

Abstract
Germinal centers (GCs) are specialized microenvironments in secondary lymphoid organs that facilitate the development of high-affinity, isotype-switched Abs, and immunological memory; consequently, many infections require GC-derived IgG for pathogen clearance. Although Ehrlichia muris infection elicits a robust expansion of splenic, IgM-secreting plasmablasts, we detected only very low frequencies of isotype-switched IgG-secreting cells in mouse spleens, until at least 3 wk postinfection. Instead, Ag-specific IgG was produced in lymph nodes, where it required CD4 T cell help. Consistent with these findings, organized GCs and phenotypically defined splenic GC B cells were found in lymph nodes, but not spleens. Ehrlichial infection also inhibited spleen IgG responses against a coadministered T cell-dependent Ag, hapten 4-hydroxy-3-nitrophenyl acetyl (NP)-conjugated chicken γ globulin in alum. NP-specific B cells failed to undergo expansion and differentiation into GC B cells in the spleen, Ab titers were reduced, and splenic IgG production was inhibited nearly 10-fold when the Ag was administered during infection. Our data provide a mechanism whereby an intracellular bacterial infection can compromise local immunity to coinfecting pathogens or antigenic challenge.
The germinal center (GC) reaction is essential for the production for high-affinity, isotype-switched Abs during the adaptive immune response (1, 2). The formation of GCs requires the interaction of B and T cells with resident follicular dendritic cells in secondary lymphoid organs (3, 4). In GCs, Ag-specific B cells undergo rapid expansion, class-switch recombination, and affinity maturation. Although many GC B cells undergo apoptosis, a select few high-affinity B cells survive the T cell-dependent (TD) selection process and differentiate into isotype-switched B cell clones (2, 5, 6).
Class-switch recombination and affinity maturation typically require as long as 2 wk (5). Therefore, to prevent pathogen dissemination during acute infection, activated B cells proliferate and differentiate into Ab-secreting cells or plasmablasts in the red pulp of the spleen, adjacent to the T cell zone (7-9), or in the medullary cords of the lymph nodes (LNs) (10). Plasmablasts secrete primarily IgM against T cell-independent and TD Ags, and in this manner provide an early, typically short-lived, component of host defense.
Our previous study documented such an IgM-producing plasmablast response during Ehrlichia muris infection in the C57BL/6 mouse.E. muris is a tick-transmitted rickettsia that is closely related to Ehrlichia chaffeensis, the etiologic agent of human monocytotropic ehrlichiosis. The plasmablasts elicited during E. muris infection had the unusual characteristic of exhibiting low surface expression of CD11c, a marker more commonly associated with dendritic cells. The CD11c-expressing extrafollicular plasmablasts accounted for >15% of mononuclear splenocytes at peak infection and were responsible for the CD4 T cell-independent production of Ag-specific IgM (11) that is likely responsible for protective immunity. In the current study, we show that the plasmablast response that we described previously is associated with an impaired GC response in the spleen, but not the LNs. The impairment of the GC responses resulted in the inhibition of the production of isotype-switched Abs, against both ehrlichial Ags and coadministered irrelevant Ags. These studies provide a mechanism whereby ehrlichial infection can compromise local immunity to a coinfecting tick-borne pathogen.
Materials and Methods
Mice
The mice used in these studies were obtained from The Jackson Laboratories (Bar Harbor, ME) or were bred in the Animal Care Facility at the Wadsworth Center under microisolator conditions, in accordance with institutional guidelines for animal welfare. Mice were gender-matched for each experiment, and were 6–12 wk old. The studies were performed in C57BL/6, B6.129S-H2dIAb-1-EaB6 (MHC class II-deficient), and B6.129P2-Tcrbtm1Mom/J (TCR b-deficient) strains of mice. The quasi-monoclonal NP-specific BCR transgenic mouse strain m+IgMxJhD/BALB/c, also described as (m+s)Ig Tg, was provided by Dr. M. Shlomchik (Yale University, New Haven, CT).
Infection and immunizations
Details regarding the bacterial strains and infection protocols have been described previously (12). Mice were infected i.p. at 6–12 wk old, with ~50,000 copies of E. muris. For the immunization studies, 4-hydroxy-3-nitrophenyl-acetyl(48)-chicken γ globulin conjugate (NP-CGG; Biosearch Technology, Novato, CA) was precipitated in alum (Imject; Sigma-Aldrich, St. Louis, MO) at a ratio of 1:5 (NP-CGG: alum) for 30 min. E. muris-infected and mock control mice were immunized i.p. or s.c. (in the left and right shoulders) with 100 μg NP-CGG/alum on the indicated day postinfection.
Abs and flow cytometry
The following Abs were used for flow cytometry: CD95-PE (clone FAS/APO-1), CD19-PerCP (clone 1D3; both from BD Biosciences, Franklin Lakes, NJ), GL7-FITC (clone Ly77), and CD38-allophycocyanin (clone 90; eBioscience, San Diego, CA); 4-hydroxy-5-iodo-3-nitrophenylacetic acid hydrosuccinimide ester-haptenated PE (NIP-PE) was used to identify NP-specific B cells. For flow cytometric analyses, spleen and LN cells were obtained by mechanical disruption in HBSS containing 2% FBS, and the tissues were disaggregated using a 70-μm pore size nylon strainer (Falcon; BD Biosciences, San Jose, CA). Erythrocytes were removed from the single-cell suspension of splenocytes by hypotonic lysis. For cell surface staining, single-cell suspensions (2 × 106 cells) were incubated with Fc blocking solution (1 μg/ml anti-CD16/CD32; FcRγ III/II; clone 2.4G2) in 10% normal goat serum/HBSS/0.1% sodium azide, prior to staining with the mAbs. The cells were incubated on ice for 20 min, washed twice, and analyzed without fixation. Negative controls were used to establish the flow cytometer voltage settings, and single-color positive controls were used to adjust compensation. Data from stained samples were acquired on a FACS Calibur with Cell Quest software (Becton Dickinson, Mountain View, CA), and were analyzed with FloJo Software (Tree Star, Ashland, OR).
ELISPOT and ELISA analyses
The detection of Ab-producing spleen and LN B cells was conducted using an ELISpot assay. Nitrocellulose plates (Multiscreen-HA; Millipore, Bedford, MA) were coated overnight at 4°C with hapten 4-hydroxy-3-nitrophenyl acetyl (NP)-conjugated BSA (NP33-BSA; used at 5 μg/ml; Biosearch Technology, Novato, CA), or purified recombinant E. muris outer membrane protein-19 (OMP-19; at 10 μg/ml) (13). The assay plates were then incubated in blocking solution (IMDM supplemented with 10% FBS), for 2–3 h at 37°C. Cells were cultured in IMDM supplemented with 2 mM l-glutamine, 100 U/ml penicillin, 100 U/ml streptomycin, 50 μM 2-ME, and 10% FBS, and seeded in 96-well plates at a concentration of 1 × 106 cells/ml, in triplicate, in a volume of 100 μl; the cells were further diluted in the microtiter plate using 2-fold doubling dilutions. After 18 h incubation at 37°C in 5% CO2, bound IgM or IgG was detected using goat anti-mouse IgM or IgG conjugated to alkaline phosphatase (Southern Biotechnology Associates, Birmingham, AL); 5-bromo-4-chloro-3-indoylphosphate/nitro blue tetrazolium (Sigma-Aldrich) was used as the substrate. Spots were enumerated with a CTL immunospot S5 Core Analyzer, and the data were analyzed by CTL ImmunoSpot software (Cellular Technology, Shaker Heights, OH). IgM and IgG serum titers were determined by ELISA using purified recombinant E. muris OMP-19, as described previously (12), or NP33-BSA (BSA; 5 μg/ml).
Immunohistochemistry
Spleen and inguinal LN sections were prepared as previously described (11). The sections were stained in succession with rat anti-mouse Thy1.2 (BD Biosciences; overnight), biotinylated rabbit anti-rat IgG (Vector Laboratories, Burlingame, CA; 60 min at room temperature [RT]), and streptavidin-Alexa Fluor-350 (Invitrogen, Carlsbad, CA; 30 min); they were washed extensively in PBS and then blocked with 10% FBS for 30 min at RT. For the detection of B cells, the sections were next incubated with biotin-conjugated rat anti-mouse B220 (BD Biosciences; 2 h at RT), and streptavidin-Alexa Fluor-647 (Invitrogen; 60 min). For the detection of GCs, the same sections were blocked with 2% BSA for 60 min at RT. Next, the sections were incubated with biotinylated peanut agglutinin (PNA) (Vector Laboratories; 1 h at RT) followed by streptavidin-Alexa Fluor-488 (Invitrogen; 30 min). When biotinylated Abs were used, a streptavidin-biotin blocking kit (Vector Laboratories) was used between each of the Ab incubations. The stained sections were mounted in anti-fading reagent (Slow Fade Gold; Invitrogen). Images were acquired using an epi-fluorescence microscope (Axioskop2; Zeiss, Peabody, MA) equipped with a Hamamatsu camera (Hamamatsu Photonic Systems, Bridgewater, NJ), and were processed with OPENLAB software (Zeiss, Peabody, MA).
Statistical analysis
The Mann-Whitney U test was used to assess statistical significance with values of p < 0.05 considered significant.
Results
Impaired IgG production in the spleens of E. muris-infected mice
Our previous study demonstrated that E. muris infection generates a large population of CD11c-expressing plasmablasts that produced the majority of total IgM, and almost all of the Ag-specific IgM within the spleen (11). That study focused primarily on IgM-producing cells, because we were unable to detect Ag-specific IgG-producing cells in the spleen on day 10 postinfection. To address the IgG response in greater detail, in this study, we infected C57BL/6 mice i.p. with E. muris (5 × 104 cells) and then measured Ab titers against the immunodominant Ag, OMP-19. OMP-19–specific IgG was detected in the sera of E. muris-infected mice beginning on day 9 postinfection, although Ab titers at this time were relatively low (<100; Fig. 1A). By day 28 postinfection, there was a 10-fold increase in OMP-19 IgG titer; OMP-19–specific IgG of the subclasses IgG2b, IgG2c, and IgG3 were detected at titers greater than 200, but IgG1 was not detected (Fig. 1B). The absence of a detectable IgG1 response was unexpected, although other infections associated with strong Th1 responses also favored switching to the IgG2a, IgG2b, and IgG3 subclasses (14). These data revealed that IgG was detectable at a low titer in the serum until at least 4 wk postinfection.
FIGURE 1.

E. muris infection inhibits the production of splenic, Ag-specific IgG-secreting cells. E. muris OMP-19–specific IgM and IgG (A), and day 28 postinfection isotype-switched IgG (B), were measured in the serum of C57BL/6 mice by ELISA. C, The frequencies and numbers of Ag-specific IgM-secreting (●) and IgG-secreting (■) cells in the spleen (top panels) and lymph nodes (bottom panels) were determined by ELI-SPOT analysis. Spots produced by cells from mock-infected control mice, which were negligible, were subtracted from the number of spots obtained from the infected mice. The data are representative of three independent experiments in which three mice were analyzed at each time point.
Given that IgG production was significantly delayed relative to the IgM response, we next used ELISPOT analysis to address whether the frequencies and numbers of Ig-producing B cells in LNs and the spleen were affected. E. muris does not exhibit any apparent LN tropism, so the studies were performed using pooled cells from the inguinal, brachial, and axillary LNs. The frequency of spleen OMP-19–specific IgM-producing cells was as high as 4% of total B cells, representing ~3 × 106 cells on day 14 postinfection, similar to our previously reported data (Fig. 1C) (11). In contrast, OMP-19–specific IgG-producing cells were found at low frequencies in the spleen and were not observed in significant numbers until day 28 postinfection, when ~1.5 ×105 cells weredetected. In contrast to the spleen, the LNs had readily detectable IgG-producing B cells between days 10 and 15 postinfection; the cells were present in LNs at a 4-fold higher frequency (0.4% and 0.1% in the LN and spleen, respectively; Fig. 1C).
CD4 T cells are required for Ag-specific IgG production in LNs
The spleen is the largest secondary lymphoid organ (15), and some IgG-secreting cells were detected in this tissue early during infection (Fig. 1C). E. muris infection causes splenomegaly, which is most pronounced at day 17 postinfection, and is associated with a 4-fold increase in mononuclear cells (11, 16). Thus, although the spleen contained IgG-producing B cells, we hypothesized that the LNs were major secondary lymphoid organs responsible for the production of high-affinity, isotype-switched IgG prior to 3–4 wk postinfection. Because IgG responses typically require CD4 T cell help, we next addressed whether T cells were required. This was determined using MHC class II-deficient mice, which lack CD4 T cells, and TCRβ-deficient mice, which lack αβT cells. In these studies, mice were infected with E. muris, and OMP-19–specific IgM and IgG serum titers were assessed on day 16 postinfection. In the MHC class II and TCRβ-deficient mice, the OMP-19 IgM titers were reduced by 2- and 3-fold, respectively, compared with C57BL/6 mock-infected mice, suggesting that the Ag-specific IgM response was partially dependent on classical T cell help (Fig. 2A). In contrast, Ag-specific IgG was detected in C57BL/6 mice, but was not detected the sera of either MHC class II-deficient or TCRβ-deficient mice (Fig. 2B). Data from ELISPOT analyses supported this conclusion: Ag-specific IgG-secreting cells were largely undetectable (<0.02% of LN B cells) in the spleens and LNs of either the MHC class II-deficient or TCR β-deficient mice (Fig. 2c). Although MHC class II-deficient mice exhibited a 2-fold decrease in the frequency and number of LN OMP-19 IgM-secreting cells, these cells were undetectable in the LNs of TCRβ-deficient mice. The spleen was the primary source of Ag-specific IgM in both the MHC class II- and TCRβ-deficient mice, but total numbers were reduced by 2- and 3-fold, respectively. Thus, T cells likely provide important helper functions that promote the development of both IgG- and IgM-producing cells in the LNs.
FIGURE 2.

CD4 T cells are required for both IgM and IgG production in the LNs. Serum titers of OMP-19 IgM (A) and IgG (B) were assessed on day 16 postinfection in MHC class II-deficient and TCRβ-deficient mouse strains and compared with titers obtained in infected C57BL/6 mice. C, An ELISPOT analysis was used to determine the frequencies and numbers of OMP-19 IgM-secreting (●) and IgG-secreting (■) cells in the spleen (top panels) and LNs (bottom panels) on day 16 postinfection. Spots produced by cells isolated from mock-infected control mice of each of the strains, which were negligible, were enumerated and subtracted from the spots measured in the infected mice. The data are representative of two independent experiments in which four mice were analyzed at each time point. *p ≤ 0.05.
Impaired GC responses in the spleen, but not the lymph nodes, during E. muris infection
To further address whether the LNs provide a better environment for the production of IgG than does the spleen, we next evaluated whether the development of GCs and GC-derived B cells differed between the two lymphoid tissues. To identify GC B cells, we first analyzed spleen and LN B cells for cell surface expression of the late activation marker GL7 (17, 18). GL7-positive B cells were detected on day 9 postinfection in the LNs, reached a maximum on day 14 postinfection, and remained at frequencies above those in mock-infected mice until at least day 28 postinfection (Fig. 3A–C). In contrast, the frequency and number of spleen GL7-expressing cells increased only moderately until day 21, when a 2–3-fold increase was observed. Because GL7 is a marker of activated B cells, we further evaluated GC-associated GL7+ CD19+ B cells for expression of CD95 (FAS) and CD38. CD95 expression is induced, and CD38 expression is downregulated on GC-associated B cells, relative to naive B cells (19-21). GC B cells were first detected in the LNs on day 14 postinfection and reached a maximum frequency of 8% on day 17 postinfection (Fig. 3D–F). In contrast, the frequency of splenic GC B cells remained low (<1.5% of total B cells), until day 21 postinfection, when a 2-fold increase in frequency was observed.
FIGURE 3.

Infection inhibits the differentiation of splenic GC B cells. A, To determine the kinetics of the onset of the GC response, cells from the spleen (top row) and lymph nodes (bottom row) of C57BL/6 mice were stained with Abs directed against GL7 and CD19, on the indicated days postinfection. The frequencies (B) and numbers (C) of GL7-positive B cells are shown. D, To further characterize GC B cells, the GL7+ CD19+ cells were analyzed for their expression of CD38 and CD95. The frequencies and numbers of GC B cells (i.e., CD95+CD38lo cells), within the GL7−, CD19-positive flow cytometry gate, are shown in E and F. The data are representative of three independent experiments in which three mice were analyzed at each time point. *p ≤ 0.05.
Although the data described above demonstrated that GC B cells in the LNs were detected earlier than in the spleen, and at much higher frequencies, the number of phenotypically-defined splenic GC B cells was higher in the spleen than in the LNs as early as day 9 postinfection. However, Ab production requires the association of Ag-specific B cells, CD4 T cells, and follicular dendritic cells, to drive the selection of high-affinity, isotype-switched GC B cells (22, 23). Therefore, we used an alternative approach to address whether ehrlichial infection influences the development of GCs. Tissue sections of spleens and LNs from mock-infected and day 16-infected C57BL/6 mice were stained with Abs against PNA, B220, and Thy1.2. GC B cells were identified by their characteristic ability to bind PNA (24), whereas B220 and Thy1.2 were used to delineate the B cell and T cells, respectively. As expected, small clusters of PNA-positive cells were detected within the follicles of mock-infected spleens and LNs (Fig. 4). On day 16 postinfection, PNA-positive B cells were dispersed throughout the splenic T cell zone, but no organized GC structures were observed in the follicles. In contrast, infected LNs exhibited multiple clusters of PNA-positive cells, and the B cells were interspersed with Thy1-positive T cells. These data suggest that ehrlichial infection inhibits the formation of GCs within the spleen, but not the LNs, and they support our hypothesis that splenic IgG production is impaired early during E. muris infection. Our previous studies have demonstrated that the degree of bacterial colonization within the spleen and LNs is similar (16), so the differing GC responses in the two secondary lymphoid organs likely reflect differences in tissue-specific GC formation. Moreover, analyses of GC B cells, and measurements of the frequencies of Ab-secreting cells, revealed no significant differences in draining and non-draining LNs on day 15 postinfection (data not shown), indicating that the LN GC responses were not simply a consequence of the infection route.
FIGURE 4.
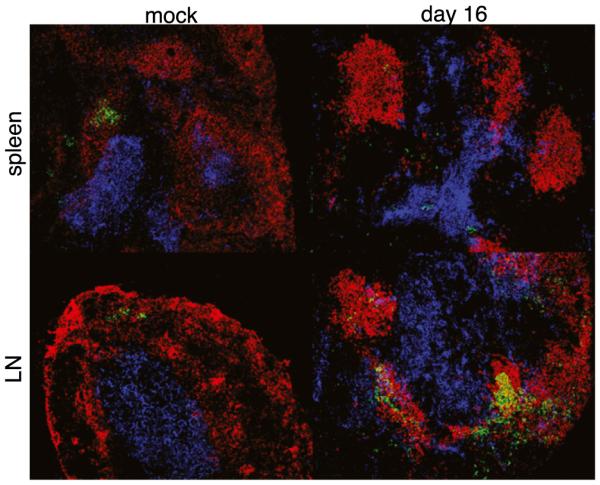
GC formation was observed in the LNs, but not in the spleens, of infected mice. Frozen sections from spleen tissue (top row) and an inguinal lymph node (bottom row) from mock-infected and day 16-infected C57BL/6 mice were stained with Abs specific for B220 (red), Thy1.2 (blue), and PNA (green). Colocalization of the PNA-positive cells with the B and T cells was detected in the LNs, but not in the spleens, of infected mice. Original magnification ×200.
Infection inhibits the splenic IgG response to an irrelevant Ag
Our findings thus far demonstrate that ehrlichial infection inhibits the IgG response in the spleen. To determine whether this phenomenon is limited to ehrlichial Ags, or whether the IgG response to other Ags is also impaired during infection, we immunized uninfected and E. muris-infected C57BL/6 mice with the TD Ag NP-CGG, in alum, at various time points after E. muris infection. Sera and spleens were harvested for analysis 12 d after NP-CGG immunization (8, 25, 26), and the production of NP-specific Abs was analyzed by ELISA and ELISPOT. Mice immunized with NP-CGG typically generated reciprocal Ab titers ranging from 2000–10,000 within 12 d of immunization. However, when immunization was performed after E. muris infection, NP-specific serum Ab titers were much reduced (Fig. 5A). The greatest suppression of NP-specific IgG was observed when the Ag was administered between 2 and 16 d postinfection, although a modest but non-significant inhibition was noted at day 23 postinfection. The reduction in NP-IgG titers was likely caused by the impaired production of IgG-secreting cells in the spleen, given that the frequency of splenic NP-specific IgG-secreting cells was reduced by ~10-fold when NP-CGG was administered on day 9 postinfection (Fig. 5B). When NP-CGG was administered at other time points relative to infection, the frequency of IgG-secreting cells was reduced by factors ranging 2–4-fold. Despite the decrease in frequency, the number of NP-specific IgG-secreting cells was significantly reduced only at days 9 and 16 postinfection, because of infection-induced splenomegaly (Fig. 5C). Thus, E. muris infection inhibited the production of spleen IgG against both ehrlichial and coadministered Ags.
FIGURE 5.

E. muris infection suppressed the IgG response to an irrelevant Ag. E. muris-infected mice and uninfected control C57BL/6 mice were immunized i.p. with NP-CGG in alum on the indicated days postinfection. To assess the NP-specific IgG response, serum and spleen tissue were harvested on day 12 postimmunization and were analyzed by ELISA (A) and ELISPOT analysis (B, C) in mice immunized with NP-CGG only (●) and in E. muris-infected, NP-CGG–immunized mice (■). The frequency (B) and number (C) of splenic NP-specific IgG-secreting cells was determined by ELISPOT analysis after subtracting background spots obtained using cells from mock-infected control mice. D, Day 9 E. muris-infected and mock-infected mice were immunized s.c. with NP-CGG in alum, and the NP-specific IgG response was analyzed at day 21 postinfection by ELISPOT analysis in the spleen (top row) and pooled brachial and axillary draining LNs (bottom row). The data are representative of three independent experiments in which three mice were analyzed at each time point. *p ≤ 0.05.
In the studies described thus far, both the pathogen and the Ag were administered i.p. We next addressed whether the NP-IgG response was affected when Ag was injected s.c., distant from the peritoneal drainage path. Mice were infected via the peritoneum with E. muris, and on day 9 postinfection, infected and mock-infected mice were immunized s.c. with NP-CGG. The anti-NP response was analyzed in the draining LNs and the spleen, 12 d after immunization. Prior E. muris infection did not affect the anti-NP IgG response in the draining LNs; instead, infection was associated with ~2-fold increase in the frequency and number of NP-specific IgG-secreting cells (Fig. 5D). In contrast to the draining lymph nodes, E. muris infection suppressed the anti-NPIgG response in the spleen by2-fold, comparedwith mice that were immunized with NP-CGG alone. Thus, the localized suppression of the IgG response is not due to the route of inoculation.
E. muris infection impairs the differentiation of NP-specific GC B cells
Based on our findings that E. muris infection inhibited the development of spleen GC B cells, we next addressed whether infection blocks the differentiation of NP-specific GC B cells following immunization. Because NP-specific B cells are found at low frequencies in normal immunized mice, we used (m+s)Ig Tg mice, a quasi-monoclonal BALB/c Ig H chain transgenic strain that encodes B cell receptors with a preferential specificity for NP (27). Pairing of the transgenic H chain with the endogenous λ1 L chain in the transgenic mice leads to the generation of a modest NP-specific B cell response (3–5%) in the spleen (28, 27). Because of the lack of endogenous H chain genes, the (m+s)Ig Tg mice are unable to secrete IgG; however, splenic architecture and the B cell compartment remain intact (28). Because NP-specific IgG serum titers were suppressed as early as day 2 postinfection, we chose this time point for immunization of E. muris-infected mice with NP-CGG. Spleens were harvested 12 d postimmunization (day 14 after E. muris infection), and NP-specific CD19-positive B cells were detected by their ability to bind the related hapten, NIP (29, 30). Consistent with the results of published studies, we observed an increase in the frequency and number of NP-specific B cells in (m+s)Ig Tg mice immunized with NP-CGG in alum, relative to unimmunized mice of the same strain (Fig. 6A). In contrast, expansion of the NP-specific B cell population was not detected in the E. muris-infected NP-CGG-immunized mice. In the infected mice, the frequency of NP-specific B cells was reduced by ~10-fold, relative to the frequencies detected in mock-infected, unimmunized mice (0.21 and 2.67%, respectively), and the number of NP-specific cells was roughly similar in both groups, indicating the lack of specific B cell expansion during infection (Fig. 6B, 6C). To determine whether the reduction of NP-specific response was due to the failure of the Ag-specific B cells to develop into GC B cells during infection, we analyzed NIP+ CD19+ cells for GL7 and CD38 expression. A large increase in the frequency of GC B cells was observed following immunization of uninfected mice, but E. muris infection reduced the frequency of GC B cells by ~10-fold (Fig. 6D, 6E). Furthermore, the total numbers of NIP+ GC B cells in the immunized–infected mice were comparable to those in the mock-infected control mice (Fig. 6F).
FIGURE 6.

The differentiation of spleen GC B cells was suppressed during infection. On day 2 after E. muris infection, infected (m+s)Ig B cell transgenic mice and mock-infected mice were immunized with NP-CGG and analyzed 12 d later. A, Splenocytes were stained for cell surface expression of NIP and CD19, to detect Ag-specific B cells. The frequencies of NIP-specific B cells among the total B cells (B) and the numbers of Ag-specific B cells (C) were determined. D, The NIP+ CD19+ B cells were analyzed for cell surface expression of GL7 and CD38 to identify GC B cells. The frequencies (E) and numbers (F) of GC B cells among the total CD19-positive B cells were determined. The data are representative of three independent experiments in which three mice were analyzed at each time point. *p ≤ 0.05.
Discussion
Our studies demonstrate that E. muris infection can suppress IgG production and inhibit the development of GC B cells within the spleen. Ag-specific IgG-secreting cells were largely absent in the spleen until 3–4 wk postinfection (i.e., 2 wk after IgG-secreting cells were first detected in the LNs). We attribute the suppression of IgG production in the spleen at least in part to the lack of proper GC formation. The inhibition was not limited to ehrlichial Ags; infection also suppressed local IgG production against an irrelevant, coadministered TD Ag. Although we cannot rule out the possibility of a defective extrafollicular response, our studies suggest that the impaired production of splenic IgG is due to an inhibition in the GC response, because we demonstrated a clear defect in GC formation. These data provide a possible mechanism whereby a bacterial infection can suppress an Ab response against a coinfecting agent. It is well established that tick-borne pathogens can be cotransmitted by the same tick or by different ticks feeding on the same host (31). Babesia microti and Borrelia burgdorferi, the respective agents of babesiosis and Lyme disease, exemplify coinfecting pathogens that are commonly transmitted with the ehrlichiae and/or other related rickettsiae (32, 33). Coinfection with Anaplasma phagocytophilum, the etiologic agent of human anaplasmosis, and B. burgdorferi was associated with increased spirochete burden and exacerbated the early onset of Lyme arthritis (34, 33). Such coinfection was proposed to impair the activation of macrophages, resulting in an increase in pathogen burden (34). Our studies suggest the impaired production of splenic IgG as another mechanism that contributes to coinfectionassociated immunosuppression.
The inhibition of IgG production during E. muris infection was associated with distinct humoral responses in various secondary lymphoid organs. Although splenic B cell function was impaired, B cell responses in LNs were largely intact. Well-developed GCs were observed in LNs by day 14 postinfection, and IgG production required T cell help, indicating that classical B cell GC responses were largely unaffected in LNs during E. muris infection. Thus, the suppression that we describe is likely relevant for understanding impaired immunity to other blood-borne pathogens that gain access to the spleen. In cases in which tick-borne pathogens first access the tissue-draining LNs, early IgG production may be unaffected (35); however, once a pathogen disseminates via the blood stream, the suppression of IgG production in the spleen could affect host defense. For example, the humoral response is required for immunity against infection with B. burgdorferi (36-38), given that the depletion of marginal zone (MZ) B cells (which are found only in the spleen) was responsible for a reduction in Borrelia-specific IgM and IgG, increased spirochete burden in the spleen, and early onset of Lyme arthritis (39). Another study demonstrated that B. burgdorferi infection caused the expansion of the MZ B cell subset, suggesting the importance of these B cells in controlling bacteremia (37). Thus, the local IgG suppression that we have observed during infection by E. muris, if it indeed occurs during other ehrlichial and/or rickettsial infections, may contribute to the suppression of IgG responses against coinfecting tick-borne pathogens, such as B. burgdorferi.
Impaired GC formation and IgG responses also have been documented in other bacterial (40), viral (41, 42), and parasitic infections (43, 44). Plasmodium berghei infection caused disruption of splenic and LN GC architecture during the first 10 d of infection (43). Impairment of the GC response was also reported during MMTV infection, although virus-specific IgG2c titers remained largely unaffected (41). A 3–4-wk delay in splenic GC formation was observed during Salmonella enterica serovar Typhimurium infection (40). Similar to our findings, systemic IgG was present during acute Salmonella infection, but the production of high-affinity isotype-switched Abs did not occur until the splenomegaly had been at least partially resolved. Several studies have also demonstrated that infections can impair the IgG response against irrelevant Ags (45-49). Although a number of mechanisms have been proposed, possible effects of infection on Ag-specific B cells and GC responses have, prior to this study, not been addressed. The findings that infections can impair IgG responses are broadly relevant to the issue of vaccination efficacy, especially in regions where pathogens are endemic (50).
Although GC B cells were detected at a low frequency in the spleen during early infection, appreciable numbers of GC B cells were detected in this tissue because of massive splenomegaly. Although it is possible that these spleen IgG-producing cells provide a significant portion of IgG produced during infection, our data suggest that the quality of the splenic IgG response is poor, given that distinct GC structures were detected only in the LNs and that PNA-binding B cells were found in low numbers and were randomly dispersed throughout the spleen. One prediction from our findings is that splenic B cells generated early during infection will fail to undergo normal TD processes, possibly including affinity maturation, although this has not yet been addressed.
We propose that the impairment of the GC responses is caused by a disruption of spleen architecture, and this loss of tissue integrity is what prevents the development of a GC response. Other studies from our laboratory have shown that the disruption of spleen architecture is temporally associated with extramedullary erythropoiesis and lymphopoiesis during acute E. muris infection (51). The latter events include the massive expansion of Ter119-positive prereticulocytes, as well as spleen extramedullary hematopoiesis. Such changes in structure and function could account for the impaired splenic IgG responses. Alternatively, suppressed T cell function, perhaps owing to impaired dendritic cell maturation, could be responsible; such suppression has been observed in other infection models (46, 48). Our data provide an argument against the possibility that the failed IgG responses are due to a paucity of Ag-specific CD4 T cells, given that we have observed an expansion of Ag-specific, IFNg-secreting T cells in the spleen during E. muris infection (16). We propose instead that Ag-specific CD4 T cells are unable to form proper associations with differentiating GC B cells, because of the loss of tissue integrity, as well as the extramedullary hematopoiesis that we have observed in the spleen.
Alteration of the inflammatory cytokine milieu may also contribute to IgG suppression. Increased production of the proinflammatory cytokine IL-12, after TD immunization of Fc receptor-associated γ-chain–deficient mice, was associated with an expansion of short-lived plasmablasts, but resulted in an impairment of GC B cell differentiation (52). Similarly, GC formation was impaired during Salmonella infection, until after the resolution of an extrafollicular plasmablast response (40). Although the role of IL-12 was not investigated in the latter study, Salmonella infection elicits a Th1 response and is associated with the production of proinflammatory cytokines (53). In our studies, IgG responses to NP-CGG immunization were most strongly affected when the Ag was administered at the peak of the plasmablast response. Both the proinflammatory response elicited during ehrlichial infection (54) and the disruption of spleen architecture probably favor the expansion of the CD11c-expressing plasmablasts and the subsequent suppression of GC production.
The origin of the CD11clo plasmablasts has not yet been resolved (11), but ongoing studies suggest that MZ B cells likely contribute to the expansion of this population (R. Racine and G.M. Winslow, unpublished data). Although MZ B cells provide an early source of IgM against blood-borne pathogens, by rapidly differentiating into plasmablasts (55), the B cell population can also participate in GC responses to produce isotype-switched Abs (56). Moreover, high cell surface levels of complement receptor 3, or CD21, on MZ B cells promotes the capture of immune complexes from the blood and transports the Ag to follicular dendritic cells in the spleen (57, 58). The Ag-immune complexes on the follicular dendritic cells are presented to follicular B cells, thereby initiating GC responses (59). Support for this hypothesis comes from Chattopadhyay et al. (60), who recently demonstrated that mice coimmunized with Streptococcus pneumoniae and soluble pneumococcal polysaccharide conjugate suppressed polysaccharide-specific IgG responses because of impaired trafficking of the conjugate from the MZ to the splenic follicles. It is also possible that defective MZ B cell trafficking of both ehrlichial and irrelevant Ags might account for the impaired GC and IgG responses reported in our studies. By day 8 after E. muris infection, both CD21 and CD23 cell surface expression was largely absent on all CD19+ splenic B cells, and surface expression was not restored until at least day 75 postinfection [(11), R. Racine and G.M. Winslow, unpublished data]. Although E. muris infection was associated with an expansion of IgM-positive B cells, the loss of CD21 expression might prevent the efficient capture and shuttling of Ag from the MZ to follicular dendritic cells, thus contributing to the localized suppression of splenic IgG. In contrast, the LNs, which lack MZ B cells, provide a more suitable environment for the formation of GCs and production of isotype-switched Abs.
Thus, E. muris infection causes, via several possible mechanisms, a localized suppression of the GC responses, and this suppression is likely responsible for the inhibition of class-switch recombination and the production of isotype-switched Abs. These finding are relevant to our understanding of how coinfections can exacerbate disease symptoms during a diverse range of microbial infections.
Acknowledgments
We thank the Wadsworth Center Immunology Core for technical assistance. We also thank Drs. Frances Lund (University of Rochester, Rochester, NY) and William Lee (Wadsworth Center) for constructive comments, Dr. Deborah Fuller (Albany Medical College, Albany, NY) for use of the CTL-Immunospot S5 Core Analyzer, and Dr. M. Shlomchik (Yale University, New Haven, CT) for providing the (m+s)Ig Tg mouse strain.
This work was supported by U.S. Public Health Service Grant R01AI064678 to G.M.W.
Abbreviations used in this paper
- GC
germinal center
- LN
lymph node
- MZ
marginal zone
- NP
4-hydroxy-3-nitrophenyl acetyl
- PNA
peanut agglutinin
- OMP-19
outermembrane protein 19
- RT
room temperature
- TD
T cell-dependent
Footnotes
Disclosures
The authors have no financial conflicts of interest.
References
- 1.MacLennan IC. Germinal centers. Annu. Rev. Immunol. 1994;12:117–139. doi: 10.1146/annurev.iy.12.040194.001001. [DOI] [PubMed] [Google Scholar]
- 2.Kelsoe G. The germinal center: a crucible for lymphocyte selection. Semin. Immunol. 1996;8:179–184. doi: 10.1006/smim.1996.0022. [DOI] [PubMed] [Google Scholar]
- 3.Klaus GG, Humphrey JH, Kunkl A, Dongworth DW. The follicular dendritic cell: its role in antigen presentation in the generation of immunological memory. Immunol. Rev. 1980;53:3–28. doi: 10.1111/j.1600-065x.1980.tb01038.x. [DOI] [PubMed] [Google Scholar]
- 4.Wu J, Qin D, Burton GF, Szakal AK, Tew JG. Follicular dendritic cell-derived antigen and accessory activity in initiation of memory IgG responses in vitro. J. Immunol. 1996;157:3404–3411. [PubMed] [Google Scholar]
- 5.Tarlinton DM, Smith KG. Dissecting affinity maturation: a model explaining selection of antibody-forming cells and memory B cells in the germinal centre. Immunol. Today. 2000;21:436–441. doi: 10.1016/s0167-5699(00)01687-x. [DOI] [PubMed] [Google Scholar]
- 6.Nossal GJ. Differentiation of the secondary B-lymphocyte repertoire: the germinal center reaction. Immunol. Rev. 1994;137:173–183. doi: 10.1111/j.1600-065x.1994.tb00664.x. [DOI] [PubMed] [Google Scholar]
- 7.Toellner KM, Gulbranson-Judge A, Taylor DR, Sze DM, MacLennan IC. Immunoglobulin switch transcript production in vivo related to the site and time of antigen-specific B cell activation. J. Exp. Med. 1996;183:2303–2312. doi: 10.1084/jem.183.5.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jacob J, Kassir R, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. I. The architecture and dynamics of responding cell populations. J. Exp. Med. 1991;173:1165–1175. doi: 10.1084/jem.173.5.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsu MC, Toellner KM, Vinuesa CG, Maclennan IC. B cell clones that sustain long-term plasmablast growth in T-independent extrafollicular antibody responses. Proc. Natl. Acad. Sci. U.S.A. 2006;103:5905–5910. doi: 10.1073/pnas.0601502103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.MacLennan IC, Toellner KM, Cunningham AF, Serre K, Sze DM, Zúñiga E, Cook MC, Vinuesa CG. Extrafollicular antibody responses. Immunol. Rev. 2003;194:8–18. doi: 10.1034/j.1600-065x.2003.00058.x. [DOI] [PubMed] [Google Scholar]
- 11.Racine R, Chatterjee M, Winslow GM. CD11c expression identifies a population of extrafollicular antigen-specific splenic plasmablasts responsible for CD4 T-independent antibody responses during intracellular bacterial infection. J. Immunol. 2008;181:1375–1385. doi: 10.4049/jimmunol.181.2.1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bitsaktsis C, Nandi B, Racine R, MacNamara KC, Winslow G. T-Cell-independent humoral immunity is sufficient for protection against fatal intracellular ehrlichia infection. Infect. Immun. 2007;75:4933–4941. doi: 10.1128/IAI.00705-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nandi B, Hogle K, Vitko N, Winslow GM. CD4 T-cell epitopes associated with protective immunity induced following vaccination of mice with an ehrlichial variable outer membrane protein. Infect. Immun. 2007;75:5453–5459. doi: 10.1128/IAI.00713-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mosmann TR, Coffman RL. TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu. Rev. Immunol. 1989;7:145–173. doi: 10.1146/annurev.iy.07.040189.001045. [DOI] [PubMed] [Google Scholar]
- 15.Cesta MF. Normal structure, function, and histology of the spleen. Toxicol. Pathol. 2006;34:455–465. doi: 10.1080/01926230600867743. [DOI] [PubMed] [Google Scholar]
- 16.Nandi B, Chatterjee M, Hogle K, McLaughlin M, MacNamara K, Racine R, Winslow GM. Antigen display, T-cell activation, and immune evasion during acute and chronic ehrlichiosis. Infect. Immun. 2009;77:4643–4653. doi: 10.1128/IAI.01433-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han S, Zheng B, Schatz DG, Spanopoulou E, Kelsoe G. Neoteny in lymphocytes: Rag1 and Rag2 expression in germinal center B cells. Science. 1996;274:2094–2097. doi: 10.1126/science.274.5295.2094. [DOI] [PubMed] [Google Scholar]
- 18.Laszlo G, Hathcock KS, Dickler HB, Hodes RJ. Characterization of a novel cell-surface molecule expressed on subpopulations of activated T and B cells. J. Immunol. 1993;150:5252–5262. [PubMed] [Google Scholar]
- 19.Martinez-Valdez H, Guret C, de Bouteiller O, Fugier I, Banchereau J, Liu YJ. Human germinal center B cells express the apoptosis-inducing genes Fas, c-myc, P53, and Bax but not the survival gene bcl-2. J. Exp. Med. 1996;183:971–977. doi: 10.1084/jem.183.3.971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.van Eijk M, Defrance T, Hennino A, de Groot C. Death-receptor contribution to the germinal-center reaction. Trends Immunol. 2001;22:677–682. doi: 10.1016/s1471-4906(01)02086-5. [DOI] [PubMed] [Google Scholar]
- 21.Oliver AM, Martin F, Kearney JF. Mouse CD38 is down-regulated on germinal center B cells and mature plasma cells. J. Immunol. 1997;158:1108–1115. [PubMed] [Google Scholar]
- 22.MacLennan IC, Gulbranson-Judge A, Toellner KM, Casamayor-Palleja M, Chan E, Sze DM, Luther SA, Orbea HA. The changing preference of T and B cells for partners as T-dependent antibody responses develop. Immunol. Rev. 1997;156:53–66. doi: 10.1111/j.1600-065x.1997.tb00958.x. [DOI] [PubMed] [Google Scholar]
- 23.Garside P, Ingulli E, Merica RR, Johnson JG, Noelle RJ, Jenkins MK. Visualization of specific B and T lymphocyte interactions in the lymph node. Science. 1998;281:96–99. doi: 10.1126/science.281.5373.96. [DOI] [PubMed] [Google Scholar]
- 24.Rose ML, Birbeck MS, Wallis VJ, Forrester JA, Davies AJ. Peanut lectin binding properties of germinal centres of mouse lymphoid tissue. Nature. 1980;284:364–366. doi: 10.1038/284364a0. [DOI] [PubMed] [Google Scholar]
- 25.Takahashi Y, Dutta PR, Cerasoli DM, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. V. Affinity maturation develops in two stages of clonal selection. J. Exp. Med. 1998;187:885–895. doi: 10.1084/jem.187.6.885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shih TA, Meffre E, Roederer M, Nussenzweig MC. Role of BCR affinity in T cell dependent antibody responses in vivo. Nat. Immunol. 2002;3:570–575. doi: 10.1038/ni803. [DOI] [PubMed] [Google Scholar]
- 27.Levine MH, Haberman AM, Sant’Angelo DB, Hannum LG, Cancro MP, Janeway CA, Jr., Shlomchik MJ. A B-cell receptor-specific selection step governs immature to mature B cell differentiation. Proc. Natl. Acad. Sci. U.S.A. 2000;97:2743–2748. doi: 10.1073/pnas.050552997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hannum LG, Haberman AM, Anderson SM, Shlomchik MJ. Germinal center initiation, variable gene region hypermutation, and mutant B cell selection without detectable immune complexes on follicular dendritic cells. J. Exp. Med. 2000;192:931–942. doi: 10.1084/jem.192.7.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wolniak KL, Noelle RJ, Waldschmidt TJ. Characterization of (4-hydroxy-3-nitrophenyl)acetyl (NP)-specific germinal center B cells and antigen-binding B220- cells after primary NP challenge in mice. J. Immunol. 2006;177:2072–2079. doi: 10.4049/jimmunol.177.4.2072. [DOI] [PubMed] [Google Scholar]
- 30.White-Scharf ME, Imanishi-Kari T. Characterization of the NPa idiotype through the analysis of monoclonal BALB/c anti-(4-hydroxy-3-nitrophenyl)acetyl (NP) antibodies. Eur. J. Immunol. 1981;11:897–904. doi: 10.1002/eji.1830111109. [DOI] [PubMed] [Google Scholar]
- 31.Swanson SJ, Neitzel D, Reed KD, Belongia EA. Coinfections acquired from ixodes ticks. Clin. Microbiol. Rev. 2006;19:708–727. doi: 10.1128/CMR.00011-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Levin ML, Fish D. Acquisition of coinfection and simultaneous transmission of Borrelia burgdorferi and Ehrlichia phagocytophila by Ixodes scapularis ticks. Infect. Immun. 2000;68:2183–2186. doi: 10.1128/iai.68.4.2183-2186.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Holden K, Hodzic E, Feng S, Freet KJ, Lefebvre RB, Barthold SW. Coinfection with Anaplasma phagocytophilum alters Borrelia burgdorferi population distribution in C3H/HeN mice. Infect. Immun. 2005;73:3440–3444. doi: 10.1128/IAI.73.6.3440-3444.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thomas V, Anguita J, Barthold SW, Fikrig E. Coinfection with Borrelia burgdorferi and the agent of human granulocytic ehrlichiosis alters murine immune responses, pathogen burden, and severity of Lyme arthritis. Infect. Immun. 2001;69:3359–3371. doi: 10.1128/IAI.69.5.3359-3371.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nithiuthai S, Allen JR. Langerhans cells present tick antigens to lymph node cells from tick-sensitized guinea-pigs. Immunology. 1985;55:157–163. [PMC free article] [PubMed] [Google Scholar]
- 36.Schaible UE, Wallich R, Kramer MD, Nerz G, Stehle T, Museteanu C, Simon MM. Protection against Borrelia burgdorferi infection in SCID mice is conferred by presensitized spleen cells and partially by B but not T cells alone. Int. Immunol. 1994;6:671–681. doi: 10.1093/intimm/6.5.671. [DOI] [PubMed] [Google Scholar]
- 37.Malkiel S, Kuhlow CJ, Mena P, Benach JL. The loss and gain of marginal zone and peritoneal B cells is different in response to relapsing fever and Lyme disease Borrelia. J. Immunol. 2009;182:498–506. doi: 10.4049/jimmunol.182.1.498. [DOI] [PubMed] [Google Scholar]
- 38.McKisic MD, Barthold SW. T-cell-independent responses to Borrelia burgdorferi are critical for protective immunity and resolution of lyme disease. Infect. Immun. 2000;68:5190–5197. doi: 10.1128/iai.68.9.5190-5197.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Belperron AA, Dailey CM, Booth CJ, Bockenstedt LK. Marginal zone B-cell depletion impairs murine host defense against Borrelia burgdorferi infection. Infect. Immun. 2007;75:3354–3360. doi: 10.1128/IAI.00422-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cunningham AF, Gaspal F, Serre K, Mohr E, Henderson IR, Scott-Tucker A, Kenny SM, Khan M, Toellner KM, Lane PJ, MacLennan IC. Salmonella induces a switched antibody response without germinal centers that impedes the extracellular spread of infection. J. Immunol. 2007;178:6200–6207. doi: 10.4049/jimmunol.178.10.6200. [DOI] [PubMed] [Google Scholar]
- 41.Luther SA, Gulbranson-Judge A, Acha-Orbea H, MacLennan IC. Viral superantigen drives extrafollicular and follicular B cell differentiation leading to virus-specific antibody production. J. Exp. Med. 1997;185:551–562. doi: 10.1084/jem.185.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pantaleo G, Graziosi C, Demarest JF, Butini L, Montroni M, Fox CH, Orenstein JM, Kotler DP, Fauci AS. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature. 1993;362:355–358. doi: 10.1038/362355a0. [DOI] [PubMed] [Google Scholar]
- 43.Carvalho LJ, Ferreira-da-Cruz MF, Daniel-Ribeiro CT, Pelajo-Machado M, Lenzi HL. Germinal center architecture disturbance during Plasmodium berghei ANKA infection in CBA mice. Malar. J. 2007;6:59. doi: 10.1186/1475-2875-6-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Achtman AH, Khan M, MacLennan IC, Langhorne J. Plasmodium chabaudi chabaudi infection in mice induces strong B cell responses and striking but temporary changes in splenic cell distribution. J. Immunol. 2003;171:317–324. doi: 10.4049/jimmunol.171.1.317. [DOI] [PubMed] [Google Scholar]
- 45.Reed SG, Roters SB, Goidl EA. Spleen cell-mediated suppression of IgG production to a non-parasite antigen during chronic Trypanosoma cruzi infection in mice. J. Immunol. 1983;131:1978–1982. [PubMed] [Google Scholar]
- 46.Millington OR, Di Lorenzo C, Phillips RS, Garside P, Brewer JM. Suppression of adaptive immunity to heterologous antigens during Plasmodium infection through hemozoin-induced failure of dendritic cell function. J. Biol. 2006;5:5. doi: 10.1186/jbiol34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cadman ET, Abdallah AY, Voisine C, Sponaas AM, Corran P, Lamb T, Brown D, Ndungu F, Langhorne J. Alterations of splenic architecture in malaria are induced independently of Toll-like receptors 2, 4, and 9 or MyD88 and may affect antibody affinity. Infect. Immun. 2008;76:3924–3931. doi: 10.1128/IAI.00372-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ostrowski M, Vermeulen M, Zabal O, Geffner JR, Sadir AM, Lopez OJ. Impairment of thymus-dependent responses by murine dendritic cells infected with foot-and-mouth disease virus. J. Immunol. 2005;175:3971–3979. doi: 10.4049/jimmunol.175.6.3971. [DOI] [PubMed] [Google Scholar]
- 49.Poudrier J, Weng X, Kay DG, Paré G, Calvo EL, Hanna Z, Kosco-Vilbois MH, Jolicoeur P. The AIDS disease of CD4C/HIV transgenic mice shows impaired germinal centers and autoantibodies and develops in the absence of IFN-gamma and IL-6. Immunity. 2001;15:173–185. doi: 10.1016/s1074-7613(01)00177-7. [DOI] [PubMed] [Google Scholar]
- 50.Radwanska M, Guirnalda P, De Trez C, Ryffel B, Black S, Magez S. Trypanosomiasis-induced B cell apoptosis results in loss of protective anti-parasite antibody responses and abolishment of vaccine-induced memory responses. PLoS Pathog. 2008;4:e1000078. doi: 10.1371/journal.ppat.1000078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.MacNamara KC, Racine R, Chatterjee M, Borjesson D, Winslow GM. Diminished hematopoietic activity associated with alterations in innate and adaptive immunity in a mouse model of human monocytic ehrlichiosis. Infect. Immun. 2009;77:4061–4069. doi: 10.1128/IAI.01550-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim SJ, Caton M, Wang C, Khalil M, Zhou ZJ, Hardin J, Diamond B. Increased IL-12 inhibits B cells’ differentiation to germinal center cells and promotes differentiation to short-lived plasmablasts. J. Exp. Med. 2008;205:2437–2448. doi: 10.1084/jem.20070731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raupach B, Kaufmann SH. Bacterial virulence, proinflammatory cytokines and host immunity: how to choose the appropriate Salmonella vaccine strain? Microbes Infect. 2001;3:1261–1269. doi: 10.1016/s1286-4579(01)01486-1. [DOI] [PubMed] [Google Scholar]
- 54.Ismail N, Soong L, McBride JW, Valbuena G, Olano JP, Feng HM, Walker DH. Overproduction of TNF-alpha by CD8+ type 1 cells and down-regulation of IFN-gamma production by CD4+ Th1 cells contribute to toxic shock-like syndrome in an animal model of fatal monocytotropic ehrlichiosis. J. Immunol. 2004;172:1786–1800. doi: 10.4049/jimmunol.172.3.1786. [DOI] [PubMed] [Google Scholar]
- 55.Martin F, Oliver AM, Kearney JF. Marginal zone and B1 B cells unite in the early response against T-independent blood-borne particulate antigens. Immunity. 2001;14:617–629. doi: 10.1016/s1074-7613(01)00129-7. [DOI] [PubMed] [Google Scholar]
- 56.Song H, Cerny J. Functional heterogeneity of marginal zone B cells revealed by their ability to generate both early antibody-forming cells and germinal centers with hypermutation and memory in response to a T-dependent antigen. J. Exp. Med. 2003;198:1923–1935. doi: 10.1084/jem.20031498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guinamard R, Okigaki M, Schlessinger J, Ravetch JV. Absence of marginal zone B cells in Pyk-2-deficient mice defines their role in the humoral response. Nat. Immunol. 2000;1:31–36. doi: 10.1038/76882. [DOI] [PubMed] [Google Scholar]
- 58.Cinamon G, Zachariah MA, Lam OM, Foss FW, Jr., Cyster JG. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat. Immunol. 2008;9:54–62. doi: 10.1038/ni1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ferguson AR, Youd ME, Corley RB. Marginal zone B cells transport and deposit IgM-containing immune complexes onto follicular dendritic cells. Int. Immunol. 2004;16:1411–1422. doi: 10.1093/intimm/dxh142. [DOI] [PubMed] [Google Scholar]
- 60.Chattopadhyay G, Chen Q, Colino J, Lees A, Snapper CM. Intact bacteria inhibit the induction of humoral immune responses to bacterial-derived and heterologous soluble T cell-dependent antigens. J. Immunol. 2009;182:2011–2019. doi: 10.4049/jimmunol.0802615. [DOI] [PubMed] [Google Scholar]