

Abstract
PKA anchoring proteins (AKAPs) optimize the efficiency of cAMP signaling by clustering interacting partners. Recently, AKAP79 has been reported to directly bind to adenylyl cyclase type 8 (AC8) and to regulate its responsiveness to store-operated Ca2+ entry (SOCE). Although AKAP79 is well targeted to the plasma membrane via phospholipid associations with three N-terminal polybasic regions, recent studies suggest that AKAP79 also has the potential to be palmitoylated, which may specifically allow it to target the lipid rafts where AC8 resides and is regulated by SOCE. In this study, we have addressed the role of palmitoylation of AKAP79 using a combination of pharmacological, mutagenesis, and cell biological approaches. We reveal that AKAP79 is palmitoylated via two cysteines in its N-terminal region. This palmitoylation plays a key role in targeting the AKAP to lipid rafts in HEK-293 cells. Mutation of the two critical cysteines results in exclusion of AKAP79 from lipid rafts and alterations in its membrane diffusion behavior. This is accompanied by a loss of the ability of AKAP79 to regulate SOCE-dependent AC8 activity in intact cells and decreased PKA-dependent phosphorylation of raft proteins, including AC8. We conclude that palmitoylation plays a key role in the targeting and action of AKAP79. This novel property of AKAP79 adds an unexpected regulatory and targeting option for AKAPs, which may be exploited in the cellular context.
Keywords: Adenylate Cyclase (Adenylyl Cyclase), Lipid Raft, Phosphatidylinositol Anchor, Protein Kinase A (PKA), Protein Palmitoylation, AKAP79
Introduction
cAMP-PKA-mediated signaling affects numerous intracellular targets in response to a wide variety of hormones, neurotransmitters, growth factors, and cytokines (1, 2). A myriad of studies over the past 40 years has identified hundreds of PKA substrates in the plasma membrane, nucleus, and cytoplasm (3–7). This pluripotency underlines a need for cellular devices to impart sensitivity and specificity. Specificity of the PKA signaling module is determined by a sophisticated subcellular targeting network that directs the spatiotemporal activation of the kinase. Key elements of this compartmentalization mechanism are the protein kinase A anchoring proteins, which recruit PKA into multiprotein complexes that may include phosphodiesterases and protein phosphatases, as well as other signaling proteins targeted to discrete subcellular microdomains (1, 8).
Human AKAP79 (and its rodent and bovine orthologues, named AKAP150 and AKAP75, respectively) is a prototypical PKA anchoring protein. In neurons, AKAP79 targets PKA to dendritic compartments (9, 10) and directly interacts with several receptors and ion channels, including the β-adrenergic receptor (11, 12), adenylyl cyclases (AC5 and AC6) (13), L-type calcium channels (10), potassium channels (14, 15), and glutamate receptors (1, 16, 17). Recently, we reported that AKAP79 also interacts with and regulates AC8 in pancreatic and neuronal tissue (18).
An additional organizing principle in signaling efficiency is provided by lipid rafts. These structures are small (10–200 nm), heterogeneous, and highly dynamic sterol- and sphingolipid-enriched membrane domains. Lipid rafts enriched in tightly packed saturated fatty acids are dispersed in the liquid-disordered phase of more unsaturated lipids (19). Signaling proteins with affinity for rafts can become concentrated in these microdomains, thus facilitating formation of protein complexes and activation of specific signaling pathways (20). A large number of studies suggest that lipid modifications such as palmitoylation, myristoylation, or glycosylphosphatidylinositol anchors provide the affinity of proteins for lipid rafts (21, 22). Several AKAP79-interacting proteins are organized in the context of lipid rafts, including β-adrenergic and glutamate receptors (11, 23, 24), as well as the Ca2+-sensitive AC1, AC3, AC5, AC6, and AC8 (25–28). We have shown that the regulation of Ca2+-stimulated AC8 and Ca2+-inhibited AC6 by store-operated Ca2+ entry (SOCE) depends on their localization in rafts, because the dissipation of lipids rafts ablates their responsiveness to SOCE (25, 28).
AKAP79 is localized to the plasma membrane (PM)3 through an N-terminal targeting domain consisting of three polybasic subdomains; the positive charges of the basic amino acids interact with the negative charge of phospholipid headgroups, particularly phosphatidylserine and phosphoinositides, on the membrane (29). The N terminus also contains three potentially palmitoylatable cysteines. Protein palmitoylation, the thioester linkage of saturated palmitic acid to cysteine, is a reversible process that is dynamically regulated by specific cellular stimuli (30, 31). Palmitoylation serves both to tether proteins to membranes and to direct their localization to membrane microdomains (32, 33).
In this study, we explored the potential palmitoylation of AKAP79 and its consequences for PM targeting and functional effects. We found that AKAP79 is indeed palmitoylated on two cysteine residues and that this palmitoylation is responsible for the recruitment of AKAP79 into lipid rafts from non-raft domains of the PM (determined by biochemical fractionation and fluorescence recovery after photobleaching (FRAP) analysis) along with the recruitment of PKA to this PM microdomain. We also showed that AKAP79 localization in lipid rafts is required for the phosphorylation of AC8 and regulation of Ca2+-dependent AC8 activity, and it allows the phosphorylation of PKA substrates associated with rafts in response to β-adrenergic receptor stimulation.
EXPERIMENTAL PROCEDURES
Cell Culture and Transfection
HEK-293 and COS-7 cells (European Collection of Cell Cultures, Porton Down, UK) were grown in minimum essential medium supplemented with 10% (v/v) fetal bovine serum, 100 units/ml penicillin, 100 μg/ml streptomycin, and 2 mm l-glutamine. Cells were maintained at 37 °C in a humidified atmosphere of 5% CO2, 95% air.
To produce stably expressing HEK-AC8 or HEK-AKAP79 cells, wild type HEK-293 cells at ∼70% confluence were transfected with 2 μg of cDNA of rat AC8, human AKAP79-YFP wild type, or the YFP-tagged AKAP79 mutants C36S,C129S and C36S,C62S,C129S using the Lipofectamine 2000 transfection method. 48 h later, the culture medium was replaced with fresh medium containing 800 μg/ml G-418 disulfate (Formedium Ltd., Hunstanton, UK) to select transfected cells. After selection, cells were maintained in medium containing 400 μg/ml G-418. Stable HEK-293 cells were established from ∼2 weeks following transfection. For transient transfection, cells at ∼90% confluence were transfected with 1.5 μg of cDNA using the Lipofectamine 2000 transfection method. The cells were used 48 h later.
Site-directed Mutagenesis
The pEC/YFPN1 (Clontech) vector encoding the C-terminal yellow fluorescent protein (YFP) AKAP79 was kindly provided by Professor Mark Dell'Acqua. The C36S, C62S, and C129S mutations of AKAP79-YFP were introduced to this vector by site-directed mutagenesis according to the QuikChange protocol (Stratagene) using the fusion high fidelity polymerase kit (FINNZYMES) according to the manufacturer's instructions.
cAMP Measurements
For single cell cAMP measurements, stable AC8 (HEK-AC8) cells were plated onto 25-mm poly-l-lysine-coated coverslips at ∼60% confluence 24 h prior to transient transfection with 0.5 μg of total cDNA per construct (Epac2-camps FRET sensor and either wild type or mutant AKAP79), using the Lipofectamine 2000 method.
Fluorescent imaging of HEK-AC8 cells expressing the FRET-based cAMP sensor, Epac2-camps, was performed as described previously (18). In brief, cells were excited at 435 nm using a monochromator (Cairn Research) and 51017 filter set (Chroma Technology Corp.) attached to a Nikon eclipse TE2000-S microscope (×40 objective). Emission images at 470 and 535 nm were collected every 3 s (250-ms integration time) using an Andor Ixon+ EMCCD camera (Andor, Belfast, UK) and Optosplit (505DC) (Cairn Research, Kent, UK). Data were analyzed using Metamorph imaging software (Molecular Devices, Berkshire, UK) and plotted as changes in background-subtracted 470 versus 535 nm (CFP/YFP) emission ratio relative to maximum FRET ratio change seen with saturating cAMP concentrations.
Lipid Raft Isolation
Lipid rafts were prepared from cell extracts as described previously with some modifications (34). Cells were homogenized in lysis buffer (150 mm NaCl, 25 mm Tris-HCl, pH 7.5, 50 mm NaF, 10 mm NaP2O7, 1 mm Na3VO4, complete protease inhibitor mixture (Roche Applied Science) and 0.5% Triton X-100). Following homogenization by sonication (5 s, 1500 Hz on ice) the sample was centrifuged for 10 min at 1500 × g, and protein concentration was determined by BCA assay. The supernatant (3.5 mg of protein in a 0.5-ml sample) was incubated for 30 min at 4 °C and centrifuged for 20 min at 16,000 × g at 4 °C, to separate a Triton-soluble extract and the insoluble pellet. The pellet was resuspended in 0.4 ml of lysis buffer and mixed with 2 m sucrose (0.8 ml), overlaid with 1 m (1.6 ml) and 0.2 m (0.8 ml) sucrose and centrifuged for 15 h at 200,000 × g at 4 °C. After centrifugation, five 0.7-ml fractions were collected from the top to the bottom of the gradient. Lipid rafts were enriched in fraction 2. The pellet was resuspended in 100 μl of lysis buffer. The proteins in the gradient fractions were pelleted by centrifugation at 200,000 × g for 45 min following dilution with 25 mm Tris-HCl, 150 mm NaCl. Where indicated, fractions 2 and 3 (lipid rafts) and fractions 4 and 5 (high density) were pooled and pelleted together. The pellets were resuspended in 100 μl of lysis buffer, and protein concentration was determined.
Lipid Raft Isolation by Detergent-free Method
A cell pellet from two 10-cm dishes was resuspended in 0.45 ml of 0.5 m sodium carbonate, pH 11.5, with protease inhibitors, and then sonicated with three 30-s bursts (1500 Hz on ice). The homogenate was adjusted to 40% sucrose by adding 0.7 ml of 60% sucrose in MBS (25 mm MES, pH 6.4, 150 mm NaCl, and 250 mm sodium carbonate), placed under a 5–30% discontinuous sucrose gradient, and centrifuged for 15 h at 200,000 × g at 4 °C. Five fractions (0.8 ml each) were harvested from the top of the tube, mixed with 9 volumes of MBS, and centrifuged for 15 h at 200,000 × g at 4 °C. Supernatants were discarded, and membrane pellets were resuspended in an adequate volume (100–150 μl) of 1% SDS (35).
Immunoprecipitation
Cells were homogenized by sonication in ice-cold lysis buffer containing 1% Nonidet P-40, 0.5% sodium deoxycholate. Samples were centrifuged (2000 × g, 5 min); the supernatants were collected, and protein concentrations were determined. Equal amounts of protein per sample were incubated overnight with 1 μl of HA antibody or 50 μl of HA affinity beads (HA antibody covalently coupled to agarose beads). Samples incubated with HA antibody (not covalently coupled to agarose) were incubated the next morning with protein G-agarose beads (Roche Applied Science) for 2 h, and the immune complexes were isolated by centrifugation at 1500 × g for 1 min. The beads (protein G-agarose and HA affinity beads) were washed five times with lysis buffer plus 0.1% SDS and once more with buffer (50 mm Tris-HCl, pH 7.5) without detergent. The proteins from HA affinity beads were eluted with 50 μl of 1% SDS. The proteins from the protein G-agarose beads were eluted with 40 μl of Laemmli buffer.
Western Blot
Samples were mixed with Laemmli buffer and heated at 90 °C for 5 min (samples from immunoprecipitations were not heated at 90 °C but were warmed at 37 °C for 30 min) and subjected to SDS-PAGE. Proteins were electrophoretically transferred to nitrocellulose membranes. Membranes were blocked for 2 h at room temperature in TBS-T buffer (10 mm Tris, 0.9% NaCl, 0.1% Tween 20, pH 7.5) containing 5% BSA and then incubated overnight at 4 °C with primary antibodies from the following sources: AKAP79 (BD Biosciences); PKA RII and catalytic subunits (Santa Cruz Biotechnology); flotillin-1 (BD Biosciences); phospho-PKA substrates RRXS and phospho-Erk (Cell Signaling); and actin, tubulin, and HA (Sigma). The membranes were washed (four times for 5 min) in TBS-T and incubated for 1 h with HRP-conjugated secondary antibody. Proteins were detected by ECL and exposed to x-ray hyperfilms, which were scanned and quantified densitometrically with the ImageJ software (National Institutes of Health, Bethesda).
Palmitoylation Detection
80 μm Azide-palmitate (Invitrogen) in DMSO was added to the culture media of HEK-293 cells transiently expressing AKAP79-YFP wild type or the indicated mutants to be metabolically incorporated into palmitoylatable proteins. 24 h later the cells were lysed by sonication in RIPA lysis buffer (150 mm NaCl, 25 mm Tris-HCl, pH 7.5, containing complete protease inhibitor mixture (Roche Applied Science) and 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% SDS) and centrifuged at 1000 × g to remove cell debris. AKAP79 antibody (1 μl) was added to the cell lysate and incubated overnight at 4 °C. Samples were then incubated with 30 μl of protein-G agarose beads (Roche Applied Science) for 2 h. The beads were washed 5 times with RIPA lysis buffer, and resuspended in Tris-HCl, pH 8, with 1% SDS. Azide-palmitate labeled proteins from the precipitate were biotinylated using the chemoselective ligation or “click“ (Invitrogen) reaction between and azide and a biotin-alkyne according to the manufacturer's instructions. The biotinylated proteins were detected by blotting with streptavidin-HRP and with anti-AKAP79 antibody.
Confocal Imaging and Spot Bleach FRAP
Confocal images were captured with a Leica SP5 TCS laser scanning confocal microscope attached to a DM16000 inverted microscope, equipped with a ×63 plan apochromatic, 1.4 NA, oil immersion objective (Leica Microsystems, Mannheim, Germany). Images were captured with LAS AF Leica software (version 1.8.2). Cells expressing AKAP79-YFP were plated onto glass coverslip inserts coated with poly-l-lysine. FRAP experiments were performed in an environmental chamber at 37 °C (Solent Scientific Ltd., Segensworth, UK). For maximum light acquisition and to defocus the bleaching light beam to effectively bleach YFP either side of the focal plane, the pinhole was fully opened to an optical section of ∼5 μm. FRAP experiments were performed according to the FRAP wizard in the LAS AF software. Images were scanned bidirectionally at maximum speed (1400 Hz) with a single line average to reduce noise, using the 488 nm line of an argon ion laser. YFP emission signal was collected between 500 and 575 nm. Images (256 × 256 pixels) were acquired at a rate of one frame per 0.675 s. 10 pre-bleach frames were acquired with a laser power of 5–8% before bleaching the fluorescent membrane proteins (100 ms, 80% argon laser at 100% transmission) using a spot bleach protocol with the laser focused on a region of the PM. Fluorescence recovery was followed for 60 s, with a laser power of ∼5–8% to ensure full recovery.
Simple and Anomalous Diffusion Analysis
For the simple diffusion analysis, mobile fraction and diffusion coefficients were calculated as described previously (36, 37). In brief, Leica image stacks from FRAP spot bleach experiments were imported into ImageJ. The 10 pre-bleach frames were averaged and used as a base line to normalize all post-bleach frames. Recovery curves of fluorescence intensity within the bleached region of the PM were determined using a region of interest with a diameter of 1.54 μm over the bleached spot for all frames. The mobile fraction was determined by the Ymax of the recovery curves.
Diffusion coefficients were determined using the segmented line tool within ImageJ to trace the bleached region across the PM and fluorescence intensity recorded in the normalized image stack for 11–30 frames (one frame/0.675 s) using the “record profile” macro. Total cellular fluorescence was adjusted for experimental photobleaching. The normalized and adjusted profiles were analyzed in Prism (GraphPad Software) and fit independently with the Gaussian function as shown in Equation 1,
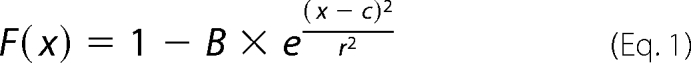 |
where F is the normalized fluorescence intensity; c represents the center of the bleached profile (of distance x, in micrometers); B represents the depth of the Gaussian profile; and r represents the Gaussian radius at e−1. Fits to these three parameters from the first time point were then used to define B0, r0, and c for the Equation 2,
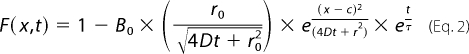 |
where B0 is the initial fraction of bleached YFP at the center of the spot; r0 is the radius of the initial bleach spot; D is the lateral diffusion coefficient within the PM (in μm2/s), and τ is the membrane dissociation time constant (in seconds) (36, 37).
Thus, this Gaussian function finds a single value of D and τ for the change in shape of all the profiles with time caused by lateral diffusion and membrane dissociation, respectively. A semi-independent check was then performed for the fitted values from Equation 2. The values of r2 calculated from those values fitted from the Gaussian function were plotted against time, and the diffusion coefficient was calculated from Equation 3,
For the anomalous diffusion analysis the values of r2 calculated from the values fitted from the Gaussian function were plotted against time, and the transport (or diffusion) coefficient (Γ or D, respectively) and the α values were calculated from Equation 4,
Or, alternatively, from Equation 5,
Here, α is defined as the anomalous exponent where 0 < α < 1 represents subdiffusion and α > 1 represents super-diffusion (38).
Inducible Depletion of PM Phosphoinositides
Inositol lipid phosphatases that could be recruited to the PM were based on a previously described construct consisting of the catalytic domain of the PI(4,5)P2 5-phosphatase (INPP5E) fused to (mammalian expression) red fluorescent protein and FK506-binding protein 12 (FKBP12) (39). To obtain a similar construct with both Sac and INPP5E catalytic activity, this original INPP5E construct was modified such that the yeast Sac1p phosphatase domain (NM_00117977, residues 2–517) replaced INPP5E. The original INPP5E domain was then re-attached to the 3′ end of this fusion via a flexible linker (GGTARGAAAGAGGAGR). To generate a construct with only Sac1 activity, a catalytic aspartate in the conserved DRVL motif of INPP5E (equivalent to residue 556 in the full-length protein) was mutated to alanine in the fusion protein. To make a completely inactive control protein, a further inactivating mutation was introduced into the catalytic CX5R(T/S) motif of the Sac domain (equivalent to residue 392 in the full-length protein). PM recruitment of these constructs was achieved using a fusion of the N-terminal 11 residues from Lyn kinase (which are myristoylated and palmitoylated) fused to a fragment of target-of-rapamycin that binds FKBP12 (FRB) and CFP, as described previously (39). COS-7 cells were transfected with 0.3 μg of each of these constructs (as indicated) using Lipofectamine 2000. Confocal imaging was performed as described above.
Statistical Analysis
Statistical differences between two groups were evaluated using the unpaired Student's t test. Experiments with two or more groups were analyzed by one-way analysis of variance with Newman-Keuls multiple comparison test. All results are expressed as the mean ± S.E.
RESULTS
Inhibition of Palmitoylation Decreases Association of AKAP79 with Lipid Rafts
AKAP79 interacts with the cell membrane by three polybasic domains in the N terminus that include three potentially palmitoylatable cysteines. As a first approach to determine whether association of AKAP79 and PKA with the plasma membrane, and specifically with lipid rafts, is dependent on palmitoylation, we incubated HEK-293 cells stably expressing AKAP79-YFP with bromopalmitate, a palmitate analogue that blocks palmitoylation of proteins (40).
Lipid raft fractions were isolated by sucrose density gradient from insoluble extracts after 0.5% Triton X-100 lysis. Five fractions, plus the pellet of the gradient and the soluble extract, were analyzed by Western blot (Fig. 1A). Strikingly, AKAP79-YFP, PKA RII, and PKA catalytic subunits were significantly concentrated in the lipid rafts (fraction 2). AKAP79 and PKA subunits were also present in the pellet and the soluble extract. Upon bromopalmitate treatment, the levels of AKAP79 and the catalytic and RII regulatory PKA subunits were significantly decreased in the lipid raft fraction (Fig. 1, A and B). No change was noted in the levels of these proteins in other fractions or in the soluble extracts. Stringent extraction conditions were used for the preparation of lipid rafts (0.7 protein to 1 detergent ratio) to ensure as unambiguously as was possible the residence of proteins in the raft preparations. As a consequence, the total amount of protein in the lipid raft fraction is ∼20 times smaller than the amount of protein in the soluble fraction. Thus, although losses from the raft fraction can be sensitively determined, an increase in protein in the non-raft fractions is unlikely to be detected.
FIGURE 1.

Bromopalmitate treatment decreases the association of AKAP79 with lipid rafts. HEK-293 cells stably expressing AKAP79-YFP were treated overnight with bromopalmitate (100 μm in DMSO or control, DMSO only). A, after bromopalmitate treatment, the Triton-soluble extract (Sol) was separated by centrifugation, and the insoluble extract was loaded on sucrose density gradients to isolate lipid rafts. Equal volumes of the fractions and 10 μg of protein from the soluble and total homogenate were immunoblotted for the proteins indicated. PKA cat, PKA catalytic subunit. B, densitometric analysis of blots (mean ± S.E.) expressed as the ratio of immunoreactivity after bromopalmitate treatment over control, for at least four independent experiments. **, p < 0.01; *, p < 0.05. C, confocal imaging of cells expressing AKAP79-YFP treated with 100 μm bromopalmitate and control (DMSO). Scale bars, 20 μm. D, cells treated with bromopalmitate and control were lysed in 0.5 m NaHCO3 buffer and loaded in a sucrose density gradient. Equal volumes of the gradient fractions and 10 μg of protein from total homogenate were immunoblotted for the proteins indicated.
Interesting, at the gross cellular level, the bromopalmitate treatment did not affect the apparent confinement of AKAP79-YFP to the plasma membrane as assessed by confocal microscopy (Fig. 1C). To confirm the effect of bromopalmitate treatment on the association of AKAP79 with lipid rafts, we isolated lipid rafts using a different nondetergent method involving resuspension in 0.5 m bicarbonate buffer, pH 11.5, and subsequent sonication (35). This procedure breaks the cellular membranes in small fragments, stripping them of cytosolic proteins, and forming sheets of membranes. The complete homogenate (without removing the soluble extracts) was fractionated by centrifugation on a discontinuous sucrose gradient to separate low density lipid rafts from high density membranes. Again, the bromopalmitate treatment decreased the association of AKAP79-YFP and PKA subunits with lipid rafts (Fig. 1D) confirming the results obtained with the Triton X-100 solubilization method.
AKAP79 Is Palmitoylated on Cysteine 36 and 129 and Both Cysteines Are Involved in the Association of AKAP79 with Triton-insoluble Membranes
We performed stepwise mutagenesis of cysteines 36, 62, and 129 in the N-terminal polybasic domains of AKAP79-YFP. As the first screening to determine the role of each cysteine in the association of AKAP79 with lipid rafts, we analyzed the effects of single, double, and triple cysteine to serine mutations on the association of AKAP79-YFP with Triton-insoluble extracts. The insoluble extracts were obtained from cells transiently expressing AKAP79-YFP wild type or the indicated mutant, lysed with 0.5% Triton X-100, and centrifuged (15,000 × g, 20 min). The soluble and insoluble extracts were analyzed by Western blot, which revealed that the association of AKAP79-YFP with insoluble membranes decreased with the single mutations C36S or C129S but not with the C62S mutation (Fig. 2, A and B). The double (C36S,C129S) and triple (C36S,C62S,C129S) palmitoylation mutants of AKAP79 displayed further loss of association with the insoluble fraction, compared with C36S or C129S mutations either alone or with C62S (p < 0.01). Association of AKAP79 with the soluble fraction was not significantly affected by any of the mutations.
FIGURE 2.

Palmitoylation on cysteine 36 and 129 of AKAP79 promotes association with Triton-insoluble membranes. A, HEK-293 cells transiently expressing AKAP79-YFP wild type (Wt), single (C36S, C62S, and C129S), double (C36S,C62S (C36,62S), C36S,C129S (C36,129S), and C62S,C129S (C62,129S)), or triple cysteine mutant (C36S,C62S,C129S (C36,62,129S)) were homogenized in lysis buffer with 0.5% Triton X-100. The soluble and insoluble fractions were isolated, and 3 or 30 μl of the extracts, respectively, were analyzed by Western blotting. B, densitometry analysis of blots expressed as the ratio of immunoreactivity of AKAP79-YFP over flotillin (loading control). Data are presented as mean ± S.E. for four independent experiments; **, p < 0.01 (compared with wild type), #, p < 0.01 between indicated groups. n.s., not significant. C, cells expressing AKAP79-YFP wild type or the cysteine mutants indicated were treated with azide-palmitate for 24 h and then lysed in RIPA buffer, and AKAP79-YFP was immunoprecipitated. The azide-palmitate incorporated in AKAP79 was biotinylated by reaction with alkyne-biotin and detected by streptavidin-HRP blotting. The right-hand blot confirms expression of AKAP79 constructs in all samples. Data are representative of three similar experiments.
To determine whether the association of AKAP79 with the insoluble extract correlates with palmitoylation, we analyzed the degree of palmitoylation of AKAP79-YFP mutants compared with wild type. Immunoprecipitation of the AKAP79 constructs was performed from cells metabolically labeled with azide-palmitate. The azide-palmitate-labeled proteins were biotinylated and detected by blotting with streptavidin-HRP and with anti-AKAP79 antibody. In support of the profile of proteins that associated with Triton-insoluble fractions above, palmitoylation was decreased in mutants C36S and C129S but was unaltered in mutant C62S compared with wild type AKAP79. No detectable palmitoylation was seen in the double C36S,C129S mutant (Fig. 2C). These results show that AKAP79 is exclusively palmitoylated on cysteines 36 and 129 and that both cysteines are required for the association of AKAP79 with Triton-insoluble membranes.
Palmitoylation on Cysteine 36 and 129 Is Required for Association of AKAP79 with Lipid Rafts Identified by Density Gradient Fractionation
Lipid rafts are insoluble membranes; however, other structures are isolated along with lipid rafts in the insoluble pellet after Triton X-100 solubilization, such as the actin cytoskeleton and tubulin. To study more specifically the association of AKAP79 with lipid rafts, we isolated rafts by sucrose density gradient fractionation after Triton X-100 solubilization from cells stably expressing AKAP79-YFP wild type and the double and triple palmitoylation mutants. Fractions 2 and 3 (representing lipid rafts, as confirmed by the co-sedimentation with flotillin) were pooled, as were the high density fractions 4 and 5. Both the double mutation C36S,C129S and triple mutation C36S,C62S,C129S abolished the association of AKAP79-YFP with lipid rafts (Fig. 3, A and B) even though these mutants remained in association with the plasma membrane (Fig. 3C). These mutations also significantly decreased the amount of PKA catalytic subunit in the lipid raft fractions (Fig. 3, A and B). Levels of AKAP79 and PKA subunit were unaffected in other fractions by the cysteine mutations. As noted earlier, the small fraction of cellular proteins associated with rafts in this protocol allow losses from the raft fraction to be sensitively determined but not corresponding increases in the non-raft fractions. These results suggest that the palmitoylation of cysteine 36 and 129 is required for the association of AKAP79 with lipid rafts and the associated recruitment of PKA to this microdomain.
FIGURE 3.

Palmitoylation of AKAP79 on cysteine 36 and 129 allows its association with lipid rafts. A, HEK-293 cells stably expressing AKAP79-YFP wild type and the triple mutant (C36S,C62S,C129S ((C36,62,129S))) were homogenized with 0.5% Triton X-100. Soluble (Sol) and insoluble extracts were isolated, and the insoluble extracts were loaded on a density gradient. The light fractions 2 and 3 (lipid rafts) and the high density fractions 4 and 5 (HD) were pooled and centrifuged to precipitate proteins. The gradient fractions, pellets (resuspended in lysis buffer), and soluble and total homogenates were analyzed by Western blot. B, densitometry analysis of blots corrected against loading control and expressed as the ratio of immunoreactivity of mutant over wild type (Wt). Data are presented as means ± S.E. for at least four independent experiments, **, p < 0.01. C, confocal imaging of cells expressing AKAP79-YFP wild type, triple (C36S,C62S,C129S (C36,62,129S)), or double (C36S,C129S (C36,129S)) mutants. Scale bars, 20 μm.
FRAP Analysis Reveals That Palmitoylation of Cysteine 36 and 129 Determines the Diffusion of AKAP79 in the PM and Its Association with Lipid Rafts
Association with lipid rafts changes the mobility of proteins in the PM (41, 42). Therefore, if AKAP79 associates with lipid rafts due to palmitoylation, the diffusion of the nonpalmitoylatable mutants might be expected to differ from the wild type. Although AKAP79 interacts with the PM via three polybasic domains (29), palmitoylation may stabilize this interaction and decrease the probability of dissociation from the membrane. To address this issue, we measured the mobility of the wild type and the nonpalmitoylatable mutant (C36S,C129S) of AKAP79-YFP using FRAP (Fig. 4). To quantify lateral diffusion and membrane dissociation at the same time, we used a bleaching paradigm developed by Oancea et al. (37). Bleaching was achieved via a brief pulse of light centered on the PM (Fig. 4B). The resultant bleached region of the PM could be fitted by Gaussian distribution. Lateral diffusion of bleached molecules along the membrane causes a widening of this profile while conserving the area under the curve; the rate of change of the square of the Gaussian radius with time is proportional to the apparent lateral diffusion coefficient, D. Conversely, dissociation of bleached molecules from the membrane and their replacement with unbleached fluorescently tagged protein causes a decrease in the area of this Gaussian profile, without a change in the radius. A simple relationship has been shown to account for simultaneous lateral diffusion (D) and dissociation (τ), which can assign values to each parameter (see under “Experimental Procedures”). The membrane dissociation time constant, τ, is equivalent to the reciprocal of the dissociation rate constant (36, 37).
FIGURE 4.

Palmitoylation on cysteine 36 and 129 determines the diffusion of AKAP79 in the membrane. Diffusion analysis by FRAP of wild type and C36S,C129S ((C36,129S)) AKAP79-YFP ± MBCD treatment. A, recovery curves of fluorescence intensity presented as means ± S.E. B, confocal images of some cells after bleaching, with arrows showing spot-bleached regions. PKA cat, PKA catalytic subunit. C, simple diffusion analysis used to calculate diffusion coefficients and mobile fractions for wild type and C36S,C129S AKAP79 mutant under control conditions and following 30 min of treatment with 100 μm MBCD. D, anomalous subdiffusion analysis of FRAP data to calculate transport coefficients and α values. Data represent means ± S.E. from at least 14 cells per condition, ***, p < 0.001; **, p < 0.01; *, p < 0.05; n.s., not significant.
Considering all the cells imaged (n = 33 cells expressing mutant AKAP79 and 24 cells expressing wild type AKAP79), an enormous range of τ values was estimated, ranging from 7 s up to 3.141 × 1017 s. The lower value suggests that in some cases diffusion from the cytoplasm contributed significantly to fluorescence recovery. However, the range of values collectively produces a mean value for τ that is essentially infinite for both the wild type and the mutant forms of AKAP79 (4.9 × 1015s and 9.5 × 1015 s, respectively). This suggests that both the wild type and double mutant AKAP79-YFP are stably associated with the membrane. However, the population of cells with a τ <100 s is bigger in cells expressing the C36S,C129S mutant AKAP79-YFP than the wild type (34 versus 50%), which suggests that under these conditions the palmitoylation of AKAP79 could contribute to the stability of the protein at the membrane.
Fig. 4A shows the fluorescence intensity recovery over time in spot-bleached areas of the PM. Using a model for simple (linear) diffusion (37), we detected no significant differences between the diffusion coefficients and mobile fractions of wild type AKAP79 compared with its nonpalmitoylatable form, C36S,C129S, when expressed in HEK-293 cells (Fig. 4C). However, methyl-β-cyclodextrin (MBCD) pretreatment (100 μm, 30 min) (which removes cholesterol from the membranes and disrupts lipid rafts (43, 44)) decreased the diffusion coefficient of the wild type AKAP79-YFP without altering the behavior of the C36S,C129S mutant (Fig. 4C, left panel, p < 0.01). The MBCD treatment did not significantly affect the mobile fraction of either form of AKAP79 (Fig. 4C, right panel).
Several experimental approaches have shown that proteins in membranes can display non-Brownian motion that is characterized by mean squared displacements that do not grow linearly with time, contrary to what is seen for normal diffusion (45). Anomalous diffusion, or subdiffusion, in cell membranes may result from obstacles or traps that transiently restrict the Brownian motion of membrane proteins, as has been reported for proteins that partition into rafts (41). Feder et al. (38) applied an anomalous diffusion model to the interpretation of FRAP experiments and showed that in some, and probably most, cases FRAP data can be fitted equally well by this model as by the conventional model. Anomalous diffusion is characterized by the transport coefficient Γ, which obeys a power law wherein mean squared displacement = Γtα (where t is the elapsed time). In simple Brownian diffusion, the exponent α = 1 (and therefore Γ = 4D), but if α becomes <1, the particle is proposed to undergo subdiffusion, and the apparent diffusion coefficient decreases with time.
We applied this anomalous diffusion model to the FRAP data presented in Fig. 4A. Interestingly, the transport diffusion coefficient of the two AKAP79-YFP constructs differed, but this difference was minimized by MBCD treatment (Fig. 4D). The α value for the wild type AKAP79-YFP was <1, suggesting that it undergoes subdiffusion. In contrast, the α value of the C36S,C129S mutant was ≈1, suggesting simple diffusion of this construct, without trapping or obstacles in that case. Following MBCD treatment, α values for both AKAP79 constructs were not significantly different from 1, indicating that lipid raft disruption eliminated any trapping in the diffusion of the AKAP79-YFP wild type. We would therefore conclude that AKAP79-YFP does diffuse with anomalous properties and that this behavior is due to an interaction with lipid rafts (and/or with proteins within rafts). In contrast, the non-raft-localized C36S,C129S AKAP79 mutant exhibits a simple pattern of diffusion.
AKAP79 Palmitoylation Allows Interaction with the PM in the Absence of Inositol Lipid Binding to the Polybasic Domains
It has previously been reported that stretches of basic residues within AKAP79 are able to interact with inositol lipids such as PI(4,5)P2 (29). We sought to test whether such interactions could explain persistent PM targeting by the palmitoylation-deficient C36S,C129S AKAP79 mutant. To this end, we employed inducible inositol lipid phosphatases in COS-7 cells. The lipid phosphatases themselves are soluble proteins that do not normally reside at the plasma membrane and hence are poorly active against PM lipids. However, fusion of these phosphatases to FKBP12 enables their recruitment to the membrane and activation upon rapamycin-induced dimerization with FRB, which is targeted to the plasma membrane via short myristoylated/palmitoylated sequences from Lyn kinase (see under “Experimental Procedures”). Because AKAP79 had been shown to interact with several inositol lipids including PI4P and PI(4,5)P2 (29), we chose to employ two inositol lipid phosphatase domains as follows: the inositol polyphosphate 5-phosphatase, 72-kDa (INPP5E) domain, which removes the 5-phosphate from PI(4,5)P2, to generate PI4P (46); and Sac1p, which dephosphorylates PI4P to phosphatidylinositol (47). Fig. 5 shows the results of an experiment where these phosphatase domains were activated for 2 min by the addition of rapamycin to the cell bathing medium, during which time robust recruitment and activation of the phosphatases had occurred (Fig. 5A). Depletion of PI(4,5)P2 or PI4P by INPP5E or Sac domains, respectively, caused a slight reduction in the membrane binding of the AKAP79 C36S,C129S mutant (Fig. 5C). However, depletion of PI4P along with PI(4,5)P2 using both phosphatases caused the complete translocation of C36S,C129S AKAP79 to the cytosol (Fig. 5C, p < 0.001). Thus, the palmitoylation-deficient AKAP79 is targeted to the plasma membrane via a nonspecific electrostatic interaction with inositol lipids. In contrast, wild type AKAP79 remained robustly bound to the plasma membrane with only a small decrease in binding upon depletion of both PI(4,5)P2 and PI4P (Fig. 5B). These results indicate that palmitoylation is sufficient to localize AKAP79 to the plasma membrane in the absence of any inositol lipid binding.
FIGURE 5.

Effect of inositol lipid depletion on plasma membrane localization of AKAP79. COS-7 cells were transfected with Lyn11-FRB-CFP and mRFP-FKBP-Sac/INPP5E domains and the indicated AKAP79-YFP construct. Images are shown before and 2 min after the addition of 1 μm rapamycin. A, recruitment of the mRFP-FKBP-Sac/INPP5E fusion proteins upon rapamycin addition. B and C, example images of wild type or C36S,C129S mutant AKAP79-YFP constructs, respectively, co-transfected with the indicated lipid phosphatase activity. Bar charts show the mean ratio of PM/cytosolic intensities before and after rapamycin (Rap) addition (n values range from 9 to 14; statistics are the result of a two-way analysis of variance with *, p < 0.05; **, p < 0.01 and ***, p < 0.001).
Recruitment of PKA to Lipid Rafts by AKAP79 Allows the Phosphorylation of Substrates after β-Adrenergic Receptor Stimulation
Because AKAP79 targets PKA to the β-adrenergic receptor (11), we considered the possibility that raft targeting of AKAP79 might affect phosphorylation of PKA substrates in lipid rafts after β-adrenergic receptor stimulation. Isoproterenol (100 μm, 4 min) was used to stimulate cAMP production via endogenous β-adrenergic receptors in HEK-293 cells stably expressing AKAP79-YFP. This stimulation induced significant (45%) increases in the phosphorylation of PKA substrates detected by an antibody directed against proteins containing a phospho-Ser/Thr residue with arginine at the −3 and −2 positions (RRX(S/T); Fig. 6).
FIGURE 6.

Recruitment of PKA to lipid rafts by AKAP79 allows the phosphorylation of substrates following β-adrenergic receptor stimulation. A, HEK-293 cells stably expressing AKAP79-YFP wild type and the double mutant (C36S,C129S (C36,129S)) were stimulated with isoproterenol (10 μm for 4 min at 37 °C), washed with cold PBS, and homogenized with Tris-HCl buffer with 0.5% Triton X-100. Soluble and insoluble extracts were isolated, and the insoluble extracts were loaded on density gradients. The resultant light fractions (2 and 3; lipid rafts) were pooled and centrifuged to precipitate proteins. The raft fractions, the pellet of the gradient, and the soluble extracts were analyzed by Western blot to detect the phosphorylation of PKA substrates and the presence of AKAP79 and PKA catalytic subunits (PKA cat). Flotillin-1 and tubulin were used as loading controls. B, densitometry analysis of blots (taking into account all the proteins detected by the antibody) with the phospho-PKA substrate antibody corrected against loading control (mean ± S.E.), for two independent experiments with cells expressing the double mutant (C36S,C129S) and three experiments with cells expressing the triple mutant (C36S,C62S,C129S) of AKAP79, **, p < 0.01; *, p < 0.05. Iso, isoproterenol; n.s., not significant.
No significant phosphorylation was achieved by the nonpalmitoylatable C36S,C129S or C36S,C62S,C129S mutants in the raft fractions (Fig. 6, A and B). The phosphorylation of proteins associated with the lipid rafts from cells expressing AKAP79-YFP wild type was ∼30% (p < 0.01) higher than in cells expressing the nonpalmitoylatable C36S,C129S or C36S,C62S,C129S mutants (Fig. 6, A and B). Such differences between wild type and mutant AKAP79 were not apparent in other cellular fractions (Fig. 6, A and B). These results strongly suggest that palmitoylated AKAP79 supports the compartmentalization of PKA signaling in lipid rafts.
AKAP79 Palmitoylation Is Required for the Phosphorylation and Regulation of the Lipid Raft-associated AC8
We have recently shown that AKAP79 interacts with AC8 and attenuates the stimulation of AC8 activity by SOCE (18), a phenomenon that depends on the integrity of lipid rafts (25, 28). Consequently, we asked whether the nonpalmitoylatable AKAP79 mutant would interact with and regulate AC8 like the wild type AKAP79.
AKAP79 was co-immunoprecipitated with AC8 from HEK-293 cells stably expressing AC8-HA and transiently transfected with either wild type or C36S,C129S mutant AKAP79-YFP. The Western blot from the HA pulldown (using agarose affinity beads) shows that mutation of cysteine residues 36 and 129 decreased the amount of AKAP79-YFP co-immunoprecipitated with AC8-HA (Fig. 7A (5th versus 6th lane) and C). This result shows that the palmitoylation is important for the interaction of AC8 with AKAP79-YFP. Western blots of total protein demonstrate similar expression levels of the C36S,C129S mutant and wild type AKAP79-YFP (Fig. 7B). In this experiment, we also analyzed cells stimulated for 5 min with the sarco(endo)plasmic reticulum Ca2+-ATPase pump inhibitor, thapsigargin (200 nm), 10 nm PGE, and 4 mm Ca2+ to determine whether the associated SOCE-mediated increase in AC8 activity would change the interaction between AC8 and AKAP79. The results showed that stimulation of AC8 did not change this interaction (Fig. 7, A and C).
FIGURE 7.

Palmitoylation of AKAP79 is required for interaction with AC8 and down-regulation of SOCE-stimulated AC8 activity. A, co-immunoprecipitation of AKAP79-YFP with AC8-HA. Mutant (C36S,C129S (C36,129S)) or wild type (Wt) AKAP79-YFP were expressed in cells stably expressing HA-AC8 (+) or wild type HEK-293 cells (−). AC8-HA pulldowns were performed using HA affinity beads, and AKAP79 and AC8-HA were detected by Western blot. Phospho-AC8 was detected using a phospho-PKA substrate-specific antibody. IB, immunoblot. B, AKAP79 inputs used to precipitate AC8-HA were blotted to measure the total expression of the proteins. C, densitometry analysis of pulldown blots in A (AKAP79 relative to AC8-HA). Data represent mean ± S.E. of four experiments. *, p < 0.05. TG, thapsigargin. D, pulldown using HA antibody not covalently coupled to agarose beads. AC8-HA and phospho-AC8 was detected by Western blot. E, inputs used to precipitate AC8-HA were blotted to measure the total expression of AC8 and AKAP79 proteins. F, densitometry analysis of pulldown blots (phospho-AC8-HA relative to AC8-HA) (mean ± S.E. of five separate experiments). n.s., not significant. G, SOCE-mediated increases in cAMP production measured with the FRET-based sensor Epac2-camps in HEK-AC8 cells expressing empty vector (control), wild type AKAP79-HA or AKAP79-YFP, or palmitoylation mutant (AKAP79-YFP C36S,C129S). Data are plotted as relative FRET ratio changes with F0 taken at 240 s, and values normalized to the maximum signal seen following stimulation with a mixture to saturate the cAMP sensor (10 μm forskolin, 10 μm isoproterenol, 10 μm PGE1, and 100 μm 3-isobutyl-1-methylxanthine). Average traces are plotted from 34 to 54 individual cells. H, input samples for AC8-HA pulldowns were also examined for ERK phosphorylation using a pERK-specific antibody. Tubulin was used a loading control. Densitometry data compares the effects of AKAP79 (Wt or double mutant) and SOCE stimulation of AC8 on pERK signals. Data are representative of mean ± S.E. from four similar experiments, **, p < 0.01; ***, p < 0.001.
We also analyzed the phosphorylation of AC8 using an antibody against PKA-phosphorylated substrates in membranes from the pulldowns (Fig. 7A, lower blot). A modest increase in phosphorylation was apparent in lysate from cells expressing the wild type AKAP79 but not in cells expressing C36S,C129S AKAP79-YFP (Fig. 7A, 7th versus 8th lane). We attempted to improve this signal using an alternative co-immunoprecipitation strategy of AC8-HA with the HA antibody not coupled covalently to protein G-agarose (see “Experimental Procedures”) to isolate AC8-HA and detect phosphorylation after SOCE stimulation with the antibody against PKA phosphosubstrates (Fig. 7, D–F). We showed that the phosphorylation of AC8 increased by 70% with the transfection of AKAP79-YFP, and the increase was significantly higher (47%; p < 0.01) in the cells transfected with the wild type compared with no significant increase in cells transfected with the nonpalmitoylatable AKAP79-YFP (Fig. 7D (6th versus 8th lane) and F). Fig. 7E shows comparable total expression levels of AC8 and AKAP79-YFP constructs in the input for the immunoprecipitation.
These data demonstrate that the palmitoylation of AKAP79 is important for the interaction of AC8 with AKAP79, which promotes the phosphorylation of AC8 after SOCE stimulation. This phosphorylation might be expected to regulate AC8 activity. The effect of the cysteine mutation of AKAP79 on the regulation of AC8 SOCE-stimulated activity was examined in live HEK-AC8 cells expressing the Epac2-camps FRET-based biosensor for cAMP (Fig. 7G). Cells were pretreated with 200 nm thapsigargin and 10 nm PGE in the absence of external Ca2+. This treatment enabled passive depletion of the endoplasmic reticulum Ca2+ stores, priming the system for sustained SOCE upon the addition of 2 mm external Ca2+ at 240 s. As seen previously (18), expression of wild type AKAP79-HA or AKAP79-YFP attenuated the peak response of AC8 to SOCE (p < 0.01 compared with control cells). However, expression of the C36S,C129S mutant AKAP79-YFP did not affect the SOCE-stimulated AC8 activity. These results indicate that the association of AKAP79 with lipid rafts by palmitoylation is required for the interaction with AC8, which underpins the ability of AKAP79 to modulate SOCE-mediated stimulation of AC8 activity.
Finally, we wondered whether the decrease in phosphorylation of AC8 due to delocalization of the mutated AKAP79 from lipid rafts might be mirrored by an increase in downstream PKA-mediated phosphorylation subsequent to the elevation of cAMP. We analyzed ERK phosphorylation after the SOCE stimulation of HEK-AC8 or wild type HEK-293 cells transfected with the wild type AKAP79-YFP, C36S,C129S AKAP79-YFP, or the empty YFP vector (Fig. 7H). The results showed that phosphorylation of ERK after SOCE stimulation was clearly detectable in cells expressing AC8 (Fig. 7H, 4th, 7th, and 8th lanes), but not in wild type HEK-293 cells (2nd lane). Transfection of the wild type AKAP79-YFP significantly affected the degree of ERK phosphorylation after SOCE stimulation (Fig. 7H, 5th versus 7th lane), whereas transfection of the C36S,C129S mutant resulted in greater phosphorylation of ERK after SOCE stimulation (Fig. 7H, 7th versus 8th lane, p < 0.01). These results suggest that delocalization of AKAP79 from lipid rafts makes more PKA available for downstream phosphorylation events.
DISCUSSION
Three linear sequences termed regions A, B, and C (residues 31–52, residues 76–101, and residues 116–145, respectively) target AKAP79 to the PM in HEK-293 cells and cortical neurons (29, 42). This study shows that AKAP79 is palmitoylated at cysteine 36 and 129 within the A and C basic regions. These palmitoylations are not necessary to localize AKAP79-YFP to the PM but instead promote association with lipid rafts. Treatment with the palmitoylation inhibitor, bromopalmitate as well as mutation of cysteines 36 and 129 decreased the amount of AKAP79-YFP isolated in lipid raft fractions. Mutation of either one of these cysteines affects the interaction of AKAP79 with insoluble membranes, although the mutation of both abolishes almost completely the recovery of AKAP79-YFP from lipid raft fractions. Note, however, that when the raft-targeted AC8 is overexpressed, significant amounts of AKAP79 can still be pulled down by AC8 (Fig. 6A). This implies that AKAP79 can still associate with lipid rafts albeit indirectly via a binding partner that is expressed in rafts. As would have been anticipated from these findings, PKA is recruited to rafts only by the wild type AKAP79 and not by the nonpalmitoylatable mutants.
A more benign live cell microscopic approach was also adopted to study the consequences of AKAP79 palmitoylation. Using FRAP analysis, we compared the mobility of wild type and the C36S,C129S mutant AKAP79 and examined how this behavior was modulated in cells treated with MBCD, which disrupts lipid rafts by extracting membrane cholesterol. The results clearly show that MBCD treatment affects only the diffusion of the wild type AKAP79 and not the nonpalmitoylatable mutant. MBCD treatment reduced the diffusion rate of the raft resident wild type AKAP79. Although this counterintuitive effect of MBCD has been widely reported for other lipid raft proteins, it may be worth speculating that the reason may lie in the destruction of lipid rafts leading to enhanced encounters of AKAP79 with other membrane targets, thereby reducing its overall mobility. Indeed, two studies have reported that MBCD treatment also decreases the diffusion of non-raft proteins (42, 48). However, we find that MBCD treatment does not affect the diffusion of the nonpalmitoylatable AKAP79 mutant. The lack of effect on the diffusion of the nonpalmitoylatable mutant may be because this protein is not immersed in the lipid bilayer but only associated with the membrane by interaction with the negatively charged phospholipid headgroups.
In this regard, although we did not detect any difference in the diffusion coefficient of the nonpalmitoylatable AKAP79 versus the wild type when modeled as simple Brownian motion, we did see an increased transport coefficient and evidence of anomalous diffusion (α < 1) of the wild type protein. This anomalous diffusion is largely determined by free diffusion coupled to transient trapping or binding, suggesting periods of temporary confinement of AKAP79 (41, 49, 50), presumably in lipid rafts. It is conceivable that AKAP79 can experience multiple types of interactions both in lipid rafts as well as in the non-raft domains. The mutant AKAP79 may undergo more interactions with the non-raft phospholipid membrane and with proteins in this region that may decrease its diffusion rate (and hence reduce its transport coefficient relative to the wild type). However, this interaction does not promote an anomalous diffusion of the nonpalmitoylatable AKAP79, because the exponent α is not significantly different from 1.
PI4P and PI(4,5)P2 depletion experiments were aimed at getting a sense of the relative importance of palmitoylation versus simple ionic attachments in the maintenance of AKAP79 at the PM. Although depletion of phosphoinositides produced only a small loss of wild type AKAP79 from the PM, the C36S,C129S mutant was rendered purely cytosolic. These data argue that palmitoylation is the predominant mechanism used to maintain AKAP79 at the PM. In terms of the association of AKAP79 with lipid rafts, it is known that PI(4,5)P2 can be sequestered within rafts as a result of association with polybasic stretches of amino acids found in raft-associated proteins (51), similar to those found in AKAP79. Because mutation of the palmitoylatable cysteines clearly results in the loss of AKAP79 from the lipid rafts, it must be concluded that palmitoylation is also the key mechanism for maintenance of AKAP79 in rafts and that the residual association of the AKAP79 with the PM relies on the association with phosphoinositides.
The functional consequence of AKAP79 association with lipid rafts is of course its recruitment of PKA and phosphorylation of proteins associated with these microdomains. Multiple independent lines of evidence have demonstrated that β-adrenergic receptors at the cell surface may occupy rafts and non-raft membranes but co-localize with their G protein and adenylyl cyclase effectors when in lipid rafts (52, 53). In cardiac myocytes, the disruption of lipid rafts (or caveolae, in that case) converts the sarcolemma-confined cAMP signal associated with β-adrenergic receptor stimulation to a global signal (54). In this context, our data showing phosphorylation in lipid rafts dependent upon wild type AKAP79 support the hypothesis that the co-localization of the β-adrenergic receptor with its effectors, in this case AKAP79 and PKA, promotes compartmentalization of the signaling.
A potentially even more acute target of PKA in lipid rafts is AC8. AC8 is highly compartmentalized (25, 27, 55), and the regulation of AC8 by Ca2+ depends on its localization in rafts (28, 35). Recently, we have shown that AC8 binds AKAP79, and its responsiveness to SOCE is attenuated by this association (18). This study strongly suggests that the residence of AKAP79 in lipid rafts is a key component of its ability to influence AC8 activity, because no modulation is seen by the nonpalmitoylatable AKAP79 mutant, and PKA-mediated phosphorylation of AC8 is only possible when the wild type AKAP79 is expressed.
Overall, we see that AKAP79 is a palmitoylated protein, whose palmitoylation dictates the targeting of AKAP79 to lipid rafts, to permit PKA-mediated phosphorylation and regulation of raft-associated targets, such as AC8. It would seem reasonable to predict that this propensity of AKAP79 to be palmitoylated could be exploited in its role to promote the selective phosphorylation of other target proteins that reside in rafts or in post-synaptic densities (which share the lipid composition of rafts). Although acylation of proteins is reportedly involved in targeting of proteins to lipid rafts, palmitoylation is a unique reversible lipid modification that can be regulated by specific extracellular signals. In mammalian systems, palmitoylation is mediated by a recently identified family of the transmembrane Asp-His-His-Cys motif protein acyltransferases and palmitoyl protein thioesterases (56–58). Recent studies have shown that palmitoylation/depalmitoylation cycles regulate the localization of specific proteins. In fact, even the subcellular localization of protein acyltransferases is regulated (59, 60). Palmitoylation cycles can therefore be expected to regulate the affinity of AKAP79 for lipid rafts and in this way regulate the interaction of AKAP79 with proteins in these microdomains. Our data demonstrate that the palmitoylation/depalmitoylation cycle of a key regulatory protein, such as AKAP79, can have important consequences for fine-tuning signaling transduction pathways mediated by key effector proteins that bind AKAP79, such as the adenylyl cyclases or numerous ion channels (1, 61).
Acknowledgments
We thank Professor Mark Dell'Acqua for the AKAP79-YFP construct. We are also grateful to Dr. Katy Everett for assistance with molecular biology.
This work was supported by Wellcome Trust Program Grant RG31760 and European Commission Grant 037189 (led by Prof. Enno Klussmann).
- PM
- plasma membrane
- Epac2-camps
- YFP/CFP-tagged Epac2-based fluorescent cAMP sensor
- FRAP
- fluorescence recovery after photobleaching
- PGE
- prostaglandin E1
- MBCD
- methyl-β-cyclodextrin
- SOCE
- store-operated Ca2+ entry
- PI(4,5)P2
- phosphatidylinositol 4,5-bisphosphate
- PI4P
- phosphatidylinositol 4-bisphosphate
- CFP
- cyan fluorescent protein
- INPP5E
- PI(4,5)P2 5-phosphatase.
REFERENCES
- 1. Dell'Acqua M. L., Smith K. E., Gorski J. A., Horne E. A., Gibson E. S., Gomez L. L. (2006) Eur. J. Cell Biol. 85, 627–633 [DOI] [PubMed] [Google Scholar]
- 2. Kim C., Vigil D., Anand G., Taylor S. S. (2006) Eur. J. Cell Biol. 85, 651–654 [DOI] [PubMed] [Google Scholar]
- 3. Budovskaya Y. V., Stephan J. S., Deminoff S. J., Herman P. K. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 13933–13938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gao X., Jin C., Ren J., Yao X., Xue Y. (2008) Genomics 92, 457–463 [DOI] [PubMed] [Google Scholar]
- 5. Gao C., Wolf M. E. (2008) J. Neurochem. 106, 2489–2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang G., Chen S., Li S., Cha J., Long C., Li L., He Q., Liu Y. (2007) Genes Dev. 21, 3283–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Neuberger G., Schneider G., Eisenhaber F. (2007) Biol. Direct.; 2:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Faruque O. M., Le-Nguyen D., Lajoix A. D., Vives E., Petit P., Bataille D., Hani el-H. (2009) Am. J. Physiol. Cell Physiol. 296, C306–C316 [DOI] [PubMed] [Google Scholar]
- 9. Glantz S. B., Amat J. A., Rubin C. S. (1992) Mol. Biol. Cell 3, 1215–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hall D. D., Davare M. A., Shi M., Allen M. L., Weisenhaus M., McKnight G. S., Hell J. W. (2007) Biochemistry 46, 1635–1646 [DOI] [PubMed] [Google Scholar]
- 11. Fraser I. D., Cong M., Kim J., Rollins E. N., Daaka Y., Lefkowitz R. J., Scott J. D. (2000) Curr. Biol. 10, 409–412 [DOI] [PubMed] [Google Scholar]
- 12. Dai S., Hall D. D., Hell J. W. (2009) Physiol. Rev. 89, 411–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bauman A. L., Soughayer J., Nguyen B. T., Willoughby D., Carnegie G. K., Wong W., Hoshi N., Langeberg L. K., Cooper D. M. F., Dessauer C. W., Scott J. D. (2006) Mol. Cell 23, 925–931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bal M., Zhang J., Hernandez C. C., Zaika O., Shapiro M. S. (2010) J. Neurosci. 30, 2311–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dart C., Leyland M. L. (2001) J. Biol. Chem. 276, 20499–20505 [DOI] [PubMed] [Google Scholar]
- 16. Gomez L. L., Alam S., Smith K. E., Horne E., Dell'Acqua M. L. (2002) J. Neurosci. 22, 7027–7044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Snyder E. M., Colledge M., Crozier R. A., Chen W. S., Scott J. D., Bear M. F. (2005) J. Biol. Chem. 280, 16962–16968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Willoughby D., Masada N., Wachten S., Pagano M., Halls M. L., Everett K. L., Ciruela A., Cooper D. M. F. (2010) J. Biol. Chem. 285, 20328–20342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pike L. J. (2006) J. Lipid Res. 47, 1597–1598 [DOI] [PubMed] [Google Scholar]
- 20. Simons M., Keller P., De Strooper B., Beyreuther K., Dotti C. G., Simons K. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 6460–6464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Simons K., Ikonen E. (1997) Nature 387, 569–572 [DOI] [PubMed] [Google Scholar]
- 22. Shenoy-Scaria A. M., Dietzen D. J., Kwong J., Link D. C., Lublin D. M. (1994) J. Cell Biol. 126, 353–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Head B. P., Patel H. H., Roth D. M., Lai N. C., Niesman I. R., Farquhar M. G., Insel P. A. (2005) J. Biol. Chem. 280, 31036–31044 [DOI] [PubMed] [Google Scholar]
- 24. Suzuki T., Ito J., Takagi H., Saitoh F., Nawa H., Shimizu H. (2001) Brain Res. Mol. Brain Res. 89, 20–28 [DOI] [PubMed] [Google Scholar]
- 25. Fagan K. A., Smith K. E., Cooper D. M. F. (2000) J. Biol. Chem. 275, 26530–26537 [DOI] [PubMed] [Google Scholar]
- 26. Ostrom R. S., Liu X., Head B. P., Gregorian C., Seasholtz T. M., Insel P. A. (2002) Mol. Pharmacol. 62, 983–992 [DOI] [PubMed] [Google Scholar]
- 27. Cooper D. M. F., Crossthwaite A. J. (2006) Trends Pharmacol. Sci. 27, 426–431 [DOI] [PubMed] [Google Scholar]
- 28. Smith K. E., Gu C., Fagan K. A., Hu B., Cooper D. M. F. (2002) J. Biol. Chem. 277, 6025–6031 [DOI] [PubMed] [Google Scholar]
- 29. Dell'Acqua M. L., Faux M. C., Thorburn J., Thorburn A., Scott J. D. (1998) EMBO J. 17, 2246–2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jackson C. S., Zlatkine P., Bano C., Kabouridis P., Mehul B., Parenti M., Milligan G., Ley S. C., Magee A. I. (1995) Biochem. Soc. Trans. 23, 568–571 [DOI] [PubMed] [Google Scholar]
- 31. Mumby S. M. (1997) Curr. Opin. Cell Biol. 9, 148–154 [DOI] [PubMed] [Google Scholar]
- 32. Anania V. G., Coscoy L. (2011) J. Virol. 85, 2288–2295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Levental I., Lingwood D., Grzybek M., Coskun U., Simons K. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 22050–22054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Delint-Ramirez I., Fernández E., Bayés A., Kicsi E., Komiyama N. H., Grant S. G. (2010) J. Neurosci. 30, 8162–8170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pagano M., Clynes M. A., Masada N., Ciruela A., Ayling L. J., Wachten S., Cooper D. M. F. (2009) Am. J. Physiol. Cell Physiol. 296, C607–C619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hammond G. R., Sim Y., Lagnado L., Irvine R. F. (2009) J. Cell Biol. 184, 297–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Oancea E., Teruel M. N., Quest A. F., Meyer T. (1998) J. Cell Biol. 140, 485–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Feder T. J., Brust-Mascher I., Slattery J. P., Baird B., Webb W. W. (1996) Biophys. J. 70, 2767–2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Várnai P., Balla T. (2008) Methods 46, 167–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Webb Y., Hermida-Matsumoto L., Resh M. D. (2000) J. Biol. Chem. 275, 261–270 [DOI] [PubMed] [Google Scholar]
- 41. Owen D. M., Williamson D., Rentero C., Gaus K. (2009) Traffic 10, 962–971 [DOI] [PubMed] [Google Scholar]
- 42. Kenworthy A. K. (2007) Methods Mol. Biol. 398, 179–192 [DOI] [PubMed] [Google Scholar]
- 43. Ohvo H., Slotte J. P. (1996) Biochemistry 35, 8018–8024 [DOI] [PubMed] [Google Scholar]
- 44. Pike L. J., Miller J. M. (1998) J. Biol. Chem. 273, 22298–22304 [DOI] [PubMed] [Google Scholar]
- 45. Saxton M. J. (2008) Biophys. J. 94, 760–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Varnai P., Thyagarajan B., Rohacs T., Balla T. (2006) J. Cell Biol. 175, 377–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Szentpetery Z., Várnai P., Balla T. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 8225–8230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Shvartsman D. E., Gutman O., Tietz A., Henis Y. I. (2006) Traffic 7, 917–926 [DOI] [PubMed] [Google Scholar]
- 49. Simson R., Sheets E. D., Jacobson K. (1995) Biophys. J. 69, 989–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Simson R., Yang B., Moore S. E., Doherty P., Walsh F. S., Jacobson K. A. (1998) Biophys. J. 74, 297–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Laux T., Fukami K., Thelen M., Golub T., Frey D., Caroni P. (2000) J. Cell Biol. 149, 1455–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Agarwal S. R., MacDougall D. A., Tyser R., Pugh S. D., Calaghan S. C., Harvey R. D. (2011) J. Mol. Cell. Cardiol. 50, 500–509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Head B. P., Patel H. H., Roth D. M., Murray F., Swaney J. S., Niesman I. R., Farquhar M. G., Insel P. A. (2006) J. Biol. Chem. 281, 26391–26399 [DOI] [PubMed] [Google Scholar]
- 54. Calaghan S., Kozera L., White E. (2008) J. Mol. Cell. Cardiol. 45, 88–92 [DOI] [PubMed] [Google Scholar]
- 55. Willoughby D., Cooper D. M. F. (2007) Physiol. Rev. 87, 965–1010 [DOI] [PubMed] [Google Scholar]
- 56. Fukata M., Fukata Y., Adesnik H., Nicoll R. A., Bredt D. S. (2004) Neuron 44, 987–996 [DOI] [PubMed] [Google Scholar]
- 57. Huang K., Yanai A., Kang R., Arstikaitis P., Singaraja R. R., Metzler M., Mullard A., Haigh B., Gauthier-Campbell C., Gutekunst C. A., Hayden M. R., El-Husseini A. (2004) Neuron 44, 977–986 [DOI] [PubMed] [Google Scholar]
- 58. Lobo S., Greentree W. K., Linder M. E., Deschenes R. J. (2002) J. Biol. Chem. 277, 41268–41273 [DOI] [PubMed] [Google Scholar]
- 59. Noritake J., Fukata Y., Iwanaga T., Hosomi N., Tsutsumi R., Matsuda N., Tani H., Iwanari H., Mochizuki Y., Kodama T., Matsuura Y., Bredt D. S., Hamakubo T., Fukata M. (2009) J. Cell Biol. 186, 147–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Iwanaga T., Tsutsumi R., Noritake J., Fukata Y., Fukata M. (2009) Prog. Lipid Res. 48, 117–127 [DOI] [PubMed] [Google Scholar]
- 61. Dessauer C. W. (2009) Mol. Pharmacol. 76, 935–941 [DOI] [PMC free article] [PubMed] [Google Scholar]