101, 176–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Singh S. V., Srivastava S. K., Choi S., Lew K. L., Antosiewicz J., Xiao D., Zeng Y., Watkins S. C., Johnson C. S., Trump D. L., Lee Y. J., Xiao H., Herman-Antosiewicz A. (2005) J. Biol. Chem. 280, 19911–19924 [DOI] [PubMed] [Google Scholar]
- 12. Xu C., Shen G., Yuan X., Kim J. H., Gopalkrishnan A., Keum Y. S., Nair S., Kong A. N. (2006) Carcinogenesis 27, 437–445 [DOI] [PubMed] [Google Scholar]
- 13. Clarke J. D., Dashwood R. H., Ho E. (2008) Cancer Lett. 269, 291–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mi L., Gan N., Cheema A., Dakshanamurthy S., Wang X., Yang D. C., Chung F. L. (2009) J. Biol. Chem. 284, 17039–17051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mi L., Gan N., Chung F. L. (2011) Carcinogenesis 32, 216–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Giaccone G., van Ark-Otte J., Scagliotti G., Capranico G., van der Valk P., Rubio G., Dalesio O., Lopez R., Zunino F., Walboomers J., Pinedo H. M. (1995) Biochim. Biophys. Acta 1264, 337–346 [DOI] [PubMed] [Google Scholar]
- 17. Chen G., Templeton D., Suttle D. P., Stacey D. W. (1999) Oncogene 18, 7149–7160 [DOI] [PubMed] [Google Scholar]
- 18. Wang Q., Zambetti G. P., Suttle D. P. (1997) Mol. Cell. Biol. 17, 389–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Smith K., Houlbrook S., Greenall M., Carmichael J., Harris A. L. (1993) Oncogene 8, 933–938 [PubMed] [Google Scholar]
- 20. Varis A., Zaika A., Puolakkainen P., Nagy B., Madrigal I., Kokkola A., Väyrynen A., Kärkkäinen P., Moskaluk C., El-Rifai W., Knuutila S. (2004) Int. J. Cancer 109, 548–553 [DOI] [PubMed] [Google Scholar]
- 21. Li T. K., Liu L. F. (2001) Annu. Rev. Pharmacol. Toxicol. 41, 53–77 [DOI] [PubMed] [Google Scholar]
- 22. Frydman B., Marton L. J., Sun J. S., Neder K., Witiak D. T., Liu A. A., Wang H. M., Mao Y., Wu H. Y., Sanders M. M., Liu L. F. (1997) Cancer Res. 57, 620–627 [PubMed] [Google Scholar]
- 23. Bandele O. J., Osheroff N. (2008) Chem. Res. Toxicol. 21, 936–943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhou N., Xiao H., Li T. K., Nur-E-Kamal A., Liu L. F. (2003) J. Biol. Chem. 278, 29532–29537 [DOI] [PubMed] [Google Scholar]
- 25. Wang H., Mao Y., Chen A. Y., Zhou N., LaVoie E. J., Liu L. F. (2001) Biochemistry 40, 3316–3323 [DOI] [PubMed] [Google Scholar]
- 26. Nakamura Y., Miyoshi N. (2010) Biosci. Biotechnol. Biochem. 74, 242–255 [DOI] [PubMed] [Google Scholar]
- 27. Tewey K. M., Rowe T. C., Yang L., Halligan B. D., Liu L. F. (1984) Science 226, 466–468 [DOI] [PubMed] [Google Scholar]
- 28. Zhang A., Lyu Y. L., Lin C. P., Zhou N., Azarova A. M., Wood L. M., Liu L. F. (2006) J. Biol. Chem. 281, 35997–36003 [DOI] [PubMed] [Google Scholar]
- 29. Wei H., Ruthenburg A. J., Bechis S. K., Verdine G. L. (2005) J. Biol. Chem. 280, 37041–37047 [DOI] [PubMed] [Google Scholar]
- 30. Soule H. D., Maloney T. M., Wolman S. R., Peterson W. D., Jr., Brenz R., McGrath C. M., Russo J., Pauley R. J., Jones R. F., Brooks S. C. (1990) Cancer Res. 50, 6075–6086 [PubMed] [Google Scholar]
- 31. Hu T., Sage H., Hsieh T. S. (2002) J. Biol. Chem. 277, 5944–5951 [DOI] [PubMed] [Google Scholar]
- 32. Schmidt B. H., Burgin A. B., Deweese J. E., Osheroff N., Berger J. M. (2010) Nature 465, 641–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lobley A., Sadowski M. I., Jones D. T. (2009) Bioinformatics 25, 1761–1767 [DOI] [PubMed] [Google Scholar]
- 34. Eswar N., Webb B., Marti-Renom M. A., Madhusudhan M. S., Eramian D., Shen M. Y., Pieper U., Sali A. (2006) Curr. Protoc. Bioinformatics, Chapter 5, Unit 5.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Fiser A., Do R. K., Sali A. (2000) Protein Sci. 9, 1753–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Martí-Renom M. A., Stuart A. C., Fiser A., Sánchez R., Melo F., Sali A. (2000) Annu. Rev. Biophys. Biomol. Struct. 29, 291–325 [DOI] [PubMed] [Google Scholar]
- 37. Sali A., Blundell T. L. (1993) J. Mol. Biol. 234, 779–815 [DOI] [PubMed] [Google Scholar]
- 38. Case D. A., Cheatham T. E., 3rd, Darden T., Gohlke H., Luo R., Merz K. M., Jr., Onufriev A., Simmerling C., Wang B., Woods R. J. (2005) J. Comput. Chem. 26, 1668–1688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J. L., Dror R. O., Shaw D. E. (2010) Proteins 78, 1950–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Onufriev A., Bashford D., Case D. A. (2004) Proteins 55, 383–394 [DOI] [PubMed] [Google Scholar]
- 41. Wang J. C. (2002) Nat. Rev. Mol. Cell Biol. 3, 430–440 [DOI] [PubMed] [Google Scholar]
- 42. Woessner R. D., Mattern M. R., Mirabelli C. K., Johnson R. K., Drake F. H. (1991) Cell Growth Differ. 2, 209–214 [PubMed] [Google Scholar]
- 43. Lyu Y. L., Kerrigan J. E., Lin C. P., Azarova A. M., Tsai Y. C., Ban Y., Liu L. F. (2007) Cancer Res. 67, 8839–8846 [DOI] [PubMed] [Google Scholar]
- 44. Liu L. F., Rowe T. C., Yang L., Tewey K. M., Chen G. L. (1983) J. Biol. Chem. 258, 15365–15370 [PubMed] [Google Scholar]
- 45. Steinmetz K. A., Potter J. D. (1996) J. Am. Diet. Assoc. 96, 1027–1039 [DOI] [PubMed] [Google Scholar]
- 46. Combs G. F., Jr. (2001) Nutr. Cancer 40, 6–11 [DOI] [PubMed] [Google Scholar]
- 47. Ip C. (1998) J. Nutr. 128, 1845–1854 [DOI] [PubMed] [Google Scholar]
- 48. Lippman S. M., Klein E. A., Goodman P. J., Lucia M. S., Thompson I. M., Ford L. G., Parnes H. L., Minasian L. M., Gaziano J. M., Hartline J. A., Parsons J. K., Bearden J. D., 3rd., Crawford E. D., Goodman G. E., Claudio J., Winquist E., Cook E. D., Karp D. D., Walther P., Lieber M. M., Kristal A. R., Darke A. K., Arnold K. B., Ganz P. A., Santella R. M., Albanes D., Taylor P. R., Probstfield J. L., Jagpal T. J., Crowley J. J., Meyskens F. L., Jr., Baker L. H., Coltman C. A., Jr. (2009) JAMA 301, 39–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gottesman M. M. (2002) Annu. Rev. Med. 53, 615–627 [DOI] [PubMed] [Google Scholar]
- 50. Asano T., An T., Zwelling L. A., Takano H., Fojo A. T., Kleinerman E. S. (1996) Oncol. Res. 8, 101–110 [PubMed] [Google Scholar]
- 51. Neder K., Marton L. J., Liu L. F., Frydman B. (1998) Cell. Mol. Biol. 44, 465–474 [PubMed] [Google Scholar]
- 52. Bandele O. J., Osheroff N. (2007) Biochemistry 46, 6097–6108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Na H. K., Surh Y. J. (2006) Mol. Carcinog. 45, 368–380 [DOI] [PubMed] [Google Scholar]
- 54. Genchi G., Olson J. A. (2001) Biochim. Biophys. Acta 1530, 146–154 [DOI] [PubMed] [Google Scholar]
- 55. Yang F., Oz H. S., Barve S., de Villiers W. J., McClain C. J., Varilek G. W. (2001) Mol. Pharmacol. 60, 528–533 [PubMed] [Google Scholar]
- 56. Gasparian A. V., Yao Y. J., Lü J., Yemelyanov A. Y., Lyakh L. A., Slaga T. J., Budunova I. V. (2002) Mol. Cancer Ther. 1, 1079–1087 [PubMed] [Google Scholar]
- 57. Na H. K., Surh Y. J. (2008) Food Chem. Toxicol. 46, 1271–1278 [DOI] [PubMed] [Google Scholar]
- 58. Shishodia S., Gutierrez A. M., Lotan R., Aggarwal B. B. (2005) Cancer Res. 65, 9555–9565 [DOI] [PubMed] [Google Scholar]
- 59. Lyu Y. L., Lin C. P., Azarova A. M., Cai L., Wang J. C., Liu L. F. (2006) Mol. Cell. Biol. 26, 7929–7941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Azarova A. M., Lyu Y. L., Lin C. P., Tsai Y. C., Lau J. Y., Wang J. C., Liu L. F. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 11014–11019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ogawa K., Futakuchi M., Hirose M., Boonyaphiphat P., Mizoguchi Y., Miki T., Shirai T. (1998) Int. J. Cancer 76, 851–856 [DOI] [PubMed] [Google Scholar]
- 62. Bender R. P., Ham A. J., Osheroff N. (2007) Biochemistry 46, 2856–2864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tsai Y.-C., Pestka S., Wang L.-H., Runnels L. W., Wan S., Liu L. F. (2011) PLoS ONE, in press [DOI] [PMC free article] [PubMed] [Google Scholar]