

Abstract
The T332I mutation in Rho guanine nucleotide exchange factor 10 (ARHGEF10) was previously found in persons with slowed nerve conduction velocities and thin myelination of peripheral nerves. However, the molecular and cellular basis of the T332I mutant is not understood. Here, we show that ARHGEF10 has a negative regulatory region in the N terminus, in which residue 332 is located, and the T332I mutant is constitutively active. An N-terminal truncated ARHGEF10 mutant, ARHGEF10 ΔN (lacking amino acids 1–332), induced cell contraction that was inhibited by a Rho kinase inhibitor Y27632 and had higher GEF activity for RhoA than the wild type. The T332I mutant also showed the phenotype similar to the N-terminal truncated mutant. These data suggest that the ARHGEF10 T332I mutation-associated phenotype observed in the peripheral nerves is due to activated GEF activity of the ARHGEF10 T332I mutant.
Keywords: Actin, Cell Contraction, Cytoskeleton, Mutant, Nerve, Neurological Diseases, Rho, RhoGEF, Schwann Cell
Introduction
The small GTPases Rho family regulates the actin cytoskeleton to influence many cellular processes, including morphological changes, migration, and cytokinesis in various cells (1). The Rho family consists of 20 members (2). Among them, RhoA, Rac1, and Cdc42 are well known to induce the formations of actin stress fibers and focal adhesions, lamellipodia, and filopodia, respectively, in fibroblasts (3–5). Rho family GTPases serve as molecular switches by cycling between an active (GTP-bound) state and an inactive (GDP-bound) state. In their active state, they can interact with a variety of downstream effectors (1). Of them, Rho kinase (ROCK),5 a serine/threonine kinase, is known to regulate actomyosin contractility by controlling the phosphorylation state of myosin light chain (6–8). The activity of the Rho family is controlled by guanine nucleotide exchange factors (GEFs), GTPase-activating proteins, and guanine nucleotide dissociation inhibitors. GEFs catalyze the replacement of GDP with GTP, GTPase-activating proteins accelerate the slow intrinsic hydrolysis activity of GTPases, and guanine nucleotide dissociation inhibitors inhibit the exchange of GDP for GTP.
RhoGEFs of the Dbl type are composed of 69 members, and they share a catalytic Dbl homology (DH) domain that is required for GEF activity (9, 10). Mutations in genes encoding several Dbl-type RhoGEFs, such as FGD4 and ARHGEF10/RhoGEF10, are implicated in motor and sensory neuropathies (11–13). FGD4 and ARHGEF10 are RhoGEFs for Cdc42 and Rho (RhoA, RhoB, and RhoC), respectively (14–17). Many mutations in FGD4 were identified, and some of them were characterized at the molecular level (11, 12). In addition, one missense mutation in ARHGEF10 identified as an amino acid substitution of threonine (T) to isoleucine (I) at codon 332 (T332I) is associated with slowed nerve conduction velocities and thin myelination of peripheral nerves in humans without any obvious clinical symptoms in the affected patients (13).
Because the molecular and cellular basis of ARHGEF10 T332I mutant is unknown, we have investigated this and shown that ARHGEF10 has a negatively regulatory region in the N terminus and that T332I mutant is a constitutively activated GEF mutant. Our results might provide the insight into the mechanism of T332I-associated phenotype observed in the peripheral nervous system.
EXPERIMENTAL PROCEDURES
Plasmid Construction
The cDNA of human ARHGEF10 (KIAA0294) was kindly provided by T. Nagase (Kazusa DNA Research Institute, Chiba, Japan). Although ARHGEF10 coding sequence used in a previous study (16) started at position 512 of the nucletotide sequence NM_014629.2 (GenBank accession number), in this study it started at position 179 of that. The full-length of ARHGEF10 wild type (wt) was amplified by PCR and subcloned into the mammalian myc- and GFP-tagged expression vectors pCMV-myc and pEGFP-C2, thus generating pCMV-myc-ARHGEF10 wt and pEGFP-C2-ARHGEF10 wt. The N-, C-, and N- and C-terminal deletion mutants were generated by PCR amplification with pCMV-myc-ARHGEF10 wt as a template and subcloned into pEGFP-C2. ARHGEF10 T332I was generated by PCR-mediated mutagenesis with pEGFP-C2-ARHGEF10 wt as a template and subcloned into pCMV-myc and pEGFP-C2. ARHGEF10 T332I ΔDH (lacking amino acids 397–583), T332I/S407A, and T332I/L547A were also generated by PCR-mediated mutagenesis with pCMV-myc-ARHGEF10 T332I as a template and subcloned into pEGFP-C2. RhoA, RhoB, and RhoC were obtained by reverse transcription PCR from mouse kidney, and they were subcloned into the HA-tagged expression vector pEF-HA. All sequences were confirmed by automatic DNA sequencers. The GST-tagged expression plasmid pGEX-5X-1-Rho-binding domain (RBD) of Rhotekin was obtained as described previously (18).
Antibodies and Reagents
Antibodies used were as follows: mouse monoclonal anti-myc antibody (American Type Culture Collection); mouse monoclonal anti-HA antibody (InvivoGen); rat monoclonal anti-GFP antibody (Nacalai Tesque); rabbit polyclonal anti-S100 antibody (DAKO); horseradish peroxidase-conjugated secondary antibodies (Jackson Laboratories); FITC-conjugated secondary antibody (Jackson ImmunoResearch). TRITC- phalloidin, cytosine β-d-arabinofuranoside (Ara-C), and a specific ROCK inhibitor Y27632 were purchased from Sigma.
Cell Culture and Transfection
HEK293T and HeLa cells were cultured in DMEM containing 10% FBS. Primary Schwann cells were obtained as described previously (19). Briefly, sciatic nerves of Wister rats at postnatal day 2 or 3 were dissected from 8–12 animals and incubated for 40 min at 37 °C in 3 ml of PBS containing 1 mg/ml collagenase, followed by incubation for 20 min after addition of 100 μl of 2.5% trypsin. The turbid suspension was passed through sterile square nylon gauze to remove debris and centrifuged at 1000 × g for 3 min. The supernatant was removed, and the pellet was resuspended in 3 ml of DMEM containing 10% FBS. The suspension was plated into the 10-cm dish and incubated for 1 day. Then, the medium was removed, and DMEM containing 10% FBS and 10 μm Ara-C was added. After 3 days, the cells were incubated in the same medium for another 7 days to select Schwann cells. Selected Schwann cells were cultured on poly-l-lysine-coated dishes in DMEM containing 10% FBS, 100 units/ml penicillin, and 0.1 mg/ml streptomycin. Schwann cells were identified by immunostaining using anti-S100 antibody. All cells were grown at 37 °C in 5% CO2.
Transfection
Transient transfections were carried out with the calcium phosphate method for HEK293T and HeLa cells except for the reporter gene assay, or with Lipofectamine 2000 (Invitrogen) for Schwann cells, according to the manufacturer's instructions.
Cell Counting
HEK293T and HeLa cells were seeded in 35-mm dishes at a density of 3.5 × 105 cells/dish, transfected, and cultured for 48 h for HEK293T cells and 24 h for HeLa cells. The fluorescence images of living cells were obtained with a digital camera (DP70; Olympus) equipped with a microscope (OPTIPHOT2-UD; Nikon), and GFP-positive cell numbers were counted. Cells floating in the culture medium were excluded from counting. For Y27632 treatment, cells seeded at a density of 3.5 × 105 cells/35-mm dish were cultured for 16 h after transfection and treated with 10 μm Y27632 for 24 h for HEK293T cells and HeLa cells for 8 h. The number of GFP-positive living cells was counted as described above. Schwann cells were seeded on poly-l-lysine-coated 24-well plates at a density of 3 × 105 cells/well and cultured for 48 h after transfection. After then, Schwann cells were stained with Hoechst33258. The fluorescence images of living cells were obtained with a digital camera (DP72; Olympus) equipped with a microscope (IX70; Olympus), and the number of GFP-positive cells was counted. Cells with the DNA fragmentation or the chromatin aggregation, and cells floating in the culture medium were excluded from counting.
Cytochemical Staining
Cells were seeded onto round 10-mm glass coverslips coated with polyethylenimine in 35-mm dishes at a density of 1 × 105 cells/dish, transfected, and cultured for 48 h for HEK293T cells and 24 h for HeLa cells. Cells on coverslips were washed with PBS and fixed with 4% paraformaldehyde in PBS for 15 min. Cells were then washed with PBS, permeabilized with 0.5% Triton X-100 in PBS, and blocked with blocking solution (5 mg/ml BSA, 50 mm glycine, 2% goat serum, 0.1% NaN3 in PBS). Cells were stained with tetramethyl rhodamine isothiocyanate-phalloidin for filamentous actin (F-actin) in blocking solution (1:1000 dilution) or primary antibodies including anti-S100 antibody (1:1000 dilution) for 1 h. Cells were then washed with PBS three times, and DNA was stained with Hoechst33258 in PBS (1:5000 dilution) for 3 min. Fluorescence images were obtained with a digital camera (DS-Qi1-U2; Nikon) equipped with a microscope (ECLIPSE E600; Nikon).
GST-fused Rhotekin-RBD Beads
GST-fused Rhotekin-RBD was purified from Escherichia coli XL1 Blue transformed with pGEX-5X-1-Rhotekin-RBD. Cells were treated with 0.1 mm isopropyl β-d-thiogalactopyranoside at room temperature for 4 h and harvested by centrifugation. Harvested cells were lysed in extraction buffer (20 mm Tris-HCl, pH 7.5, 1 mm EDTA, 150 mm NaCl, 0.1% Triton X-100, 2 mm DTT, 10 mm MgCl2, 0.1 mm PMSF) and centrifuged. The supernatants were mixed with glutathione-Sepharose beads (Amersham Biosciences) and incubated at 4 °C for 60 min. The beads were washed with extraction buffer six times. Concentration of purified protein was quantified by SDS-PAGE and Coomassie Brilliant Blue staining.
Immunoblotting
Proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride membrane (Millipore Corp.). The membranes were blocked with 3% low fat milk in Tris-buffered saline with Tween 20 (20 mm Tris-HCl, pH 7.5, 150 mm NaCl, 0.05% Tween 20) and then incubated with primary antibodies. The bound antibodies were detected with HRP-conjugated secondary antibodies and the ECL detection kit (Amersham Biosciences).
Detection of Active RhoA, RhoB, and RhoC
Detection of active RhoA, RhoB, and RhoC was performed as described previously (18, 20). Briefly, HEK293T cells were co-transfected with plasmids encoding GFP- or myc-tagged ARHGEF10 and HA-tagged RhoA, RhoB, or RhoC. After 48 h, cells were washed with ice-cold Tris-buffered saline (20 mm Tris-HCl, pH 7.5, 150 mm NaCl), lysed with lysis buffer (50 mm Tris-HCl, pH 7.5, 1% Nonidet P-40, 150 mm NaCl, 10 mm MgCl2, 1 mm EDTA, 10% glycerol, 25 mm NaF, 1 mm Na3VO4, 0.1 mm PMSF), and centrifuged. The supernatants were mixed with GST-fused Rhotekin-RBD beads and incubated at 4 °C for 45 min. The beads were washed with lysis buffer four times, and bound proteins were eluted by SDS-sample buffer and subjected to SDS-PAGE and immunoblotting. The band intensity in the immunoblot with anti-HA antibody was quantified with National Institutes of Health ImageJ software, and the amounts of RhoA, RhoB, or RhoC-GTP were normalized to the total amounts of them, respectively.
Reporter Gene Assay
Reporter gene assay was performed with Dual-Luciferase Reporter Assay System (Promega) according to the manufacturer's protocol. HEK293T cells and HeLa cells were seeded in 96-well plates at a density of 1.5 × 104 cells/well and in 24-well plates at a density of 7.5 × 104 cells/well, respectively. Cells were transiently co-transfected with the plasmid encoding GFP-ARHGEF10 (150 ng), pSRE.l-luciferase reporter plasmid (Promega) (43 ng), and pRL-TK control vector (Promega) (7 ng). After 48 h, HEK293T cells or HeLa cells were washed with ice-cold PBS and lysed with 40 or 100 μl of Passive Lysis Buffer (Promega), respectively. Five μl (for HEK293T cells) or 10 μl (for HeLa cells) of the supernatants was mixed with 50 μl of luciferase substrate (Promega), and luciferase activities were determined by luminescence intensity. Luciferase activities of the reporter plasmid were normalized to those of the control vector.
Statistics
Data are expressed as means ± S.E. derived from at least three independent experiments. Statistical significance was calculated with a Student's t test. A value of p < 0.05 was taken to be statistically significant.
RESULTS
An N-terminal Truncation Mutant of ARHGEF10 Induces Cell Contraction
The N-terminal regions of several RhoGEFs often regulate negatively their GEF activities (21–26); however, little is known about the role of the N-terminal region of ARHGEF10 in which Thr-332 is located. Until now, at least two start codons at different position in ARHGEF10 have been selected in studies about the function of ARHGEF10. Mohl et al. selected position 437 of the nucletotide sequence NM_014629.2 (GenBank accession number) as a start codon (16), whereas Aoki et al. selected position 179 of that as a start codon (17). In this study we chose position 179 as Aoki et al. did. Thus, when our initiating methionine is at residue Met-1, that selected by Mohl et al. is at residue Met-87, and they studied an ARHGEF10 protein starting at residue Ala-112 (16). The N-terminal region of ARHGEF10 was first analyzed by focusing on its regulatory role on GEF activity. An N-terminal truncation mutant (ΔN) that lacks 1–332 amino acids existing in the upstream region of DH domain was generated in this study (Fig. 1A). To investigate the effect of the N-terminal region of ARHGEF10 for cell morphology, HEK293T cells were transfected with the plasmid encoding GFP-tagged ARHGEF10 wt, GFP-ARHGEF10 ΔN, or GFP as a control. GFP images of living cells were obtained using fluorescence microscopy. Expression of ARHGEF10 ΔN significantly induced cell contraction compared with wt or GFP (Fig. 1, B and C). To characterize the cell contraction, cells were co-stained with TRITC-phalloidin for F-actin and Hoechst33258 for DNA. The cell morphology represented by fluorescence of F-actin was similar to that represented by the fluorescence of GFP (Fig. 1D). F-actin was often observed in the periphery in the cells overexpressing GFP-ARHGEF10 wt or ΔN. In most contracted cells, the DNA fragmentation, the chromatin aggregation, or a metaphase plate of aligned chromosomes was not observed, suggesting that these cells are neither apoptotic nor in metaphase of mitosis. Because a recent study (17) indicated that ARHGEF10 is involved in the regulation of centrosome duplication, the effect of overexpression of ARHGEF10 ΔN was examined on centrosome duplication by using the fluorescence microscopy. No effect was observed on the number and localization of centrosome of HEK293T cells (data not shown).
FIGURE 1.

Cell contraction induced by ARHGEF10 ΔN in HEK293T cells. A, domain structures of ARHGEF10 are presented. Full-length ARHGEF10 (wt) and an N-terminal truncation mutant lacking N-terminal 332 amino acids of ARHGEF10 (ΔN) are shown. B, HEK293T cells were transiently transfected with the plasmid encoding GFP, GFP-tagged ARHGEF10 wt, or GFP-ARHGEF10 ΔN. After 48 h, fluorescence images of living cells were observed. Scale bar, 10 μm. C, quantitative analysis of cell contraction of HEK293T cells is shown. HEK293T cells were transiently transfected with the plasmid encoding GFP, GFP-ARHGEF10 wt or ΔN. The proportion of cell contraction was scored as a percentage of the rounded cells of GFP-positive cells. Cells floating in the culture medium were excluded from counting. The data represent the mean ± S.E. (error bars) from three independent experiments. *, p < 0.05 compared with empty. For each experiment, >100 cells were counted. D, HEK293T cells were transiently transfected with the plasmid encoding GFP-ARHGEF10 wt or ΔN. After 48 h, cells were fixed and co-stained with TRITC-phalloidin (F-actin) and Hoechst33258 (DNA). Transfected cells are shown by the fluorescence of GFP. Scale bar, 10 μm.
ARHGEF10 ΔN-induced Cell Contraction Is Mediated by ROCK
Rho-ROCK signaling is implicated in the cellular morphological change (6–8). We next examined whether ARHGEF10 ΔN-induced cell contraction is mediated through this signaling or not. HEK293T cells were transfected with the plasmid encoding GFP-ARHGEF10 ΔN and treated with a specific ROCK inhibitor Y27632 for 1 day. As shown in Fig. 2, A and B, ARHGEF10 ΔN-induced cell contraction was significantly inhibited by this treatment. This result indicates that ARHGEF10 ΔN-induced cell contraction is mediated by ROCK, suggesting that this cell contraction is mediated through Rho-ROCK signaling.
FIGURE 2.

ARHGEF10 ΔN-induced cell contraction mediated through Rho-ROCK signaling. A, HEK293T cells were transiently transfected with the plasmid encoding GFP-ARHGEF10 ΔN and treated with or without 10 μm Y27632 for 24 h. After that, fluorescence images of living cells were observed. Arrowheads indicate rounded cells. Arrows indicate cells floating in the culture medium. Scale bar, 10 μm. B, quantitative analysis of cell contraction of HEK293T cells. HEK293T cells were transiently transfected with the plasmid encoding GFP-ARHGEF10 ΔN and treated with or without 10 μm Y27632 for 24 h. The proportion of cell contraction was scored as a percentage of the rounded cells of GFP-positive cells. Cells floating in the culture medium were excluded from counting. The data represent the mean ± S.E. (error bars) from three independent experiments. *, p < 0.05. For each experiment, >100 cells were counted. C and D, HEK293T cells were transiently co-transfected with the plasmids encoding GFP-ARHGEF10 wt or ΔN, and HA-tagged RhoA. After 48 h, cells were lysed with lysis buffer. The supernatants were mixed with GST-fused Rhotekin-RBD, and an active form of RhoA (RhoA-GTP) was pulled down. The amounts of active form of RhoA were determined by immunoblotting with anti-HA antibody. Expressions of HA-RhoA and GFP-ARHGEF10 in cell lysates were also detected by immunoblotting with anti-HA and anti-GFP antibodies, respectively. The representative image of three independent experiments is shown. Relative RhoA activity is indicated by the amounts of RhoA-GTP normalized to total amounts of RhoA in cell lysates, and values are expressed as -fold value of cells transfected with wt. The data are the mean ± S.E. from three independent experiments. *, p < 0.05.
ARHGEF10 ΔN Activates RhoA, RhoB, and RhoC
To test whether the ARHGEF10 ΔN-induced cell contraction is dependent on Rho activity, active RhoA, RhoB, and RhoC were evaluated by a pulldown assay using GST-tagged Rhotekin-RBD (20). There was significantly more active RhoA in lysates from cells expressing ARHGEF10 ΔN than in lysates from cells expressing wt (Fig. 2, C and D). This result revealed that ARHGEF10 ΔN has higher GEF activity for RhoA compared with wt. Previously, Mohl et al. showed that ARHGEF10 activates RhoA, RhoB, and RhoC (16). RhoB and RhoC activities were also enhanced in the cells expressing ARHGEF10 ΔN (supplemental Fig. S1). In contrast, we could not detect GEF activity for Rac1 and Cdc42 of ARHGEF10 ΔN (data not shown). These data suggest that the N-terminal region of ARHGEF10 functions as the domain required for repressing its GEF activity for RhoA, RhoB, and/or RhoC.
Identification of Negatively Regulatory and Essential Regions for GEF Activity of ARHGEF10
To investigate the negatively regulatory region of GEF activity of ARHGEF10 in detail, we generated another N-terminal truncation mutant (ΔN′), which lacks 211 amino acids from the N terminus of ARHGEF10 wt (Fig. 3A). To examine the effect of expression of this mutant for cell morphology, HEK293T cells were transfected with the plasmid encoding GFP-ARHGEF10 wt, ΔN, or ΔN′, and GFP images of living cells were analyzed using fluorescence microscopy. Cells expressing ARHGEF10 ΔN′ did not significantly show cell contraction in contrast to ARHGEF10 ΔN (Fig. 3, B and C). To determine whether or not this mutant has lower GEF activity for RhoA compared with ΔN, we evaluated the active RhoA by a pulldown assay using GST-Rhotekin-RBD. There was significantly less active RhoA in lysates from cells expressing ARHGEF10 ΔN′ as well as wt than in lysates from cells expressing ΔN (Fig. 3D). These data demonstrate that the region containing amino acids 212–332 of ARHGEF10 is the region necessary to repress its GEF activity. To investigate whether this region inhibits GEF activity of ARHGEF10 ΔN, ARHGEF10 ΔN-induced cell contraction was examined in HEK293T cells co-transfected with this region. The co-expression of residues 212–332 could not inhibit ΔN-induced cell contraction (data not shown). This observation suggests that the region containing amino acids 212–332 is necessary but not sufficient for the inhibition of GEF activity of ARHGEF10. Previously, Mohl et al. reported that both full-length ARHGEF10 and ARHGEF10short, which is a splicing variant of ARHGEF10 and lacks residues 282–320, have a similar level of GEF activity toward Rho (16). To investigate whether ARHGEF10 282–320 is involved in the negative regulatory effect on GEF activity, HEK293T cells were transfected with the plasmid encoding GFP-ARHGEF10 ΔN or 265–1344 (lacking amino acids 1–264). Both ARHGEF10 ΔN and 265–1344 induced cell contraction at a similar level, suggesting that they have a similar level of GEF activity for Rho (data not shown). Our data support the idea that ARHGEF10 282–320 is not a negative regulatory region for GEF activity.
FIGURE 3.

Cell contraction and activation of RhoA by various deletion mutants of ARHGEF10 in HEK293T cells. A, domain structures of ARHGEF10. Full-length ARHGEF10 (wt), N-terminal truncation mutants lacking N-terminal 332 and 211 amino acids of ARHGEF10 (ΔN and ΔN′, respectively), and C-terminal truncation mutants lacking C-terminal 2 and 41 amino acids of ARHGEF10 ΔN (ΔN/ΔC and ΔN/ΔC′, respectively) are shown. B, HEK293T cells transiently transfected with the plasmid encoding GFP-ARHGEF10 wt, ΔN, ΔN′, ΔN/ΔC, or ΔN/ΔC′. After 48 h, fluorescence images of living cells observed. Scale bar, 10 μm. C, quantitative analyses of cell contraction of HEK293T cells. HEK293T cells were transiently transfected with the plasmid encoding GFP-ARHGEF10 wt, ΔN, ΔN′, ΔN/ΔC, or ΔN/ΔC′. The proportion of cell contraction was scored as a percentage of the rounded cells of GFP-positive cells. Cells floating in the culture medium were excluded from counting. The data represent the mean ± S.E. (error bars) from three independent experiments. *, p < 0.05 compared with wt. For each experiment, >100 cells were counted. D, HEK293T cells transiently co-transfected with the plasmids encoding GFP-ARHGEF10 wt, ΔN, ΔN′, ΔN/ΔC, or ΔN/ΔC′, and HA-RhoA. After 48 h, cells were lysed with lysis buffer. The supernatants were mixed with GST-Rhotekin-RBD, and an active form of RhoA was pulled down. The amounts of active form of RhoA were determined by immunoblotting with anti-HA antibody. Expressions of HA-RhoA and GFP-ARHGEF10 in cell lysates were also detected by immunoblotting with anti-HA and anti-GFP antibodies, respectively. The representative image of three independent experiments is shown. Relative RhoA activity is indicated by the amounts of RhoA-GTP normalized to total amounts of RhoA in cell lysates, and values are expressed as -fold value of cells transfected with wt. The data are the mean ± S.E. from three independent experiments. *, p < 0.05 compared with wt.
It was reported that the C-terminal truncation mutant of ARHGEF10 showed lower GEF activity for Rho compared with full-length ARHGEF10 (16). To investigate in detail whether the C-terminal region is involved in GEF activity, two C-terminal truncation mutants, each of which lacks 2 amino acids (ΔN/ΔC) or 41 amino acids (ΔN/ΔC') from the C terminus of ARHGEF10 ΔN were generated (Fig. 3A). To examine the effect of expressions of these mutants on GEF activity including cell contraction, HEK293T cells were transfected with the plasmid encoding GFP-ARHGEF10 wt, ΔN, ΔN/ΔC, or ΔN/ΔC′, and GFP images of living cells were analyzed using fluorescence microscopy. ARHGEF10 ΔN and ΔN/ΔC but not ΔN/ΔC' significantly induced cell contraction compared with wt (Fig. 3B, C). Then the levels of active RhoA of cells expressing ARHGEF10 wt, ΔN, ΔN/ΔC and ΔN/ΔC' were evaluated by the pull-down assay using GST-Rhotekin-RBD. The levels of active RhoA were higher in lysates from cells expressing ARHGEF10 ΔN or ΔN/ΔC but not ΔN/ΔC' than in lysates from cells expressing wt (Fig. 3D). The results of active RhoA assay coincided with the results of the cell contraction assay. ARHGEF10 ΔN/ΔC as well as ARHGEF10 ΔN showed higher GEF activity than wt, whereas ARHGEF10 ΔN/ΔC' showed lower GEF activity compared with ARHGEF10 ΔN and ΔN/ΔC. These data indicate that the region containing amino acids 1304–1342 of ARHGEF10 is required for its GEF activity, but the last 2 amino acids of the C terminus of ARHGEF10 is not necessary for the GEF activity.
ARHGEF10 T332I Induces Cell Contraction Similar to ΔN
Our data described above indicate that the region including amino acids 212–332 of ARHGEF10 contains negatively regulatory domain for its GEF activity. From these data, we speculated that the substitution of threonine to isoleucine at residue 332 might affect GEF activity of ARHGEF10. To investigate this hypothesis, the plasmid encoding GFP-ARHGEF10 T332I was transfected into HEK293T cells and GFP images of living cells were analyzed by fluorescence microscopy (Fig. 4A). Similar to ARHGEF10 ΔN, ARHGEF10 T332I significantly induced cell contraction compared with wt (Fig. 4, B and C). Similar results were also obtained using HeLa cells (supplemental Fig. S2, A and B). To characterize ARHGEF10 T332I-induced cell contraction, cells were co-stained with TRITC-phalloidin and Hoechst33258. Morphology represented by the GFP fluorescence of GFP-ARHGEF10 wt and T332I was similar to those represented by the TRITC fluorescence for F-actin (Fig. 4D). F-actin was particularly concentrated in the periphery of HEK293T cells expressing GFP-ARHGEF10 T332I, which show round and contracted morphology (Fig. 4D). Similar results were obtained using HeLa cells (supplemental Fig. S2C). In most contracted cells, the DNA fragmentation, the chromatin aggregation, or a metaphase plate of aligned chromosomes was not observed, suggesting that these cells are neither apoptotic nor in metaphase of mitosis. No effect was observed on the number or localization of centrosome of HEK293T cells, in which ARHGEF10 T332I was overexpressed (data not shown).
FIGURE 4.

Cell contraction induced by ARHGEF10 T332I in HEK293T cells. A, domain structures of ARHGEF10. Full-length ARHGEF10 (wt) and the T332I mutant of full-length ARHGEF10 are shown. B, HEK293T cells transiently transfected with the plasmid encoding GFP, GFP-ARHGEF10 wt, or GFP-ARHGEF10 T332I. After 48 h, fluorescence images of living cells were observed. Scale bar, 10 μm. C, quantitative analysis of cell contraction of HEK293T cells. HEK293T cells were transiently transfected with the plasmid encoding GFP, GFP-ARHGEF10 wt, or T332I. The proportion of cell contraction was scored as a percentage of the rounded cells of GFP-positive cells. Cells floating in the culture medium were excluded from counting. The data represent the mean ± S.E. (error bars) from three independent experiments. *, p < 0.05 compared with empty. For each experiment, >100 cells were counted. D, HEK293T cells transiently transfected with the plasmid encoding GFP-ARHGEF10 wt or T332I. After 48 h, cells were fixed and co-stained with TRITC-phalloidin (F-actin) and Hoechst33258 (DNA). Transfected cells are shown by the fluorescence of GFP. Scale bar, 10 μm.
ARHGEF10 T332I Induces Cell Contraction through Rho-ROCK Signaling
Expression of ARHGEF10 ΔN induced cell contraction in a Rho-ROCK-dependent manner. We examined whether or not ARHGEF10 T332I-induced cell contraction is also mediated through Rho-ROCK signaling. HEK293T cells were transfected with the plasmid encoding GFP-ARHGEF10 T332I and treated with Y27632. This treatment significantly inhibited ARHGEF10 T332I-induced cell contraction (Fig. 5, A and B). Similar results were obtained using HeLa cells (supplemental Fig. S3). These results indicate that ARHGEF10 T332I-induced cell contraction is mediated by ROCK. Next, we evaluated the amount of active RhoA in cells expressing ARHGEF10 wt or T332I by a pulldown assay using GST-Rhotekin-RBD. There was significantly more active RhoA in lysates from cells expressing ARHGEF10 T332I than in lysates from cells expressing wt (Fig. 5, C and D). Moreover, we measured serum-response element (SRE)-mediated transcriptional activity using the luciferase reporter assay system to analyze Rho activity enhanced by ARHGEF10 wt and T332I. It is known that active Rho enhances SRE-driven gene expression through the activation of serum response factor (27). HEK293T cells were co-transfected with the plasmid encoding GFP-ARHGEF10 wt or T332I, and pSRE.l-luciferase reporter plasmid, in which expression of luciferase is under control of SRE. ARHGEF10 T332I significantly induced higher levels of luciferase activity than wt (Fig. 5E). These data revealed that ARHGEF10 T332I activates Rho more strongly than wt and suggested that ARHGEF10 T332I-induced cell contraction is mediated through Rho-ROCK signaling.
FIGURE 5.

ARHGEF10 T332I-induced cell contraction mediated through Rho-ROCK signaling. A, HEK293T cells transiently transfected with the plasmid encoding GFP-ARHGEF10 T332I and treated with or without 10 μm Y27632 for 24 h. Then, fluorescence images of living cells were observed. Arrowheads indicate rounded cells. Arrows indicate cells floating in the culture medium. Scale bar, 10 μm. B, quantitative analysis of cell contraction of HEK293T cells. HEK293T cells were transiently transfected with the plasmid encoding GFP-ARHGEF10 T332I and treated with or without 10 μm Y27632 for 24 h. The proportion of cell contraction was scored as a percentage of the rounded cells of GFP-positive cells. Cells floating in the culture medium were excluded from counting. The data represent the mean ± S.E. (error bars) from three independent experiments. *, p < 0.05. For each experiment, >100 cells were counted. C and D, HEK293T cells transiently co-transfected with the plasmids encoding myc-tagged ARHGEF10 wt or T332I, and HA-RhoA. After 48 h, cells were lysed with lysis buffer. The supernatants were mixed with GST-Rhotekin-RBD, and an active form of RhoA was pulled down. The amounts of active form of RhoA were determined by immunoblotting with anti-HA antibody. Expressions of HA-RhoA and myc-ARHGEF10 in cell lysates were also detected by immunoblotting with anti-HA and anti-myc antibodies, respectively. A representative image of three independent experiments is shown. Relative RhoA activity is indicated by the amounts of RhoA-GTP normalized to total amounts of RhoA in cell lysates, and values are expressed as -fold value of cells transfected with wt. The data are the mean ± S.E. from three independent experiments. *, p < 0.05. E, HEK293T cells transiently co-transfected with the plasmid encoding GFP-ARHGEF10 wt or T332I, pSRE.l-luciferase reporter plasmid encoding firefly luciferase, and pRL-TK control vector encoding Renilla luciferase. The firefly luciferase activities were normalized to the Renilla luciferase activities, and values are expressed as -fold induction compared with wt. The data are the mean ± S.E. from three independent experiments. *, p < 0.05.
ARHGEF10 T332I-induced Cell Contraction Corresponds to Its GEF Activity
A series of GFP-tagged ARHGEF10 T332I mutants (T332I ΔDH (a DH domain-deleted mutant) and T332I/S407A and T332I/L547A (mutants in the conserved catalytic residues of DH domain)) (28) were generated to confirm whether cell contraction activity induced by various mutants in DH domain of ARHGEF10 T332I corresponds to its GEF activity (Fig. 6A). RhoGEF activity was determined by SRE-driven gene expression using the Dual-Luciferase Reporter Assay System. Deletion of DH domain and L547A substitution almost completely lowered the level of luciferase activity increased by T332I to the normal level of wt (Fig. 6B). S407A substitution also moderately lowered the level of luciferase activity increased by T332I, but did not lower its level to the normal level of wt. These results indicate that GEF activity of ARHGEF10 T332I is strongly reduced by deletion of DH domain and L547A substitution and moderately by S407A substitution. Then, we examined the effect of these mutants for cell morphology using HEK293T cells. T332I ΔDH, T332I/S407A, and T332I/L547A had significantly weaker activity to induce cell contraction than T332I, although the inhibitory effect of S407A substitution on T332I-induced cell contraction was significantly weaker than that of deletion of DH domain or L547A substitution (Fig. 6, C and D). Induction of cell contraction by ARHGEF10 T332I mutants correlated well with their level of GEF activity. Similar results were obtained using HeLa cells (supplemental Fig. S2, A–C). Protein expression of ARHGEF10 wt, T332I, T332I ΔDH, T332I/S407A, or T332I/L547A is shown in Fig. 6E. These results indicate that ARHGEF10 T332I-induced cell contraction is dependent on its GEF activity of DH domain and that ARHGEF10 T332I mutant is a constitutively active ARHGEF10 mutant.
FIGURE 6.

Effect of DH mutations of ARHGEF10 T332I on activation of Rho and cell contraction. A, domain structures of ARHGEF10. Full-length ARHGEF10 (wt) and the T332I mutant of full-length ARHGEF10, a DH domain truncation mutant lacking amino acids 397–583 of ARHGEF10 T332I (T332I ΔDH), and the S407A and L547A mutants of ARHGEF10 T332I (T332I/S407A and T332I/L547A, respectively) are shown. B, HEK293T cells transiently co-transfected with the plasmid encoding GFP-ARHGEF10 wt, T332I, T332I ΔDH, T332I/S407A, or T332I/L547A, pSRE.l-luciferase reporter plasmid encoding firefly luciferase, and pRL-TK control vector encoding Renilla luciferase. The firefly luciferase activities were normalized to the Renilla luciferase activities, and values are expressed as -fold induction compared with wt. The data are the mean ± S.E. (error bars) from three independent experiments. *, p < 0.05. C, HEK293T cells transiently transfected with the plasmid encoding GFP-ARHGEF10 wt, T332I, T332I ΔDH, T332I ΔS407A, or T332I/L547A. After 48 h, fluorescence images of living cells were observed. Arrowheads indicate rounded cells. Scale bar, 10 μm. D, quantitative analysis of cell contraction of HEK293T cells. HEK293T cells were transiently transfected with the plasmid encoding GFP-ARHGEF10 wt, T332I, T332I ΔDH, T332I/S407A, or T332I/L547A. The proportion of cell contraction was scored as a percentage of the rounded cells of GFP-positive cells. Cells floating in the culture medium were excluded from counting. The data represent the mean ± S.E. from three independent experiments. *, p < 0.05. For each experiment, >100 cells were counted. E, protein expressions of GFP-ARHGEF10 wt, T332I, T332I ΔDH, T332I/S407A, and T332I/L547A were confirmed by immunoblotting with anti-GFP antibody.
ARHGEF10 T332I Also Induces Cell Contraction in Schwann Cells
Our results show that ARHGEF10 T332I is a constitutively active ARHGEF10 mutant and induces changes in cellular morphology by its enhanced GEF activity. Because the ARHGEF10 T332I mutant is implicated in aberrant myelination of peripheral nerves (13), we finally examined whether ARHGEF10 T332I can induces changes in cell morphology of Schwann cells. Before the experiment, Schwann cells derived from rat sciatic nerves were confirmed by immunostaining with anti-S100 antibody which is known as a marker for Schwann cells (Fig. 7A) (19). All cells were S100-immunoreactive and showed bipolar morphology. Schwann cells were transfected with the plasmid encoding GFP-ARHGEF10 wt or T332I, and the GFP images of living cells were analyzed by fluorescence microscopy. Similar to HEK293T and HeLa cells, Schwann cells expressing ARHGEF10 T332I also showed cell contraction more frequently than those expressing wt (Fig. 7, B and C). These results strongly suggest that ARHGEF10 T332I affects Schwann cell morphology via its activated GEF activity.
FIGURE 7.
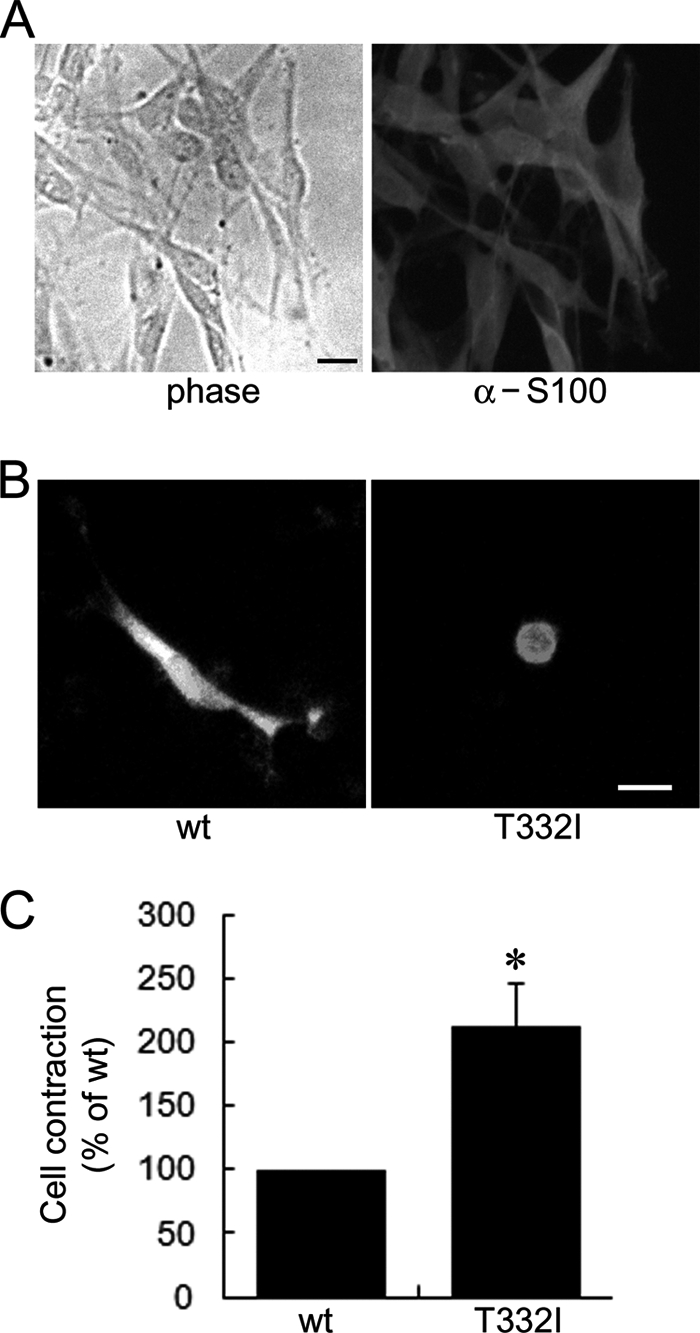
Cell contraction induced by ARHGEF10 T332I in Schwann cells. A, left, phase contrast image of Schwann cells derived from rat sciatic nerves. Right, Schwann cells fixed and stained with anti-S100 antibody. Scale bar, 10 μm. B, Schwann cells transiently transfected with the plasmid encoding GFP-ARHGEF10 wt or T332I. After 48 h, fluorescence images of living cells were observed. Scale bar, 10 μm. C, quantitative analysis of cell contraction of Schwann cells. Schwann cells were transiently transfected with the plasmid encoding GFP-ARHGEF10 wt or T332I. The proportion of cell contraction was scored as a percentage of the rounded cells of GFP-positive cells, and values are expressed as -fold change compared with wt. Cells floating in the culture medium were excluded from counting. The data represent the mean ± S.E. (error bars) from three independent experiments. *, p < 0.05. For each experiment, >30 cells were counted.
DISCUSSION
In this study, we showed that ARHGEF10 has a negatively regulatory region in the N terminus and that the T332I mutant is a constitutively active ARHGEF10 mutant for RhoA, B, and C. ARHGEF10 ΔN and T332I induced cell contraction through Rho-ROCK signaling. Furthermore, the mutants that induced cell contraction had higher GEF activity than the wt, suggesting that cell contraction observed in this study reflects the GEF activity of ARHGEF10 mutants. In agreement with our result, Banerjee and Wedegaertner reported by using various mutants of PSD-95/Dlg/ZO-1 homology (PDZ)-RhoGEF that PDZ-RhoGEF induced cell process retraction and contraction of HEK293T and Neuro2a cells corresponding to its GEF activity and its ability to activate Rho (29). It is known that active ROCK phosphorylates myosin light chain and stimulates actomyosin contraction (6–8). If this contraction occurs near the cell cortex, the cell may become the rounded shape due to the cortical retraction. On the other hand, ROCK phosphorylates LIM kinase, and then phosphorylated LIM kinase phosphorylates cofilin. Although cofilin promotes depolymerization of F-actin, its function is inhibited when phosphorylated by LIM kinase (30). Thus, phosphorylated cofilin serves as the stabilization of actin filaments. This would stabilize the actin cytoskeleton in the cell cortex and contribute to cell contraction. A recent study reported the involvement of ARHGEF10 in centrosome duplication of HeLa cells because its knockdown resulted in multipolar mitotic spindle formation. However, no effect on the number or localization of centrosome was observed in cells overexpressing ARHGEF10 ΔN or T332I, each of which is an active ARHGEF10 mutant; alternatively, the morphological change of cell contraction occurred in cells transfected with these active ARHGEF10 mutants. These data indicate that ARHGEF10 is implicated in the regulation of cell morphology mediated by actomyosin contraction at the cell cortex as well as in the centrosome biogenesis.
We selected position 179, but not 437, of the nucletotide sequence NM_014629.2 (GenBank accession number) as a start codon as Aoki et al. did previously (17). We chose this position according to the nucleotide sequence NM_014629.2 and the protein sequence linked to this sequence by NCBI, NP_055444.2. However, A179TG may not be the true initiation codon. In another database, a start codon of ARHGEF10 was selected in the 5′ region upstream from the position which we selected as a start codon (UniProtKB: O15013). Of course, at present we are not able to exclude the possibility that there is a start codon in the further upstream region of ARHGEF10.
We demonstrated that ARHGEF10 ΔN has higher level of GEF activity for RhoA than wt. Mohl et al. reported that the N-terminal truncation mutant of ARHGEF10 has lower GEF activity than wt (16). Because ARHGEF10 ΔN used in this study is 72 amino acids longer than the N-terminal truncation mutant of ARHGEF10 used by Mohl et al., these extra 72 amino acids, which contains a part of predicted DH domain, may contribute to GEF activity or stability of ARHGEF10. GEF activity of several RhoGEFs of the Dbl type is regulated by the intramolecular autoinhibition. For example, Vav, Dbl, Asef, Tim, and PDZ-RhoGEF are negatively regulated by their own N-terminal regions (21–26). Our results demonstrated that the region containing amino acids 212–332 of ARHGEF10 is a negatively regulatory region for its GEF activity. Similar to the intramolecular interaction of Vav etc., ARHGEF10 may also be negatively regulated by this region because the deletion of N-terminal 212–332 residues from ARHGEF10 induced higher GEF activity than wt. However, we could not observe an inhibitory effect of the overexpression of residues 212–332 on GEF activity of ARHGEF10 ΔN. These results suggest that the region containing amino acids 212–332 is necessary but not sufficient to down-regulate GEF activity of ARHGEF10. In addition to this region, involvement of other factors may be required for down-regulation of GEF activity of ARHGEF10. A secondary structure prediction shows that Thr-332 of ARHGEF10 might reside in the turn structure. Because threonine and isoleucine are different in nature, we hypothesize that this predicted turn structure is disturbed in ARHGEF10 T332I. This disturbance might reduce the autoinhibitory effect of its N-terminal region on GEF activity, resulting in the increased GEF activity of ARHGEF10. The region including Thr-332 is conserved among species, suggesting that this region somehow has important roles in the regulation of the exchange activity, although this region is not found in generally known domains or motifs so far.
In accordance with a previous report (16), our results suggested that the C-terminal region containing amino acids 1304–1342 is required for GEF activity of ARHGEF10 by using its active mutant ARHGEF10 ΔN. Although how this C-terminal region contributes to the GEF activity is not clear, any activators of ARHGEF10 might interact with this region and then activate GEF activity of ARHGEF10. ARHGEF10 has a putative PDZ-binding motif at the C terminus, and this putative PDZ-binding motif is proposed to play an important role in the function of ARHGEF10 (31). However, deletion of 2 amino acids from the C terminus of ARHGEF10 ΔN had no effect on GEF activity for RhoA, suggesting that this motif is not required for activation of its GEF activity.
The T332I mutation in ARHGEF10 is proposed to cause slowed nerve conduction velocities and thin myelination in the peripheral nervous system, although why this ARHGEF10 mutation can cause aberrant myelination is not known (13). One possible explanation is that ARHGEF10 T332I mutant affects extension of Schwann cell processes. The process extension of Schwann cells is supposed to have an important role in radial sorting, which is required for myelination of axons by Schwann cells (32). In radial sorting, Schwann cells extend cytoplasmic processes into axon bundles to establish 1:1 relationships with individual axons. Recently, the functions of small GTPases Rho family in Schwann cells have been uncovered. Schwann cell-specific deletion of Rac1, which is known as a protein involved in β1-integrin-mediated signaling, results in deficient radial sorting, suggesting that Rac1 is required in radial sorting in β1-integrin-mediated signaling (33, 34). On the other hand, pharmacological inhibition of ROCK promotes aberrant process extension of Schwann cells, such as aberrant branches and multiple and small myelin segments (35). Integrin-linked kinase negatively regulates Rho-ROCK signaling, resulting in promotion of Schwann cell process extension and initiation of radial sorting (36). These data suggest that Rho-ROCK signaling negatively regulates Schwann cell process extension. In this study, we demonstrated that ARHGEF10 T332I mutant is constitutively active, and this mutant shows stronger activity to induce cell contraction of Schwann cells than wt. We observed that contracted Schwann cells expressing ARHGEF10 T332I have shortened processes. This suggests that ARHGEF10 T332I mutant may abnormally shorten Schwann cell processes through activated Rho-ROCK signaling, resulting in impairment of myelination. Schwann cells migrate along axons before radial sorting. The functions of the small GTPases Rho family, its regulators, and its effectors in this process have become obvious in detail (37–40). However, in other processes including the process extension and radial sorting, the regulators of the Rho family have not yet been elucidated. Our study about characterization of ARHGEF10 T332I mutant suggests that ARHGEF10 serves as the regulator of Rho and regulates the process extension in Schwann cells.
Supplementary Material
This work was supported in part by a grant-in-aid for general scientific research from the Japan Ministry of Education, Culture, Sports, Science, and Technology (to S. I.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Figs. S1–S3.
- ROCK
- Rho kinase
- DH
- Dbl homology
- F-actin
- filamentous actin
- GEF
- guanine nucleotide exchange factor
- RBD
- Rho-binding domain
- SRE
- serum response element.
REFERENCES
- 1. Etienne-Manneville S., Hall A. (2002) Nature 420, 629–635 [DOI] [PubMed] [Google Scholar]
- 2. Burridge K., Wennerberg K. (2004) Cell 116, 167–179 [DOI] [PubMed] [Google Scholar]
- 3. Ridley A. J., Hall A. (1992) Cell 70, 389–399 [DOI] [PubMed] [Google Scholar]
- 4. Ridley A. J., Paterson H. F., Johnston C. L., Diekmann D., Hall A. (1992) Cell 70, 401–410 [DOI] [PubMed] [Google Scholar]
- 5. Nobes C. D., Hall A. (1995) Cell 81, 53–62 [DOI] [PubMed] [Google Scholar]
- 6. Kimura K., Ito M., Amano M., Chihara K., Fukata Y., Nakafuku M., Yamamori B., Feng J., Nakano T., Okawa K., Iwamatsu A., Kaibuchi K. (1996) Science 273, 245–248 [DOI] [PubMed] [Google Scholar]
- 7. Amano M., Ito M., Kimura K., Fukata Y., Chihara K., Nakano T., Matsuura Y., Kaibuchi K. (1996) J. Biol. Chem. 271, 20246–20249 [DOI] [PubMed] [Google Scholar]
- 8. Riento K., Ridley A. J. (2003) Nat. Rev. Mol. Cell Biol. 4, 446–456 [DOI] [PubMed] [Google Scholar]
- 9. Schmidt A., Hall A. (2002) Genes Dev. 16, 1587–1609 [DOI] [PubMed] [Google Scholar]
- 10. Rossman K. L., Der C. J., Sondek J. (2005) Nat. Rev. Mol. Cell Biol. 6, 167–180 [DOI] [PubMed] [Google Scholar]
- 11. Delague V., Jacquier A., Hamadouche T., Poitelon Y., Baudot C., Boccaccio I., Chouery E., Chaouch M., Kassouri N., Jabbour R., Grid D., Mégarbané A., Haase G., Lévy N. (2007) Am. J. Hum. Genet. 81, 1–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Stendel C., Roos A., Deconinck T., Pereira J., Castagner F., Niemann A., Kirschner J., Korinthenberg R., Ketelsen U. P., Battaloglu E., Parman Y., Nicholson G., Ouvrier R., Seeger J., De, Jonghe P., Weis J., Krüttgen A., Rudnik-Schöneborn S., Bergmann C., Suter U., Zerres K., Timmerman V., Relvas J. B., Senderek J. (2007) Am. J. Hum. Genet. 81, 158–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Verhoeven K., De Jonghe P., Van de Putte T., Nelis E., Zwijsen A., Verpoorten N., De Vriendt E., Jacobs A., Van Gerwen V., Francis A., Ceuterick C., Huylebroeck D., Timmerman V. (2003) Am. J. Hum. Genet. 73, 926–932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Obaishi H., Nakanishi H., Mandai K., Satoh K., Satoh A., Takahashi K., Miyahara M., Nishioka H., Takaishi K., Takai Y. (1998) J. Biol. Chem. 273, 18697–18700 [DOI] [PubMed] [Google Scholar]
- 15. Umikawa M., Obaishi H., Nakanishi H., Satoh-Horikawa K., Takahashi K., Hotta I., Matsuura Y., Takai Y. (1999) J. Biol. Chem. 274, 25197–25200 [DOI] [PubMed] [Google Scholar]
- 16. Mohl M., Winkler S., Wieland T., Lutz S. (2006) Naunyn Schmiedebergs Arch. Pharmacol. 373, 333–341 [DOI] [PubMed] [Google Scholar]
- 17. Aoki T., Ueda S., Kataoka T., Satoh T. (2009) BMC Cell Biol. 10, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yamada T., Ohoka Y., Kogo M., Inagaki S. (2005) J. Biol. Chem. 280, 19358–19363 [DOI] [PubMed] [Google Scholar]
- 19. Brockes J. P., Fields K. L., Raff M. C. (1979) Brain Res. 165, 105–118 [DOI] [PubMed] [Google Scholar]
- 20. Ren X. D., Schwartz M. A. (2000) Methods Enzymol. 325, 264–272 [DOI] [PubMed] [Google Scholar]
- 21. Yu B., Martins I. R., Li P., Amarasinghe G. K., Umetani J., Fernandez-Zapico M. E., Billadeau D. D., Machius M., Tomchick D. R., Rosen M. K. (2010) Cell 140, 246–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bi F., Debreceni B., Zhu K., Salani B., Eva A., Zheng Y. (2001) Mol. Cell. Biol. 21, 1463–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Murayama K., Shirouzu M., Kawasaki Y., Kato-Murayama M., Hanawa-Suetsugu K., Sakamoto A., Katsura Y., Suenaga A., Toyama M., Terada T., Taiji M., Akiyama T., Yokoyama S. (2007) J. Biol. Chem. 282, 4238–4242 [DOI] [PubMed] [Google Scholar]
- 24. Mitin N., Betts L., Yohe M. E., Der C. J., Sondek J., Rossman K. L. (2007) Nat. Struct. Mol. Biol. 14, 814–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Yohe M. E., Rossman K., Sondek J. (2008) Biochemistry 47, 6827–6839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zheng M., Cierpicki T., Momotani K., Artamonov M. V., Derewenda U., Bushweller J. H., Somlyo A. V., Derewenda Z. S. (2009) BMC Struct. Biol. 9, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hill C. S., Wynne J., Treisman R. (1995) Cell 81, 1159–1170 [DOI] [PubMed] [Google Scholar]
- 28. Zhu K., Debreceni B., Li R., Zheng Y. (2000) J. Biol. Chem. 275, 25993–26001 [DOI] [PubMed] [Google Scholar]
- 29. Banerjee J., Wedegaertner P. B. (2004) Mol. Biol. Cell 15, 1760–1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maekawa M., Ishizaki T., Boku S., Watanabe N., Fujita A., Iwamatsu A., Obinata T., Ohashi K., Mizuno K., Narumiya S. (1999) Science 285, 895–898 [DOI] [PubMed] [Google Scholar]
- 31. García-Mata R., Burridge K. (2007) Trends Cell Biol. 17, 36–43 [DOI] [PubMed] [Google Scholar]
- 32. Jessen K. R., Mirsky R. (2005) Nat. Rev. Neurosci. 6, 671–682 [DOI] [PubMed] [Google Scholar]
- 33. Benninger Y., Thurnherr T., Pereira J. A., Krause S., Wu X., Chrostek-Grashoff A., Herzog D., Nave K. A., Franklin R. J., Meijer D., Brakebusch C., Suter U., Relvas J. B. (2007) J. Cell Biol. 177, 1051–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nodari A., Zambroni D., Quattrini A., Court F. A., D'Urso A., Recchia A., Tybulewicz V. L., Wrabetz L., Feltri M. L. (2007) J. Cell Biol. 177, 1063–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Melendez-Vasquez C. V., Einheber S., Salzer J. L. (2004) J. Neurosci. 24, 3953–3963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pereira J. A., Benninger Y., Baumann R., Gonçalves A. F., Ozçelik M., Thurnherr T., Tricaud N., Meijer D., Fässler R., Suter U., Relvas J. B. (2009) J. Cell Biol. 185, 147–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yamauchi J., Chan J. R., Shooter E. M. (2004) Proc. Natl. Acad. Sci. U.S.A. 101, 8774–8779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yamauchi J., Miyamoto Y., Tanoue A., Shooter E. M., Chan J. R. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 14889–14894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yamauchi J., Chan J. R., Miyamoto Y., Tsujimoto G., Shooter E. M. (2005) Proc. Natl. Acad. Sci. U.S.A. 102, 5198–5203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Yamauchi J., Miyamoto Y., Chan J. R., Tanoue A. (2008) J. Cell Biol. 181, 351–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.