

Abstract
The DNA binding activity of the transcriptional regulator activator protein-1 shows considerable promise as a target in cancer therapy. A number of different strategies have been employed to inhibit the function of this protein with promise having been demonstrated both in vitro and in vivo. Peptide-based therapeutics have received renewed interest in the last few years, and a number of 37-amino acid peptides capable of binding to the coiled coil dimerization domain of Jun and Fos have been derived. Here, we demonstrate how truncation and semi-rational library design, followed by protein-fragment complementation, can be used to produce a leucine zipper binding peptide by iterative means. To this end, we have implemented this strategy on the FosW peptide to produce 4hFosW. This peptide is truncated by four residues with comparably favorable binding properties and demonstrates the possibility to design progressively shorter peptides to serve as leucine zipper antagonists while retaining many of the key features of the parent peptide. Whether or not the necessity for low molecular weight antagonists is required from the perspective of druggability and efficacy is subject to debate. However, antagonists of reduced length are worthy of perusal from the point of view of synthetic cost as well as identifying the smallest functional unit that is required for binding.
Keywords: AP-1 Transcription Factor, Circular Dichroism (CD), Peptide Interactions, Protein Stability, Protein-Protein Interactions, Library Design, Coiled Coil, Protein Design
Introduction
The basic region leucine zipper transcription factors are a family of dimeric DNA-binding proteins containing a transactivation domain, a DNA binding basic region, and a leucine zipper region that mediates interaction of the two proteins. Dimeric complexes of the Fos and Jun family combine to form the transcriptional regulator, activator protein 1 (AP-1). AP-1 can be composed of a range of dimeric complexes (1) that have an abundance of roles in gene regulation and tissue development (2); however, their key universal function is in binding to the 12-O-tetradecanoylphorbol-13-acetate (TPA)2-response element (TRE) sequence (3) resulting in the activation of a number of genes. However, pathological events can lead to the overexpression of AP-1 causing unregulated proliferation or differentiation and therefore cancer. This well established central role in oncogenesis has positioned AP-1, which is one of the final steps involved in signal transduction, as an exciting target in cancer therapy (4). In particular, c-Jun has been widely studied and was initially linked to cancer due to initial discovery as a homologue of the v-Jun viral oncoprotein (5, 6). The leucine zipper region that drives dimerization, and on which our antagonists are based, consists of supercoiled helices. These helices interact such that there are 3.5 residues per turn in each helix, so forming a heptad repeat (described by the nomenclature abcdefg). In the heptad repeat a and d residues are found at the dimer interface and provide a hydrophobic core. The a position is found to contain a variety of hydrophobic residues (7) with occasional selection of polar Asn/Lys residues to aid with specificity constraints (8), and the d positions are almost exclusively restricted to Leu (9). Positions e and g form electrostatic inter-helical contacts (10), which help to mediate specificity and are thought to constitute a late folding event that predominantly affects the dissociation rate of the complex (11). Finally, the b, c, and f positions of the heptad repeat make up the backbone of the coiled coil structure. These residues are positioned away from the dimer interface and are thought to play a more subtle role in coiled coil stability, for example by assisting with intra-helical stability by forming electrostatic interactions between flanking residues within the same helix and consequently, by improving helical propensity as has been used in other coiled coil design strategies (12).
To investigate the potential for reducing the size and length of the peptide antagonist while retaining c-Jun-antagonist interaction stability, we have performed a cycle of truncation of the parent peptide, followed by re-randomization and screening of the library using a protein fragment complementation approach. We have used the FosW as our framework for design, which is a peptide that was previously selected to bind to c-Jun with high affinity (Tm = 63 °C) and that is based on the parallel and dimeric wild-type Jun-Fos coiled coil interaction (13). The work presented here will describe the rationale behind truncation of the parent peptide and the library design. In addition, we will use a range of biophysical techniques to demonstrate that the helices interact and will provide a measure of the affinity of this interaction.
EXPERIMENTAL PROCEDURES
Library Design and Creation
Library design and cloning have been described elsewhere (13–15). Briefly, megaprimers were synthesized, including relevant degenerate codons for library residue options, and a fill-in reaction was performed, resulting in 111-bp double-stranded oligonucleotides. These were cloned via NheI and AscI sites into a pQE16 derivative (Qiagen) containing a G/S linker tagged to fragment 1 (pES230d; library; ampicillin resistance (AmpR)) and fragment 2 (pES300d; c-Jun target chloramphenicol resistance (CmR)) of murine DHFR, respectively, resulting in pES230d-4hFosW-library-DHFR1 and pES300d-c-Jun-DHFR2, respectively.
Winner Selection
The 4hFosW winner sequence was selected by PCA from a 49,152 member library (see Fig. 1) designed from the truncation of the final four residues of FosW (13). The PCA protocol is described in detail elsewhere (8). Briefly, the library and target peptides are genetically fused to two halves of a split of murine dihydrofolate reductase (DHFR) (16). Interaction between a library member and the target peptide reconstitutes the DHFR activity (reduction of dihydrofolate and subsequent synthesis of nucleotides). This reconstitution of activity is relative to the interaction affinity. Therefore, in media deficient in complex nutrients and containing trimethoprim (which selectively inhibits bacterial DHFR), only those bacterial cells that contain a library member that interacts with the target peptide will grow in the media. Transformed cells containing library members with the highest interaction affinity for the target protein will grow most rapidly and after successive passages in liquid media will predominate in the bacterial pool. Expression of target and library proteins were under control of a lac promoter, with expression induced using 1 mm isopropyl β-d-1-thiogalactopyranoside.
FIGURE 1.

Helical wheel representation and peptide sequence comparison. A, helical wheel representation highlighting the interaction patterns for the FosW-c-Jun and 4hFosW-c-Jun heterodimers. Residues for FosW (left) and c-Jun (right) are colored black. Library options for the 4hFosW library that differ from the FosW template are shown in gray. Residues present in FosW that have been truncated in 4hFosW are shown in parentheses. Library design and PCA selection (circled) residues in the 49,152 member 4hFosW library are also shown. B represents a linear arrangement of the interacting helices, as well as the position of residues within the 4.5 (c-Jun and FosW) or 4 (4hFosW) heptads of the individual helices. The arrows indicate potential g/e′ interactions between the helices in the heterodimer. The three library selected positions of 4hFosW that differ from FosW are underlined. These are Ala to Arg at f1, Arg to Leu at g2, and Arg to Gln at e3. Also shown are N- and C-terminal capping motifs to aid helix stability.
Confirmation of Winner by Growth Curve Assay
Those colonies with the strongest antagonist-target interaction allowed the most rapid growth under PCA conditions. Therefore, bacterial growth rates resulting from bacteria expressing 4hFosW and c-Jun were compared with those expressing wild-type c-Fos and c-Jun. Cell lines for this assay were prepared from single colonies taken from plates under PCA conditions and corresponded to the correctly sequenced plasmids.
Peptide Synthesis and Purification
Peptides were synthesized by Protein Peptide Research Ltd. (Fareham, UK) and subsequently purified to >98% purity using RP-HPLC with a Jupiter Proteo column (4-μm particle size, 90-Å pore size, 250 × 10-mm (Phenomenex)) and a gradient from 5 to 50% acetonitrile (0.1% TFA) over 50 min at a rate of 1.5 ml/min. Correct masses were verified by electrospray mass spectrometry. The following peptides were synthesized as amidated and acetylated and generally contained N- and C-capping motifs (underlined) for improved helix stability and solubility: c-Jun ASIARLEEKVKTLKAQNYELASTANMLREQVAQLGAP; c-Fos ASTDTLQAETDQLEDEKYALQTEIANLLKEKEKLGAP; FosW ASLDELQAEIEQLEERNYALRKEIEDLQKQLEKLGAP; 4hFosW ASLDELQREIEQLEELNYALQKEIEDLQKQGAP; basic c-Jun RIKAERKRMRNRIAASKCRKRKLERIARLEEKVKTLKAQNYELASTANMLREQVAQLGAP; and basic c-Fos EEKRRIRRERNKMAAAKCRNRRRELTDTLQAETDQLDEKYALQTEIANLLKEKEKLGAP. Peptide concentrations were determined by weighing and were confirmed in water using the absorbance at 280 nm with an extinction coefficient of 1209 m−1 cm−1 (17) corresponding to a Tyr residue inserted into a solvent-exposed b3 heptad position.
Circular Dichroism
Circular dichroism (CD) was carried out using an Applied Photophysics Chirascan CD apparatus (Leatherhead, UK) using a 200-μl sample in a CD cell with a 1-mm path length. Samples contained 150 μm total peptide (Pt) concentration suspended in 10 mm potassium phosphate and 100 mm potassium fluoride at pH 7. The CD spectra of the homodimeric c-Jun and 4hFosW complexes and the heterodimeric complex were scanned between 300 and 190 nm at 20 °C (for both pre- and postmelt samples) and at −8 °C to assess helical levels and coiled coil structure (see Fig. 2). All data have been converted from raw ellipticity to molar residue ellipticity according to Equation 1,
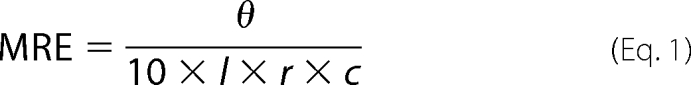 |
where θ is the CD signal of the sample in millidegrees; l is the path length of the cell in centimeters; r is the number of residues in the peptide; and c is the total peptide concentration of the sample in molar.
FIGURE 2.

CD spectra of 4hFosW homodimeric (A) and 4hFosW-c-Jun heterodimeric (B) complexes. Peaks at 222 and 208 nm are indicative of a helical structure with the 222/208 ratio providing evidence for the formation of a coiled coil structure (see Table 1). Overlapping of the spectra at 20 °C in both cases shows the structure is able to reconstitute following thermal denaturation. Increased stability of the structure at −8 °C is consistent with further stabilization at lower temperatures. The greater magnitude of the peak at 222 nm in the heterodimeric complex (B) (approximately −112 millidegrees) compared with the homodimeric complex (A) (approximately −87 millidegrees) at 20 °C suggests that the heterodimeric complex is more stable than the homodimeric 4hFosW and that 4hFosW will preferentially form a heterodimer with c-Jun.
Thermal Melts
Spectra and thermal melts were performed at 150 μm Pt in 10 mm potassium phosphate, 100 mm potassium fluoride, pH 7, using an Applied Photophysics Chirascan CD instrument (Leatherhead, UK). The temperature ramp was set to stepping mode using 1 °C increments and paused for 30 s at each temperature before measuring ellipticity at 222 nm. For all temperature denaturation experiments, data collection was started at −8 °C, and at this temperature, the peptide solutions remained aqueous. Data points for thermal denaturation profiles represent the averaged signal after 4 s of data collection. Samples were identical in composition to the CD buffer samples. Melting profiles (see Fig. 3) were ≥95% reversible with equilibrium denaturation curves fitted to a two-state model, derived via modification of the Gibbs-Helmholtz equation (18, 19), to yield the melting temperature (Tm) (Equation 2).
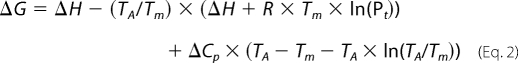 |
where ΔH is the change in enthalpy; TA is the reference temperature in kelvin; R is the ideal gas constant (1.9872 cal·mol−1·K−1); Pt the total peptide concentration (150 μm); and ΔCp the change in heat capacity. Melting profiles for heterodimers are clearly distinct from averages of constituent homodimeric melts (shown in Fig. 3 and via dimer exchange in Fig. 4), indicating that helices are heterodimerizing. The cooperative nature of the melting profiles suggests an apparent two-state process. Tm values were determined by least squares fitting of the denaturation assuming a two-state folding model that is widely used for coiled coils (19) and provided an excellent fit to our data.
FIGURE 3.

Thermal denaturation profiles. Denaturation profiles for homodimeric c-Jun (closed circles) as well as 4hFosW (closed triangles) and c-Jun-4hFosW (open squares) are shown (see also Table 1). All experiments were undertaken in 10 mm potassium phosphate, 100 mm potassium fluoride at pH 7. The total peptide concentration for all experiments was 150 μm. Data were fitted to the two-state model (Equation 2). FU, fraction unfolded.
FIGURE 4.

ITC analysis of leucine zipper domain interactions between c-Jun and c-Fos (A) as well as the interaction of c-Jun with FosW (B) and 4hFosW (C) antagonists. The top and bottom panels show raw data after base-line correction. During ITC experiments, ∼200–600 μm peptide A was injected in 30–40× 5-μl batches from the injection syringe into the cell, which contained 10–40 μm of peptide B. Both partners were in a 10 mm potassium phosphate buffer, 100 mm potassium fluoride at pH 7. Experiments were undertaken at 20 °C. The solid lines represent the fit of the data to the function based on the binding of a ligand to a macromolecule using the MicroCal (GE Healthcare) Origin software (20).
ITC Measurements
ITC measurements were made using a MicroCal VP-ITC instrument with data collected and processed using the Origin 7.0 software package. All measurements were carried out at least two times. Briefly, all peptides were studied at 20 °C in 10 mm potassium phosphate and 100 mm potassium fluoride at pH 7. 600 μl of peptide 1 was loaded into the syringe at between 175 and 250 μm peptide concentration. 1800 μl of peptide 2 was loaded into the cell at 10–40 μm. The peptide in the syringe and cell was reversed to check that the results were unaffected by this change. The experiment was undertaken by injecting 5 μl ×25 injections of peptide 1 into the calorimetric cell. The change in thermal power as a function of each injection was automatically recorded using the MicroCal Origin software (20), and the raw data were integrated to yield ITC isotherms of heat release per injection as a function of Fos to Jun molar ratio. In general, the concentration of peptide 2 loaded into the cell was 30 times the anticipated protein-protein interaction KD value, and the concentration of peptide 1 in the syringe was at least 20 times the concentration of peptide 2. No precipitation of protein was observed in any of the experiments undertaken. Following ITC measurements, the data were fit to a one-site model (Equation 3),
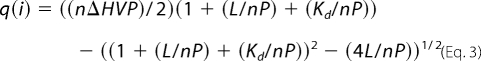 |
where q(i) is the heat release (kcal/mol) for the ith injection; n is the stoichiometry of heterodimerization; V is the effective volume of protein sample loaded into the calorimetric cell (1.46 ml); P is the total Jun concentration in the calorimetric cell (in micromolars), and L is the total Fos concentration in the calorimetric cell at the end of each injection (in micromolars). This model is derived from the binding of a ligand to a macromolecule using the law of mass action (assuming a 1:1 stoichiometry of 4hFosW to c-Jun) to extract the various thermodynamic parameters (20, 21), namely the apparent equilibrium constant (Kd) and the enthalpy change (ΔH) associated with heterodimerization. By iteratively fitting the binding isotherms to the above, one is able to generate values for Kd and ΔH. The free energy change (ΔGbind) upon ligand binding can then be calculated from the relationship shown in Equation 4,
where R is the universal molar gas constant (1.9872 cal·K−1·mol−1); T is the absolute temperature in kelvin (293.15 K); and KD is the dissociation constant of binding with units of mol·liter−1. Finally, the entropic contribution (TΔS) to the free energy of binding was calculated using the derived values of ΔH and ΔGbind.
Electrophoretic Mobility Shift Assay (EMSA)
Pure basic c-Jun and basic c-Fos proteins were mixed at a ratio of 1:1 with 70 μm of each protein and either the presence or absence of a 1:2 or 1:7 molar ratio of 4hFosW before being added to 100 ng of DNA (14 μm) containing the AP-1 TRE consensus sequence (5′-CGCTTGATGACTCAGCCGGAA-3′). Protein-DNA samples were then incubated on ice for 30 min in 1× binding buffer (700 mm KCl, 2.5 mm DTT, 2 mm EDTA, 25 mm Tris-HCl, 10 mm MgCl2, and 2% glycerol). The samples were run on a 6.5% TBE nondenaturing polyacrylamide gel in 0.5× TBE buffer before being stained with SYBR® Green for 30 min to stain for DNA and imaged on a transilluminator followed by 3 h of staining with SYPRO® Ruby followed by destaining and imaging on a transilluminator to monitor for the colocalization of DNA and protein in shifted bands.
Free Energy Changes from the Literature
Calculated differences between coiled coil values at 37 °C (Table 3) have been previously reported in Krylov et al. (22) for electrostatic gi − e′i+1 and ei+1 − g′i contributions and Acharya et al. (7) for hydrophobic a-a′ contributions. In these publications, free energies were calculated relative to Ala-Ala pairs. It should be noted that these values have been averaged from the data reported for g/ei+1 and ei+/g.
RESULTS
We have previously derived a range of peptides capable of binding to c-Jun (13, 15, 23). c-Jun is a particularly interesting target because sequestering it will release the Fos homologue, with which it predominantly heterodimerizes. Fos members are thought to be unable to homodimerize in vivo and are therefore less likely to give rise to an oncogenic phenotype (24). All previous peptide antagonists have been 37 residues in length, consisting of 4.5 heptads (32 residues) and flanked by an N-capping and a C-capping motif. We are now seeking to reduce the length and molecular weight of our derived antagonists while retaining a suitably high free energy of binding to the target peptide. The c-Jun target has been kept at 37 residues as this incorporates the entire coiled coil region of the peptide and therefore represents the target zipper as it would be found in vivo. Consequently, truncation of c-Jun would unnecessarily destabilize the interaction with the antagonist. Here, we describe the truncation of four amino acids from the C terminus of the FosW peptide (while retaining the C-capping motif; see Fig. 1) followed by a re-randomization of key positions within the 33-residue 4hFosW library peptide scaffold. Shown in Fig. 1A is the FosW and 4hFosW helix as would be found in a coiled coil interaction with c-Jun. The four truncated residues in FosW are shown in Fig. 1A in parentheses. Fig. 1B displays complete sequence alignments and includes capping motifs and predicted e-g interactions.
4hFosW Library Randomization
Library design was through a semi-rational approach, because of the need to explore residue selections that may enhance affinity, while maintaining the library to a manageable size and sampling useful space where possible. LZ residue preferences have been widely explored with regard to inter-helical contacts (7, 10), and helical propensity scales have been developed to allow structural prediction (25, 26), which allow libraries to be developed to test the trade offs of different factors in coiled coil stability. Through this approach, the Arg residue at position g2 was randomized using the codon vkh to give options of Leu, Ile, Val, Arg, Ser, and Gly, which is a library including long chain hydrophobic Leu, Ile, and Val residues while retaining the Arg option in the parent molecule (and consequentially including the unfavorable Ser and Gly residues, which were not expected to be selected). Leu, Ile, and Val were specifically included to test for a preference for long chain hydrophobic residues situated to interact with the adjacent Ala residue at e3 and would also be predicted to pack into the hydrophobic pocket formed by the ethyl groups of adjacent e2 and e4 Glu residues. Positions f1 and c3 have been randomized using the codon ssn to generate Ala, Arg, Gly, and Pro options. Inclusion of Ala generates the possibility of retaining high levels of helicity, although Arg is a logical option given the negative charge on residues that flank both of these positions. Inclusion of Gly/Pro is a by-product of Ala/Arg options but would be predicted to be helix-destabilizing and therefore not favored at these positions. Finally, positions g1, e3, and e4 were randomized using the codon svm to generate options for Arg, Glu, Ala, Pro, Gly, Asp, His, and Gln options at these typically electrostatic positions. This codon therefore generates both positively charged (Arg), negatively charged (Glu/Asp), and polar (Gln) electrostatic options, helical options (Ala) as well as His and the Gly/Pro that would not be predicted to be selected.
4hFosW Library Selection
Following construction of the libraries, single step PCA was undertaken followed by sequential liquid growths. This process identified a single sequence by the 5th passage of liquid growth competition. Of the six randomizations described above, we observed that three positions (g1, c3, and e4) re-selected the same residue found in the FosW parent peptide. For position e4, Gln arose from a smaller subset of options at this position in the selection of the original FosW peptide (13) demonstrating that the original selection is also favored over an extended range of residues in this library. The remaining three positions, f1, g2, and e3, constituted Ala → Arg, Arg → Leu, and Arg → Gln selections, respectively. The first of these changes occurs at a solvent-exposed position. Ala at this position is flanked by Asp and Glu at positions b1 and c1 as well as Glu at position f2 of the next heptad, and so a preference for this change would be anticipated to improve intrahelical interactions within the 4hFosW helix and therefore the stability of the coiled coil complex. The second of these changes occurs at a g2 position that has the potential to make a favorable interaction with e′3 residue on the c-Jun target helix. In this target helix, these positions are usually occupied by a polar residue of opposite charge to facilitate favorable electrostatic interactions. Because Ala occupies this position in c-Jun, the Arg option present in FosW was included with Leu, Ile, and Val, options additionally included to generate branched hydrophobic options that can make contact with the short Ala side chain and generate a favorable hydrophobic interaction. This hydrophobic association is then able to assist with desolvation of the core residues and perhaps providing a favorable a′-g interaction, a contributory factor that has been speculated to play a role in specificity determination (27). The last of the three changes from the FosW parent peptide is an Arg to Gln change found at position e3, which is predicted to interact with another Gln at position g′2 of c-Jun. For this electrostatic interaction, the contribution to the free energy of binding has been experimentally determined and is predicted to be 0.5 kcal/mol more stable for Gln-Gln (−1.2 kcal/mol) than Arg-Gln (−0.7 kcal/mol) (22).
Circular Dichroism Experiments
CD spectra taken of both 4hFosW in isolation and in heterodimeric form with c-Jun indicate the presence of a coiled coil structure. Minima are present at 222 and 208 nm, indicating the presence of a helical structure, and in particular, the ratio between these two values is ∼1.0 (see Table 1). Typically a value of less than 0.9 indicates the presence of an α-helix in isolation, as is found with c-Jun-c-Fos, which is found as a mixture of coiled coil and monomeric helices at values close to this temperature (28, 29). Indeed, thermal melt analysis indicates that this coiled coil containing dimerization region for the wild-type c-Jun-c-Fos has a Tm of 16 °C (homodimeric c-Jun Tm = 24 °C) (13). In contrast, homodimeric 4hFosW displays a Tm of 36 °C. Thus, the observed Tm of 49 °C for the designed c-Jun·4hFosW complex far exceeds the average of the c-Jun and 4hFosW constituent homodimeric melts that would be predicted if no heterodimeric interaction were observed ((24 °C + 36 °C)/2 = 30 °C).
TABLE 1.
Thermodynamic data from thermal melt analysis, ITC, and θ222/θ208 ratios and measured helicity taken from CD spectra
The helicity of c-Jun, FosW, and 4hFosW in isolation is calculated to be 12.9, 43.7, and 50.6%, respectively. Tm has been estimated from a fit to thermal melting data using Equation 2, from which values for KD and ΔGbind(Thermal) were obtained. ITC data were obtained at 20 °C with a MicroCal VP-ITC using a fit to Equation 3. The θ222/θ208 ratios provide information on the likelihood of the α-helix being in isolation or found within a coiled coil structure (28, 29, 47). A ratio greater than 1.0 typically indicates the latter, and a ratio of approximately 0.9 or less indicates the presence of a helix in isolation. With the exception of c-Jun-c-Fos, both c-Jun-antagonist interactions are greater than 0.9, thus supporting the formation of a coiled coil structure. Fraction helicity (fH) can be calculated as fH = (θ222 − qc)/(θ222∞ − qc), where θ222∞ = (−44,000 + 250T)·(1 − k/Nr) and θc = 2220 − (53·T). In these equations, the wavelength-dependent constant k = 2.4 (at 222 nm), Nr = the number of residues, and T = 20 °C (293 K). Agadir (48, 49) severely underestimates helicity for many of the dimeric pairs, most likely because it does not take into account that interhelical interactions assist with helix integrity in the dimeric pairs; it only considers the helicity of individual helices in isolation. Thus, the measured helicity is often higher than the values predicted from the average of the two constituent helices by Agadir.
| c-Jun-c-Fos | c-Jun-FosW | c-Jun-4hFosW | |
|---|---|---|---|
| Tm | 16 | 63 | 49 |
| KD (thermal) | 26.6 μm | 45 nM | 487 nM |
| ΔGbind (thermal) | −5.5 kcal/mol | −11.4 kcal/mol | −8.5 kcal/mol |
| N | 1.1 ± 0.01 | 0.65 ± 0.01 | 1.05 ± 0.01 |
| KB(20 °C) m−1 (ITC) | 3.8e4 ± 2.5e4 | 2.2e7 ± 4.3e6 | 2.1e6 ± 2.0e5 |
| ΔGbind (ITC) | −6.1 ± 0.4 kcal/mol | −9.9 ± 0.1 kcal/mol | −8.8 ± 0.1 kcal/mol |
| ΔH | −0.8 ± 0.4 kcal/mol | −10.6 ± 0.2 kcal/mol | −14.1 ± 0.2 kcal/mol |
| TΔS (ΔH − ΔG) | 5.3 ± 0.5 kcal/mol | −0.7 ± 0.2 kcal/mol | −5.3 ± 0.2 kcal/mol |
| θ222/θ208 (20 °C) | 0.75 | 1.0 | 0.99 |
| Fraction helical (fH) | 3.6% | 35.8% | 60.4% |
Predicted and Measured Helicity
The PCA winner sequence is predicted to give a Tm of 32 °C according to bCIPA (13). This rather large underestimation in stability may stem from a stronger helical component than bCIPA calculates based on the propensity scale of Williams et al. (26). Indeed, all helical predictors consider helices in isolation and do not account for increases attributable to intermolecular interactions. The best example of this is for c-Jun(Arg)-FosW(Glu), which have both modest predicted and measured helicities in isolation, but when mixed are hugely stabilized by a favorable Glu-Arg electrostatic complement (11). This gives rise to a measured fractional helicity of 0.88 but would be predicted to have a low helical content if this calculation was made based on averaged individual helices. bCIPA reports a negligible loss in electrostatic contribution and a 25% loss in the hydrophobic contribution. This is attributable to the loss of a5 and d5 interactions but almost no loss in the helicity of the truncated interaction. Measured helicity based on θ222 in this study indicated that c-Jun-4hFosW is 60.4% helical compared with 35.8% for c-Jun-FosW (see Table 1). Indeed, despite truncation, 4hFosW is actually more helical than FosW (50.6 versus 43.7%).
Dimer Exchange Experiments
In an experiment to verify the interaction between 4hFosW and c-Jun, we performed a dimer exchange experiment (see Fig. 5). In the first of two experiments, the spectra for the c-Fos·c-Fos homodimeric complex (Fig. 5A, closed circles, Tm −1 °C) and c-Jun-4hFosW (open squares, Tm 49 °C) were recorded using 200 μl of sample at 150 μm total peptide concentration. Next, 100 μl of each solution was mixed, and the spectra were taken such that the final total peptide concentration was also 150 μm. In this case, the observed spectra (Fig. 5A, closed triangles) exactly matched the average of the previous two spectra, indicating that no exchange of dimer has occurred. This experiment therefore indicates that 4hFosW retains preference for c-Jun and that c-Fos is consequently left remaining in its homodimeric form. In the second experiment (see Fig. 5B), c-Jun-c-Jun (closed circles, Tm 24 °C) is mixed with c-Fos-4hFosW (open squares) such that the final composition of the mixed solution is identical to that of the previous experiment. Thus, as predicted, the helices exchange such that 4hFosW binds to c-Jun and the c-Fos peptide is free to homodimerize. Therefore a large increase in spectra magnitude over the average of the two premixed samples is observed (Fig. 5B, closed triangles). Finally, it is shown (Fig. 5C) that the spectra for both postmixed samples superimpose, signifying that the same species is being populated in both samples. Collectively, this experiment indicates that 4hFosW binds and sequesters the c-Jun peptide from c-Fos and in doing so is specific for c-Jun in the presence of c-Fos.
FIGURE 5.

Circular dichroism dimer exchange experiments. A, homodimeric c-Jun mixed with c-Fos-4hFosW clearly causes dimer exchange to give rise to spectra that not only far exceed the average of the two components but that is more helical than either dimer. B, c-Fos is mixed with c-Jun-4hFosW, and the averaged spectra are exactly what is observed, indicating that no helix exchange occurs. C, we observe that the mixed spectra in both A and B are completely superimposable, indicating that the same species is being populated in both instances. In all experiments, Pt is held constant at 150 μm.
Isothermal Titration Calorimetry Experiments
To provide a second measure of the binding affinity (KB) between target and antagonist, ITC experiments were undertaken. This additionally permitted the free energy of binding to be split into its entropic and enthalpic components as well as providing a guide on the stoichiometry of binding. The KB values are in reasonable agreement with those derived from the thermal melting data. It appears, however, that c-Jun-4hFosW has a greater enthalpic contribution to the binding energy relative to c-Jun-FosW (30), but this is more strongly opposed by the entropic term. This is perhaps due to the fact that although the chain has become more ordered (as is evident from increased measured helicity) and that this may contribute to a moderately more favorable enthalpy of binding, it is not matched by improved desolvational entropy. Thus, overall, the c-Jun-4hFosW is around 1 kcal/mol less stable than c-Jun-FosW.
Growth Competition Experiments
Under selective conditions, Escherichia coli growth rates for cells containing either (i) c-Jun-c-Fos, (ii) c-Jun-FosW, or (iii) c-Jun-4hFosW were monitored under PCA conditions in M9 minimal media containing ampicillin, chloramphenicol, kanamycin, trimethoprim (for bacterial inhibition of DHFR), and isopropyl β-d-1-thiogalactopyranoside (to induce protein expression). This experiment was carried out with c-Jun expressed on the pES300d plasmid as a DHFR2 fragment fusion and the Fos peptide expressed on the pES230d plasmid as a DHFR1 fragment fusion (see Fig. 6). Thus, E. coli growth rates are dictated by the binding affinity of the two expressed helices. The higher the affinity, the greater the DHFR activity and the faster cells are able to grow. In broad agreement with thermal melting studies (Fig. 3), ITC experiments (Fig. 4), and dimer exchange studies (Fig. 5), we observe that FosW and 4hFosW both bind c-Jun with much improved affinity over c-Fos, such that the order of fastest growth is c-Jun-FosW ∼ c-Jun-4hFosW > c-Jun-c-Fos.
FIGURE 6.

Bacterial growth data for E. coli BL21 Gold expressing c-Jun-DHFR2 (CmR) and pREP4 (KanR) for lac repression and either c-Fos-DHFR1, FosW-DHFR1, or 4hFosW-DHFR1 (AmpR). Cells were grown under PCA conditions to monitor growth mediated by DHFR reconstitution as a function of the formation of the interacting dimers. Thus, more rapid growth is indicative of a higher interaction affinity between the Fos-DHFR1 constructs and c-Jun-DHFR2 construct. Starting cultures were grown overnight in M9 media, inoculated in duplicate to A600 = 0.01, and grown. Growth was monitored hourly by measuring the A600. The rapid rate of growth for the 4hFosW-c-Jun (open triangles) and FosW-c-Jun (open squares) cultures relative to the c-Fos-c-Jun (closed circles) indicates a high interaction affinity relative to the native complex, suggesting that despite the loss of some hydrophobic core residues, this does not greatly diminish interaction affinity. The result supports thermal melting and ITC data in suggesting that these interactions are of higher affinity than the native c-Fos-c-Jun interaction, which displays an extended growth period indicating a lower affinity interaction. This result suggests that 4hFosW would displace c-Fos from the c-Fos-c-Jun complex due to having higher affinity for c-Jun, as is observed in the dimer exchange assays and EMSA experiments.
Electrophoretic Mobility Shift Assay
Finally, EMSA was undertaken using an assay kit (Invitrogen) that contained two fluorescent dyes to stain sequentially for the presence of DNA (SYBR® Green) and protein (SYPRO® Ruby). Using this technique, shifts in DNA could be correlated to the presence of AP-1 protein bands, thus verifying that the protein and DNA are contained within the same shifted band. EMSAs were performed to demonstrate that 4hFosW cannot only prevent dimerization of the leucine zipper regions of c-Jun but can also antagonize the DNA binding ability of the c-Jun/c-Fos AP-1 protein by sequestering c-Jun. In this experiment, AP-1 consisting of equimolar amounts of basic c-Jun and basic c-Fos was mixed with DNA containing the TPA-response element, either in the absence or presence of 1:2 or 1:7 molar ratios of 4hFosW (see Fig. 7). In the absence of 4hFosW, DNA binding is clearly demonstrated by a loss of intensity relative to the free DNA band (Fig. 7, 1st lane) and by the observed migration of the protein·DNA complex (2nd lane). However, in the presence of 4hFosW, DNA binding is significantly reduced as can be observed by a decrease in intensity of the migrated DNA·protein complex (Fig. 7, 3rd and 4th lanes) and an increase in intensity of the nonshifted free DNA relative that is similar to DNA in isolation.
FIGURE 7.

EMSA demonstrates that 4hFosW is able to prevent AP-1 from binding to the TRE consensus sequence. Left panel, gel, stained with SYBR® Green to detect DNA, shows TRE-DNA in isolation (1st lane), with the addition of the wild-type basic c-Jun·basic c-Fos AP-1 complex in the presence of DNA corresponding to a shift in the observed DNA band (2nd lane). Upon addition of 4hFosW at either 1:2 or 1:7 molar ratio (3rd and 4th lanes), the amount of observed free DNA relative to the 2nd lane is increased. Right panel, when the same gel is stained for protein using SYPRO® Ruby, no protein is observed in the 1st lane, whereas the co-localized DNA-protein complex is observed in the 2nd lane. Upon addition of 4hFosW at 1:2 (3rd lane) or 1:7 molar ratio (4th lane), the intensity of the free stained DNA in A is seen to increase. Moreover, at the increased molar ratio of 1:7, the intensity of the protein stained DNA·protein complex is significantly lower, demonstrating that 4hFosW has an inhibitory effect on the DNA binding ability of AP-1.
DISCUSSION
The concept of peptides and their mimetics as the starting point as druggable agents is an area of growing interest in the pharmaceutical industry (31–33). One of the main issues that has traditionally dampened the appeal has been the size of the molecule and consequent implications associated with peptide drugs. In particular, it was thought that molecules of greater than ∼500 Da would hamper potential efficacy during clinical trials. In contrast, small molecules that met the required criteria at the preclinical stage would have a higher likelihood of success at later more involved stages and would therefore be a safer investment as a starting point for research. In particular, because of their size, many peptides are unable to traverse membranes. They can also be susceptible to proteases and can be quickly eliminated from the body. Although these last two problems can potentially be addressed via increasingly viable nonpeptidic modification of the chain, the first is intrinsic. More recently, techniques such as conjugation to a “protein transduction domain” to form “cell-penetrating peptides” (34, 35), production of stabilized “cyclic peptides” (36, 37), creation of “retro-inverso peptides” (38), or introduction of hydrocarbon staples (39–41) into peptide backbones have all been attempted to circumvent these problems. Therefore the potential of peptide therapeutics is now a growing area, with peptides harboring the potential for chemical and biological diversity while maintaining high specificity and affinity for a protein target.
In our own studies, we are undergoing iterations of producing increasingly smaller peptides that are able to bind to AP-1 components via their coiled coil region and antagonize their natural function. We ultimately seek to identify the smallest functional region and further investigate and develop its therapeutic value. To this end, we have applied one iteration of “Truncation, Randomization, and Selection” (TRaSe). Previously selected peptides were 37 residues in length and therefore comparable in size to commercially available peptide-based drugs such as enfuvirtide (42), which is administered for patients suffering from multidrug-resistant forms of HIV. However, in the interest of identifying the smallest region required for efficient binding to c-Jun, we truncated the previously selected FosW by four residues at the C-terminal end. This culminated in the loss of one a and one d residue from the core region of the fifth heptad in the coiled coil interaction (see Fig. 1). This represents a loss of ∼11% of the parent peptide, and it is worthy of note that even the smallest designed coiled coil was 15 residues in length, equating to two heptad repeats (43). In that case, both helices in the coiled coil interaction were free to be modified to maximize binding affinity. However, designing coiled coils that deviate from peptide-Velcro theory of protein design (44), whereby the target helix cannot be modified, presents numerous obstacles in truncating the antagonizing helix while maintaining interaction stability, because only one half of the interaction can be optimized. Consequently, we have used the TRaSe strategy to partially randomize key residues at and away from the interface and which offer the potential for further improvement of the chain in binding to c-Jun. The ΔTm between c-Jun-FosW and the c-Jun-4hFosW interaction is only 14 °C (see Table 1) despite the loss of the a5/d5 hydrophobic residues from the chain. Strikingly, inspection of the measured helicity by CD demonstrates that c-Jun-4hFosW is almost 25% more helical than c-Jun-FosW despite the loss of these four C-terminal residues. Of the three changes made to the chain, it is most likely that improved helicity is the result of two changes; the Ala to Arg mutation at position f1 results in improved intramolecular electrostatic interactions with flanking Glu/Asp residues, and the Arg to Leu mutation at g2 results in a branched hydrophobic residue capable of forming an intermolecular interaction with Ala on the flanking c-Jun helix by generating a favorable hydrophobic interaction and assisting with desolvation of buried core residues. Consistent with this, additional helicity and stability compensating for the loss in truncation is that 4hFosW is also marginally more helical in isolation (∼7%) relative to FosW. The 4hFosW peptide demonstrates that leucine zipper peptides of reduced length relative to the target region are able to inhibit functional complexes formed by the target upon interaction in full-length parent molecules. Thus, it is shown here that truncated leucine zippers as opposed to full-length zippers (13), acidic zippers (45), or c-Jun lacking the transactivation domain (27) are able to bind to c-Jun and inhibit the DNA binding activity of AP-1, as has been demonstrated by EMSA. In addition, the ITC results demonstrate that the TRaSe strategy has optimized the enthalpy of binding despite a decrease in antagonist size. Optimizing for favorable enthalpy has been described as the key parameter in lead compound design, but its optimization during lead discovery is considered difficult because of the correct engineering of correct bond angles and lengths (46).
These results show that by using intelligent options during the semirational design process, it may in the future be possible to apply additional iterations of TRaSe to further reduce the size of our antagonists while gaining added insight into the smallest known functional region required to generate an interaction.
Acknowledgments
We thank the Department of Biological Sciences, University of Essex, RCIF funding for the purchase of an Isothermal Titration Calorimeter. We thank Dr. Miao Yu for technical assistance.
This work was supported in part by Wellcome Trust Grant WT090184MA.
- TPA
- 12-O-tetradecanoylphorbol-13-acetate
- ITC
- isothermal titration calorimetry
- PCA
- protein-fragment complementation assay
- TRE
- TPA-response element
- DHFR
- dihydrofolate reductase
- TRaSe
- truncation, randomization, and selection
- bCIPA
- basic leucine zipper protein coiled coil interaction prediction algorithm.
REFERENCES
- 1. Darnell J. E., Jr. (2002) Nat. Rev. Cancer 2, 740–749 [DOI] [PubMed] [Google Scholar]
- 2. Jochum W., Passegué E., Wagner E. F. (2001) Oncogene 20, 2401–2412 [DOI] [PubMed] [Google Scholar]
- 3. Angel P., Imagawa M., Chiu R., Stein B., Imbra R. J., Rahmsdorf H. J., Jonat C., Herrlich P., Karin M. (1987) Cell 49, 729–739 [DOI] [PubMed] [Google Scholar]
- 4. Matthews C. P., Colburn N. H., Young M. R. (2007) Curr. Cancer Drug Targets 7, 317–324 [DOI] [PubMed] [Google Scholar]
- 5. Vogt P. K. (2001) Oncogene 20, 2365–2377 [DOI] [PubMed] [Google Scholar]
- 6. Maki Y., Bos T. J., Davis C., Starbuck M., Vogt P. K. (1987) Proc. Natl. Acad. Sci. U.S.A. 84, 2848–2852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Acharya A., Rishi V., Vinson C. (2006) Biochemistry 45, 11324–11332 [DOI] [PubMed] [Google Scholar]
- 8. Pelletier J. N., Arndt K. M., Plückthun A., Michnick S. W. (1999) Nat. Biotechnol. 17, 683–690 [DOI] [PubMed] [Google Scholar]
- 9. Moitra J., Szilák L., Krylov D., Vinson C. (1997) Biochemistry 36, 12567–12573 [DOI] [PubMed] [Google Scholar]
- 10. Krylov D., Mikhailenko I., Vinson C. (1994) EMBO J. 13, 2849–2861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mason J. M. (2009) FEBS J. 276, 7305–7318 [DOI] [PubMed] [Google Scholar]
- 12. Kwok S. C., Hodges R. S. (2004) Biopolymers 76, 378–390 [DOI] [PubMed] [Google Scholar]
- 13. Mason J. M., Schmitz M. A., Müller K. M., Arndt K. M. (2006) Proc. Natl. Acad. Sci. U.S.A. 103, 8989–8994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mason J. M., Müller K. M., Arndt K. M. (2007) Biochemistry 46, 4804–4814 [DOI] [PubMed] [Google Scholar]
- 15. Mason J. M., Hagemann U. B., Arndt K. M. (2009) Biochemistry 48, 10380–10388 [DOI] [PubMed] [Google Scholar]
- 16. Pelletier J. N., Campbell-Valois F. X., Michnick S. W. (1998) Proc. Natl. Acad. Sci. U.S.A. 95, 12141–12146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Dixon J. M., Taniguchi M., Lindsey J. S. (2005) Photochem. Photobiol. 81, 212–213 [DOI] [PubMed] [Google Scholar]
- 18. Elwell M. L., Schellman J. A. (1977) Biochim. Biophys. Acta 494, 367–383 [DOI] [PubMed] [Google Scholar]
- 19. Mason J. M., Hagemann U. B., Arndt K. M. (2007) J. Biol. Chem. 282, 23015–23024 [DOI] [PubMed] [Google Scholar]
- 20. Wiseman T., Williston S., Brandts J. F., Lin L. N. (1989) Anal. Biochem. 179, 131–137 [DOI] [PubMed] [Google Scholar]
- 21. Seldeen K. L., McDonald C. B., Deegan B. J., Farooq A. (2008) Arch. Biochem. Biophys. 473, 48–60 [DOI] [PubMed] [Google Scholar]
- 22. Krylov D., Barchi J., Vinson C. (1998) J. Mol. Biol. 279, 959–972 [DOI] [PubMed] [Google Scholar]
- 23. Mason J. M., M, Müller K., Arndt K. M. (2008) Biochem. Soc. Trans. 36, 1442–1447 [DOI] [PubMed] [Google Scholar]
- 24. O'Shea E. K., Rutkowski R., Kim P. S. (1992) Cell 68, 699–708 [DOI] [PubMed] [Google Scholar]
- 25. Pace C. N., Scholtz J. M. (1998) Biophys. J. 75, 422–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Williams R. W., Chang A., Juretiæ D., Loughran S. (1987) Biochim. Biophys. Acta 916, 200–204 [DOI] [PubMed] [Google Scholar]
- 27. Grigoryan G., Reinke A. W., Keating A. E. (2009) Nature 458, 859–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lau S. Y., Taneja A. K., Hodges R. S. (1984) J. Biol. Chem. 259, 13253–13261 [PubMed] [Google Scholar]
- 29. Kwok S. C., Hodges R. S. (2004) J. Biol. Chem. 279, 21576–21588 [DOI] [PubMed] [Google Scholar]
- 30. Worrall J. A., Mason J. M. (2011) FEBS J. 278, 663–672 [DOI] [PubMed] [Google Scholar]
- 31. Otvos L., Jr. (2008) Methods Mol. Biol. 494, 1–8 [DOI] [PubMed] [Google Scholar]
- 32. Mason J. M. (2010) Future Med. Chem. 2, 1813–1822 [DOI] [PubMed] [Google Scholar]
- 33. Vlieghe P., Lisowski V., Martinez J., Khrestchatisky M. (2010) Drug. Discov. Today 15, 40–56 [DOI] [PubMed] [Google Scholar]
- 34. Derossi D., Chassaing G., Prochiantz A. (1998) Trends Cell Biol. 8, 84–87 [PubMed] [Google Scholar]
- 35. Vivès E., Brodin P., Lebleu B. (1997) J. Biol. Chem. 272, 16010–16017 [DOI] [PubMed] [Google Scholar]
- 36. Bernal F., Tyler A. F., Korsmeyer S. J., Walensky L. D., Verdine G. L. (2007) J. Am. Chem. Soc. 129, 2456–2457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Guo Z., Mohanty U., Noehre J., Sawyer T. K., Sherman W., Krilov G. (2010) Chem. Biol. Drug Des. 75, 348–359 [DOI] [PubMed] [Google Scholar]
- 38. Taylor M., Moore S., Mayes J., Parkin E., Beeg M., Canovi M., Gobbi M., Mann D. M., Allsop D. (2010) Biochemistry 49, 3261–3272 [DOI] [PubMed] [Google Scholar]
- 39. Walensky L. D., Kung A. L., Escher I., Malia T. J., Barbuto S., Wright R. D., Wagner G., Verdine G. L., Korsmeyer S. J. (2004) Science 305, 1466–1470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Harrison R. S., Shepherd N. E., Hoang H. N., Ruiz-Gómez G., Hill T. A., Driver R. W., Desai V. S., Young P. R., Abbenante G., Fairlie D. P. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 11686–11691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bird G. H., Madani N., Perry A. F., Princiotto A. M., Supko J. G., He X., Gavathiotis E., Sodroski J. G., Walensky L. D. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 14093–14098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lalezari J. P., Henry K., O'Hearn M., Montaner J. S., Piliero P. J., Trottier B., Walmsley S., Cohen C., Kuritzkes D. R., Eron J. J., Jr., Chung J., DeMasi R., Donatacci L., Drobnes C., Delehanty J., Salgo M. (2003) N. Engl. J. Med. 348, 2175–2185 [DOI] [PubMed] [Google Scholar]
- 43. Burkhard P., Meier M., Lustig A. (2000) Protein Sci. 9, 2294–2301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. O'Shea E. K., Lumb K. J., Kim P. S. (1993) Curr. Biol. 3, 658–667 [DOI] [PubMed] [Google Scholar]
- 45. Olive M., Krylov D., Echlin D. R., Gardner K., Taparowsky E., Vinson C. (1997) J. Biol. Chem. 272, 18586–18594 [DOI] [PubMed] [Google Scholar]
- 46. Ladbury J. E., Klebe G., Freire E. (2010) Nat. Rev. Drug Discov. 9, 23–27 [DOI] [PubMed] [Google Scholar]
- 47. Shepherd N. E., Hoang H. N., Abbenante G., Fairlie D. P. (2005) J. Am. Chem. Soc. 127, 2974–2983 [DOI] [PubMed] [Google Scholar]
- 48. Muñoz V., Serrano L. (1995) J. Mol. Biol. 245, 297–308 [DOI] [PubMed] [Google Scholar]
- 49. Muñoz V., Serrano L. (1995) J. Mol. Biol. 245, 275–296 [DOI] [PubMed] [Google Scholar]