

Abstract
The early growth response (EGR) family of transcription factors has been implicated in control of lipid biosynthetic genes. Egr1 is induced by insulin both in vitro and in vivo and is the most highly expressed family member in liver. In this study, we investigated whether Egr1 regulates cholesterol biosynthetic genes in liver. Using an insulin-sensitive liver cell line, we show that localization of Egr1 to cholesterol biosynthetic genes is induced by insulin treatment and that this localization precedes the induction of the genes. Reduction in Egr1 expression using targeted siRNA blunted the insulin-dependent induction of cholesterol genes. A similar reduction in squalene epoxidase expression was also observed in Egr1 null mice. In addition, application of chromatin immunoprecipitation (ChIP) samples to tiled gene microarrays revealed localization of Egr1 in promoter regions of many cholesterol gene loci. In vivo ChIP assays using liver tissue show that Egr1 localization to several cholesterol biosynthetic gene promoters is induced by feeding. Finally, analysis of plasma cholesterol in Egr1−/− mice indicated a significant decrease in serum cholesterol when compared with wild-type mice. Together these data point to Egr1 as a modulator of the cholesterol biosynthetic gene family in liver.
Keywords: Cholesterol, Cholesterol Regulation, Chromatin Immunoprecipitation (ChIP), Gene Expression, Gene Regulation, Hepatocyte, High Density Lipoprotein (HDL), Insulin, Lipid Synthesis, Transcription Factors
Introduction
Elevated serum cholesterol is associated with an increased risk of atherosclerosis, heart disease, and stroke. The dramatic success of the statin class of cholesterol-reducing drugs has shown that even small changes in serum cholesterol can have significant effects in the prevention of such diseases (1). The production of cholesterol is regulated in part by transcriptional regulation of genes involved in cholesterol biosynthesis. Among the most important transcriptional regulators of cholesterol and fatty acid production are the sterol-response element-binding proteins (SREBPs).2 There are three SREBP isoforms: SREBP-1a induces both cholesterol and fatty acid biosynthetic genes but is not highly expressed in vivo, SREBP-1c preferentially induces fatty acid biosynthetic genes and SREBP-2 preferentially induces cholesterol biosynthetic genes. These transcriptional activators are initially tethered to the endoplasmic reticulum in an inactive form and then transported to the Golgi in response to cholesterol depletion where proteolytic cleavage frees the N-terminal portion of SREBPs to translocate to the nucleus to activate transcription (reviewed in Ref. 2).
Upon high carbohydrate feeding or insulin treatment in vitro, expression of many fatty acid and cholesterol biosynthetic genes is induced. Insulin signaling has been shown to increase SREBP-1 nuclear localization and binding to promoters of fatty acid and triglyceride biosynthetic genes (3–6). In contrast, induction of cholesterol biosynthetic genes by insulin may involve increased SREBP-2 levels (7, 8), but activation of SREBP-2 does not always accompany the insulin-induced increase in cholesterol biosynthetic genes (5). Recently, in vivo binding analyses have suggested relatively little insulin-induced change in SREBP-2 binding to genes involved in cholesterol biosynthesis (9, 10). Such studies have led to a proposed model in which SREBP-2 may promote insulin regulation of cholesterol biosynthetic genes by maintaining target promoters in a receptive state for binding of additional, insulin-dependent transcription factors (5).
Several recent lines of evidence have suggested that cholesterol biosynthesis is regulated at the transcriptional level by members of the early growth response (EGR) family. EGRs are zinc finger transcription factors produced from immediate early genes that are induced by a variety of physiological stimuli. EGR family members recognize and bind the same GC-rich sequence, rendering these regions potentially responsive to EGR activation in multiple tissues (11). Appropriate Egr2/Krox20 expression is integral to the formation of the cholesterol-rich myelin sheath by Schwann cells in the peripheral nervous system (12, 13). Accordingly, we have shown that Egr2 can act synergistically with SREBP-2 to activate transcription of the promoters of certain cholesterol biosynthetic genes (e.g. Hmgcr) (14). Additionally, in the Krox20/Egr2 null mouse, the expression of Hmgcr and Cyp51 was significantly reduced (>80%) in peripheral nerve despite little change in SREBP levels, suggesting a role for EGR factors in the regulation of cholesterol metabolism. Egr1 (also known as krox24/Zif268/NGFI-A) is the predominantly expressed EGR family member in liver and is also induced by insulin in vivo (15) as well as liver-derived cell lines (16, 17), but the physiological significance of this induction has not been explored. Studies using chemical inhibitors have shown that this induction of Egr1 in liver cells depends on the MEK1/ERK1/2 pathway, putting it directly downstream of the insulin receptor (17). Interestingly, a modulatory effect of Egr1 on cholesterol levels is supported by the identification of a polymorphism in the human EGR1 promoter linked to lower serum cholesterol levels (18).
These studies suggest that EGR factors may modulate expression of cholesterol biosynthetic genes not only in peripheral nerve myelin but also in liver. Based upon the previously described induction of Egr1 by insulin (17), we performed chromatin immunoprecipitation followed by microarray (ChIP-chip) in an insulin-sensitive cell line to identify potential Egr1-regulated genes and the putative Egr1 binding sites in insulin-treated cells. The localization of Egr1 precedes induction of Hmgcr and other cholesterol biosynthetic genes; however insulin did not affect SREBP binding to these loci. Using Egr1-specific siRNA, we found that Egr1 was required for appropriate induction of Hmgcr and other cholesterol biosynthetic genes by insulin. In vivo ChIP showed that Egr1 localizes to cholesterol biosynthetic gene promoters in liver using a protocol of fasting followed by high carbohydrate refeeding. However, Egr1−/− mice induce the expression of a cholesterol biosynthetic gene, Sqle, to a lesser degree than wild types. Finally, Egr1 null mice have significantly less serum cholesterol than age-matched wild-type controls. Overall, these data identify a new role for Egr1 in the direct regulation of systemic cholesterol levels via regulation of cholesterol synthesis in the liver. Moreover, we suggest that Egr1 acts in concert with SREBP factors to mediate the insulin-dependent induction of cholesterol biosynthesis in liver.
MATERIALS AND METHODS
H4IIE Tissue Culture
H4IIE cells were maintained in Dulbecco's modification of Eagle's medium (DMEM) (Mediatech) supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin at 37 °C and 5% CO2 and passaged twice weekly. To perform expression analysis, cells were plated in 12-well plates in growth medium and allowed to attach overnight. The cells were then washed once with sterile PBS and placed in serum-free DMEM for ∼16 h to reduce expression of Egr1 and other stimulating factors to background. Insulin (Sigma) was then added to the medium to a final concentration of 0.1 μm. After the prescribed length of time had passed, the cells were washed once with sterile PBS and harvested with TRI Reagent, and RNA was purified by the manufacturer's instructions (Ambion). 1 μg of RNA was used to make cDNA as described previously (19). Quantitative RT-PCR was performed using the Power SYBR Green PCR master mix and the TaqMan 7000 sequence detection system (Applied Biosystems). The comparative CT method was employed to determine relative changes in gene expression when compared with 18 S rRNA (20). Primer sets used are listed in supplemental Table 1. Protein lysate cells were analyzed by immunoblotting for Egr1 (Santa Cruz Biotechnology, sc-189) and actin as described previously (21).
Promoter Analysis
Using rVISTA analysis, highly homologous (rat, mouse, and human) promoter regions of cholesterol biosynthetic genes were identified (22). Within these regions of high homology, EGR binding sites were identified based upon the consensus EGR binding sequence GCGGGGGCG (11). Primers were designed to flank these putative EGR binding sites in an effort to detect enrichment of DNA in the Egr1 immunoprecipitations when compared with DNA recovered from IgG precipitations.
H4IIE Chromatin Immunoprecipitation
Confluent H4IIE cultures were trypsinized, pooled, and plated in growth medium on 15-cm plates. When cells had reached 90% confluency, the cells were washed with PBS, placed in serum-free media, and starved for 16 h. The cells were then treated with 0.1 μm insulin for the prescribed time, washed once with PBS, and placed in PBS containing 1% formaldehyde at 37 °C for 20 min to covalently cross link any DNA-protein complexes. The cells were then washed twice with PBS to remove formaldehyde, suspended in 15 ml of PBS, and harvested using a cell scraper. Cells were spun down in a 15-ml conical flask at 1000 × g for 3 min, and the pellets were frozen until processing. Cross-linked cell pellets were resuspended in 2 ml lysis of buffer (150 mm NaCl, 10% glycerol, 0.3% Triton X-100, 50 mm Tris 8.0, 0.5% protease inhibitor solution (Sigma)) and sonicated by repeating a protocol of 30 s on and 30 s off on the maximum setting for 20 min using the Bioruptor (Diagenode). The samples were centrifuged for 30 min at 12,000 × g to pellet cell debris, and the soluble chromatin was harvested. The Bradford method was performed to determine the quantity of protein in the chromatin sample (Bio-Rad). Immunoprecipitations were prepared with ∼1 mg of chromatin and 2 μg of antibody (sc-189 Egr1 antibody, Santa Cruz Biotechnology; sc-8984 SREBP-1 antibody, Santa Cruz Biotechnology; 7D4 SREBP-2 antibody, ATCC; and normal rabbit IgG, Upstate Biotech Millipore). Immunoprecipitations were processed as described previously (23). Primer sets used in quantitative PCR for ChIP are listed in supplemental Table 1.
ChIP-Chip Analysis
Insulin treatment of H4IIE cells and standard ChIP were performed as described above. 10 ng of purified DNA from ChIP samples were amplified by the non-biased GenomePlex whole genome amplification kit per the manufacturer's instructions (Sigma). Reactions were purified using QIAquick columns (Qiagen) and concentrated by centrifugation under vacuum. Samples were sent to Roche NimbleGen (Reykjavík, Iceland), where labeling of experimental (Cy5) and reference samples (Cy3) was performed followed by hybridization to tiled microarrays with each gene locus represented by probes extending 20–50 kb from the 5′ and 3′ end of the gene. In parallel, an Egr1 immunoprecipitation from H4IIEs not treated with insulin was labeled with Cy3 and used as the reference sample, whereas an Egr1 immunoprecipitation from insulin-stimulated cells was labeled with Cy5 and used as the experimental sample. Using the same antibody for experimental and control samples reduces apparent peaks due to fluctuations in the IgG signal. To reduce noise and normalize for single probe error, the signal from each probe was averaged with the four neighboring probes. After putative binding sites were identified using ChIP-chip, the abovementioned promoter analysis using rVISTA was performed to identify likely EGR binding sites. Primer sets were designed to determine Egr1 binding to these regions. All raw data sets for the custom tiled array are available from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus website (accession number: GSE30823).
H4IIE siRNA Transfection
H4IIE cells were transfected with siRNA using the Amaxa Nucleofection transfection system (Lonza Cologne AG, Cologne, Germany). Transfections were conducted per Amaxa protocols on normally passaged H4IIEs. Briefly, 5 million cells were used in each transfection using Amaxa Nucleofection Solution V and program T-020 and 200 nm siRNA. Egr1-specific siRNA pools were purchased from Invitrogen, as were the appropriate negative controls. Cells were recovered in supplemented RPMI medium and split into multiple wells containing H4IIE growth medium. Cells were allowed to recover in growth medium for 48 and stimulated with 0.1 μm insulin for 1.5 h. RNA was harvested, and cDNA was synthesized as described in “Methods.”
In Vivo Gene Expression Analysis
Experiments on mice were performed with strict adherence to animal protocols approved by the Animal Care and Use Committee, the Research Animal Resource Center, and the University of Wisconsin-Madison Graduate School. Age-matched wild-type and Egr1 knock-out mice were fasted for 12 h and fed high carbohydrate chow for 12 h. This protocol was repeated for five consecutive days, at which point the animals were sacrificed after feeding for 12 h. A cohort of wild-type control animals was treated identically but was sacrificed after 12 h of fasting to establish baseline expression of target genes. Mice were sacrificed, RNA was purified, and cDNA was synthesized as described in “Methods.”
In Vivo Liver ChIP
Fasting and refeeding were performed as described (5, 8), in which mice were fasted for 24 h before refeeding with a high carbohydrate and low fat chow (Harlan TD99252) for 12 h before harvesting the liver (5, 24). The liver was flushed with cold PBS via the portal vein to remove blood. The liver was perfused with 10 ml of cold PBS containing 1% formaldehyde to cross-link chromatin. A lobe of treated liver was removed, shredded with a mincing scissors, and transferred to a 1.5-ml tube and further homogenized using a Tissue Master (Omni International). The sample was incubated in cross-linking buffer for 20 min at room temperature, spun down at 5000 rpm for 10 min at 4 °C, washed once with cold PBS, aspirated, and frozen at −80 °C. The pellet was resuspended in 2 ml of ChIP lysis buffer and mixed with 100 mg of glass beads. Using a Microson tip sonicator (Misonix, Farmingdale, NY) on setting 13, the sample was sonicated by repeating a protocol of a 10-s pulse followed by a 50-s rest on ice for 20 min. The sample was spun down at 15,000 rpm for 30 min at 4 °C to remove cell debris. The supernatant, containing soluble chromatin, was used for immunoprecipitations as described previously (23).
Mouse Serum Cholesterol
Age-matched Egr1 null mice were purchased from Taconic. Assessment of mouse serum cholesterol levels from fasted wild-type and Egr1 mice was performed by the Mouse Metabolic Phenotyping Center (MMPC) (University of Cincinnati) in accordance with standard procedures. Briefly, total serum cholesterol was measured, and then samples were analyzed by FPLC to determine the amount of cholesterol in the HDL associated fractions. Complete protocols are available from the MMPC. All statistics were performed using analysis of variance.
RESULTS
Insulin Induces Egr1 Localization to Cholesterol Biosynthetic Gene Promoters
To test whether Egr1 regulates cholesterol biosynthetic genes in liver, we utilized the H4IIE rat hepatoma cell line, which responds to insulin treatment. It has been reported that insulin activates Egr1 expression in this cell line (17), and our quantitative PCR analysis confirmed that Egr1 mRNA is highly induced by insulin in H4IIE cells, maximizing 30 min after insulin treatment at 180-fold greater than untreated cells (Fig. 1A). Expression of Egr1 mRNA returned to near background levels 2 h after insulin. Consistent with published findings (17), we found that the induction of Egr1 protein was similar to the mRNA, albeit somewhat delayed relative to the peak of Egr1 mRNA (Fig. 1B). However, even after 4 h of insulin treatment, the Egr1 protein was still detectable. To measure nascent HMG-CoA reductase (Hmgcr) expression, we designed RT-qPCR primers to an intron region of the Hmgcr gene, which would measure pre-mRNA levels. Hmgcr, the rate-limiting enzyme in cholesterol biosynthesis, was also induced in H4IIEs following insulin treatment, and this induction coincided with the induction of Egr1 (Fig. 1A). Expression of Hmgcr was reduced later in the time course, which coincides with the reduction in Egr1.
FIGURE 1.

Expression of Egr1 and Hmgcr is induced by insulin in H4IIE cells. The H4IIE insulin-sensitive rat hepatoma cell line was treated with 0.1 μm insulin for time points indicated on the x axis. In A, expression of Egr1 mRNA (left y axis) and Hmgcr pre-mRNA (right y axis) were assayed by RT-qPCR, as described under “Materials and Methods,” and normalized to the level of 18 S rRNA. These data show the average and S.D. of four independent experiments. Data points identified with asterisks indicate points statistically significant from untreated cells, and those identified with number signs are significantly different from the maximum expression at 30 min (p < 0.05, Welch's t test). B, shows an immunoblot of Egr1 protein levels during the insulin stimulation time course with an actin loading control. The lower band in the Egr1 immunoblot is a nonspecific band that did not change under any conditions tested; the upper band is Egr1.
To determine whether Egr1 localizes to regulatory elements of cholesterol biosynthetic promoters in response to insulin, we performed chromatin immunoprecipitation (ChIP) assays to detect Egr1 binding at various time points after insulin stimulation of H4IIE cells. Immunoprecipitation of formaldehyde-cross-linked samples was performed with an Egr1 antibody previously used for ChIP analysis (25–28), and quantitative PCR using promoter-specific primer sets was used to determine the recovery of input DNA relative to a control immunoprecipitation using purified rabbit IgG. The initial studies were performed on the promoter of the Hmgcr gene, which is an SREBP-2 target gene activated by insulin (29, 30). In addition, promoter analysis identified several conserved EGR binding sites in this promoter (see Fig. 2C).
FIGURE 2.

Localization of Egr1, but not SREBP-1 or SREBP-2, to the Hmgcr promoter is induced by insulin in H4IIE cells. H4IIE cells were treated with insulin for indicated time points and then processed for chromatin immunoprecipitation assays, as described under “Materials and Methods,” using antibodies against Egr1 and IgG (A) or SREBP-1, SREBP-2, and IgG (B). The percentage of recovery relative to input DNA was measured using qPCR with primers designed to detect putative EGR binding sites. Y axis values indicate average observations of two independent ChIP assays, and error bars represent S.D. Egr1 binding after 1 h of insulin was found to be statistically distinct from background Egr1 binding (asterisks indicate p < 0.05, Welch's t test). C, a diagram of the Hmgcr promoter shows the characterized cAMP-response element (CRE) and sterol response-element (SRE) indicated as well as putative EGR binding sites. The location of the primers used for ChIP analysis is indicated with arrows.
As shown in Fig. 2A, there is a robust induction of Egr1 localization to the Hmgcr promoter after 1 h of insulin treatment when compared with untreated cells. This induction of binding to Hmgcr corresponds to greater than 0.62% DNA recovery or 14-fold greater than the recovery with an IgG control. Although Egr1 binding was still high after 2 h of insulin treatment, the total recovery had decreased to nearly 3-fold over background Egr1 binding or 0.28% DNA recovery. Therefore, insulin treatment of H4IIE cells is accompanied by localization of Egr1 at the Hmgcr promoter.
ChIP assays detected significant localization of both SREBP-1 and SREBP-2 to the Hmgcr promoter in H4IIE cells, but surprisingly little induction of either SREBP-1 or SREBP-2 localization after insulin treatment was observed (Fig. 2B). Overall, these data indicate that insulin treatment of H4IIE cells results in induced localization of Egr1 to the Hmgcr promoter, whereas SREBP binding is relatively unaffected.
Egr1 Localizes to the Promoters of Several Genes in the Cholesterol Biosynthetic Pathway
Based upon our initial determination that Egr1 binds promoter elements of Hmgcr in response to insulin in a rat liver cell line, we aimed to determine whether Egr1 binding is observed in other cholesterol biosynthetic genes. Regions of 100 kb surrounding each cholesterol biosynthetic gene were tiled on a custom microarray, where each gene locus was represented by overlapping tiled probes with an average spacing of 17 bp. Gaps in the tiling represent repetitive DNA regions for which unique, optimal probes could not be designed. Immunoprecipitations were performed using the Egr1 antibody with chromatin from insulin-treated and untreated H4IIE cells. The samples were applied to the tiled microarray, and the resulting ChIP-chip data were plotted in a ratio of Cy5 (experimental + insulin) to Cy3 (control) on a log2 scale. The NimbleScan software was used to identify peaks and assign false discovery rates for each peak (see supplemental Fig. 3 for example analysis). All genes identified in Table 1 had Egr1 peaks of binding with false discovery rates <0.05 in at least two of the three independent ChIP-chip experiments.
TABLE 1.
Cholesterol biosynthetic genes bound by Egr1 in response to insulin
| Gene name(s) | Symbol | ChIP-chip binding |
|---|---|---|
| HMG-CoA synthase | Hmgcs | + |
| HMG-CoA reductase | Hmgcr | + |
| Phosphomevalonate kinase | Pmvk | - |
| Mevalonate diphosphate decarboxylase | Mvd | − |
| Isopentenyl diphosphate isomerase | Idi1 | − |
| Farnesyl diphosphate synthase | Fdps | + |
| Farnesyl diphosphate farnesyltransferase 1 | Fdft1 | + |
| Squalene Epoxidase | Sqle | + |
| Lanosterol synthase/lanosterol cyclase | Lss | + |
| Lanosterol 14α demethylase | Cyp51 | + |
| NAD(P)-dependent steroid dehydrogenase-like/NAD(P)H steroid dehydrogenase | Nsdhl | + |
| Sterol C5-desaturase | Sc5d | − |
| 7-Dehydrocholesterol reductase | Dhcr7 | − |
| Malic enzyme | Me1/Mod1 | + |
As shown in Fig. 3, specific peaks of Egr1 binding were detected at several cholesterol biosynthetic promoters including not only Hmgcr but also lanosterol demethylase (Cyp51) and squalene epoxidase (Sqle). Peaks of binding were localized to the transcription start site and extended into the proximal promoter and sometimes into the first exon of the gene. Cyp51 has been previously shown to be responsive to EGR regulation (14), as has Me1 (31). In addition to these genes, Egr1 binding was identified in the promoters of the HMG-CoA synthase (Hmgcs), farnesyl diphosphate synthase (Fdps), farnesyl-diphosphate farnesyltransferase 1 (Fdft1), lanosterol synthase (Lss), sterol-4α-carboxylate 3-dehydrogenase (Nsdhl), and malic enzyme (Me1) genes (see supplemental Fig. 1 for representative data). The results of the ChIP-chip analysis are summarized in Table 1 and show widespread localization of Egr1 in proximal promoter regions of cholesterol biosynthetic genes in response to insulin. However, not all cholesterol biosynthetic promoters were positive for Egr1 as no prominent peak was observed in the phosphomevalonate kinase gene, for example.
FIGURE 3.

ChIP-chip shows that Egr1 selectively binds promoters of cholesterol biosynthetic genes. The binding of Egr1 to many cholesterol biosynthetic genes was determined using ChIP-chip assays of H4IIE cells treated with insulin. The ChIP samples were labeled with Cy5 (Egr1 + insulin) or Cy3 (Egr1 − insulin control) for hybridization to the genomic tiling array. The enrichment ratio of Cy5 to Cy3 was plotted on a log2 scale and further processed to display a five-point moving average. Genomic location of peaks is displayed on the x axis. Peaks of Egr1 binding coincide with transcription start sites of cholesterol biosynthetic genes such as Hmgcr, Cyp51, and Sqle. Arrows indicate gene location and transcriptional direction. These data are representative of three independent ChIP-chip experiments, and false discovery rates values for these identified peaks are <0.05 using NimbleScan analysis.
To determine whether this localization was specific or an artifact of the ChIP-chip procedure, ChIP primer sets for qPCR were designed to independently test putative Egr1 binding sites within the ChIP-chip peaks. Verification of ChIP-chip binding was performed on the Hmgcr, Cyp51, Sqle, and Me1 promoters, as shown in Fig. 4. These data show an insulin-dependent induction of Egr1 binding to these promoters that is significantly above the negative control IgG immunoprecipitation. In addition, analysis of a negative control site at −1.5 kb upstream of Hmgcr showed no localization of Egr1 to this site and no change with insulin treatment. Using the samples from the insulin time course ChIP experiment, a similar temporal pattern of Egr1 localization was observed for Nsdhl, Hmgcs, and Fdps (data not shown). All show an induction of Egr1 binding with insulin at least 2-fold above background (see supplemental Fig. 2 for representative data), and most have been shown to be insulin-sensitive in liver tissue (8, 32). Table 1 displays the results of those genes interrogated for Egr1 binding by ChIP-chip; in each case, binding of Egr1 was validated by two independent ChIP assays. These findings indicate that Egr1 binds many of the cholesterol biosynthetic promoters in response to insulin in liver cells.
FIGURE 4.

Validation of newly identified Egr1 binding sites by ChIP. Gene elements identified as putative Egr1 binding sites were interrogated for Egr1 binding following insulin treatment for 1 h using ChIP assays. The percentage of recovery relative to input DNA was measured using qPCR with primers designed to detect putative EGR binding sites. The treatment of the cells is indicated on the x axis (SF = serum free, Ins = 1 h of insulin), as is the promoter being tested, and darker bars indicate the percentage of recovery of Egr1 at the locus, whereas lighter bars indicate the level observed with nonspecific IgG immunoprecipitation. The Hmgcr −1.5-kb primer set was used as a locus-specific negative control. Graphs represent average values from two separate experiments, data are representative of five independent experiments, and error bars indicate S.D. Bars labeled with different letters indicate statistically significant inductions of Egr1 binding (Welch's t test, p < 0.05).
Loss of Egr1 Reduces the Insulin-dependent Induction of Cholesterol Genes
To test whether Egr1 was necessary for the insulin-dependent induction of Hmgcr, we transfected H4IIE cells with Egr1-specific siRNAs or control siRNAs and stimulated them with insulin. We observed a greater than 50% reduction in Egr1 mRNA in cells transfected with Egr1 siRNA and also observed a large reduction in Egr1 protein following insulin treatment, indicating successful knockdown of Egr1 expression (supplemental Fig. 4).
In cells transfected with control siRNA, we observed an average of 2.3-fold induction in the expression of the Hmgcr gene after 90 min of insulin treatment. This value was set as 100% induction. However, in cells transfected with Egr1 siRNAs, the induction of Hmgcr was blunted over 35% (Fig. 5, n = 4). We did not observe a change in the expression of the SREBPs in cells transfected with Egr1 siRNA (data not shown). Thus, the induction of Egr1 is necessary for the insulin-dependent induction of Hmgcr expression. We extended this analysis to other cholesterol biosynthetic genes we had identified as putative Egr1 target genes by ChIP-chip and found a similar effect including a reduction of Sqle (35%) and Me1 (42%) expression in response to insulin (Fig. 5).
FIGURE 5.
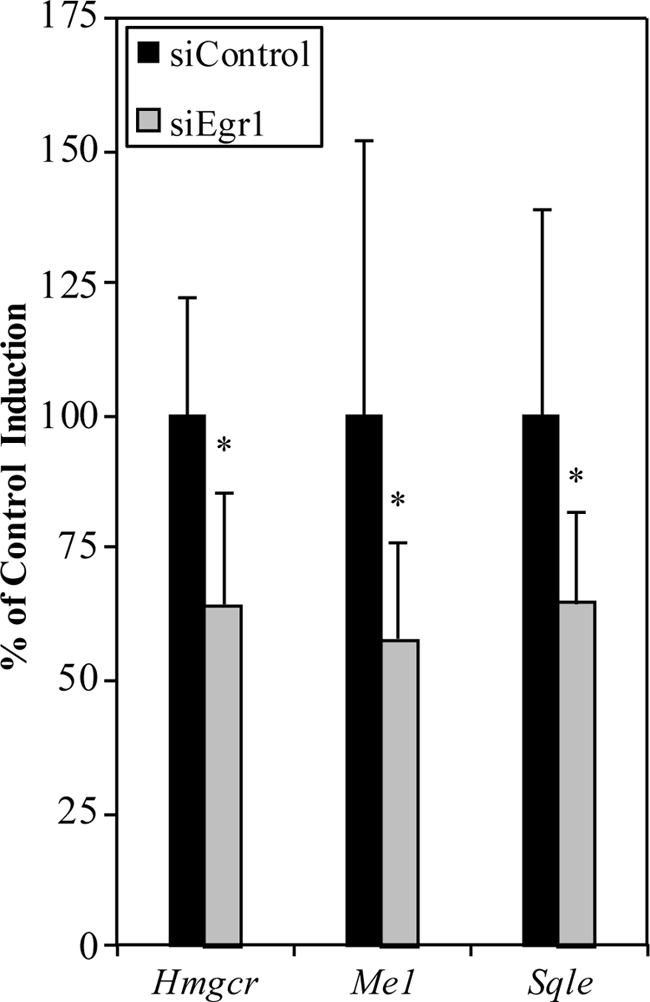
Egr1 siRNA blunts the insulin-dependent induction of cholesterol biosynthetic genes. H4IIE cells were transfected with either control siRNA or Egr1-specific siRNAs. Data are displayed relative to gene inductions in control transfected cells. RT-qPCR indicated a 35% reduction in the response of Hmgcr to insulin in cells transfected with siRNA for Egr1 after treatment with insulin for 1.5 h. Similar reductions were observed for other cholesterol biosynthetic genes such as Me1 and Sqle. Average values from four independent experiments are shown, and error bars represent S.D. Asterisks indicate p < 0.05 (Welch's t test).
Refeeding Stimulates Egr1 Localization to Cholesterol Biosynthetic Promoters in Liver
Previous work had indicated that Egr1 is the primary EGR transcript in liver (33); therefore, we performed a fasting-refeeding experiment to test whether Egr1 expression is induced by feeding in vivo. A pilot experiment indicated that the peak of Egr1 mRNA expression was 8 h, while the peak of Egr1 binding was 12 h postprandial (data not shown). Therefore, mice were either fasted for 24 h or fasted for 24 h and fed high carbohydrate chow for 12 h. A similar strategy was recently used to measure SREBP occupancy in lipid biosynthetic promoters (10). After fasting for 24 h and feeding for 12 h, mice were sacrificed, and the livers were processed for chromatin immunoprecipitation assays using an antibody against Egr1. Localization of Egr1 to the Hmgcr promoter was determined in both fasted and refed animals. Although we observed Egr1 occupancy of 0.34% at the Hmgcr promoter in fasted animals, the binding of Egr1 was induced 4-fold above basal (1.34%) after 12 h of feeding (Fig. 6). Analysis of a negative control site was performed using a second Hmgcr primer set located 4 kb downstream of the transcription start site. In contrast to the refeeding-induced binding of Egr1 to the promoter, we observed only background levels of Egr1 localization to the +4 kb site in both the fasted and the fed mice, showing that Egr1 binding is specific for its target regulatory elements.
FIGURE 6.

Egr1 binds to cholesterol biosynthetic promoters in liver in response to feeding. In vivo ChIP, described under “Materials and Methods,” was used to assess Egr1 binding to putative binding sites in cholesterol biosynthetic gene promoters after 24 h of fasting or 24 h of fasting and 12 h of high carbohydrate feeding. The treatment of the mice is indicated on the x axis, where Fast denotes average values from three mice deprived of food for 24 h and Fed denotes average values from four mice fed for 12 h. Dark bars represent Egr1 binding, light bars represent IgG or background binding, and bars labeled with different letters indicate statistical significance, and error bars represent S.D. (p < 0.05, Welch's t test).
To determine whether this induction of Egr1 localization in response to feeding was extended to other cholesterol biosynthetic genes, as shown in the in vitro experiments, we investigated Egr1 binding to three other genes: Cyp51, Sqle, and Me1 (Fig. 6). The greatest induction of binding was observed at the Me1 promoter with nearly 5-fold induction of Egr1 localization representing a change from 1.22 to 5.80%. A similar increase in Egr1 localization was observed at Sqle, which is consistent with the insulin-dependent binding of Egr1 to the same gene in H4IIE cells. We observed a 3-fold induction of Egr1 binding to Cyp51 in mouse liver after feeding, corresponding to a change in binding from 0.17 to 0.50%. However, although the Egr1 binding at Cyp51 was statistically different from the IgG background binding in both fasted and fed animals; due to experimental variation, the induction of Egr1 localization did not reach statistical significance (Fig. 6). ChIP assays performed in fasted and fed Egr1 null mice detected only background levels of binding and no change with feeding when compared with fed wild-type mice, supporting the specificity of the Egr1 antibody used in our assays (data not shown).
Egr1−/− Mice Have Reduced Expression of Squalene Epoxidase
To test whether Egr1 plays a role in the induction of cholesterol gene expression following feeding, we performed a modified fasting and refeeding study. Age-matched wild-type and Egr1 knock-out mice were fasted for 12 h and fed high carbohydrate chow for 12 h. This protocol was repeated for five consecutive days, at which point the animals were sacrificed after feeding for 12 h. A cohort of wild-type control animals was treated identically but was sacrificed after 12 h of fasting to establish baseline expression of target genes. Cholesterol gene expression was induced after feeding (Fig. 7), consistent with previous work showing increased expression and cholesterol synthesis following feeding (3–6). Interestingly, Egr1 null animals showed a reduced response to high carbohydrate feeding when the expression of the cholesterol biosynthetic gene Sqle expression was examined (Fig. 7). The expression of Sqle was reduced 55% when compared with wild-type fed animals (p < 0.05, Welch's t test). Hmgcr expression was reduced 35%, but the reduction did not reach significance. However, Egr2 expression was 50% higher in the Egr1 null animals, which may indicate compensation of Egr2 for loss of Egr1.
FIGURE 7.

Egr1−/− mice have reduced Sqle gene expression. RT-qPCR was used to measure the response of wild-type and Egr1−/− mice to a protocol of fasting and refeeding as described under “Materials and Methods.” Average results are shown relative to gene expression in fed wild-type mice. In wild-type mice, the expression of Egr1, Egr2, Hmgcr, and Sqle was induced in fed mice when compared with fasted mice. However, Egr1−/− mice showed a significantly reduced induction of Sqle after feeding. Asterisks indicate p < 0.05. (n = 6 wild-type fasted mice, 6 wild-type fed mice, 6 Egr1−/− fed mice).
Egr1−/− Mice Have Reduced Serum Cholesterol
To determine how loss of Egr1 affects cholesterol levels, we measured fasted serum cholesterol levels in wild-type and Egr1 null mice. The targeted disruption of Egr1 is grossly normal, although it does have specific reproductive deficits (34). We used 14 age-matched Egr1−/− and 14 wild-type mice and observed that the Egr1−/− mice had on average 40% less serum cholesterol than wild-type mice (p < 0.0012) (Fig. 8A). 10 mice from each cohort were further analyzed to better pinpoint the role of Egr1. The knock-out mice had 53% less serum HDL (Fig. 8B), the lipoprotein containing the most serum cholesterol in rodents (p < 0.005). FPLC analysis of serum lipoprotein profiles is shown in supplemental Fig. 5. Four Egr1 knock-out mice were found to have HDL quantities below the detection level of this assay; these mice presumably lack detectable serum HDL molecules. Western blots for apoA-I of the FPLC fractions indicated that HDL apolipoproteins were synthesized but were not well lipidated in these knock-out mice (data not shown). In contrast, a moderate increase in serum triglycerides was observed, with the Egr1−/− mice having 27% higher levels (p < 0.01, data not shown). Based upon these data, we conclude that Egr1 is an important factor in the regulation of plasma cholesterol levels.
FIGURE 8.

Egr1−/− mice have reduced serum cholesterol. Total cholesterol levels were measured from the serum of 14 age-matched Egr1−/− and wild-type mice, and FPLC was performed to determine the amount of cholesterol in the HDL fraction. Each dot indicates the cholesterol level from a single mouse, with dashed lines indicating average values for each cohort. A, Egr1 null mice were found to have nearly 40% less serum cholesterol than age-matched wild-type controls (p < 0.0012, Welch's t test). B, HDL, the serum fraction containing the majority of murine cholesterol, was also reduced in Egr1 null mice by more than 50% (p < 0.01). Four knock-out mice had HDL levels below the detection limit of this assay and were assigned a value of 0.
DISCUSSION
The synthesis of lipids is controlled at multiple levels and is responsive to various physiological stimuli. Although SREBPs play a central role in transcriptional regulation of lipid biosynthetic genes (35), there is substantial evidence that other factors also contribute to regulation of this gene network by hormonal signals such as insulin. While genes involved in fatty acid metabolism appear to be induced primarily by insulin-dependent induction of SREBP-1 (3, 36), experiments in different systems have not consistently shown the induction of cholesterol biosynthetic genes to be due to a similar increase in SREBP-2 binding (5, 7, 9, 10). We and others (9) have found little change in SREBP occupancy in cholesterol genes in response to insulin (Fig. 2). Moreover, a fragment of the HMGCR promoter containing the sterol-response element was not found to be insulin-sensitive, whereas a fragment containing putative EGR sites was responsive to insulin (37). Recent ChIP analysis showed little change in SREBP-2 binding at the Hmgcr promoter in response to refeeding, although there was a significant increase in binding of SREBP-1 (10). Therefore, it is possible that SREBP-1 could mediate some of the induction of cholesterol biosynthetic promoters, but there are probably additional mechanisms because mice lacking SREBP-1c exhibit unimpaired induction of cholesterol gene expression in response to refeeding (5). Such considerations have formed the basis of one proposed model in which SREBP-2 acts as a permissive factor to provide a receptive environment for activation of target promoters by other insulin-responsive transcription factors (5).
This study tested whether Egr1 is such an accessory factor involved in cholesterol biosynthetic gene regulation. Egr1 was one of the first transcription factors found to be inducible by insulin (16), but a precise physiological role for Egr1 in liver has not been defined. Some hints regarding the physiological function of EGR factors have emerged from analysis of myelin formation in peripheral nerves, which has indicated that the large induction of lipid biosynthetic genes accompanying myelination by Schwann cells is dependent on Egr2 expression (13, 14, 38). Both functional analysis (14) and ChIP assays (39) are consistent with direct regulation of Hmgcr and other cholesterol biosynthetic genes by Egr2 in myelinating peripheral nerve. Interestingly, Egr1 knock-out mice have been shown to be resistant to ethanol-induced fatty liver and show much reduced lipogenesis. Although initial studies suggested that Egr1 may play a role in the inflammatory pathway by regulating TNF-α production by Kupffer cells, recent studies suggest that Egr1 may play a more intrinsic role in hepatocytes by regulating lipid synthesis (40, 41). Our data provide a potential mechanistic explanation for the reduced lipid synthesis in Egr1 null mice in response to ethanol. In addition, Egr1 was previously implicated in the induction of the LDLR gene by oncostatin M, specifically through an interaction with a sterol-independent regulatory element in the LDLR promoter (42–44). Finally, our data are consistent with a bioinformatics analysis implicating Egr1 in induction of cholesterol biosynthetic genes by growth factors (45).
Given that Egr1 is the predominant member of this family expressed in liver, we have tested whether Egr1 may play a similar regulatory role in hepatic cholesterol gene regulation. Here we have demonstrated an insulin-dependent induction of Egr1 in a liver cell line, and this induction of Egr1 expression is accompanied by an increase in Egr1 binding to the Hmgcr promoter. Additionally, we have shown that siRNA-directed knockdown of Egr1 transcript resulted in a reduced response of the Hmgcr gene to insulin induction. In addition, ChIP-chip analysis showed extensive localization of Egr1 to the proximal promoters of cholesterol biosynthetic genes in response to insulin, suggesting that Egr1 regulates a number of genes in this pathway (Table 1).
We have also demonstrated an induction of Egr1 binding to cholesterol biosynthetic promoters following high carbohydrate feeding. Our analysis revealed significantly lower levels of cholesterol in the serum of Egr1 null mice. This observation was not entirely unexpected as subtle reductions in serum cholesterol in Egr1 null mice had been reported in an ApoE knock-out background, albeit without reaching statistical significance (46). Consistent with the hypothesis of Egr1 as a modifier of cholesterol gene expression, a polymorphism in the human EGR1 promoter is associated with reduced serum cholesterol as well as a higher ratio of HDL to LDL (18). Here, we provide a potential mechanism explaining these observations.
Finally, we observed a reduced gene expression response to feeding in Egr1−/− animals. Squalene epoxidase (Sqle) oxidizes squalene to produce a reactive intermediate, which is then cyclized by lanosterol synthase into the four carbon rings characteristic of sterols. Interestingly, the expression of Sqle was significantly reduced in fed Egr1−/− animals. Although we observed a downward trend in the response of other lipogenic genes to feeding in Egr1−/− mice (e.g. Hmgcr, Cyp51, and Fdps), these changes did not reach statistical significance. The SREBPs have been shown to regulate expression of Sqle (47) along with nuclear transcription factor Y (48, 49). Chemical inhibition of Sqle has been shown to reduce cholesterol synthesis and result in devastating consequences in the nervous system due to demyelination (50, 51).
However, we also observed an induction of Egr2 expression in the Egr1−/− animals. Egr2 has been previously implicated in lipid gene regulation during sciatic nerve development (13, 38), as well as reporter assays testing promoter elements of Hmgcr (14). In vitro studies have also identified Egr2 as sensitive to insulin in the H4IIE cell line (17). The unexpected induction of Egr2 in the absence of Egr1 may indicate a compensatory mechanism present in vivo. Indeed, our Egr1 siRNA experiments in H4IIE cells indicate that Egr1 is a direct regulator of Sqle expression, as well as Hmgcr and other cholesterol biosynthetic genes. Other insulin-inducible factors with known binding sites in promoters of cholesterol genes include transcription factors Sp1 and cAMP-response element-binding protein (30, 37, 52), and recent genome-wide studies have documented widespread colocalization of SREBP binding sites with Sp1 consensus sites (6, 53). It should be noted that EGR factors have a binding specificity that overlaps with Sp1, such that many GC-rich sites can be bound by either Egr1 or Sp1. However, it is difficult to directly compare the relative physiological roles of Egr1 and Sp1 because the Sp1 null mouse is embryonic lethal (54). Another study has implicated another close EGR relative, Wilms tumor 1 (WT1), in regulation of cholesterol genes, and it is likely that common binding sites are used by these transcription factors (55).
One of the best studied cholesterol biosynthetic promoters is that of the Hmgcr gene (56, 57). Hmgcr activity is the rate-limiting step for cholesterol synthesis; consequently, many regulatory molecules affect this gene and gene product as a means of regulating the cholesterol biosynthetic pathway. There is substantial regulation of the activity of this key enzyme at the post-transcriptional level; however, studies using either SREBP knock-out or constitutively active SREBP studies in transgenic rodents have also shown substantial regulation at the transcriptional level (5, 8, 58–63). Several groups have published data supporting a role for an Sp1-like transcription factor to regulate this locus (9, 37). Using reporters containing deletion mutants of the Hmgcr promoter, Osborne et al. (37) identified a GC-rich site at −130 necessary for maximal transcription of Hmgcr. This site is distinct from known cAMP-response elements and sterol-response elements in the promoter and highly conserved in the human mouse and rat genomes (diagrammed in Fig. 2C). Interestingly, this site lies within a segment of the Hmgcr promoter that is required for optimal insulin induction of the Hmgcr gene (30). Finally, an in vivo footprinting analysis showed that the −119 to −140 region of the Hmgcr promoter is protected in normal rodent liver but susceptible in diabetic rodent liver, and it was concluded that an Sp1-like protein binds this region in normal tissue (9). Because Egr1 and Sp1 share binding specificity, our ChIP analysis using primers designed to detect enrichment of this insulin-sensitive region would suggest that Egr1 is the insulin-dependent factor binding to this regulatory site in the Hmgcr promoter.
The use of ChIP-chip analysis in this study allowed us to interrogate numerous target genes simultaneously and compare the binding at different promoters directly. One notable aspect of the experimental design is that the robust insulin induction of Egr1 allowed us to use DNA from an Egr1 immunoprecipitation from untreated cells as the reference sample to detect Egr1 binding in insulin-treated cells, which provides additional evidence for the specificity of the peaks that were detected. In parallel, ChIP-chip analysis using a control IgG ChIP sample as a reference identified the same peaks (data not shown).
We propose that liver and peripheral nerve tissues use the same family of transcription factors, the early growth response genes, as modulators of intracellular cholesterol biosynthesis. We and others have shown that Egr2 is required for induction of cholesterol biosynthetic genes during peripheral nerve myelination by Schwann cells (13, 14). Interestingly, formation of the cholesterol-rich myelin sheath relies on endogenous synthesis by Schwann cells (64). The direct role of EGR proteins in cholesterol synthesis is supported by the marked reduction of Hmgcr and other cholesterol biosynthetic genes in the Egr2 null mouse (14) as well as our data showing direct binding and activation of these genes by Egr2 (39). These same Egr2 binding sites utilized in peripheral nerve are bound by Egr1 in liver tissue in response to insulin. Overall, these data are consistent with utilization of EGR transcription factors to modulate cholesterol gene expression in both peripheral nerve and liver in response to developmental as well as hormonal cues.
Supplementary Material
Acknowledgments
We thank Anju Singh for assistance in the mouse liver perfusions and David Hui, Dana Lee, and Linda Keller at the Mouse Metabolic Phenotyping Center at the University of Cincinnati for performing assays and analysis of cholesterol levels. The MMPC is supported by National Institutes of Health Grant DK59630.
This work was supported by Grant-in-aid 0650102Z from the American Heart Association.

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table 1 and Figs. 1–5.
- SREBP
- sterol-response element-binding protein
- EGR
- early growth response
- qPCR
- quantitative PCR.
REFERENCES
- 1. Grundy S. M., Cleeman J. I., Merz C. N., Brewer H. B., Jr., Clark L. T., Hunninghake D. B., Pasternak R. C., Smith S. C., Jr., Stone N. J. (2004) J. Am Coll. Cardiol. 44, 720–732 [DOI] [PubMed] [Google Scholar]
- 2. Goldstein J. L., DeBose-Boyd R. A., Brown M. S. (2006) Cell 124, 35–46 [DOI] [PubMed] [Google Scholar]
- 3. Shimomura I., Bashmakov Y., Ikemoto S., Horton J. D., Brown M. S., Goldstein J. L. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 13656–13661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Foretz M., Pacot C., Dugail I., Lemarchand P., Guichard C., Le Lièpvre X., Berthelier-Lubrano C., Spiegelman B., Kim J. B., Ferré P., Foufelle F. (1999) Mol. Cell. Biol. 19, 3760–3768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liang G., Yang J., Horton J. D., Hammer R. E., Goldstein J. L., Brown M. S. (2002) J. Biol. Chem. 277, 9520–9528 [DOI] [PubMed] [Google Scholar]
- 6. Seo Y. K., Chong H. K., Infante A. M., Im S. S., Xie X., Osborne T. F. (2009) Proc. Natl. Acad. Sci. U.S.A 106, 13765–13769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Xie X., Liao H., Dang H., Pang W., Guan Y., Wang X., Shyy J. Y., Zhu Y., Sladek F. M. (2009) Mol. Endocrinol. 23, 434–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Matsuda M., Korn B. S., Hammer R. E., Moon Y. A., Komuro R., Horton J. D., Goldstein J. L., Brown M. S., Shimomura I. (2001) Genes Dev. 15, 1206–1216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lagor W. R., de Groh E. D., Ness G. C. (2005) J. Biol. Chem. 280, 36601–36608 [DOI] [PubMed] [Google Scholar]
- 10. Bennett M. K., Seo Y. K., Datta S., Shin D. J., Osborne T. F. (2008) J. Biol. Chem. 283, 15628–15637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Swirnoff A. H., Milbrandt J. (1995) Mol. Cell. Biol. 15, 2275–2287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Topilko P., Schneider-Maunoury S., Levi G., Baron-Van Evercooren A., Chennoufi A. B., Seitanidou T., Babinet C., Charnay P. (1994) Nature 371, 796–799 [DOI] [PubMed] [Google Scholar]
- 13. Le N., Nagarajan R., Wang J. Y., Araki T., Schmidt R. E., Milbrandt J. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 2596–2601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Leblanc S. E., Srinivasan R., Ferri C., Mager G. M., Gillian-Daniel A. L., Wrabetz L., Svaren J. (2005) J. Neurochem. 93, 737–748 [DOI] [PubMed] [Google Scholar]
- 15. Jee S., Hwang D., Seo S., Kim Y., Kim C., Kim B., Shim S., Lee S., Sin J., Bae C., Lee B., Jang M., Kim M., Yim S., Jang I., Cho J., Chae K. (2007) Int. J. Mol. Med. 20, 829–835 [PubMed] [Google Scholar]
- 16. Sukhatme V. P., Kartha S., Toback F. G., Taub R., Hoover R. G., Tsai-Morris C. H. (1987) Oncogene Res. 1, 343–355 [PubMed] [Google Scholar]
- 17. Keeton A. B., Bortoff K. D., Bennett W. L., Franklin J. L., Venable D. Y., Messina J. L. (2003) Endocrinology 144, 5402–5410 [DOI] [PubMed] [Google Scholar]
- 18. Brand E., Herrmann S. M., Nicaud V., Evans A., Ruidavets J. B., Arveiler D., Luc G., Cambien F., Soubrier F. (2000) J. Mol. Med. 78, 81–86 [DOI] [PubMed] [Google Scholar]
- 19. Lee S. L., Wang Y., Milbrandt J. (1996) Mol. Cell. Biol. 16, 4566–4572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Livak K. J., Schmittgen T. D. (2001) Methods 25, 402–408 [DOI] [PubMed] [Google Scholar]
- 21. Jones E. A., Lopez-Anido C., Srinivasan R., Krueger C., Chang L. W., Nagarajan R., Svaren J. (2011) J. Neurosci. 31, 4242–4250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Loots G. G., Ovcharenko I., Pachter L., Dubchak I., Rubin E. M. (2002) Genome Res. 12, 832–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jang S. W., LeBlanc S. E., Roopra A., Wrabetz L., Svaren J. (2006) J. Neurochem. 98, 1678–1687 [DOI] [PubMed] [Google Scholar]
- 24. Flowers M. T., Groen A. K., Oler A. T., Keller M. P., Choi Y., Schueler K. L., Richards O. C., Lan H., Miyazaki M., Kuipers F., Kendziorski C. M., Ntambi J. M., Attie A. D. (2006) J. Lipid Res. 47, 2668–2680 [DOI] [PubMed] [Google Scholar]
- 25. Nakanishi M., Tomaru Y., Miura H., Hayashizaki Y., Suzuki M. (2008) Nucleic Acids Res. 36, 3443–3454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Figliola R., Busanello A., Vaccarello G., Maione R. (2008) J. Mol. Biol. 380, 265–277 [DOI] [PubMed] [Google Scholar]
- 27. Mayer S. I., Dexheimer V., Nishida E., Kitajima S., Thiel G. (2008) Endocrinology 149, 6311–6325 [DOI] [PubMed] [Google Scholar]
- 28. Thyss R., Virolle V., Imbert V., Peyron J. F., Aberdam D., Virolle T. (2005) EMBO J. 24, 128–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Brown M. S., Goldstein J. L. (1997) Cell 89, 331–340 [DOI] [PubMed] [Google Scholar]
- 30. Osborne A. R., Pollock V. V., Lagor W. R., Ness G. C. (2004) Biochem. Biophys. Res. Commun. 318, 814–818 [DOI] [PubMed] [Google Scholar]
- 31. Barroso I., Santisteban P. (1999) J. Biol. Chem. 274, 17997–18004 [DOI] [PubMed] [Google Scholar]
- 32. Tobin K. A., Ulven S. M., Schuster G. U., Steineger H. H., Andresen S. M., Gustafsson J. A., Nebb H. I. (2002) J. Biol. Chem. 277, 10691–10697 [DOI] [PubMed] [Google Scholar]
- 33. Kang J., Holland M., Jones H., Kaysen G. A. (1999) Kidney Int. 56, 452–460 [DOI] [PubMed] [Google Scholar]
- 34. Wolfe M. W., Call G. B. (1999) Mol. Endocrinol. 13, 752–763 [DOI] [PubMed] [Google Scholar]
- 35. Horton J. D., Goldstein J. L., Brown M. S. (2002) J. Clin. Invest. 109, 1125–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Foretz M., Guichard C., Ferré P., Foufelle F. (1999) Proc. Natl. Acad. Sci. U.S.A. 96, 12737–12742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Osborne T. F., Gil G., Goldstein J. L., Brown M. S. (1988) J. Biol. Chem. 263, 3380–3387 [PubMed] [Google Scholar]
- 38. Nagarajan R., Svaren J., Le N., Araki T., Watson M., Milbrandt J. (2001) Neuron 30, 355–368 [DOI] [PubMed] [Google Scholar]
- 39. Jang S. W., Srinivasan R., Jones E. A., Sun G., Keles S., Krueger C., Chang L. W., Nagarajan R., Svaren J. (2010) J. Neurochem. 115, 1409–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McMullen M. R., Pritchard M. T., Wang Q., Millward C. A., Croniger C. M., Nagy L. E. (2005) Gastroenterology 128, 2066–2076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kishore R., Hill J. R., McMullen M. R., Frenkel J., Nagy L. E. (2002) Am. J. Physiol. Gastrointest. Liver Physiol. 282, G6–15 [DOI] [PubMed] [Google Scholar]
- 42. Zhang F., Ahlborn T. E., Li C., Kraemer F. B., Liu J. (2002) J. Lipid Res. 43, 1477–1485 [DOI] [PubMed] [Google Scholar]
- 43. Zhang F., Lin M., Abidi P., Thiel G., Liu J. (2003) J. Biol. Chem. 278, 44246–44254 [DOI] [PubMed] [Google Scholar]
- 44. Zhou Y., Zhang F., Abidi P., Lin M., Thiel G., Liu J. (2006) Biochem. J. 397, 101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Seifert M., Scherf M., Epple A., Werner T. (2005) Trends Genet 21, 553–558 [DOI] [PubMed] [Google Scholar]
- 46. Harja E., Bucciarelli L. G., Lu Y., Stern D. M., Zou Y. S., Schmidt A. M., Yan S. F. (2004) Circ. Res. 94, 333–339 [DOI] [PubMed] [Google Scholar]
- 47. Sakakura Y., Shimano H., Sone H., Takahashi A., Inoue N., Toyoshima H., Suzuki S., Yamada N., Inoue K. (2001) Biochem. Biophys. Res. Commun. 286, 176–183 [DOI] [PubMed] [Google Scholar]
- 48. Murphy C., Ledmyr H., Ehrenborg E., Gåfvels M. (2006) Biochim. Biophys. Acta 1761, 1213–1227 [DOI] [PubMed] [Google Scholar]
- 49. Nagai M., Sakakibara J., Nakamura Y., Gejyo F., Ono T. (2002) Biochem. Biophys. Res. Commun. 295, 74–80 [DOI] [PubMed] [Google Scholar]
- 50. Toews A. D., Roe E. B., Goodrum J. F., Bouldin T. W., Weaver J., Goines N. D., Morell P. (1997) Brain Res. Mol. Brain Res. 49, 113–119 [DOI] [PubMed] [Google Scholar]
- 51. Toews A. D., Jurevics H., Hostettler J., Roe E. B., Morell P. (1996) J. Lipid Res. 37, 2502–2509 [PubMed] [Google Scholar]
- 52. Dooley K. A., Bennett M. K., Osborne T. F. (1999) J. Biol. Chem. 274, 5285–5291 [DOI] [PubMed] [Google Scholar]
- 53. Reed B. D., Charos A. E., Szekely A. M., Weissman S. M., Snyder M. (2008) PLoS Genet 4, e1000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Marin M., Karis A., Visser P., Grosveld F., Philipsen S. (1997) Cell 89, 619–628 [DOI] [PubMed] [Google Scholar]
- 55. Rae F. K., Martinez G., Gillinder K. R., Smith A., Shooter G., Forrest A. R., Grimmond S. M., Little M. H. (2004) Oncogene 23, 3067–3079 [DOI] [PubMed] [Google Scholar]
- 56. Ness G. C., Chambers C. M. (2000) Proc. Soc. Exp. Biol. Med. 224, 8–19 [DOI] [PubMed] [Google Scholar]
- 57. Naito M., Bomsztyk K., Zager R. A. (2009) Am. J. Pathol. 174, 54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Shimano H., Horton J. D., Hammer R. E., Shimomura I., Brown M. S., Goldstein J. L. (1996) J. Clin. Invest. 98, 1575–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Shimano H., Shimomura I., Hammer R. E., Herz J., Goldstein J. L., Brown M. S., Horton J. D. (1997) J. Clin. Invest. 100, 2115–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Shimano H., Horton J. D., Shimomura I., Hammer R. E., Brown M. S., Goldstein J. L. (1997) J. Clin. Invest. 99, 846–854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Horton J. D., Shimomura I., Brown M. S., Hammer R. E., Goldstein J. L., Shimano H. (1998) J. Clin. Invest. 101, 2331–2339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Korn B. S., Shimomura I., Bashmakov Y., Hammer R. E., Horton J. D., Goldstein J. L., Brown M. S. (1998) J. Clin. Invest. 102, 2050–2060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Yang J., Goldstein J. L., Hammer R. E., Moon Y. A., Brown M. S., Horton J. D. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 13607–13612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Jurevics H. A., Morell P. (1994) J. Lipid Res. 35, 112–120 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.