

Abstract
The minichromosome maintenance (MCM) complex, a replicative helicase, is a heterohexamer essential for DNA duplication and genome stability. We identified Schizosaccharomyces pombe mcb1+ (Mcm-binding protein 1), an apparent orthologue of the human MCM-binding protein that associates with a subset of MCM complex proteins. mcb1+ is an essential gene. Deletion of mcb1+ caused cell cycle arrest after several generations with a cdc phenotype and disrupted nuclear structure. Mcb1 is an abundant protein, constitutively present across the cell cycle. It is widely distributed in cytoplasm and nucleoplasm and bound to chromatin. Co-immunoprecipitation suggested that Mcb1 interacts robustly with Mcm3–7 but not Mcm2. Overproduction of Mcb1 disrupted the association of Mcm2 with other MCM proteins, resulting in inhibition of DNA replication, DNA damage, and activation of the checkpoint kinase Chk1. Thus, Mcb1 appears to antagonize the function of MCM helicase.
Keywords: Chromosomes, DNA Replication, DNA Synthesis, Yeast, Yeast Genetics, Checkpoint, MCM Complex
Introduction
DNA replication requires a series of tightly coordinated events to ensure that each daughter cell receives one complete copy of genetic information (1, 2). In response to damage generated by mutations in the replication machinery or by exogenous damaging agents, eukaryotic cells activate checkpoint responses that arrest S phase progression and activate DNA repair (3, 4). Defects either in replication or checkpoint responses generate genome instability and increase cancer susceptibility (5, 6).
The minichromosome maintenance (MCM)3 complex is a replicative helicase conserved in eukaryotes and archaea (for reviews, see Refs. 7 and 8). The complex consists of six distinct yet structurally related subunits, Mcm2–7, assembled into a heterohexameric ring. MCM proteins are members of the AAA ATPase family sharing several distinctive protein sequences that define the family. In metazoa and plants, there are several additional MCM family members, Mcm8 and Mcm9 (9–12), as well as developmentally specific versions of the MCMs (13), all of which contain the characteristic MCM-specific protein sequence motifs. The fission yeast genome encodes just the six core MCMs, which assemble into a complex that is constitutively located in the nucleus throughout the cell cycle (14–17). In late M and early G1 phases, the MCM complex is recruited onto chromatin as part of the prereplication complex and onto unreplicated DNA; this chromatin localization is dislodged as replication proceeds (18). MCM proteins are abundant and exceed the number of replication origins (19–21). Each of the six MCM proteins is essential for viability with a similar deletion phenotype (16, 22–27). Reduction of MCM protein levels causes genome instability in fission yeast due to replication fork collapse and DNA damage (25, 28).
A novel component of the human MCM complex was discovered in human cells using tandem affinity purification (29). Human MCM-binding protein (hMCM-BP) shares no homology to MCM proteins or AAA ATPases. Biochemical analysis suggests that MCM-BP replaces Mcm2 and forms an “alternative” MCM complex with Mcm3–7. Similar to the MCM proteins, hMCM-BP localizes primarily in the nucleus and associates with chromatin in most of the cell cycle except early M phase. Recently, an hMCM-BP orthologue, ETG1, was isolated from plants (30, 31). Depletion of ETG1 activates a G2 cell cycle checkpoint, resulting in a late G2 cell arrest, and also plays a role in establishing cohesion. Interestingly, hMCM-BP orthologues have been found in fruit flies, frogs, zebrafish, and two fission yeasts but not in budding yeast.
Here, we report the identification and characterization of the hMCM-BP orthologue in Schizosaccharomyces pombe, mcb1+ (Mcm-binding protein 1). We show that mcb1+ encodes an essential gene. Spores lacking mcb1+ arrest after several divisions with a G2 DNA content and a cdc phenotype similar to MCM deletion mutants. We epitope-tagged Mcb1 and showed that Mcb1 is an abundant protein constitutively expressed through the cell cycle. Mcb1 is distributed in all cellular compartments, including a substantial chromatin-bound fraction. Mcb1 associated robustly with Mcm3–7 but not Mcm2. Overproduction (OP) of Mcb1 was toxic to cells, creating a dominant negative phenotype that resembles the initiation defect observed in cdc18-shutoff cells. In OP-Mcb1 cells, Rad22 foci accumulated, and Chk1 kinase was activated, indicating that DNA damage had occurred. Mutant analysis indicated that only full-length Mcb1 and a truncated form lacking the N terminus are capable of dissociating Mcm2 from other MCMs. Our data suggest that high levels of Mcb1 inhibit Mcm2 from interacting with other MCM proteins and disrupt normal MCM function during replication initiation. We propose that Mcb1 contributes to MCM regulation possibly by controlling the accessibility of MCM complex to chromatin.
EXPERIMENTAL PROCEDURES
Fission Yeast Strains, Plasmids, and Manipulation
All S. pombe strains (supplemental Table 1) were constructed and maintained in yeast extract plus supplement medium or under selection in Edinburgh minimal medium (EMM) with appropriate supplements using standard techniques (32–34). Transformation was performed by electroporation. Unless noted, asynchronous cultures were grown at 32 °C. In cell cycle block and release experiment, cells were grown at 25 °C (permissive temperature) to early exponential phase and shifted to 36 °C for 4 h (restrictive temperature). HA-tagged Mcb1 from the endogenous locus was generated by using the pFA6a series of plasmids with primers 5′-CGAAGAGTTTCGGTCGTCAACTGGTTTCAAGAATTGATTTTGAGGCTGCCCGTAGTCTAATCAATCATTGGACTGTCAACCGGATCCCCGGGTTAATTAA-3′ and 5′-CTTGGAAATTCCAAAAAGACATGAAAAGTAATTTCTAACATTGGTTAAATGATGTTGATTATAAGAAAATATGCGATCAAGAATTCGAGCTCGTTTAAAC-3′ (35). Doubly tagged strains were isolated by mating and from tetrad analysis. The mcb1+ gene was cloned using cDNA as template and was inserted into pREP-based expression plasmids to generate pLD10 (nmt1-mcb1HA) and pLD18 (nmt1-mcb1V5), which were used for ectopic expression (36). The mcb1g gene was amplified using genomic DNA. To generate stable Mcb1HA-overproducing cells (OP-Mcb1), we made pLD14 by inserting the nmt1-mcb1HA fragment from pLD10 into pJK210. NruI-linearized pLD14 was integrated at leu1-32 locus as described (37). The strains for the mutation analysis were generated with the same approach. The nmt1 promoter-containing strains were maintained on yeast extract plus supplement agar (for integrants) or EMM with supplements and thiamine. To perform overproduction/induction experiments, liquid cultures grown in the presence of 2.5 μg/ml thiamine to early exponential phase were washed twice with an equal volume of EMM before inoculating in the absence of thiamine (overproduction state) or in the presence of 5 μg/ml thiamine (strong repression state) (38, 39).
Construction of mcb1+ Deletion
To delete the mcb1+ gene (SPAC1687.04), we removed the entire coding sequence according to a gene deletion protocol using upstream primers 5′-GAGATCTAGACAGGACGATTGGACGATACT-3′ and 5′-GAGACTCGAGATTATAAATATATAATTTTATCCTTTAAACC-3′ and downstream primers 5′-GAGAGCGGCCGCTTGATCGCATATTTTCTTATAATC-3′ and 5′-GAGATCTAGAGTCGCTTTAGTACATTCTAAAC-3′. The resulting plasmid (pLD21) was amplified, linearized at XbaI, and transformed into a fresh mated wild-type diploid strain. Stable Ura+ integrated diploids were selected and confirmed by PCR. The deletion was also confirmed by tetrad analysis and complementation. Bulk spore germination was performed as described previously (23).
Complementation
A heterozygous diploid strain (mcb1+/Δmcb1::ura4+) was transformed with linearized leu1+ integration plasmids that express HA-tagged mcb1 deletion mutants cloned from cDNA and plated on thiamine-containing selective medium. Random spore analysis (34) was used to recover haploids that were Ura+ and Leu+. The resulting haploids were confirmed by PCR and Western blot.
Flow Cytometry
Flow cytometry was performed as described (40, 41) with minor modifications. Briefly, cells were fixed in 70% ice-cold ethanol, rehydrated with 50 mm sodium citrate, and treated with 0.1 mg/ml RNase A. Cells were stained with 1 μm SYTOX Green (Invitrogen) in 50 mm sodium citrate. Macintosh BD CellQuestTM Pro 5.2.1 software (BD Biosciences) was used to analyze and organize the data acquired by the FACScan cytometer (BD Biosciences).
Cell Fractionation Assay
The cell fractionation protocol was derived from Refs. 42 and 43. Cells were washed with ice-cold stop buffer (0.9% NaCl, 10 mm EDTA, 0.2% NaN3). The pellet was incubated at 36 °C for 15 min in CSE buffer (20 mm citric acid, 20 mm Na2HPO4, 40 mm EDTA, 1.2 m sorbitol, pH 5.6) with the addition of 7.2 mm β-mercaptoethanol and 12.5 mg/ml zymolyase-20T. The protoplast cells were washed twice with ice-cold CSE buffer with 1:100 (v/v) protease inhibitor mixture (P-8215, Sigma) and resuspended in ice-cold nucleus buffer (20 mm Tris, pH 7.0, 20 mm potassium acetate, 1 mm magnesium chloride) with the addition of 18% Ficoll, 1 mm ATP, 0.05% Nonidet P-40, and 1:100 (v/v) protease inhibitor mixture. Glass bead lysates were cleared twice by spinning at 2,700 × g for 3 min at 4 °C. The cytoplasm fraction and the whole nucleus fraction were separated by spinning at 21,000 × g for 20 min. To permeabilize the nuclear envelope, the pelleted whole nucleus fraction was resuspended in ice-cold nucleus buffer with the addition of 0.2 m sorbitol, 1 mm ATP, 1 mm dithiothreitol (DTT), 0.5% Nonidet P-40, and 1:100 (v/v) protease inhibitor mixture. The nucleoplasm fraction and chromatin-bound fraction were separated by spinning at 21,000 × g for 20 min. The chromatin-bound fraction was resuspend in ice-cold nucleus buffer with the addition of 0.2 m sorbitol, 1 mm ATP, 1 mm DTT, 0.5% Nonidet P-40, and 1:100 (v/v) protease inhibitor mixture. An equal volume of each fraction was boiled in 2× sample buffer (100 mm Tris, pH 6.8, 20% glycerol, 4% SDS, 200 mm DTT, 0.02% bromphenol blue) and loaded on 8 or 15% SDS-polyacrylamide gels for separation.
Protein Extracts, Immunoprecipitation, and Immunoblotting
Total protein extracts were prepared either by glass bead lysis using trichloroacetic acid (TCA) extraction as described (44) or alkaline (NaOH) lysis protein extraction (45). The concentrations of TCA-extracted protein samples were quantified by BCA protein assay (Pierce). Twenty micrograms of total protein were separated by 8% SDS-PAGE. For alkaline lysis protein extraction, equal numbers of cells were collected, resuspended in 0.3 m NaOH, and incubated at room temperature for 10 min. Permeabilized cells were centrifuged at 1,700 × g for 3 min. The cells pellets were resuspended in 30 μl of 2× sample buffer and boiled for 10 min. Ten microliters were loaded on an SDS-polyacrylamide gel for separation.
Soluble lysates and immunoprecipitates were prepared as described (17). Cell cultures were grown to midlog phase, collected by centrifugation, and lysed in cell lysis (B88) buffer (50 mm HEPES, pH 7.0, 50 mm potassium acetate, 5 mm magnesium acetate, 100 mm sorbitol with freshly added 1 mm ATP, 1 mm DTT, 1:100 (v/v) protease inhibitor mixture (P-8215, Sigma), 1:100 (v/v) phosphatase inhibitor mixture set II (524625, Calbiochem)). The soluble protein concentration was determined by Bradford protein assay (Bio-Rad).
Immunoprecipitations were performed with 750 μg of precleared soluble protein overnight at 4 °C. Fifty microliters of immobilized rProtein A (IPA300, RepliGen; 1:1 in lysis buffer) were added and incubated for 2 h at 4 °C. Beads were spun down and washed four times with 1 ml of cold lysis buffer. After the final wash, beads were resuspended in 2× sample buffer and boiled for 5 min. An equal volume was loaded on an 8% SDS-polyacrylamide gel for separation.
For immunoblotting, samples separated by SDS-PAGE were transferred to Immobilon-P membrane (Millipore). ECL Western blotting substrate (Pierce) and Blue Ultra autoradiography film (BioExpress) were used to detect signals.
Antibodies
Mcm-specific antibodies were purified from rabbit antisera using the methods described previously (16, 17, 24): Mcm3 from serum 6178, Mcm4 (Cdc21) from serum 5898, Mcm5 (Nda4) from serum 5897, Mcm6 (Mis5) from serum 5899, and Mcm7 from serum 6184. We also used the following commercially available primary antibodies: anti-HA (16B12, Covance), anti-GFP (abcam290, Abcam), mouse anti-V5 (R960-25, Invitrogen), rabbit anti-V5 (abcam15828, Abcam), anti-Nop1 (28F2, EnCor Biotech), anti-α-tubulin (T5168, Sigma), anti-proliferating cell nuclear antigen (PC10, Delta Biolabs). We used the following secondary antibodies: anti-mouse-HRP (Millipore) and anti-rabbit-HRP (BD Biosciences). Most primary and secondary antibodies were diluted 1:2,000. Mouse anti-V5 and anti-Nop1 antibodies were diluted 1:1,000.
Viability Assays
Cells (FY11, FY838, FY4594, and FY4596) were grown overnight at 32 °C to early exponential phase (A595 ∼ 0.2–0.3) in EMM lacking leucine (EMM-L) with 2.5 μg/ml thiamine. Cells were washed twice with EMM-L and inoculated into EMM-L. At each time point, cultures were serially diluted in yeast extract plus supplement (1:100, 1:1,000, and 1:10,000). Equal volumes of each dilution were plated on yeast extract plus supplement plates for viability testing and incubated at 30 °C for 4 days. The number of colonies was averaged from different dilutions. Relative viability at time T was calculated as follows: (averaged number of colonies at time T)/(averaged number of colonies at time 0). At each time point, cells were also fixed in 70% ethanol. Cellular DNA content was analyzed by flow cytometry. Rehydrated cells were counted twice for cell growth analysis. Relative growth at time T was defined as follows: (averaged number of cells at time T)/(averaged number of cells at time 0).
In Situ Chromatin Binding Assay and Fluorescence Microscopy
An in situ chromatin binding assay was performed as described (18) with modifications as described (46). Proteins were detected using rabbit anti-V5 (1:300 volume) or rabbit anti-GFP (1:200 volume) and chicken anti-rabbit Alexa Fluor 488 (Invitrogen). Cells were mixed with poly-l-lysine (Sigma), heat-fixed on microscope slides, and mounted in 50% glycerol, phosphate-buffered saline (PBS) for visualization.
DAPI staining for rehydrated cells was performed by mounting the heat-fixed cells with 1 μg/ml DAPI containing antifade mounting solution (50% glycerol in water with 0.1% p-phenylenediamine dihydrochloride).
To examine Rad22-YFP foci in live cells, cells were washed twice in EMM containing 10 μg/ml DAPI, air-dried on ColorFrost Plus Microscope slides (Fisher Scientific), and mounted in 50% glycerol, PBS. All pictures were taken on a Leica DMR florescence microscope using a 63× oil immersion objective (Leica Plan Apochromat; numerical aperture, 1.32) and recorded with OpenLab software (Improvision).
Digital Image Manipulation
All the plates and films were electronically scanned using a ScanJet IIcx scanner (Hewlett-Packard). Digitized pictures/photos were analyzed and contrast-enhanced by NIH ImageJ software and assembled in Canvas software (ACD Systems).
Sequence Alignments
Sequence alignments were done in BioEdit version 7.0.9.0 software with the ClustalW function.
RESULTS
Identification and Deletion of S. pombe mcb1+
hMCM-BP was first identified as a novel component of MCM (29). Although there is no obvious Saccharomyces cerevisiae orthologue (see yeast orthology table version 2.15 (Sanger)), we found a putative orthologue of hMCM-BP gene in S. pombe, SPAC1687.04, and named it mcb1+ (supplemental Fig. S1). The mcb1+ gene is highly conserved in eukaryotes. It has two exons and one intron and encodes a protein with 501 amino acids and a predicted molecular mass of 56.6 kDa. Similar to the hMCM-BP protein, Mcb1 has no obvious sequence motifs and shares no obvious homology with S. pombe MCMs or AAA ATPases (supplemental Fig. S2).
We created a heterozygous diploid mcb1+/Δmcb1::ura4+ for tetrad analysis. All tetrads displayed a 2:2 segregation of viable:inviable spores, and all viable colonies were Ura− (supplemental Fig. S3A). The inviable colonies were Δmcb1::ura4+ spores, which managed to complete a few cell divisions and form microcolonies of 8–16 cells (supplemental Fig. S3B). Ectopic expression of mcb1+ under its own promoter from a plasmid was able to rescue the lethality in the spores, confirming that the phenotype arises from disruption of this gene (data not shown).
We analyzed a population of Δmcb1 spores by a bulk spore germination assay using Δmcb1::ura4+/mcb1+ diploid cells and wild-type ura4+/ura4-D18 diploid cells as a control. The spores were inoculated into liquid medium lacking uracil, which ensures that only the Δmcb1::ura4+ or ura4+ spores can germinate. In contrast to Δmcm spores (23, 24), Δmcb1::ura4+ spores showed no obvious delays in their first S phase compared with the wild type (Fig. 1A). However, similar to Δmcm spores, the terminal phenotype of germinated Δmcb1::ura4+ spores was an elongated cdc morphology and in most cases a single nucleus. About 10% of cells had an abnormally shaped nucleus, either teardrop-shaped or cut (Fig. 1B, 16 h). Thus, mcb1+ is an essential gene.
FIGURE 1.
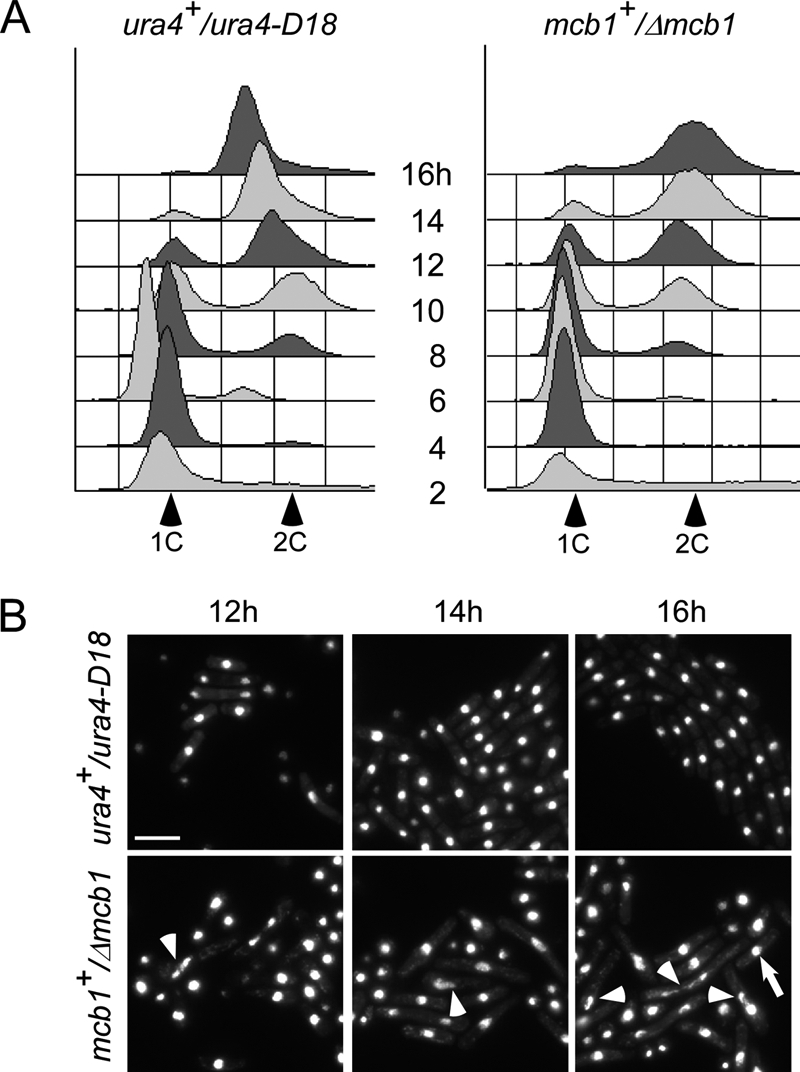
Spore germination of Δmcb1 spores. A, spores prepared from wild-type ura4+/ura4D-18 (FY261x11) and heterozygous disruption mutant mcb1+/Δmcb1::ura4+ (FY3747) were inoculated into medium lacking uracil at 32 °C. The populations were sampled every 2 h for 16 h and analyzed by flow cytometry to monitor S phase entry and DNA replication progression. B, photomicrographs of DAPI-stained spores after 12, 14, and 16 h at 32 °C. Arrowheads indicate cells with an abnormal nucleus. An arrow indicates cells with a normal nucleus. Scale bar, 10 μm.
Characterization of Mcb1 Protein
In fission yeast, MCM proteins are localized in the nucleus throughout the cell cycle by nuclear localization sequences on Mcm2 and Mcm3 (15). Human MCM-BP is also a nuclear protein (29). We constructed a C-terminally HA-tagged Mcb1 (mcb1HA) to replace the wild-type copy in the genome. Mcb1HA cells showed normal growth, indicating that the tagged copy is functional. When compared with a strain expressing Mcm2HA, we observed that Mcb1HA is expressed at a higher level, indicating that it is a very abundant protein (supplemental Fig. S4).
Because human MCM-BP is chromatin-associated (29), we examined Mcb1HA localization by immunofluorescence using an mcb1HA mcm2V5 strain for an in situ chromatin binding assay (supplemental Fig. S5). Consistent with previous studies (15, 47), Mcm2V5 stayed in the nucleus (supplemental Fig. S5A) and remained chromatin-bound in binucleated (S phase) cells (supplemental Fig. S5B). However, we were unable to detect Mcb1HA cytologically (supplemental Fig. S5), suggesting that the epitope tag is inaccessible or occluded. Similar results were observed for N-terminally HA-tagged Mcb1 (HAmcb1; data not shown). We also tagged Mcb1 with GFP and mCherry at the C terminus. Although these tags were easily detected on Western blots, we were not able to detect any fluorescence in live cells (data not shown).
Therefore, we took a biochemical approach to examine localization and performed a cell fractionation assay. Asynchronous cells were treated to release different cellular compartments in subsequent fractions (supplemental Fig. S6). Equal volumes of each fraction were separated by SDS-PAGE and blotted for Mcm2V5, Mcm7, and Mcb1HA (Fig. 2A). Fib1 (Nop1) is involved in pre-rRNA processing and is a marker for the chromatin fraction, whereas α-tubulin is a predominantly but not exclusively cytoplasmic (48, 49). Consistent with previous studies, Mcm2V5 and Mcm7 are nuclear proteins. Mcm2V5 was mainly in the nucleoplasm, whereas the majority of Mcm7 was chromatin-bound. In contrast, Mcb1HA was present throughout the cells but strongly enriched in the nuclear fractions.
FIGURE 2.

Mcb1 localization and its association to MCM. A, cellular fractions were prepared from mcb1HA mcm2V5 (FY4122) according to Fig. S6A. An equal volume (10 μl) of each fraction was separated by 8% or 15% SDS-PAGE and immunoblotted for Mcb1HA, Mcm2V5, Mcm7, α-tubulin (marker for cytosol and nucleoplasm), and Fib1 (Nop1; marker for chromatin). B, Mcb1 interacts with all other MCM proteins except Mcm2. Lysate was prepared from asynchronous mcb1HA mcm2V5 (FY4122) cells with B88 buffer. Twenty micrograms of soluble protein were loaded as input (lane 1). Identical amounts of lysate were precleared and immunoprecipitated (IP) with the antibodies shown. Ten microliters of immunoprecipitated sample (1:7, v/v) were used for each immunoblot. Samples were separated by 8% SDS-PAGE. Soluble lysate was immunoprecipitated with no antibody, anti-HA, anti-V5, anti-Mcm4, and anti-Mcm7 and immunoblotted for Mcm2V5, Mcb1HA, Mcm4, Mcm6, Mcm7, Mcm3, or Mcm5.
The levels of mcm2+ mRNA (23, 50) and the protein level (51) are constant throughout the cell cycle. Studies suggest that all MCM subunit levels are comparable (17, 47). To investigate whether levels of Mcb1 protein level fluctuate in the cell cycle, we used a cdc25–22 mcb1HA mcm2V5 strain to synchronize and release the cells by controlling the temperature. We observed no cell cycle-dependent change in total protein level or mobility (supplemental Fig. S7). Mcb1HA migrated as a doublet in this experiment, but that was not apparent in soluble lysate (Fig. 2B, lane 1).
Mcb1 Complex Formation
The six MCM subunits form a heterohexameric protein complex (for reviews, see Refs. 7 and 8). The relative affinities among members vary (14, 17, 24, 52), suggesting that there are several subcomplexes: Mcm3 and Mcm5 form a dimer; Mcm4, -6, and -7 form a high affinity core complex; and Mcm2 connects the two subcomplexes (15). This is consistent with observations of MCM organization in other systems that suggest that Mcm2 forms a “gate” to open the MCM complex possibly to encircle DNA (8, 53, 54). Human MCM-BP is proposed to replace the Mcm2 subunit (29). Using strain mcb1HA mcm2V5, we performed separate immunoprecipitations of Mcb1HA, Mcm2V5, Mcm4, and Mcm7 and blotted for other MCM proteins. Mcb1HA immunoprecipitated Mcm4, -6, -7, and -3 but not Mcm2 (Fig. 2B, lane 3). Conversely, Mcm2V5 associated with Mcm4, -6, -7, and -3 but not Mcb1 (Fig. 2B, lane 4). This suggests that there are at least two MCM complexes, one with Mcm2 and one with Mcb1. We found no significant association between Mcb1HA and Mcm2V5 in this experiment (Fig. 2B, lanes 3 and 4). Antibodies to Mcm4 or Mcm7 precipitated both Mcb1 and Mcm2 as well as the other MCMs. Interestingly, the Mcb1 association was extremely robust, and Mcb1 was immunoprecipitated at higher levels than Mcm2 in the Mcm4 and Mcm7 experiments. We performed chromatin immunoprecipitation (ChIP) to see whether Mcb1 was located at replication origins but were unable to detect it under conditions where we could observe Mcm2 (data not shown).
Isolation of Mcb1 Mutants
To identify the functional regions of Mcb1, we constructed a series of deletions within the protein, arbitrarily defining five domains: Exon 1, amino acids 2–54; A region, amino acids 55–230; B region, amino acids 231–414; and C region, amino acids 415–501 (Fig. 3A). Constructs lacking these domains were cloned into episomal plasmids under the high strength nmt1 promoter (38). The resulting plasmids were transformed into wild-type cells. We controlled expression by the levels of thiamine. In the presence of thiamine (low amount of protein expression from the nmt1+ promoter (55)), all the transformed cells were viable (Fig. 3B). However, in the absence of the thiamine (overexpression), we found that cells transformed with full-length Mcb1 (mcb1 and mcb1g) were unable to form colonies (Fig. 3B). Interestingly, cells that overexpressed mcb1-D2 and mcb1-D22 were viable but generated notably smaller colonies than the vector control. Both of these mutants lack the N-terminal exon, and D22 has an additional point mutation introduced during PCR (E423G). This suggests that D2 and D22 have residual Mcb1 activity.
FIGURE 3.

Structure and function analysis of Mcb1. A, a schematic of Mcb1 truncations (1–10) made with a summary of their toxicity to wild-type cells, complementation of Δmcb1, and interaction with Mcm4. B, wild-type cells (FY254) transformed with plasmids encoding Mcb1 truncations (1–10) were streaked on EMM-leucine plates with (+T; left) or without (−T; right) thiamine to repress or induce nmt1 promoter, respectively. aa, amino acids; ND, not determined.
We tested each mutant for complementation of mcb1Δ by integrating the constructs into the leu1-32 locus under the nmt1 promoter in the diploid Δmcb1::ura4+/mcb1+. Following sporulation, we screened haploids for Ura+ Leu+ clones containing both the deletion and the insertion alleles in the presence of thiamine.
Full-length mcb1+ and mcb1-D2 and mcb1-D22 rescued mcb1Δ with comparable levels of expression (Figs. 3A and 4A). None of the other mutants were recovered, indicating that they are non-functional. We observed that both mcb1-D2 and mcb1-D22 cells showed elongated cells with a 2C DNA content (Fig. 4B), suggesting that they are hypomorphs of mcb1+. Their elongated cell morphology suggests that they have activated a checkpoint that delays the cell cycle. We crossed these mutants into strains lacking the S phase checkpoint (cds1Δ), the damage checkpoint (chk1Δ), or the upstream regulator of both checkpoints (rad3Δ) and determined that deletion of either chk1 or rad3 relieved the cell elongation phenotype. Thus, we conclude that the damage checkpoint is responsible for their cell cycle delay.
FIGURE 4.

N-terminal deletion mutants (mcb1D2 and mcb1D22) are hypomorphic. A, wild-type (FY11), mcb1HA (FY4041), mcm2V5 mcb1HA (FY4122), Δmcb nmt1-mcb1HA (FY4596), Δmcb nmt1-mcb1gHA (FY5417), Δmcb nmt1-mcb1D2HA (FY5419), and Δmcb nmt1-mcb1D22HA (FY5421) cells were grown asynchronously in medium containing thiamine (+T). An equal number of cells were collected and alkaline lysed. An equal volume of total protein was loaded on an 8% SDS-polyacrylamide gel for separation and immunoblotted for Mcb1HA and proliferating cell nuclear antigen (PCNA) (a loading control). B, photomicrographs 1–5 of DAPI-stained asynchronous wild-type (FY11), Δmcb nmt1-mcb1HA (FY4596), Δmcb nmt1-mcb1gHA (FY5417), Δmcb nmt1-mcb1D2HA (FY5419) and Δmcb nmt1-mcb1D22HA (FY5421) cells. Scale bar, 10 μm. DNA content and cell length were monitored by flow cytometry. C, photomicrographs 1–8 of DAPI-stained wild-type (FY11); Δmcb nmt1-mcb1HA (FY4596); Δmcb nmt1-mcb1D2HA in a Δchk1, Δcds1, or Δrad3 background (FY5499, -5501, or -5505); and Δmcb nmt1-mcb1D22HA in a Δchk1, Δcds1, or Δrad3 background (FY5500, -5503, or -5506) cells. Scale bar, 10 μm. DNA content and cell length were monitored by flow cytometry.
Overexpression of Mcb1 in Cells Inhibits S Phase and Activates DNA Damage Checkpoint
We next investigated the lethality associated with overexpression of full-length mcb1+. We expressed nmt1-mcb1HA integrated at the leu1-32 locus in an mcb1+ or mcb1Δ background. In the presence of thiamine, both strains were healthy and produced colonies with normal sizes (supplemental Fig. S8A, top left). In the absence of thiamine, the overproducing strain in mcb1Δ was completely inviable (supplemental Fig. S8A, bottom, d). Surprisingly, the overproducing strain that also contains one copy of wild-type mcb1+ had a few surviving colonies (supplemental Fig. S8A, bottom, c).
We followed the cells during promoter induction and found that the overall cell number was similar in both strains; however, the viability (plating efficiency) of the mcb1 overproducers began to drop by 9 h (supplemental Fig. S8B). Although the number of Δmcb1 nmt1-mcb1HA survivors continued to drop, the viability of mcb1+ nmt1-mcb1HA cells plateaued at 13 h and started to increase again around 17 h. The survivor class that emerged by 26 h in mcb1+ nmt1-mcb1HA showed normal 2C DNA content (Fig. 5A, panel c), whereas Δmcb1 nmt1-mcb1HA was lethal at the same time point (Fig. 5A, panel d). The levels of ectopically produced Mcb1HA protein dropped significantly at 26 h (supplemental Fig. S9). We conclude that under the selective pressure of toxic overproduction, the survivors escaped by down-regulating the nmt1 promoter. Because the endogenous mcb1+ gene was still intact, the cells could survive repression of the dominant negative (nmt1-mcb1HA) transgene. For the strain in which the transgene is the only source of mcb1+, there were no survivors.
FIGURE 5.

Overproduction of Mcb1 causes DNA damage. A, wild-type, negative control, and Mcb1 overproducing cells (a–d) at each time point were processed for flow cytometry analysis. Arrowheads indicate a starved 1C population. An arrow indicates a 2C population. Unfilled histograms outlined with dotted lines indicate FACS profiles of standard S. pombe cells processed at the same time. B, left, representative pictures of samples at each time point stained with DAPI. Arrowheads indicate short cells with abnormal nuclear morphology. Arrows indicate elongated cells with or without abnormal nuclear morphology. Scale bar, 10 μm. Right, quantification of cells with different nuclear morphology. Cells were counted for mononucleate, binucleate, and abnormal nucleus categories. C, Rad22-YFP foci formation in Mcb1-overproducing cells. rad22-YFP leu1-32::nmt1-mcb1HA-leu1+ cells (FY4591) were grown in −leucine with low thiamine to low OD, washed, and inoculated into −leucine with −thiamine (−T) or +thiamine (+T) medium. 14 and 16 h after thiamine removal, cells were harvested and washed twice in DAPI-containing medium. Left, representative photomicrographs of FY4591 at 14 h with or without thiamine. Scale bar, 10 μm. Cells were counted for Rad22-YFP foci (zero foci, one focus, and more than one focus). Right, quantification of cells counts averaged from two experiments.
However, at early time points there was no difference in the phenotypes or behavior of the overproducing strains. By 13 h, both strains accumulated significant numbers of cells with DNA contents less than 1C (Fig. 5A). This phenotype is reminiscent of cells with defects in replication initiation. For example, shutting off the cdc18+ replication initiation gene leads to accumulation of cells with less than 1C DNA and abnormal nuclear morphology (56). Indeed, consistent with the behavior of a cdc18-shutoff allele (see below and Ref. 56), we observed a mixture of long and short cells in nmt1-mcb1HA (Fig. 5B). By 14 h, 26% of mcb1+ nmt1-mcb1HA cells and 33% of Δmcb1 nmt1-mcb1HA cells had abnormal nuclear morphology, including anucleate, fragmented nuclei or cut cells. At 17 h, the abnormal cells increased to 64 and 66%, respectively (Fig. 5B). A similar phenotype was observed in strains overexpressing untagged Mcb1 from plasmids (data not shown).
The presence of elongated cells in the population suggested that some sort of checkpoint was activated in Mcb1HA overproducers (OP-Mcb1). DNA damage during replication can be visualized by the formation of repair foci containing the homologous recombination protein Rad22 (ScRad52) (e.g. Ref. 28). We observed an increase in the formation of Rad22 foci in cells overproducing Mcb1HA (Fig. 5C). Cells with Rad22-YFP foci increased to 37% by 14 h, and 13% of cells had more than one focus compared with about 10% for the cells grown in repressing (thiamine) conditions.
We reasoned that these repair foci might accompany activation of the DNA damage checkpoint, so we combined the overproducer strain with cds1Δ, chk1Δ, or rad3Δ. After growing in thiamine-free medium for 16 h, only the Δcds1 nmt1-mcb1HA cells showed elongated cells, whereas the other strains had small cells and abnormal nuclear structure (Fig. 6A, bottom row). This suggests that the DNA damage checkpoint is activated in some of the cells. Finally, we examined the phosphorylation status of Chk1 as a measure of Chk1 activation (57). Because detection of Chk1 relies on an HA epitope tag, we transformed a plasmid (36) overexpressing Mcb1V5 under the nmt1 promoter into a strain containing chk1HA integrated at the native locus. Induction of Mcb1V5 was detectable 12 h after induction and continued to increase until the last time point, 17 h (Fig. 6B). Phosphorylation of Chk1HA was observed at 14 h after induction. Together, these results suggest that Mcb1 overproduction causes DNA damage that activates Chk1 kinase.
FIGURE 6.

Chk1 is activated in Mcb1-overproducing cells. A, photomicrographs of DAPI-stained mcb1HA (FY4041), nmt1-mcb1HA (FY4594), Δcds1 nmt1-mcb1HA (FY4734), Δchk1 nmt1-mcb1HA (FY4736), Δcds1 Δchk1 nmt1-mcb1HA (FY4739), and Δrad3 nmt1-mcb1HA (FY4740) cells 0 and 16 h after inoculation into −thiamine (−T) medium. Scale bar, 10 μm. B, chk1HA (FY4610) cells were transformed with plasmid expressing Mcb1V5 (pLD18) or empty vector (pSLF972). An equal number of cells were collected at the indicated time points after inoculation into −thiamine medium and alkaline lysed. An equal volume of protein was loaded on an SDS-polyacrylamide gel for separation and immunoblotted for Chk1HA, Mcb1V5, and α-tubulin (a loading control). Lane 1, total lysate of untreated chk1HA cells; lanes 2 and 3, total lysates of chk1HA cells treated with 0.1% methyl methanesulfonate (MMS) for 4 and 1 h (phosphorylated Chk1HA migrates slower), respectively; lanes 4-8, total lysates of pLD18-transformed chk1HA cells from different time points after thiamine removal; and lanes 9-13, total lysates of empty vector (pSLF972)-transformed chk1HA cells from different time points after thiamine removal.
Overproduction of Mcb1 Disrupts Mcm2 from MCM Complex
Because Mcb1 and Mcm2 form alternative MCM complexes, we speculated that overproduced Mcb1 sequesters Mcm3–7 away from Mcm2 and thus blocks replication. To test our hypothesis, we examined MCM complex formation in the overproducer (Fig. 7A). In these experiments, we examined the structure of the MCM complex by immunoprecipitating Mcm2V5 or Mcm4GFP and detected Mcb1 or other MCM subunits.
FIGURE 7.

Overproduction of Mcb1 causes dissociation of Mcm2 from other MCM proteins. A, nmt1-mcb1HA mcm2V5 mcm4GFP (FY4961) overnight culture grown in low thiamine was inoculated into −thiamine (−T) and +thiamine (+T) media and grown at 32 °C for 14 h. Cells were harvested and lysed in B88 buffer. Soluble lysates were immunoprecipitated (IP) with anti-GFP (lanes 3 and 4) and anti-V5 (lanes 5 and 6). Immunoprecipitated samples were separated by SDS-PAGE gel and blotted for Mcm2V5, Mcm4GFP, Mcm7, Mcm6, and Mcb1HA. B, wild-type (FY254), nmt1-mcm4HA (FY1602), and nmt1-mcm2HA (FY861) cells were transformed with plasmids overexpressing Mcb1 (pLD18 or pLD10) and empty vector (pSLF972 or pSGP72). Transformants (top, a–d, and bottom, a–d) were restreaked on −thiamine and +thiamine plates and incubated at 32 °C. C, mcm2V5 mcm4GFP cells carrying full-length mcb1 (FY4961 and FY5407) and mcb1 deletion mutants (FY5408–5415) at the leu1-32 locus were grown in thiamine-containing medium, then harvested, and lysed in B88 buffer. Soluble lysates were immunoprecipitated with anti-GFP. Immunoprecipitated samples were separated by 12% SDS-PAGE. We used an 8% gel to separate bigger Mcb1 truncations from IgG (bottom panel). We blotted for Mcm4GFP and Mcb1HA. The data are summarized in Fig. 3A. D, mcm2V5 mcm4GFP cells carrying six mcb1 deletion mutants at the leu1-32 locus (nmt1-mcb1D2HA (FY5408), nmt1-mcb1D5HA (FY5411), nmt1-mcb1D6HA (FY5412), nmt1-mcb1D78HA (FY5413), nmt1-mcb1D9HA (FY5414), and nmt1-mcb1D22HA (FY5415)) were grown in −thiamine medium, then harvested, and lysed in B88 buffer. Soluble lysates were immunoprecipitated with anti-GFP. Fifteen micrograms of soluble proteins (lanes 1-8) and immunoprecipitated samples (lanes 9-14) were separated by SDS-PAGE and blotted for Mcm4GFP, Mcm2V5, Mcm6, Mcm7, and Mcb1HA.
As we predicted, when we immunoprecipitated Mcm4GFP, we observed increased association between Mcm4GFP and Mcb1HA and decreased interaction between Mcm4GFP and Mcm2V5 (Fig. 7A, compare lanes 3 and 4). Interestingly, however, Mcm4GFP also showed reduced association with Mcm6 and Mcm7, suggesting that the MCM complex overall is disrupted by this level of Mcb1 expression. If excess Mcb1 sequesters Mcm4 away from the other MCMs, an increased dosage of Mcm4 might attenuate this phenotype. We transformed nmt1-mcm4HA cells with pLD18 (nmt1-mcb1V5) or empty vector. We found that overproduction of Mcm4HA, but not Mcm2HA, partially rescued the lethality of Mcb1 accumulation (Fig. 7B).
When we precipitated Mcm2V5 in the overproducers, we were unable to detect other MCM proteins (Fig. 7A, compare lanes 5 and 6). This suggests that the normal MCM complex is disrupted. Surprisingly, under these conditions, we detected an interaction between Mcb1HA and Mcm2V5 (Fig. 7A, lane 6). This suggests that Mcb1 and Mcm2 are capable of interacting directly although not as efficiently as Mcb1 with the other subunits. Thus, high levels of Mcb1 do not simply replace Mcm2 in the complex, but the Mcb1 protein may interact with individual MCM subunits.
Next, we examined the effect of our deletion mutants on MCM complex formation using the same strategy. In the presence of thiamine, protein levels of most Mcb1 truncation mutants were comparable with that of full-length Mcb1 (supplemental Fig. S10). Surprisingly, we observed that Mcb1-D5, Mcb1-D6, and Mcb1-D9 associated with Mcm4 when expressed at a near normal level, although they were unable to complement the null and were not toxic when overproduced (Fig. 3A, 7C). However, when overproduced, these mutants did not dissociate the interaction between Mcm4 and the other MCMs (Fig. 7D), leading us to conclude that they do not replace Mcm2. In contrast, the two N-terminal truncation mutants (Mcb1-D2 and Mcb1-D22) did reduce association of Mcm4 with other MCMs, including Mcm2 (Fig. 7D, lanes 9 and 14). Thus, there is a clear correlation between three phenotypes: Mcb1, Mcb1-D2, and Mcb1-D22 are the only constructs that were able to complement mcb1Δ, were toxic when overproduced, and disrupted association between Mcm2 and Mcm4 upon overproduction.
In fission yeast as in most eukaryotes, MCM proteins are found predominantly in the nucleus throughout the cell cycle, but their chromatin association is cell cycle-regulated (for a review, see Ref. 7). Chromatin binding depends on activation of the prereplication complex, including the initiator protein Cdc18 (18). Previously, we showed that mutations that disrupt the MCM complex such as mcm4ts cause all the subunits to exit the nucleus (15). Thus, complex assembly is required for nuclear retention. We hypothesized that the overexpression of Mcb1, which disrupts the normal MCM complex, therefore would also disrupt chromatin binding and nuclear localization of the MCMs.
We used indirect immunofluorescence in an in situ chromatin binding assay (18, 47). Proteins located on the chromatin are resistant to Triton, whereas proteins located in the nucleoplasm but not on the chromatin are removed by Triton treatment. This method allows the S phase subset of chromatin-bound MCMs (on the binucleate cells) to be distinguished from the abundant unbound MCM protein in the nucleus (18, 47). As a control, we used a cdc18-shutoff strain that blocks MCM binding to the chromatin. Importantly, both of these phenotypes depend on thiamine although in opposite directions. For Mcb1 overproducers (OP-Mcb1), lethality is caused by removing thiamine from the medium to induce nmt1-mcb1HA (minus thiamine condition). In the case of the cdc18-shutoff cells, lethality is caused by adding thiamine to the medium to repress nmt1-cdc18+ expression (56).
As shown in Fig. 8, under permissive conditions, both Mcm4GFP and Mcm2V5 were found in the nucleus (−Triton) and were chromatin-bound in S phase (binucleates +Triton). When Mcb1HA was overexpressed in the absence of thiamine, nuclear localization of both MCM proteins was reduced, and chromatin binding (+Triton) was abolished. Similar results were observed if cdc18+ was shut off by addition of thiamine. These data suggest that the toxic effect associated with Mcb1 expression results in delocalization of the MCM subunits to the cytoplasm and inhibition of replication.
FIGURE 8.

Overproduction of Mcb1 causes dissociation of chromatin-bound MCM proteins. nmt1-cdc18 mcm2V5 mcm4GFP (FY4958), nmt1-mcb1HA mcm2V5 mcm4GFP (FY4961), and wild-type (FY11) overnight cultures were grown in low thiamine, then inoculated into −thiamine (−T) or +thiamine (+T) medium, grown at 32 °C for 14 h, and harvested for an in situ chromatin binding assay. Mcb1HA and Mcm2V5 localization was detected in untreated cells and Triton-treated cells with specific antibodies. Scale bar, 10 μm.
DISCUSSION
MCM proteins are members of the AAA+ ATPase family and share a unique motif, the MCM box, which is important for MCM complex formation and ATP hydrolysis (for reviews, see Refs. 7 and 8). Recent studies identified a novel component of the MCM complex in human as well as plants, MCM-BP (29–31). Evidence from these systems suggests that MCM-BP is a component of the replisome and replaces the Mcm2 subunit. Recent work suggests that MCM-BP may contribute to sister chromatid cohesion and DNA repair (31). Although no orthologue has been found in budding yeast, we identified a putative orthologue of human MCM-BP that we named Mcb1. Similar to other MCM-BPs, Mcb1 shares no homology to S. pombe MCM proteins and lacks the MCM box.
Disruption of mcb1+ was lethal. However, disrupted spores managed to germinate and complete several cell cycles before arresting with an elongated, cdc morphology. Most of the cells had a single nucleus of normal appearance; a few had a disordered nucleus or evidence of mitosis. This delayed lethality is likely to reflect the abundance of the maternal protein packaged in the spores. For example, mcm4Δ cells complete S phase prior to arresting with a 2C DNA content; the mcm4Δ cells arrest prior to S phase only if the residual maternal Mcm4 protein is inactivated with a temperature-sensitive mutation (25). Therefore, although we can conclude that Mcb1 is essential for viability and cell cycle progression, we cannot conclude at what stage(s) of the cell cycle it works.
We found Mcb1 to be amenable to epitope tagging and detection by Western blot. Comparison of Mcb1HA with Mcm2HA with the same antibody showed that the proteins are expressed at similar levels with Mcb1 somewhat more abundant even than Mcm2. MCM proteins are estimated at around 104 molecules per cell (47), so Mcb1 is a very abundant protein. Mcb1 levels are constant throughout the cell cycle with no evidence for periodic modifications. Curiously, we were unable to visualize either Mcb1GFP in live cells or HA-tagged Mcb1 in fixed cells using immunofluorescence, although they are readily detected by Western blot. It is possible that the tags are blocked in some way in its normal environment and only available upon denaturation. We used cell fractionation to examine Mcb1 localization and found that Mcb1 is ubiquitously distributed through the entire cell but enriched in the nucleus.
Similar to observations in humans (29), we found that Mcb1 normally associates with Mcm3, -4, -6, and -7, but not Mcm2, thus forming an alternative MCM complex. The canonical MCM complex consists of Mcm2–7 with 1:1:1:1:1:1 stoichiometry (52, 58–60). Mcm4, -6, and -7 form a trimeric subcomplex known as “the MCM core.” Mcm2 binds to the core and a dimer formed by Mcm3 and Mcm5. The MCMs interdigitate with one another in a ring structure in which the arginine finger of one MCM meets with the P-loop in the Walker A motif of its neighbor to form an ATP binding site (53, 58). Coordinated ATP hydrolysis occurs at a subset of sites (53, 58, 60). Mcm2 is thought to be the gate of the ring and the site at which the ring opens to encircle chromatin (53). Mcb1 lacks these sequences, so it is unlikely to contribute to the ATP-dependent structure. It is possible that Mcb1 bound to Mcm3–7 forms an open structure, not a ring.
We were not able to detect Mcb1 at replication origins using ChIP under conditions that detect Mcm2, suggesting that Mcb1 is not a component of the core replisome or at least is not as closely bound as the MCMs. We hypothesize that the Mcm3–7 bound to Mcb1 is not active as a helicase in vivo. There is good biochemical evidence that all six canonical MCMs participate as a helicase in vivo with Cdc45 and GINS complex as cofactors (53, 61, 62). Mcm2 is required to recruit Mcm4, -6, and -7 into the nucleus where an intact MCM complex is necessary to retain them (15). Moreover, the phenotypes associated with mcm2 mutations are indistinguishable from mutations in other MCM subunits; if it were not a core constituent of the helicase, this would not be expected. We found no evidence that Mcb1 expression can substitute for Mcm2. Therefore, the interaction between Mcb1 and the other MCMs is likely to have some other, possibly regulatory function.
In the absence of a tight conditional allele, we used two approaches to examine Mcb1 activity. We constructed a series of deletion/truncation mutations in Mcb1 and assessed their ability to function. Most of these mutants were unable to complement an mcb1Δ mutant. However, two mutants containing a truncation of the N terminus were apparent hypomorphs; the growing cells were elongated, and this elongation depended upon the damage checkpoint. DNA content, however, was normal, and cells formed colonies with timing similar to wild type. We conclude that attenuating Mcb1 function leads to some genome instability, which is similar to the phenotypes associated with attenuation of MCM function (e.g. Refs. 24 and 25).
We also found that overexpression of Mcb1 generates a dominant lethal phenotype. OP-Mcb1 cells showed evidence of an initiation defect characterized by an increase of cells with a sub-1C DNA content. This phenotype is reminiscent of cells with mutations in the essential replication initiation factors orc1 (63, 64), cdc18 (56), and rad4/cut5 (65). The general model is that cells that do not initiate replication have no way to activate a checkpoint or register that S phase has not occurred. Thus, the cells proceed through mitosis and tear apart the unreplicated genome. We observed that OP-Mcb1 cells show a modest increase in Rad22 foci, which is indicative of DNA damage, and activation of the Chk1 damage checkpoint kinase, which has been seen in many mutants defective in DNA replication initiation (66). This may occur in the subset of cells in the population that are elongated.
This initiation-defective phenotype is likely to result from inactivation of the MCM complex. In strains overproducing Mcb1, association between the canonical MCM proteins was disrupted. This was most strikingly observed by the failure of Mcm4 to associate with Mcm2, but interaction between Mcm4 and the core MCMs was also reduced. Interestingly, the overproduced Mcb1 was able to bind to Mcm2, suggesting that when expressed at high enough levels this protein can interact with all MCM subunits.
When we examined the truncation/deletion mutants for overproduction phenotypes, we found that only the two hypomorphic mutants, which contain a short N-terminal truncation, were toxic upon overexpression. Although they were still able to form small colonies (unlike expression of the full-length protein which is lethal), they also reduced association between the canonical MCM proteins.
Several of the non-functional mutants were able to bind the MCMs but showed no evidence for complex disruption (Mcm2 and Mcm4 remained associated, for example). We conclude that there are three modes of interaction between Mcb1 and the MCM complex (supplemental Fig. S11). The first is a normal “functional” mode in which Mcb1 replaces the Mcm2 subunit. The second interaction is a “sticky” mode in which Mcb1 appears to bind nonspecifically to all the MCM subunits. This is not toxic, does not replace Mcm2, and does not disrupt the MCM complex. Finally, the third is the overproduction toxicity. Only proteins capable of functional interactions can disrupt the complex when overproduced, leading to toxicity and an apparent arrest of DNA replication initiation. Although it remains a formal possibility that the inhibitory effect Mcb1 has on the MCM complex is exacerbated by overexpression and not a representation of its true phenotype, we consider this unlikely given the correlation of functional Mcb1 with complex disassembly. We conclude that overproduction of Mcb1 causes a dramatic inhibition of replication initiation similar to that caused by mutations in the genes required for formation of the prereplication complex.
MCMs in most eukaryotes exist in three populations within the nucleus: 1) the replisome MCMs bound to chromatin at the replication fork that are detectable by ChIP, 2) “remote” MCMs bound on unreplicated DNA during S phase that can be visualized cytologically (18, 47, 67, 68) but not by ChIP, and 3) a soluble pool not bound to chromatin. The large amount of remote MCMs creates a puzzle known as “the MCM paradox” (19, 21). These “remote MCMs” are very important for distributing origins, marking unreplicated chromatin for replication, and reserving dormant replication origins to complete replication under replication stress (69–73). Formally, Mcb1 could contribute to the formation of any of these pools possibly by disrupting the intact hexamer to change the distribution between them. This could occur by promoting removal of MCMs from the chromatin, particularly the remote MCMs. Because the Arabidopsis ETG1 protein has been linked to sister chromatid cohesion (31), another possibility is that Mcb1 changes the composition of the MCM complex to facilitate binding of cohesin assembly proteins at the fork.
It is interesting that budding yeast does not have an obvious orthologue of Mcb1, which is readily identified in other eukaryotes. In this regard, it may be worth noting that a significant difference between budding yeast behavior in S. cerevisiae compared with other systems is regulated nuclear localization. In budding yeast, MCMs cycle in and out of the nucleus during the cell cycle in a cyclin-dependent kinase-dependent pathway (74, 75). Newly synthesized MCMs are preferentially transported into the budding yeast nucleus (76). By contrast, in other eukaryotes, including S. pombe, MCMs are located constitutively in the nucleus, and only their chromatin association is regulated. Dissociation of the MCM complex causes chromatin dissociation and crm1+-dependent nuclear export (15). We saw reduced MCM on chromatin in OP-Mcb1 cells, and there was a reduction in the overall nuclear signal of Mcm4 compared with Mcm2. It is possible that Mcb1 functions to regulate the MCMs at the level of chromatin association to prevent binding or activation outside of S phase. This could be a regulator function that is not needed in budding yeast. This is consistent with data from other systems. In the process of preparing this work, a new study has shown that Xenopus MCM-BP unloads MCM complex from the chromatin in late S phase to prevent rereplication by dissociating Mcm2–7 from chromatin (77). Our data support a model in which the abundant Mcb1 protein contributes to redistribution of the MCM proteins at the conclusion of S phase.
Supplementary Material
Acknowledgments
We thank past and present members of the Forsburg and Aparicio laboratories for discussion and technical support. We thank PaoChen Li, Matthew Michael, Rebecca Nugent, Ruben Petreaca, and Sarah Sabatinos for critical reading of the manuscript.
This work was supported, in whole or in part, by National Institutes of Health Grant R01 GM059321 (to S. L. F.) and the National Institutes of Health Cellular, Biochemical, and Molecular Sciences Training Program at the University of Southern California Keck School of Medicine (to L. D.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S11 and Table 1.
- MCM
- minichromosome maintenance
- MCM-BP
- MCM-binding protein
- hMCM-BP
- human MCM-binding protein
- Mcb1
- Mcm-binding protein 1
- nmt
- no message in thiamine
- OP
- overproduction
- EMM
- Edinburgh minimal medium
- Ura
- uracil
- AAA
- ATPase associated with various cellular activities.
REFERENCES
- 1. Masai H., Matsumoto S., You Z., Yoshizawa-Sugata N., Oda M. (2010) Annu. Rev. Biochem. 79, 89–130 [DOI] [PubMed] [Google Scholar]
- 2. Kelly T. J., Nurse P., Forsburg S. L. (1993) Cold Spring Harb. Symp. Quant. Biol. 58, 637–644 [DOI] [PubMed] [Google Scholar]
- 3. Hartwell L. H., Weinert T. A. (1989) Science 246, 629–634 [DOI] [PubMed] [Google Scholar]
- 4. Branzei D., Foiani M. (2008) Nat. Rev. Mol. Cell Biol. 9, 297–308 [DOI] [PubMed] [Google Scholar]
- 5. Jackson S. P., Bartek J. (2009) Nature 461, 1071–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hartwell L., Weinert T., Kadyk L., Garvik B. (1994) Cold Spring Harb. Symp. Quant. Biol. 59, 259–263 [DOI] [PubMed] [Google Scholar]
- 7. Forsburg S. L. (2004) Microbiol. Mol. Biol. Rev. 68, 109–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bochman M. L., Schwacha A. (2009) Microbiol. Mol. Biol. Rev. 73, 652–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gozuacik D., Chami M., Lagorce D., Faivre J., Murakami Y., Poch O., Biermann E., Knippers R., Bréchot C., Paterlini-Bréchot P. (2003) Nucleic Acids Res. 31, 570–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lutzmann M., Maiorano D., Méchali M. (2005) Gene 362, 51–56 [DOI] [PubMed] [Google Scholar]
- 11. Shultz R. W., Tatineni V. M., Hanley-Bowdoin L., Thompson W. F. (2007) Plant Physiol. 144, 1697–1714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yoshida K. (2005) Biochem. Biophys. Res. Commun. 331, 669–674 [DOI] [PubMed] [Google Scholar]
- 13. Sible J. C., Erikson E., Hendrickson M., Maller J. L., Gautier J. (1998) Curr. Biol. 8, 347–350 [DOI] [PubMed] [Google Scholar]
- 14. Adachi Y., Usukura J., Yanagida M. (1997) Genes Cells 2, 467–479 [DOI] [PubMed] [Google Scholar]
- 15. Pasion S. G., Forsburg S. L. (1999) Mol. Biol. Cell 10, 4043–4057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sherman D. A., Forsburg S. L. (1998) Nucleic Acids Res. 26, 3955–3960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sherman D. A., Pasion S. G., Forsburg S. L. (1998) Mol. Biol. Cell 9, 1833–1845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kearsey S. E., Montgomery S., Labib K., Lindner K. (2000) EMBO J. 19, 1681–1690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hyrien O., Marheineke K., Goldar A. (2003) BioEssays 25, 116–125 [DOI] [PubMed] [Google Scholar]
- 20. Lei M., Kawasaki Y., Tye B. K. (1996) Mol. Cell. Biol. 16, 5081–5090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Laskey R. A., Madine M. A. (2003) EMBO Rep. 4, 26–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Coxon A., Maundrell K., Kearsey S. E. (1992) Nucleic Acids Res. 20, 5571–5577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Forsburg S. L., Nurse P. (1994) J. Cell Sci. 107, 2779–2788 [DOI] [PubMed] [Google Scholar]
- 24. Liang D. T., Forsburg S. L. (2001) Genetics 159, 471–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liang D. T., Hodson J. A., Forsburg S. L. (1999) J. Cell Sci. 112, 559–567 [DOI] [PubMed] [Google Scholar]
- 26. Miyake S., Okishio N., Samejima I., Hiraoka Y., Toda T., Saitoh I., Yanagida M. (1993) Mol. Biol. Cell 4, 1003–1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Takahashi K., Yamada H., Yanagida M. (1994) Mol. Biol. Cell 5, 1145–1158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bailis J. M., Luche D. D., Hunter T., Forsburg S. L. (2008) Mol. Cell. Biol. 28, 1724–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sakwe A. M., Nguyen T., Athanasopoulos V., Shire K., Frappier L. (2007) Mol. Cell. Biol. 27, 3044–3055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Takahashi N., Lammens T., Boudolf V., Maes S., Yoshizumi T., De Jaeger G., Witters E., Inzé D., De Veylder L. (2008) EMBO J. 27, 1840–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Takahashi N., Quimbaya M., Schubert V., Lammens T., Vandepoele K., Schubert I., Matsui M., Inzé D., Berx G., De Veylder L. (2010) PLoS Genet. 6, e1000817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moreno S., Klar A., Nurse P. (1991) Methods Enzymol. 194, 795–823 [DOI] [PubMed] [Google Scholar]
- 33. Sabatinos S. A., Forsburg S. L. (2010) Methods Enzymol. 470, 759–795 [DOI] [PubMed] [Google Scholar]
- 34. Forsburg S. L., Rhind N. (2006) Yeast 23, 173–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bähler J., Wu J. Q., Longtine M. S., Shah N. G., McKenzie A., 3rd, Steever A. B., Wach A., Philippsen P., Pringle J. R. (1998) Yeast 14, 943–951 [DOI] [PubMed] [Google Scholar]
- 36. Maundrell K. (1993) Gene 123, 127–130 [DOI] [PubMed] [Google Scholar]
- 37. Keeney J. B., Boeke J. D. (1994) Genetics 136, 849–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Maundrell K. (1990) J. Biol. Chem. 265, 10857–10864 [PubMed] [Google Scholar]
- 39. Basi G., Schmid E., Maundrell K. (1993) Gene 123, 131–136 [DOI] [PubMed] [Google Scholar]
- 40. Sazer S., Sherwood S. W. (1990) J. Cell Sci. 97, 509–516 [DOI] [PubMed] [Google Scholar]
- 41. Sabatinos S. A., Forsburg S. L. (2009) Methods Mol. Biol. 521, 449–461 [DOI] [PubMed] [Google Scholar]
- 42. Liang C., Stillman B. (1997) Genes Dev. 11, 3375–3386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Mason J. A., Mellor J. (1997) Nucleic Acids Res. 25, 4700–4701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Catlett M. G., Forsburg S. L. (2003) Mol. Biol. Cell 14, 4707–4720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Matsuo Y., Asakawa K., Toda T., Katayama S. (2006) Biosci. Biotechnol. Biochem. 70, 1992–1994 [DOI] [PubMed] [Google Scholar]
- 46. Gómez E. B., Catlett M. G., Forsburg S. L. (2002) Genetics 160, 1305–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Namdar M., Kearsey S. E. (2006) Exp. Cell Res. 312, 3360–3369 [DOI] [PubMed] [Google Scholar]
- 48. Radcliffe P., Hirata D., Childs D., Vardy L., Toda T. (1998) Mol. Biol. Cell 9, 1757–1771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Matsuyama A., Arai R., Yashiroda Y., Shirai A., Kamata A., Sekido S., Kobayashi Y., Hashimoto A., Hamamoto M., Hiraoka Y., Horinouchi S., Yoshida M. (2006) Nat. Biotechnol. 24, 841–847 [DOI] [PubMed] [Google Scholar]
- 50. Rustici G., Mata J., Kivinen K., Lió P., Penkett C. J., Burns G., Hayles J., Brazma A., Nurse P., Bähler J. (2004) Nat. Genet. 36, 809–817 [DOI] [PubMed] [Google Scholar]
- 51. Forsburg S. L., Sherman D. A., Ottilie S., Yasuda J. R., Hodson J. A. (1997) Genetics 147, 1025–1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee J. K., Hurwitz J. (2000) J. Biol. Chem. 275, 18871–18878 [DOI] [PubMed] [Google Scholar]
- 53. Bochman M. L., Schwacha A. (2010) Nucleic Acids Res. 38, 6078–6088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hingorani M. M., O'Donnell M. (1998) Curr. Biol. 8, R83–R86 [DOI] [PubMed] [Google Scholar]
- 55. Forsburg S. L. (1993) Nucleic Acids Res. 21, 2955–2956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kelly T. J., Martin G. S., Forsburg S. L., Stephen R. J., Russo A., Nurse P. (1993) Cell 74, 371–382 [DOI] [PubMed] [Google Scholar]
- 57. Walworth N. C., Bernards R. (1996) Science 271, 353–356 [DOI] [PubMed] [Google Scholar]
- 58. Davey M. J., Indiani C., O'Donnell M. (2003) J. Biol. Chem. 278, 4491–4499 [DOI] [PubMed] [Google Scholar]
- 59. Lee J. K., Hurwitz J. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 54–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schwacha A., Bell S. P. (2001) Mol. Cell 8, 1093–1104 [DOI] [PubMed] [Google Scholar]
- 61. Bauerschmidt C., Pollok S., Kremmer E., Nasheuer H. P., Grosse F. (2007) Genes Cells 12, 745–758 [DOI] [PubMed] [Google Scholar]
- 62. Ilves I., Petojevic T., Pesavento J. J., Botchan M. R. (2010) Mol. Cell 37, 247–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Grallert B., Nurse P. (1996) Genes Dev. 10, 2644–2654 [DOI] [PubMed] [Google Scholar]
- 64. Muzi-Falconi M., Kelly T. J. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 12475–12479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Saka Y., Yanagida M. (1993) Cell 74, 383–393 [DOI] [PubMed] [Google Scholar]
- 66. Yin L., Locovei A. M., D'Urso G. (2008) Mol. Biol. Cell 19, 4374–4382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Krude T., Musahl C., Laskey R. A., Knippers R. (1996) J. Cell Sci. 109, 309–318 [DOI] [PubMed] [Google Scholar]
- 68. Madine M. A., Khoo C. Y., Mills A. D., Musahl C., Laskey R. A. (1995) Curr. Biol. 5, 1270–1279 [DOI] [PubMed] [Google Scholar]
- 69. Blow J. J., Ge X. Q. (2009) EMBO Rep. 10, 406–412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Chuang C. H., Wallace M. D., Abratte C., Southard T., Schimenti J. C. (2010) PLoS Genet. 6, e1001110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Edwards M. C., Tutter A. V., Cvetic C., Gilbert C. H., Prokhorova T. A., Walter J. C. (2002) J. Biol. Chem. 277, 33049–33057 [DOI] [PubMed] [Google Scholar]
- 72. Ge X. Q., Jackson D. A., Blow J. J. (2007) Genes Dev. 21, 3331–3341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Woodward A. M., Göhler T., Luciani M. G., Oehlmann M., Ge X., Gartner A., Jackson D. A., Blow J. J. (2006) J. Cell Biol. 173, 673–683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Labib K., Diffley J. F., Kearsey S. E. (1999) Nat. Cell Biol. 1, 415–422 [DOI] [PubMed] [Google Scholar]
- 75. Nguyen V. Q., Co C., Irie K., Li J. J. (2000) Curr. Biol. 10, 195–205 [DOI] [PubMed] [Google Scholar]
- 76. Braun K. A., Breeden L. L. (2007) Mol. Biol. Cell 18, 1447–1456 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nishiyama A., Frappier L., Méchali M. (2011) Genes Dev. 25, 165–175 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.