

Abstract
WW domain-containing E3 ubiquitin protein ligase 1 (WWP1) plays an important role in the proliferation of tumor cells and the lifespan of Caenorhabditis elegans. However, the role of WWP1 in cellular senescence is still unknown. Here, we show that the expression patterns of p27Kip1 and WWP1 are inversely correlated during cellular senescence. Moreover, the overexpression of WWP1 delayed senescence, whereas the knockdown of WWP1 led to premature senescence in human fibroblasts. Furthermore, we demonstrate that WWP1 repressed endogenous p27Kip1 expression through ubiquitin-proteasome-mediated degradation. Additionally, WWP1 had a strong preference for catalyzing the Lys-48-linked polyubiquitination of p27Kip1 in vitro. Finally, we demonstrate that WWP1 markedly inhibited the replicative senescence induced by p27Kip1 by promoting p27Kip1 degradation. Therefore, our study provides a new molecular mechanism for the regulation of cellular senescence.
Keywords: Cellular Senescence, Proteasome, Protein Degradation, Ubiquitin Ligase, Ubiquitination, WWP1, p27
Introduction
The concept of replicative senescence is based on the inability of cells to divide indefinitely in culture. Instead, the cells enter an irreversible state of proliferative arrest with specific associated changes in cellular morphology and gene expression. The senescent phenotype is characterized by growth inhibition with enlarged and flattened cellular morphology (1), highly active senescence-associated β-galactosidase (SA-β-gal)2 (2), and the accumulation of senescence-associated heterochromatin foci (SAHF) (3, 4). In a sense, cellular senescence is a reflection of organism aging, and investigation into replicative senescence may thus provide information about the molecular mechanisms of organism senescence and aging-related diseases. In recent years, increasing numbers of molecular mechanisms of cellular senescence have been studied. It is well accepted that there are three pathways that play key roles in replicative senescence, namely the p16INK4a pathway, the p53/p21Cip1 pathway, and the p27Kip1 pathway (5). However, in comparison with the first two pathways, relatively few studies have focused on the p27Kip1 pathway.
The majority of proteins involved in replicative senescence are modified by polyubiquitin, which directs proteins for degradation by the 26 S proteasome. For example, cyclin-dependent kinase inhibitors, which are of special relevance to senescence, are mainly regulated by the ubiquitin-proteasome system. The ubiquitination cascade operates by the sequential action of the E1, E2, and E3 enzymes. E3 ubiquitin ligases are significant in cellular regulation because E3 enzymes specifically recognize the substrate to be modified. It is well established that the E3 enzymes control substrate specificity.
WW domain-containing E3 ubiquitin protein ligase 1 (WWP1) is an E3 ubiquitin ligase first identified by its WW domain (6). WWP1 belongs to the Nedd4-like family of E3 ubiquitin ligases, a subfamily of HECT, which contains Nedd4, Itch, WWP2, AIP4, and RPF1 (7, 8). Members of this family have been shown to be involved in the regulation of cellular signaling and protein sorting (9). Although WWP1 has been shown to function as an E3 ubiquitin ligase, only a few of its substrates have been identified. Among these are Kruppel-like factors (10–12), Smad4 (13), p53 (14), and p63 (15, 16), which have all been reported to be regulated by WWP1. Functional analysis has shown that WWP1 promotes prostate and breast epithelial cell proliferation and survival (17, 18). In Caenorhabditis elegans, WWP1 is a positive regulator of lifespan, acting through diet restriction and the DAF-2 insulin/insulin-like growth factor-1 signaling pathway (19, 20). In addition, WWP1 is essential for embryonic development in C. elegans (21). However, it is still unknown whether WWP1 influences cellular senescence in humans.
In this study, we evaluated the role of WWP1 in cellular senescence by assessing the senescent phenotypes associated with WWP1 overexpression and small hairpin RNA (shRNA)-mediated WWP1 silencing. Moreover, we demonstrated the mechanisms that underlie the WWP1-mediated degradation of p27Kip1. Our findings provide valuable new insights into the mechanism of p27Kip1 regulation and cellular senescence.
EXPERIMENTAL PROCEDURES
Cell Lines, Cell Culture, and Treatment
Human embryonic lung diploid fibroblast 2BS cells (National Institute of Biological Products, Beijing, China) are considered to be young at PD30 or below and to be senescent at PD55 or above. Human embryonic lung diploid fibroblast WI38 cells (Chinese Academy of Sciences, Shanghai, China) are considered to be young at PD25 or below and to be fully senescent at PD50 or above. HeLa cells were from our laboratory, and ψnx packaging cells were gifts from Dr. M. Narita (Cancer Research UK, Cambridge Research Institute). All cells were cultured in DMEM (Invitrogen) supplemented with 10% fetal bovine serum at 37 °C in 5% CO2. Cultures at 80% confluence were detached from the dish with a PBS solution containing 0.25% trypsin and then split at a ratio of 1:2.
Carbobenzoxy-l-leucyl-l-leucyl-l-leucinal (MG132; Sigma) and acetyl-Leu-Leu-norleucinal (Sigma) were dissolved in DMSO (Sigma) and added to the culture medium at 15 μm for 4–6 h. A corresponding volume of DMSO was added to untreated control cells. Cycloheximide (Sigma) was also dissolved in DMSO and added to cells at 15 μg/ml for the indicated time periods.
Generation of Plasmids and Transfection
Full-length WWP1 was kindly provided by Dr. Ceshi Chen and cloned into pWZL-Hygro, pcDNA3.1-FLAG, and pGEX-4T2-GST. The constructs CD1 (HECT domain deleted), CD2 (both HECT and WW domains deleted), and MD1 (WW domains deleted) were cloned into pcDNA3.1-FLAG, and all constructs expressed fusion proteins with FLAG. The p27Kip1 fragment was cloned from the cDNA of young 2BS cells using a PCR-based approach and was placed into pBabe-neo, pcDNA3.1-Myc, and pGEX-4T2-GST. All clones were confirmed by DNA sequencing. Deletions in p27Kip1 were generated using overlapping PCR and were cloned into pGEX-4T2-GST and pcDNA3.1-Myc. Nucleotide base pairs 250–264, 271–282, and 250–282 of p27Kip1 cDNA were deleted.
The shRNA was designed according to the pMSCV instruction manual (Clontech). The template oligonucleotides were chemically synthesized with a 5′-phosphate and two base overhangs on each strand. The double-stranded DNA was inserted into the EcoRI and XhoI sites of the pMSCV-puro-miR30 vector. The following gene-specific sequences were used successfully: WWP1, 5′-GAGTTGATGATCGTAGAAG-3′ (11); p21Cip1, 5′-GACAGATTTCTATCACTCCAA-3′ (22).
For transient transfection, HeLa cells at 70–80% confluence were transfected with Lipofectamine 2000 (Invitrogen) following the manufacturer's instructions. For stable transfection, retroviral infection was used (3). After ψnx cells reached 60–70% confluence, the retroviral plasmids were transfected with CaCl2 reagent (M&C Gene Technology Ltd., Beijing, China) according to the manufacturer's instructions. The retrovirus supernatants were collected 48 h after transfection and then filtered. Young 2BS and WI38 cells were infected with retrovirus in the presence of 8 μg/ml Polybrene (Invitrogen). Pools of stable transformants were obtained by sustained selection with 200 μg/ml neomycin (Invitrogen), 1.5 μg/ml puromycin (Invitrogen), or 100 μg/ml hygromycin (Invitrogen), starting 1 day after infection.
siRNA Transfection
To transiently silence WWP1, siRNA targeting WWP1 (11) and control siRNA (5′-TTCTCCGAACGTGTCACGT-3′) (11) were synthesized (Genechem). Both siRNAs were transfected with Oligofectamine (Invitrogen) following the manufacturer's recommendations. Cells were collected 48 h after transfection for further analysis.
Immunoblot Analysis
Cells were washed with PBS, collected, and lysed on ice for 30 min with modified radioimmune precipitation assay buffer (Applygen Technologies Inc., Beijing, China) containing a protease inhibitor mixture (Fermentas). Cell lysates were sonicated and then centrifuged for 10 min at 15,000 × g at 4 °C. The supernatant was collected, and the protein concentration was determined using the BCA Protein Assay Reagent (Pierce). Total protein (50–80 μg) was subjected to 10–15% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and was transferred to nitrocellulose membranes (Millipore). After blocking in 5% nonfat dry milk in TBST (10 mm Tris-Cl, pH 7.5, 150 mm NaCl, 0.05% Tween 20), the membranes were incubated with primary antibodies overnight at 4 °C. The membranes were then washed four times with TBST and then incubated with HRP-conjugated secondary antibodies (Zhongshan Biotechnologies Inc., China) for 1 h at room temperature. Proteins were visualized using chemiluminescent substrate (Millipore) according to the manufacturer's instructions. The following antibodies were used for the Western blot analysis: anti-WWP1 (Abnova), anti-p27Kip1 (MBL), anti-multiubiquitin (MBL), anti-FLAG (Sigma), anti-PTEN (Santa Cruz Biotechnology), anti-p16INK4a (Santa Cruz Biotechnology), anti-p21Cip1 (Santa Cruz Biotechnology), and anti-tubulin (Santa Cruz Biotechnology).
Reverse Transcription-PCR
Total RNA was isolated from cells using the RNeasy minikit (Qiagen) according to the manufacturer's instructions. First strand cDNA was synthesized using the StarScript first strand cDNA synthesis kit (GenStar Biosolutions Co. Ltd., Beijing, China). For the RT-PCR analysis of WWP1, p27Kip1, p21Cip1, and GAPDH expression, specific primers were used. PCR was performed with the 2× Taq PCR StarMix kit (GenStar Biosolutions Co. Ltd.). Levels of WWP1, p27Kip1, p21Cip1, and GAPDH mRNA were assessed by staining gels with ethidium bromide.
SA-β-gal and Analysis of SAHF
For SA-β-gal staining, cells were washed twice in PBS, fixed for 3–5 min at room temperature in 3% formaldehyde, and washed twice with PBS. The cells were incubated overnight at 37 °C without CO2 in a freshly prepared SA-β-gal-staining solution as described (23). To determine formation of SAHF, cells were cultured on glass coverslips and fixed using 4% paraformaldehyde. After washing with PBS, cells were permeabilized with 0.2% Triton X-100 in PBS for 10 min. DNA was stained with 1 mg/ml DAPI for 5 min followed by three PBS washes. Coverslips were mounted in a 90% glycerol, PBS solution. Coverslips were examined using a Leica confocal TCS SP2 microscope.
Cell Cycle Analysis and Growth Curves
When cells reached 70–80% confluence, they were washed with PBS, detached with 0.25% trypsin, and fixed with 75% ethanol overnight. After treatment with 1 mg/ml RNase A (Sigma) at 37 °C for 30 min, cells were resuspended in 0.5 ml of PBS and stained with propidium iodide in the dark for 30 min. Fluorescence was measured with a FACScan flow cytometry system (BD Biosciences). Growth curves were assayed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method (24). Cells were seeded into 96-well plates with 2 × 103 cells/well and cultured for periods ranging from 1 to 7 days. Every 24 h, an aliquot of cells was stained with 20 ml of MTT (10 mg/ml in PBS; Sigma) for 3 h and then treated with DMSO for 10 min. The optical density was measured at 570 nm.
Co-immunoprecipitation Assays
Cells were scraped from the plate and lysed on ice for 30 min with 300 μl of radioimmune precipitation assay buffer containing a protease inhibitor mixture. Lysates were precleared by incubation with 20 μl of 50% protein A-Sepharose beads (Sigma) for 10 min. Lysates were then centrifuged for 5 min at 3,000 × g at 4 °C. The supernatant and antibody were gently rocked overnight at 4 °C. A 50% slurry of protein A-Sepharose beads (30 μl) was then added and incubated for 5 h at 4 °C. Precipitates were washed four times with 800 μl of radioimmune precipitation assay buffer and then resuspended in 50 μl of 2× SDS loading buffer. The samples were boiled for 5 min and analyzed by Western blotting.
GST Pulldown Assays
Escherichia coli strain BL21 was used to produce GST or GST fusion proteins. Expression of fusion proteins was induced with isopropyl thio-β-d-galactopyranoside, and the protein was purified with glutathione-conjugated Sepharose beads (GE Healthcare) following the manufacturer's instructions. Proteins transcribed/translated in vitro using the TnT T7 Quick Coupled Transcription/Translation System (Promega) were used as another source of purified protein. The GST pulldown assay was performed as described previously (25). In brief, 1–5 mg of purified bacterial recombinant protein immobilized on glutathione-conjugated Sepharose beads was preblocked with 1% bovine serum albumin and then incubated with in vitro transcribed/translated proteins for 4 h on a rotator at 4 °C. After extensive washing with lysis buffer, the bound proteins were subjected to Western blot analysis.
In Vitro Ubiquitination Assay
Purified GST-p27Kip1 (20 μg) was incubated in a reaction volume of 20 μl of reaction buffer (Boston Biochem) containing 100 nm E1 enzyme (Boston Biochem), 5 μm UbcH7 (Boston Biochem), 100 μm ubiquitin, and 5 μg of either purified WWP1 or HECT-deleted CD1. Wild-type ubiquitin and all mutant forms of ubiquitin were from Boston Biochem. This mixture was incubated at 37 °C for 90 min. The reaction was terminated by the addition of SDS sample buffer. The sample was then boiled, separated by 12% SDS-PAGE, and analyzed by Western blotting.
RESULTS
Expression of WWP1 Is Decreased during Replicative Senescence in Human Fibroblasts
In view of the positive effect of WWP1 on the proliferation of prostate and breast epithelial cancer cells and the role of WWP1 in increasing the lifespan of C. elegans, we speculated that WWP1 might also be involved in cellular senescence. Therefore, we first evaluated WWP1 expression patterns in young, middle-aged, and senescent 2BS and WI38 cells. As expected, Western blot analysis revealed that the expression of WWP1 was high in young cells but decreased significantly during cellular senescence, whereas the expression of p16INK4a, p21Cip1, and p27Kip1 increased in senescent cells (Fig. 1A). RT-PCR analysis demonstrated the down-regulation of the WWP1 transcript in aged fibroblasts (Fig. 1B). This passage-dependent reduction suggested that WWP1 might be involved in the process of cellular senescence.
FIGURE 1.
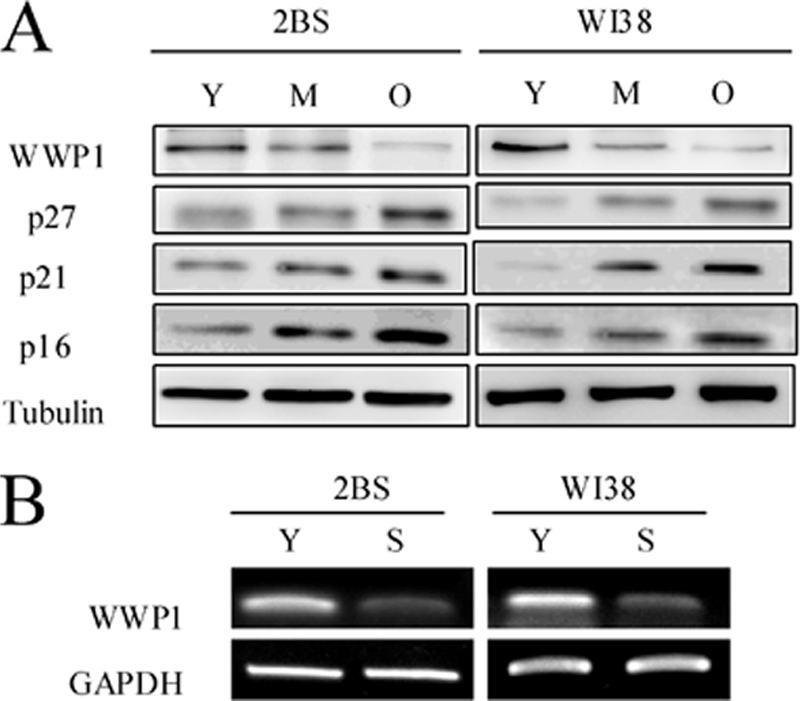
Expression patterns of WWP1 in young, middle-aged, and senescent cells. A, Western blot analysis of WWP1, p16 INK4a, p21Cip1, and p27Kip1 expression in young (Y), middle-aged (M), and senescent (O) 2BS and WI38 cells. Total protein was extracted, and immunoblotting was performed using specific antibodies against WWP1, p16INK4a, p21Cip1, and p27Kip1 as indicated. Tubulin served as a loading control. B, RT-PCR analysis of WWP1 in young (Y) and senescent (S) 2BS and WI38 cells. Total mRNA was extracted and assessed by RT-PCR using specific primers. GAPDH was used as a loading control.
WWP1 Overexpression Delays Cellular Senescence, whereas WWP1 Silencing Leads to Premature Senescence in Human Fibroblasts
Senescent human diploid fibroblast cells characteristically show irreversible growth arrest with enlarged and flattened cellular morphology and accumulation of prominent lipofuscin granules. To determine the effect of WWP1 on cellular senescence, WWP1 was overexpressed and silenced, respectively, with a retrovirus expression system in young 2BS and WI38 cells. Senescence markers for WWP1-overexpressing and WWP1 shRNA-infected cells were then monitored at the same time points as their corresponding control cells (Fig. 2). We found that WWP1 affected the process of cellular senescence.
FIGURE 2.

WWP1 repressed senescence-associated features in 2BS and WI38 cells. The stable transformants were passaged until senescence was achieved. They were then analyzed for the related senescence markers. A, Western blot analysis of WWP1 expression levels in WWP1-infected or WWP1 shRNA-infected cells. B, WWP1-transfected and WWP1 shRNA-transfected 2BS and WI38 cells were stained for SA-β-gal activity (upper panel) and formation of SAHF (middle and lower panels). C, growth curves of WWP1-transfected and WWP1 shRNA-transfected cells were determined by the MTT assay. Values are the mean ± S.D. of triplicate points from a representative experiment (n = 3), which was repeated three times with similar results. D, flow cytometry analysis of WWP1-transfected and WWP1 shRNA-transfected 2BS cells. Values represent the means ± S.E. of triplicate points from a representative experiment (n = 3), which was repeated three times. sh, shRNA.
We first observed morphological changes in cells with diverse WWP1 expression. The WWP1 shRNA-transfected 2BS cells displayed enlargement, flattening, and the accumulation of vacuolated cytoplasmic inclusions. However, no significant morphological changes were observed in the WWP1-transfected cells, which were similar to young 2BS cells (Fig. 2B).
We further detected staining with SA-β-gal, which is considered a specific senescence marker. Most of the WWP1 shRNA-transfected cells showed strong blue SA-β-gal staining similar to senescent cells (Fig. 2B). In contrast, only scattered SA-β-gal-positive cells were found in WWP1-transfected cells compared with the corresponding control cells (Fig. 2B). The same results were observed in WI38 cells (Fig. 2B).
Another hallmark of senescent cells is the formation of SAHF. We observed that 2BS cells and WI38 cells overexpressing WWP1 did not display marked changes in chromatin structure (Fig. 2B). However, WWP1 shRNA-infected cells showed prominent heterochromatin foci, which were virtually indistinguishable from those in senescent 2BS cells or WI38 cells (Fig. 2A). In summary, these results were consistent with our SA-β-gal staining studies and morphological phenotype observations.
It has been reported that WWP1 promotes the proliferation and survival of prostate and breast epithelial cancer cells (18, 19). To define the effect of WWP1 on human fibroblast proliferation, the growth rates of cells expressing different transformants were measured using the MTT assay. As shown in Fig. 2C, 2BS and WI38 cells overexpressing WWP1 exhibited higher cell proliferation rates than control cells. However, when WWP1 expression was silenced by shRNA targeting, cells showed marked growth retardation compared with the control cells.
To determine the mechanisms underlying this WWP1-promoted growth rate increase, the effects of WWP1 on cell cycle distribution were examined by flow cytometry. 2BS cells overexpressing WWP1 showed increased S and reduced G1 compartments. It was obvious that WWP1 shRNA-infected 2BS cells entered G1 cell cycle arrest (Fig. 2D). Thus, WWP1 may promote human fibroblast proliferation by stimulating cell cycle progression.
WWP1 Decreases Protein Level of p27Kip1
Our results show that WWP1 overexpression promotes cell growth, whereas knockdown of WWP1 leads to growth inhibition. We therefore examined the molecular mechanism by which WWP1 delays cellular senescence. Considering the importance of p16INK4a, p53/p21Cip1, and p27Kip1 in cellular proliferation, it was essential to determine the influences of WWP1 on these key effectors of cellular senescence. A recent report revealed that WWP1-mediated monoubiquitination of p53 promotes its nuclear export, which results in diminished p53 transcriptional activity and a decrease in p21Cip1 expression (14). Therefore, the effect of WWP1 on p21Cip1 can serve as a positive control in these experiments. We first analyzed the expression patterns of p16INK4a, p21Cip1, and p27Kip1 using Western blot analysis and RT-PCR. As shown in Fig. 3, the protein levels of p27Kip1 were markedly depressed in WWP1-transfected HeLa cells as compared with the vector-transfected cells, whereas the protein levels of p16INK4a and PTEN were invariable. In addition, we silenced WWP1 in HeLa cells. Similarly, there was no detectable change in the protein levels of p16INK4a or PTEN, but a marked increase in the p27Kip1 protein level was found. Moreover, the RT-PCR analysis demonstrated no alteration in p27Kip1 mRNA levels in both WWP1-overexpressing and knockdown cells, which indicated that WWP1 might normally decrease the p27Kip1 protein level but not the mRNA level. Similar results were observed in 2BS cells.
FIGURE 3.

WWP1 decreased p27Kip1 protein levels. A, Western blot analysis of WWP1, p21Cip1, p27Kip1, and p16INK4a expression was carried out in WWP1-overexpressing or knockdown cells compared with control cells. 2BS cells and HeLa cells were used for these experiments. B, RT-PCR analysis of WWP1, p21Cip1, and p27Kip1 was performed in cells as in A. Specific RT-PCR primers for WWP1, p21Cip1, and p27Kip1 were used. C, the half-life of p27Kip1 was evaluated in HeLa cells with altered WWP1 expression. Forty-eight hours post-transfection, cells were treated with cycloheximide (CHX) (15 μg/ml) for different time periods. Blots were evaluated with WWP1, p27Kip1, and tubulin antibodies. sh, shRNA; si, siRNA.
To determine whether WWP1 mediates the decrease in p27Kip1 levels post-transcriptionally, we performed a cycloheximide chase experiment in HeLa cells. As shown in Fig. 3C, overexpression of WWP1 in HeLa cells significantly decreased the half-life of p27Kip1 to less than 1.5 h. As expected, in WWP1 siRNA-transfected cells, the half-life of p27Kip1 was markedly prolonged to more than 6 h. These data suggest that WWP1 increases p27Kip1 levels by prolonging its protein half-life. Taken together, these results suggest that WWP1 decreases p27Kip1 protein levels and that p27Kip1 may play a key regulatory role in WWP1-mediated cellular senescence.
WWP1 Interacts with p27Kip1 Directly
We found that WWP1 decreased p27Kip1 protein levels but did not affect the protein level of PTEN, which is upstream of p27Kip1 in cellular senescence. We next sought to determine whether this down-regulation depends on a direct interaction between WWP1 and p27Kip1. First, we co-immunoprecipitated endogenous p27Kip1 from a HeLa cell lysate and then performed Western blotting with p27Kip1 and WWP1 antibodies. Fig. 4C shows that WWP1 co-immunoprecipitated with p27Kip1. This in vivo association also existed in 2BS and WI38 cells and was confirmed by co-immunoprecipitation and immunoblotting experiments. These data indicate that p27Kip1 interacts with WWP1 in cells. To further confirm the interaction of p27Kip1 with WWP1, we performed a GST pulldown assay using in vitro transcribed and translated WWP1 and purified GST-p27Kip1. This assay showed that WWP1 displays a strong association with p27Kip1 (Fig. 4D). Hence, the direct interaction between WWP1 and p27Kip1 was demonstrated.
FIGURE 4.

WWP1 interacted with p27Kip1in vitro and in vivo. A and B, co-immunoprecipitation (IP) of endogenous WWP1 and p27Kip1 was performed in 2BS and WI38 cells. C, immunoprecipitation of WWP1 and p27Kip1 in WWP1-transfected HeLa cells using the p27Kip1 antibody. D, GST pulldown assay using in vitro transcribed and translated WWP1 and purified GST-p27. Blots were evaluated with WWP1 and GST antibodies. IB, immunoblot.
The full-length WWP1 protein contains several functional domains. At the N terminus, there is a proline-rich domain and a polyserine motif with an adjacent C2 domain. The central portion of WWP1 also contains four WW domains, and the signature ubiquitin ligase domain, also known as the HECT domain, resides at the C terminus (6). To determine the domain of WWP1 that is required for down-regulating p27Kip1, we overexpressed full-length, HECT domain-deleted (CD1), HECT-WW domain-deleted (CD2), and WW domain-deleted (MD1) constructs of WWP1 in HeLa cells and analyzed the expression patterns of p27Kip1, p21Cip1, and p16INK4a (Fig. 5, A and B). The Western blot analysis results indicate that only full-length WWP1 was able to decrease p27Kip1 expression levels. In other words, both the HECT domain and WW domain were necessary for down-regulating p27Kip1. In this case, we also evaluated the co-immunoprecipitation of WWP1, CD1, and CD2 with p27Kip1 in HeLa cells. As shown in Fig. 5C, WWP1 and CD1 interacted with p27Kip1 in vivo, suggesting that only the WW domain is required for the interaction of WWP1 with p27Kip1. Thus, this interaction is necessary for WWP1 to down-regulate p27Kip1.
FIGURE 5.

WWP1 associated with p27Kip1 directly through WW domain of WWP1 and LPEFY motif of p27Kip1. A, schematic representation of the WWP1 protein sequence and the fragments used in further assays. B, HeLa cells were transfected with FLAG-tagged WWP1, CD1, CD2, and MD1 and a vector control. After 48 h, cell lysates were processed for Western blot analysis of FLAG, p16INK4a, p21Cip1, and p27Kip1. C, cells were transfected with WWP1, CD1, and CD2 and treated with MG132 (15 μm) for 5 h before collection. Cell lysates were then used for co-immunoprecipitation (IP) with the p27Kip1 antibody or FLAG antibody. Blots were evaluated with FLAG and p27Kip1 antibodies. D, schematic representation of wild-type p27Kip1 and its deletions. E and F, GST pulldown assay of WWP1 and p27Kip1 or three deletions of p27Kip1, including M1, M2, and M3. Blots were evaluated with WWP1, p27Kip1, and GST antibodies. IB, immunoblot.
As previously noted, the WW domain recognizes substrate proteins. It has been reported that WW domains mediate protein-protein interactions in a number of different cellular functions by recognizing proline-containing peptide sequences (27). We found two potential binding motifs, LPEEY (amino acids 84–88) and PPRP (amino acids 91–94), in the protein sequence of p27Kip1. To identify the motif of p27Kip1 required for its interaction with WWP1, we constructed three deletions of p27Kip1 as described under “Experimental Procedures” (Fig. 5D) and performed a GST pulldown assay. WWP1 was found to associate with the LPEEY motif (amino acids 84–88) of p27Kip1 (Fig. 5, E and F) but not with the proline-rich PPRP motif (amino acids 91–94).
WWP1 Induces Ubiquitination of p27Kip1
WWP1 is known to function as an E3 ubiquitin ligase and decreases p27Kip1 levels via a direct protein interaction, suggesting that WWP1 may induce p27Kip1 polyubiquitination to promote p27Kip1 degradation. To further test this hypothesis, we performed several experiments. First, a dose-dependent assay showed that different levels of overexpression of wild-type WWP1 led to a dose-dependent decrease in the p27Kip1 protein level, which was blocked by an addition of the proteasome inhibitor MG132 (Fig. 6A). Second, the WWP1-mediated down-regulation of the p27Kip1 protein was prevented by two different proteasome inhibitors, MG132 and acetyl-Leu-Leu-norleucinal (Fig. 6B). These results suggest that the WWP1-mediated decrease in p27Kip1 might be proteasome-dependent. Third, we tested the potential role of WWP1 in the polyubiquitination of p27Kip1 in HeLa cells. The cells were transfected with WWP1, CD1, CD2, and MD1; collected 48 h post-transfection; and subjected to co-immunoprecipitation using the p27Kip1 antibody. Blotting showed that the overexpression of WWP1 induced the ubiquitination of p27Kip1, and this effect was dependent on both the HECT domain and WW domain (Fig. 6C). Removal of the HECT domain or WW domain of WWP1 completely abolished the function of polyubiquitin p27Kip1. Fourth, an in vitro ubiquitination assay was performed. Affinity-purified GST-p27Kip1 was incubated with purified GST-WWP1 or GST-CD1 together with biotin-ubiquitin, E1, and E2 (UbcH7). As demonstrated in Fig. 6D, p27Kip1 was dramatically ubiquitinated by WWP1 in the presence wild-type ubiquitin, E1, and UbcH7. Finally, to establish the chain type specificities of WWP1, in vitro ubiquitination reactions were performed with p27Kip1, E1, UbcH7, and WWP1 in the presence of wild-type or variant forms of ubiquitin. Ubiquitin variants contained either a single lysine-to-arginine mutation (K29R, K48R, and K63R) or six lysine-to-arginine mutations so that only a single lysine remained (Lys-29(1), Lys-48(1), and Lys-63(1)). As shown in Fig. 6E, p27Kip1 was efficiently ubiquitinated by WWP1 in the presence of wild-type ubiquitin, K29R, K63R, and Lys-48(1). This result reveals that WWP1 has a strong preference for catalyzing the Lys-48-linked polyubiquitination of p27Kip1, which can be recognized and degraded by 26 S proteasomes. These results indicate that WWP1 induces the polyubiquitination of p27Kip1 and promotes its degradation.
FIGURE 6.

WWP1 polyubiquitinated p27Kip1. A, different amounts of WWP1 were expressed in HeLa cells. Twenty-four hours after transfection, cells were treated with MG132 (15 μm) for 5 h before collection. Proteins were analyzed by Western blotting. B, HeLa cells were transfected with WWP1 and vector. Twenty-four hours after transfection, WWP1-transfected cells were treated with MG132 (15 μm) or acetyl-Leu-Leu-norleucinal (ALLN) (20 μm) 5 h before harvesting. Proteins were analyzed by Western blotting. C, an in vivo ubiquitination assay was performed with WWP1 and p27Kip1. HeLa cells were transiently transfected with WWP1, CD1, CD2, and MD1 constructs. Forty-two hours later, cells were treated with MG132 (15 μm) for 5 h. Cells were lysed and processed for co-immunoprecipitation (IP) using the p27Kip1 antibody. Blots were analyzed with ubiquitin, p27Kip1, and FLAG antibodies. D, an in vitro ubiquitination assay was performed. Affinity-purified p27Kip1 was incubated with purified WWP1, E1 enzyme, UbcH7, and ubiquitin. The reaction mixture was incubated at 37 °C for 90 min and analyzed by Western blotting. E, in vitro ubiquitination of p27Kip1 was performed in the presence of human E1, UbcH7, and WWP1 with wild-type ubiquitin or the indicated ubiquitin (Ub) mutant proteins. Following a 90-min reaction, the products were analyzed by Western blotting. IB, immunoblot.
WWP1 Expression Positively Correlates with p27Kip1 Ubiquitination Level during Cellular Senescence
Considering the influence of WWP1 on cellular senescence and the role of WWP1 in the ubiquitination of p27Kip1, we further evaluated the expression patterns of WWP1 and the ubiquitination levels of p27Kip1 in young and senescent 2BS and WI38 cells. An immunoblot analysis revealed that the protein levels of WWP1 were significantly decreased in senescent cells compared with young cells (Fig. 7A). As expected, young cells possessed low p27Kip1 levels, which were greatly increased by MG132. We then determined whether there was an increase in the ubiquitination of p27Kip1 in young cells. To this end, in vivo ubiquitination assays were performed in young and old cells. Young cells were found to have higher p27Kip1 ubiquitination levels than old cells (Fig. 7B). As a decline in WWP1 expression is accompanied by decreasing levels of p27Kip1 ubiquitination during cellular senescence, it is conceivable that WWP1 may function by antagonizing p27Kip1-induced cellular senescence.
FIGURE 7.

WWP1 expression positively correlated with p27Kip1 ubiquitination level during cellular senescence. A, Western blot analysis of the expression patterns of WWP1 and p27Kip1 in young cells (treated with MG132 or DMSO) and old cells (treated with DMSO). Tubulin served as a loading control. B, young human fibroblasts (22PD 2BS and 25PD WI38) and old human fibroblasts (58PD 2BS and 46PD WI38) were treated with MG132 (15 μm) for 5 h. Cells were lysed and processed for co-immunoprecipitation (IP) using the p27Kip1 antibody. Blots were analyzed with ubiquitin, p27Kip1, and WWP1 antibodies. IB, immunoblot.
WWP1 Inhibits Cellular Senescence Induced by p27Kip1
On the basis of all of our results, we concluded that p27Kip1 likely plays a key role in WWP1-mediated cellular senescence. We therefore investigated whether WWP1 delays the cellular senescence induced by p27Kip1. Because WWP1 regulates p21Cip1, which is a defined regulator of senescence, we performed this experiment with cells that were infected with p21Cip1 shRNA. As shown in Fig. 8, 2BS cells co-expressing pWZL, p27Kip1, and p21Cip1 shRNA exhibited decreased proliferation rates, higher SA-β-gal activity, and a more uniform DAPI staining pattern compared with the WWP1-p27Kip1-p21Cip1 shRNA-infected 2BS cells. At the same time, the senescence markers of WWP1-p27Kip1-p21Cip1 shRNA-infected cells appeared to be comparable with that of pWZL-pBabe-p21Cip1 shRNA-infected 2BS cells (Fig. 8, B and C). These findings strongly established that WWP1 inhibits p27Kip1-induced senescence. Thus, the decrease in p27Kip1 mediated by WWP1 is required for the restriction of cellular senescence.
FIGURE 8.

WWP1 repressed cellular senescence induced by p27Kip1 in the presence of p21Cip1 inhibitor. A, the expression of WWP1, p27Kip1, and p21Cip1 protein levels in the stable transformants was analyzed by Western blotting. B, cells from A were stained for SA-β-gal activity (upper panel) and formation of SAHF (middle and lower panels). C, growth curves for cells from A were determined using the MTT assay. Values represent the mean ± S.D. of triplicate points from a representative experiment (n = 3), which was repeated three times with similar results. sh, shRNA.
DISCUSSION
Previous studies have demonstrated that knockdown of WWP1 markedly restricts the proliferation of MCF7, PC-3, and HCC1500 cancer cells and HEK293T cells (18, 28). Moreover, the overexpression of WWP1 accelerates tumor cell proliferation and survival (17, 18). In addition, WWP1 is a positive regulator of lifespan in C. elegans where it acts through diet restriction and the DAF-2 insulin/IGF-1 signaling pathway (19, 20). However, the function of WWP1 in human cellular senescence has not been described. Cellular senescence represents an irreversible form of cell proliferation arrest that can be triggered by a variety of insults. The p21Cip1, p16INK4a, and p27Kip1 signaling pathways are common key downstream effectors in the induction of senescence mediated by different types of stress (5). However, the role of p27Kip1 in cellular senescence is incompletely characterized, and there have only been a few studies that have shown that p27Kip1 is a key factor in the induction of senescence (29–32). In this study, we provide several lines of evidence to support the concept that WWP1 can delay cellular senescence by targeting p27Kip1 for ubiquitin-mediated proteasomal degradation. Our investigation showed that the patterns of p27Kip1 and WWP1 expression are inversely correlated during cell aging. Furthermore, we evaluated the role of WWP1 in the regulation of cellular senescence and demonstrated that overexpression of WWP1 delayed cellular senescence, whereas silencing of WWP1 led to premature senescence in human fibroblasts (Fig. 2). Collectively, this evidence shows that WWP1 regulates the process of replicative senescence.
In addition, we found that overexpressing WWP1 markedly reduced p27Kip1 protein expression and that silencing WWP1 led to an up-regulation of p27Kip1 protein levels. However, there was no associated change in the mRNA levels of p27Kip1. Considering that WWP1 decreased the endogenous p27Kip1 protein levels, we further evaluated how WWP1 regulates p27Kip1 in a dose-dependent manner. First, we found that WWP1 bound to p27Kip1 through a direct interaction between the WW domain and the LPEFY motif. WWP1 did not associate with the PPRP motif, which is a proline-rich region, but rather with the LPEFY motif as might be expected because it is similar to the conventional (L/P)PXY motif. Moreover, we demonstrated for the first time that p27Kip1 is a substrate of WWP1. WWP1 increased the polyubiquitination of p27Kip1, which depended on both the HECT and WW domains. Accordingly, we found that the WWP1-dependent ubiquitination of p27Kip1 occurred through the formation of Lys-48-linked polyubiquitin chains, which can be recognized by 26 S proteasomes. It is important to note that the different shapes of ubiquitin conjugates have the potential to signal different biochemical fates. For example, lysine 29 (Lys-29)- and Lys-48-linked polyubiquitin chains are associated with proteasomal degradation, whereas Lys-63-linked polyubiquitin chains are involved in trafficking and signaling pathways. Notably, our result not only indicates that the WWP1-mediated p27Kip1 degradation is proteasome-dependent but also explains why WWP1 targets some of its protein substrates (KLF5, KLF2, Smad4, and ErbB4) for direct degradation (10–13, 33). Because WWP1 inhibited endogenous p27Kip1 expression and delayed cellular senescence, we then determined whether p27Kip1 is required for WWP1-regulated senescence. Experiments shown in Fig. 8 demonstrate that WWP1 inhibits p27Kip1-induced senescence. Therefore, WWP1 delays cellular senescence by promoting p27Kip1 degradation.
The protein p27Kip1, a cyclin-dependent kinase inhibitor, is important for arrest in the G0/G1 phase of the cell cycle, and its accumulation triggers the onset of cellular senescence (5). The degradation of p27Kip1 is a key event in the G1/S transition and operates through ubiquitination and subsequent degradation by the 26 S proteasome (34). However, there have been relatively few studies on the irreversible change in the p27Kip1 protein level during cell aging, which is the onset of cellular senescence (5, 30, 35). Here, we show that young cells express a high level of WWP1. However, WWP1 expression declines with an irreversible increase in the level of p27Kip1 and the induction of senescence. It is well known that the PTEN/p27Kip1 pathway is important for controlling the replicative senescence and life span of normal human fibroblasts (5, 35). PTEN is upstream of p27Kip1 in the PTEN/p27Kip1 pathway. However, our results show that PTEN is not required for the regulation of p27Kip1 by WWP1. Moreover, we verified that WWP1 mediated the delay of cellular senescence by promoting p27Kip1 degradation (Fig. 8). We therefore conclude that the WWP1/p27Kip1 pathway bypasses the p27Kip1 pathway in cell aging. However, p27Kip1 degradation might not be the only method by which WWP1 regulates cellular senescence by WWP1 because WWP1 has other cellular proliferation-related substrate proteins (13–16).
In summary, it is well accepted that the process of cellular senescence is complicated and involves a signaling network. Here, we demonstrate that WWP1 is a member of this network. Our studies showed that the high protein level of WWP1 in young fibroblasts decreases the level of p27Kip1, thereby promoting cellular proliferation. Blocking WWP1 expression increased p27Kip1 in 2BS cells, which in turn led to cellular senescence. Therefore, WWP1 plays an important role in the regulation of cellular senescence. Moreover, we established for the first time that WWP1 promotes p27Kip1 degradation through the ubiquitin-proteasome pathway. Because only a few studies on E3 ligase-mediated senescence exist (26, 30), our results may offer an opportunity to investigate the interactions of the ubiquitin system that are associated with cellular senescence in aging-related diseases.
Acknowledgments
We greatly appreciate the gift of the retroviral infection system from Dr. M. Narita (Cancer Research UK, Cambridge Research Institute). We also thank Dr. Ceshi Chen (The Center for Cell Biology and Cancer Research, Albany Medical College) for providing the lenti-hWWP1 vector.
This work was supported by grants from the Special Funds for Major State Basis Research of China (Grant 2007CB507400) and the National Natural Science Foundation of China (Grant 30973146).
- SA
- senescence-associated
- WWP1
- WW domain-containing E3 ubiquitin protein ligase 1
- SAHF
- senescence-associated heterochromatin foci
- PD
- postnatal day
- MG132
- carbobenzoxy-l-leucyl-l-leucyl-l-leucinal
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- HECT
- homologous to the E6-associated protein carboxyl terminus
- PTEN
- phosphatase and tensin homolog.
REFERENCES
- 1. Bayreuther K., Rodemann H. P., Hommel R., Dittmann K., Albiez M., Francz P. I. (1988) Proc. Natl. Acad. Sci. U.S.A. 85, 5112–5116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dimri G. P., Lee X., Basile G., Acosta M., Scott G., Roskelley C., Medrano E. E., Linskens M., Rubelj I., Pereira-Smith O., Et A. (1995) Proc. Natl. Acad. Sci. U.S.A. 92, 9363–9367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Narita M., Nũnez S., Heard E., Narita M., Lin A. W., Hearn S. A., Spector D. L., Hannon G. J., Lowe S. W. (2003) Cell 113, 703–716 [DOI] [PubMed] [Google Scholar]
- 4. Schulz L., Tyler J. (2005) Mol. Cell 17, 168–170 [DOI] [PubMed] [Google Scholar]
- 5. Bringold F., Serrano M. (2000) Exp. Gerontol. 35, 317–329 [DOI] [PubMed] [Google Scholar]
- 6. Verdecia M. A., Joazeiro C. A., Wells N. J., Ferrer J. L., Bowman M. E., Hunter T., Noel J. P. (2003) Mol. Cell 11, 249–259 [DOI] [PubMed] [Google Scholar]
- 7. Sudol M., Hunter T. (2000) Cell 103, 1001–1004 [DOI] [PubMed] [Google Scholar]
- 8. Ingham R. J., Gish G., Pawson T. (2004) Oncogene 23, 1972–1984 [DOI] [PubMed] [Google Scholar]
- 9. Galinier R., Gout E., Lortat-Jacob H., Wood J., Chroboczek J. (2002) Biochemistry 41, 14299–14305 [DOI] [PubMed] [Google Scholar]
- 10. Zhang X., Srinivasan S. V., Lingrel J. B. (2004) Biochem. Biophys. Res. Commun. 316, 139–148 [DOI] [PubMed] [Google Scholar]
- 11. Chen C., Sun X., Guo P., Dong X. Y., Sethi P., Cheng X., Zhou J., Ling J., Simons J. W., Lingrel J. B., Dong J. T. (2005) J. Biol. Chem. 280, 41553–41561 [DOI] [PubMed] [Google Scholar]
- 12. Conkright M. D., Wani M. A., Lingrel J. B. (2001) J. Biol. Chem. 276, 29299–29306 [DOI] [PubMed] [Google Scholar]
- 13. Morén A., Imamura T., Miyazono K., Heldin C. H., Moustakas A. (2005) J. Biol. Chem. 280, 22115–22123 [DOI] [PubMed] [Google Scholar]
- 14. Laine A., Ronai Z. (2007) Oncogene 26, 1477–1483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li Y., Zhou Z., Chen C. (2008) Cell Death Differ. 15, 1941–1951 [DOI] [PubMed] [Google Scholar]
- 16. Peschiaroli A., Scialpi F., Bernassola F., El Sherbini el S., Melino G. (2010) Biochem. Biophys. Res. Commun. 402, 425–430 [DOI] [PubMed] [Google Scholar]
- 17. Chen C., Zhou Z., Ross J. S., Zhou W., Dong J. T. (2007) Int. J. Cancer 121, 80–87 [DOI] [PubMed] [Google Scholar]
- 18. Chen C., Sun X., Guo P., Dong X. Y., Sethi P., Zhou W., Zhou Z., Petros J., Frierson H. F., Jr., Vessella R. L., Atfi A., Dong J. T. (2007) Oncogene 26, 2386–2394 [DOI] [PubMed] [Google Scholar]
- 19. Carrano A. C., Liu Z., Dillin A., Hunter T. (2009) Nature 460, 396–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen C. S., Bellier A., Kao C. Y., Yang Y. L., Chen H. D., Los F. C., Aroian R. V. (2010) PLoS One 5, e9494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang K., Johnson K. D., Petcherski A. G., Vandergon T., Mosser E. A., Copeland N. G., Jenkins N. A., Kimble J., Bresnick E. H. (2000) Gene 252, 137–145 [DOI] [PubMed] [Google Scholar]
- 22. Yew T. L., Chiu F. Y., Tsai C. C., Chen H. L., Lee W. P., Chen Y. J., Chang M. C., Hung S. C. (2011) Aging Cell 10, 349–361 [DOI] [PubMed] [Google Scholar]
- 23. Zhou R., Han L., Li G., Tong T. (2009) Nucleic Acids Res. 37, 5183–5196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gan Q., Huang J., Zhou R., Niu J., Zhu X., Wang J., Zhang Z., Tong T. (2008) J. Cell Sci. 121, 2235–2245 [DOI] [PubMed] [Google Scholar]
- 25. McGlade C. J., Ellis C., Reedijk M., Anderson D., Mbamalu G., Reith A. D., Panayotou G., End P., Bernstein A., Kazlauskas A. (1992) Mol. Cell. Biol. 12, 991–997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zhang H., Cohen S. N. (2004) Genes Dev. 18, 3028–3040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Otte L., Wiedemann U., Schlegel B., Pires J. R., Beyermann M., Schmieder P., Krause G., Volkmer-Engert R., Schneider-Mergener J., Oschkinat H. (2003) Protein Sci. 12, 491–500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Martin-Serrano J., Eastman S. W., Chung W., Bieniasz P. D. (2005) J. Cell Biol. 168, 89–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Young A. P., Schlisio S., Minamishima Y. A., Zhang Q., Li L., Grisanzio C., Signoretti S., Kaelin W. G., Jr. (2008) Nat. Cell Biol. 10, 361–369 [DOI] [PubMed] [Google Scholar]
- 30. Majumder P. K., Grisanzio C., O'Connell F., Barry M., Brito J. M., Xu Q., Guney I., Berger R., Herman P., Bikoff R., Fedele G., Baek W. K., Wang S., Ellwood-Yen K., Wu H., Sawyers C. L., Signoretti S., Hahn W. C., Loda M., Sellers W. R. (2008) Cancer Cell 14, 146–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lin H. K., Chen Z., Wang G., Nardella C., Lee S. W., Chan C. H., Chan C. H., Yang W. L., Wang J., Egia A., Nakayama K. I., Cordon-Cardo C., Teruya-Feldstein J., Pandolfi P. P. (2010) Nature 464, 374–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zeng J., Wang L., Li Q., Li W., Björkholm M., Jia J., Xu D. (2009) J. Pathol. 218, 419–427 [DOI] [PubMed] [Google Scholar]
- 33. Li Y., Zhou Z., Alimandi M., Chen C. (2009) Oncogene 28, 2948–2958 [DOI] [PubMed] [Google Scholar]
- 34. Pagano M., Tam S. W., Theodoras A. M., Beer-Romero P., Del Sal G., Chau V., Yew P. R., Draetta G. F., Rolfe M. (1995) Science 269, 682–685 [DOI] [PubMed] [Google Scholar]
- 35. Alexander K., Hinds P. W. (2001) Mol. Cell. Biol. 21, 3616–3631 [DOI] [PMC free article] [PubMed] [Google Scholar]