

Abstract
Angiogenesis is heavily influenced by VEGF-A and its family of receptors, particularly VEGF receptor 2 (VEGF-R2). Like most cell surface proteins, VEGF-R2 is glycosylated, although the function of VEGF-R2 with respect to its glycosylation pattern is poorly characterized. Galectin-3, a glycan binding protein, interacts with the EGF and TGFβ receptors, retaining them on the plasma membrane and altering their signal transduction. Because VEGF-R2 is glycosylated and both galectin-3 and VEGF-R2 are involved with angiogenesis, we hypothesized that galectin-3 binds VEGF-R2 and modulates its signal transduction as well. Employing a Western blot analysis approach, we found that galectin-3 induces phosphorylation of VEGF-R2 in endothelial cells. Knockdown of galectin-3 and Mgat5, an enzyme that synthesizes high-affinity glycan ligands of galectin-3, reduced VEGF-A mediated angiogenesis in vitro. A direct interaction on the plasma membrane was detected between galectin-3 and VEGF-R2, and this interaction was dependent on the expression of Mgat5. Using immunofluorescence and cell surface labeling, we found an increase in the level of internalized VEGF-R2 in both Mgat5 and galectin-3 knockdown cells, suggesting that galectin-3 retains the receptor on the plasma membrane. Finally, we observed reduced suture-induced neovascularization in the corneas of Gal3−/− and Mgat5−/− mice. These findings are consistent with the hypothesis that, like its role with the EGF and TGFβ receptors, galectin-3 contributes to the plasma membrane retention and proangiogenic function of VEGF-R2.
Keywords: Carbohydrate Binding Protein, Cell Surface Protein, Glycoprotein, Protein Cross-linking, Receptor Endocytosis, Mgat5, VEGF, VEGF-R2, Angiogenesis, Galectin-3
Introduction
Angiogenesis, or the growth of new blood vessels from preexisting vasculature, is involved in an array of normal processes like embryonic development, wound healing, and reproductive function and in many pathological processes like tumor growth and metastasis, age-related macular degeneration, ischemia, diabetic microvascular disease, and rheumatoid arthritis (1–3). Angiogenesis is promoted by the loosening of endothelial cells from the basement membrane and periendothelial cells, allowing them to migrate, proliferate, and ultimately form a new capillary lumen (4). Although facilitated by a number of cytokine receptors and signaling factors, VEGF-A and its interaction with VEGF receptor 2 (VEGF-R2),3 has emerged as the major positive signal transduction pathway for both physiological and pathological angiogenesis (5–9). The binding of VEGF-A to VEGF-R2 promotes receptor dimerization, kinase activation, and autophosphorylation of multiple tyrosine residues within the dimeric complex. Autophosphorylation activates a number of intracellular signaling pathways, such as the p42/44 MAP kinase pathway, that are involved in endothelial cell proliferation, migration, and survival (10).
Previous studies have shown that galectin-3, a member of the galectin family of β-galactoside binding proteins, promotes endothelial cell migration and capillary tubule formation in vitro and enhances vascularization in vivo (11). Studies in our laboratory revealed that VEGF-A-mediated angiogenesis is reduced in vitro by the addition of a dominant negative galectin-3, a pan-galectin inhibitor (β-lactose), and by galectin-3 knockdown (12). Furthermore, VEGF-A induced angiogenesis was significantly reduced in Gal3−/− mice in vivo. Although our previous work described an important role for galectin-3 in VEGF signaling, in that study our focus was primarily on establishing the angiogenic potential of galectin-3. Here, in an effort to better characterize the mechanism of galectin-3-induced angiogenesis, we demonstrate a direct effect of galectin-3 on VEGF signaling by binding a VEGF receptor.
Galectin-3 is a ubiquitously expressed, 30-kDa protein present in the cytoplasm, nucleus, and extracellular space and bound to the cell surface (13). Galectin-3 is composed of a highly conserved N-terminal domain that facilitates its multimerization (14) and a C-terminal carbohydrate recognition domain that preferentially interacts with glycoproteins that have been modified by the glycosyltransferase Mgat5 (β1,6-N-acetylglucosaminyl transferase 5) (15). Because of its ability to multimerize via its N-terminal domain and bind specific carbohydrate branches by the C-terminal domain, galectin-3 is thought to cross-link glycoproteins and form a cell surface molecular lattice that influences cellular function (14). Galectin-3 has been shown to interact with the Mgat5-modified N-glycans of the EGF and TGFβ receptors in a carbohydrate-dependent manner and delay their constitutive endocytic removal from the plasma membrane (16). Furthermore, it was shown that Mgat5-modified N-glycans promote EGF and TGFβ signaling, suggesting that galectin-3 plays a vital role in the signal transduction of these cytokines.
As previous work in our laboratory demonstrated a clear role of galectin-3 in VEGF-mediated angiogenesis, here we hypothesize that galectin-3 affects VEGF-A-driven angiogenesis by directly interacting with the N-glycan branches of VEGF-R2, influencing its cell surface retention and thereby modulating its signaling. To test this, a cell surface biotinylation and cross-linking approach was used in human umbilical vein endothelial (HUVE) cells. We demonstrate an Mgat5-dependent interaction between galectin-3 and VEGF-R2, suggesting that Mgat5-modified N-glycans mediate this interaction. Using confocal microscopy and cell surface biotinylation, we found that galectin-3 delays the endocytic removal of VEGF-R2 in a carbohydrate-dependent manner, as disruption of either galectin-3 or Mgat5 reduces the cell surface localization of VEGF-R2. We also demonstrate that the addition of exogenous galectin-3 results in time-dependent phosphorylation of VEGF-R2 (Tyr-1175) and that galectin-3 expression is rate-limiting for VEGF-mediated capillary sprouting, a key early indicator of angiogenesis. Furthermore, we found reduced suture-induced neovascularization in the corneas of galectin-3 and Mgat5 null animals.
EXPERIMENTAL PROCEDURES
Materials and Cell Culture
HUVE cells, obtained from Lonza (Basel, Switzerland), were maintained up to passage eight in endothelial cell growth medium (EGM-2). Endothelial basal medium (EBM-2) was also purchased from Lonza. Recombinant VEGF-A was obtained from PeproTech, Inc. (Rocky Hill, NJ). EZ-Link Sulfo-NHS-LC-Biotin, EZ-Link Sulfo-SS-Biotin, DTSSP, and a SuperSignal West Femto maximum sensitivity substrate detection kit were purchased from Pierce. Anti-VEGF-R2 and anti-phospho-VEGF-R2 rabbit monoclonal antibodies (#2479 and #2478) were obtained from Cell Signaling Technology, Inc. (Danvers, MA). Mouse anti-human EEA1 was purchased from Abcam, Inc. (Cambridge, MA). Hybridoma M3/38-expressing monoclonal anti-galectin-3 antibody was acquired from the ATCC. FITC-BS1, anti-mouse IgG, anti-rabbit IgG secondary antibodies, rhodamine-conjugated L-PHA, and Con-A were obtained from Vector Laboratories (Burlingame, CA). Alexa Fluor 488 goat anti-mouse and Alexa Fluor 596 goat anti-rabbit antibodies, M199 medium, and anti-rat Dynal paramagnetic beads were obtained from Invitrogen. Collagen type I was purchased from BD Biosciences. The RNEasy kit was from Qiagen (Valencia, CA). The high-capacity cDNA archive kit, qPCR master mix, and qPCR primers were from Applied Biosystems, Inc. (Foster City, CA). The chemiluminescence detection system was purchased from PerkinElmer Life Sciences. The 11–0 nylon sutures were manufactured by Sharpoint Angiotech Pharmaceuticals (Vancouver, Canada). All other reagents were obtained from Sigma-Aldrich. Confocal images were obtained using a Leica DM IRE confocal laser scanning microscope (Leica Lasertechnik, Heidelberg, Germany) equipped with a ×40/0.75 (f/1.25) objective. Fluorescent images were acquired using an Eclipse E400 microscope (Nikon, Tokyo, Japan) with a CFI Plan Fluor ×4 (f/0.13) or CFI Plan Fluor ×20 (f/0.50) objective. The images of cell sprouts were photographed with a Nikon Eclipse TE200 phase contrast microscope with a CFI Plan Fluor ×4 and CFI Plan Fluor ×10 (f/0.30) objective. Images were acquired at room temperature with a SPOT RT Color digital camera (Diagnostic Instruments, Sterling Heights, MI) and the SPOT Acquisition software version 4.0.6 (Diagnostic Instruments).
Animals
In these experiments, 8-week- to 3-month-old age-matched Gal3+/+, Gal3−/−, Mgat5−/−, and Mgat5+/+ mice were used. Gal3−/− mice were generated by targeted interruption of the galectin-3 gene (34) and were a gift from Drs. Dan Hsu and Fu-Tong Liu (University of California Davis). Gal3+/+ and Gal3−/− mice were obtained by interbreeding Gal3+/− mice and carried as separated lineages. Mgat5−/− animals, generated by Dennis and coworkers (17), were provided by the Consortium for Functional Glycomics (La Jolla, CA).
Galectin-3-mediated VEGF-R2 Phosphorylation
HUVE cells, serum-starved overnight in EBM-2 media, were stimulated with 0, 2.5, 5, 10, 20, and 50 μg/ml galectin-3 for 0, 2, 5, or 10 min. After stimulation, the cells were washed with PBS and lysed with TNTE lysis buffer (50 mm Tris-HCl (pH 7.4), 150 mm NaCl, 1% Triton-X100, 1 mm EDTA). Cell lysates were precleared by incubation with 20 μl of a 40% suspension of Sepharose 4B beads, incubated with monoclonal VEGF-R2 (clone 55B11) antibody, and immunoprecipitated with protein A-Sepharose. Bound proteins were eluted by boiling the beads in Laemmli sample buffer, separated by 4–20% SDS-PAGE, transferred to nitrocellulose, and analyzed using anti-phospho-VEGF-R2 (1:1000) and HRP-conjugated anti-rabbit IgG (1:20000) antibodies. ECL substrate was used to detect immobilized products. Total VEGF-R2 expression in whole cell lysates was evaluated using monoclonal VEGF-R2 (clone 55B11, 1:1000) as the primary antibody and HRP-conjugated anti-rabbit IgG as a secondary antibody.
Analysis of Carbohydrate-dependent VEGF-R2 Binding to Galectin-3
Analysis of carbohydrate-dependent binding was performed as described previously (12). Briefly, HUVE cell lysates were chromatographed on a galectin-3 affinity column. The unbound components were removed by washing the column, and bound proteins were sequentially eluted using PBS plus 0.1% tween 20 containing 0.1 m sucrose and 0.1 m β-lactose. Eluted fractions were dialyzed with water, lyophilized, and resolved by SDS-PAGE and immunoblot analysis using monoclonal anti-VEGF-R2 and HRP-conjugated anti-rabbit IgG antibodies.
Transfection of HUVE Cells and Evaluation
Stable shRNA cell lines targeting Mgat5 (TRCN0000036030) and galectin-3 (TRCN0000029305) were generated using Mission-TRC lentiviral particles, obtained from Sigma-Aldrich, following the manufacturer's protocol. The efficacy of Mgat5 and galectin-3 knockdown at the mRNA level was assessed by RT qPCR using gene-specific primers (Mgat5, Hs00159136_m1; Gal3, Hs00173587_m1; and GAPDH, Hs99999905_ml). The knockdown of β1,6GlcNAc-branched chains was evaluated by staining with rhodamine-conjugated L-PHA lectin. HUVE cells transfected with either control or Mgat5 shRNA were plated in eight-chamber slides, fixed with 4% paraformaldehyde, and stained with 10 μg/ml rhodamine-conjugated L-PHA or a control lectin, ConA, in PBS. After washing, the slides were mounted and subjected to fluorescence microscopy. The knockdown of galectin-3 protein was evaluated by Western blot analysis (clone M3/38) and β-actin (clone AC-74).
Analysis of VEGF-R2 Glycans
Control shRNA and Mgat5 shRNA-transfected HUVE cell lysates were pre-cleared by incubation with 20 μl of Sepharose 4B (1 h at 4 °C) to reduce nonspecific binding, and then incubated with 30 μl of agarose-conjugated L-PHA or ConA for 1 h at 4 °C. The beads were washed, and bound proteins were eluted by boiling in Laemmli sample buffer, separated by SDS-PAGE, transferred to nitrocellulose membranes, and analyzed by Western blot analysis using an anti-VEGF-R2 antibody as described above.
Analysis of the Galectin-3 Cell Surface Interaction with VEGF-R2
To investigate whether a galectin-3 and VEGF-R2 interaction occurs on the plasma membrane, cell surface proteins were labeled with biotin and chemically cross-linked with a homo-bifunctional, thiol-cleavable, and membrane-impermeable cross-linker, DTSSP, following the methods described by Altin et al. (18) with the following modifications. Cell lysates were incubated with anti-human galectin-3 antibodies bound to anti-rat Dynal paramagnetic beads that were prepared following the manufacturer's protocol. An immunoblot analysis was performed using anti-VEGF-R2, anti-galectin-3, and anti-β-actin as primary antibodies and HRP-conjugated anti-rabbit, anti-mouse, and anti-rat IgG as secondary antibodies. In some experiments to show that galectin-3 associates with unphosphorylated and phosphorylated VEGF-R2, serum-starved HUVE cells were stimulated with 50 ng/ml VEGF-A for 5 min prior to cross-linking and cell lysis.
Confocal Microscopy
The localization of VEGF-R2 in early endosomal vesicles was evaluated by confocal microscopy. HUVE cells plated on gelatin-coated eight-chamber slides were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton-X100 in PBS. To reduce nonspecific interactions, the slides were blocked with 10% goat serum. The slides were then probed with rabbit anti-human VEGF-R2 (clone 55B11, 1:100) and/or mouse anti-human EEA1 (clone 1G11, 1:100) and incubated with anti-rabbit Alexa Fluor 596 (1:200) and anti-mouse Alexa Fluor 488 (1:200). The slides were mounted using Vectashield containing DAPI. Images of the two fluoroprobes were captured simultaneously to exclude artifacts from sequential acquisition. To determine the antibody specificity, primary antibodies were omitted in control experiments. Colocalization was evaluated by Costes analysis using the JACoP plug-in in ImageJ. The Costes coefficient was normalized to total EEA1 staining.
VEGF-R2 Internalization Assay
Internalization of VEGF-R2 was performed as described previously with some modifications (19). Cell surface proteins were labeled by incubation with 0.5 mg/ml EZ-Link Sulfo-SS-Biotin for 2 h on ice. After the incubation, excess biotin was removed by washing the cells with PBS, and VEGF-R2 internalization was initiated by stimulation with 80 ng/ml VEGF-A in EBM-2. The cells were then washed again with PBS and incubated for 20 min on ice with L-glutathione-containing media (45 mm GSH, 75 mm NaCl, 75 mm NaOH, 1% BSA) to remove cell surface-bound biotin and lysed with TNTE buffer. Cell lysates were precleared with 25 μL of 40% packed Sepharose 4B beads, and the internalized, biotinylated proteins were isolated by incubation with streptavidin-agarose CL-4B. The streptavidin-bound proteins were released by boiling the beads in Laemmli sample buffer, separated on 4–20% SDS-PAGE gels in reducing conditions, transferred to nitrocellulose membranes, and probed for VEGF-R2 and β-actin as described above.
Endothelial Cell Sprouting Assay
The endothelial cell sprouting assay was performed as described previously (20).
Mouse Model of Suture-induced Corneal Neovascularization
All animal procedures were approved by the Institutional Animal Care and Use Committee at Tufts University. The investigation conformed to the Association for Research in Vision and Ophthalmology Resolution on the Use of Animals in Vision Research. Suture-induced corneal neovascularization was performed as described previously (21). On day 7 post-surgery, the animals were anesthetized and injected intracardially with 100 μl of 5 mg/ml of FITC-BS1 to visualize the newly formed vessels. After 30 min, the animals were sacrificed, the eyes were fixed with 0.1% paraformaldehyde, and flat mounts of the dissected corneas were prepared and evaluated by fluorescence microscopy as described previously (22). The total area of corneal neovascularization was calculated using ImageJ. Statistical significance was evaluated by the rank sum test.
Statistical Analysis
Data are expressed as mean ± S.E. Unless otherwise noted, the comparisons were made by the two-tailed Student's t test.
RESULTS
Galectin-3 Promotes VEGF-R2 Phosphorylation
Earlier studies by Nangia-Makker et al. (11) demonstrated that addition of up to 20 μg/ml (0.67 μm) of exogenous galectin-3 to HUVE cells induces the formation of capillary tubes. Considering this, we wondered whether similar concentrations of exogenous galectin-3 added to serum-starved HUVE cells would also induce phosphorylation of VEGF-R2. To test the hypothesis that galectin-3 modulates VEGF-R2 activation, we tested both dose- and time-dependant phosphorylation of VEGF-R2 by galectin-3. First, we added 10 μg/ml (0.34 μm) of exogenous galectin-3 to serum-starved HUVE cells. After a zero- to 10-min incubation, whole cell lysates were prepared, and phosphorylation of VEGF-R2 was evaluated by Western blot analysis using phospho-specific antibodies directed against VEGF-R2. Incubation with galectin-3 resulted in time-dependant phosphorylation of VEGF-R2 (Fig. 1A). Next, the assay was performed using increasing concentrations of exogenous galectin-3 up to 50 μg/ml (1.6 μm) with serum-starved HUVE cells. After a 10-min incubation, increasing levels of phosphorylated VEGF-R2 were observed with higher concentrations of galectin-3 (Fig. 1B). These results suggest that exogenous, extracellular galectin-3 is sufficient to activate VEGF-R2.
FIGURE 1.
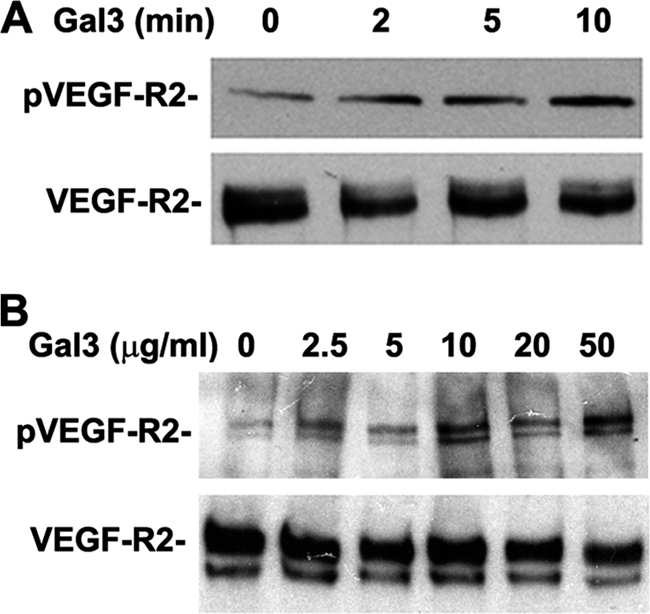
Galectin-3 induces VEGF-R2 phosphorylation. HUVE cells were incubated with galectin-3 prior to lysis. Samples were separated by SDS-PAGE and analyzed by Western blot analysis with phospho-specific antibodies directed against tyrosine residue 1175 of VEGF-R2. Samples were probed for expression of VEGF-R2 with antibodies against VEGF-R2. A, HUVE cells were incubated with 10 μg/ml galectin-3 for 0, 2, 5, and 10 min prior to lysis. B, HUVE cells were incubated with 0, 2.5, 5, 10, 20, and 50 μg/ml of galectin-3 for 10 min prior to lysis.
Galectin-3 Interacts with VEGF-R2 on the Plasma Membrane
Considering that exogenous galectin-3 activates VEGF-R2, we wondered whether galectin-3 modulates VEGF-R2 activation by directly interacting with the receptor in a carbohydrate-dependent manner. To test this, lysates from HUVE cells were incubated with galectin-3-conjugated Sepharose beads, and bound proteins were sequentially eluted with 0.1 m sucrose, a non-competing disaccharide, and β-lactose, a competing disaccharide that binds the carbohydrate recognition domain of galectin-3. The eluted proteins were separated by SDS-PAGE and analyzed by Western blot analysis using monoclonal antibodies directed against VEGF-R2. A 250-kDa band was detectable in the fraction eluted with β-lactose but not in the fraction eluted with sucrose, indicating that VEGF-R2 and galectin-3 interact in a carbohydrate-dependent manner (Fig. 2A).
FIGURE 2.

Galectin-3 interacts with VEGF-R2 on the plasma membrane in an Mgat5-dependent manner. A, carbohydrate-dependent binding of galectin-3 to VEGF-R2. HUVE cell lysates were incubated with a galectin-3 affinity column, and bound proteins were sequentially eluted with a non-competing saccharide, 0.1 m sucrose (Suc) and a competing saccharide, 0.1 m β-lactose (Lac). Samples were separated by SDS-PAGE and probed for VEGF-R2 by Western blot analysis. B and C, knockdown of Mgat5. HUVE cells were transfected with lentiviral particles expressing shRNA directed against Mgat5 (shMgat5) or non-targeting shRNA (shCtrl). B, the knockdown efficiency of mRNA was evaluated by RT qPCR. *, p < 0.01 compared with control. C, the expression of β1,6 branched glycans was evaluated by staining with rhodamine-conjugated L-PHA (red) and DAPI (blue). ConA (red) was used as a lectin control. D, VEGF-R2 contains Mgat5-modified N-glycans. Whole cell lysates (WCL) of Mgat5 knockdown (shMgat5) and control HUVE cells (shCtrl) were incubated with either L-PHA-agarose or ConA-agarose. Bound proteins were eluted, separated on SDS-PAGE, and analyzed by Western blot for VEGF-R2. E, disruption of Mgat5 reduces VEGF-R2 at the cell surface. Mgat5 knockdown (shMgat5) or control HUVE cells (shCtrl) were biotinylated, cross-linked, and lysed. Samples were immunoprecipitated with anti-galectin-3 antibodies and streptavidin (Gal3/Strep) or with streptavidin alone (Strep). Bound proteins were analyzed by Western blot analysis (Blot). Results are representative of three independent experiments. PD, pull-down. F, knockdown of galectin-3. HUVE cells were transfected with lentiviral particles expressing shRNA directed against galectin-3 (shGal3) or a non-targeting shRNA control (shControl). Knockdown of galectin-3 was evaluated at the mRNA level by RT qPCR and at the protein level by Western blot analysis (inset). *, p < 0.01 compared with shControl cells. G, disruption of galectin-3 reduces VEGF-R2 at the cell surface. Cell surface proteins of HUVE cells transfected with lentiviral particles directed against galectin-3 (shGal3) or non-targeting control (shCtrl) were biotinylated and cross-linked, and lysates were incubated with streptavidin-coated agarose beads (Strep). The bound proteins, along with whole cell lysates (WCL), were separated by SDS-PAGE and analyzed by Western blot analysis (panels 1 and 2) (Blot). Whole cell lysates from shCtrl and shGal3 cells were probed with anti-VEGF-R2 antibody to test for changes in receptor expression (panel 3), and blots were probed with for β-actin to ensure equal loading (panel 4). H, galectin-3 interacts with phosphorylated and unphosphorylated VEGF-R2. HUVE cells were incubated with either 50 ng/ml of VEGF-A in 0.1% BSA (+) or with 0.1% BSA alone (-) for 5 min and then lysed. Lysates were incubated with galectin-3-conjugated Sepharose beads, eluted with sample buffer, separated by SDS-PAGE, and analyzed by Western blot analysis using antibodies against phosphorylated tyrosine 1175 of VEGF-R2 (panel 1), or VEGF-R2 (panel 2). As controls, whole cell lysates (WCL) were analyzed by Western blot analysis with antibodies against VEGF-R2 or phosphorylated tyrosine 1175 of VEGF-R2 (panels 3 and 4).
Early studies suggest that VEGF-R2 is N-glycosylated (23). To determine whether VEGF-R2 bears Mgat5-modified N-glycans, the high-affinity ligands of galectin-3, the effect of Mgat5 knockdown on the glycosylation branching of VEGF-R2 was assessed. Knockdown cells were generated by transfection of HUVE cells with lentiviral particles expressing shRNA directed against the Mgat5 gene. The knockdown resulted in an approximately 89% reduction of expression of Mgat5 at the mRNA level as evaluated by RT-qPCR and 95% at the protein level as evaluated by immunocytochemistry (Fig. 2, B and C). Control cells were generated by transfection of HUVE cells with lentiviral particles expressing non-targeting shRNA.
Previous studies have shown that leukoagglutinin of Phaseolus vulgaris (L-PHA) specifically interacts with β1,6 N-glycans (24). To test whether glycosylation of VEGF-R2 is modified by Mgat5, lysates of control and knockdown cells were incubated with L-PHA-coated Sepharose beads, and bound proteins were analyzed by Western blot analysis using anti-VEGF-R2 antibodies. VEGF-R2 was only detected in the control shRNA cells and not in Mgat5 knockdown cells (Fig. 2D), suggesting that VEGF-R2 is modified by Mgat5 and therefore contains the preferred N-glycans for galectin-3 binding. In contrast, binding of VEGF-R2 to a control lectin, ConA, was similar in both control and knockdown cells (Fig. 2D).
To determine whether the interaction between VEGF-R2 and galectin-3 occurs on the plasma membrane and whether this interaction is dependent on the β1,6 N-glycan branching of VEGF-R2, cell surface biotinylation and cross-linking experiments were conducted. Mgat5 knockdown and control cells were labeled with biotin and chemically cross-linked with DTSSP, a homo-bifunctional, thiol-cleavable cross-linker, and then lysed. As neither biotin nor DTSSP are membrane-permeable, biotin labeling and cross-linking were restricted to proteins on the cell surface. The cell lysates were sequentially incubated with beads coated with anti-galectin-3 antibodies and then streptavidin coated beads to isolate the cell surface fraction of galectin-3-cross-linked products. The samples were then analyzed by Western blot analysis using antibodies directed against VEGF-R2. A 250-kDa band that reacted with anti-VEGF-R2 antibodies was detected in the control samples but not in the Mgat5 knockdown cells (Fig. 2E, panel 1), indicating that an interaction occurs between galectin-3 and VEGF-R2 on the cell surface and that this interaction is dependent upon the N-glycan branching generated by Mgat5.
To determine whether galectin-3 and VEGF-R2 cell surface localization is dependent on Mgat5, Mgat5 knockdown cell lysates were incubated with streptavidin-coated beads alone, eluted, and analyzed by Western blot analysis with antibodies against VEGF-R2 and galectin-3. The presence of a robust band in the control sample probed for both VEGF-R2 and galectin-3, but not in the Mgat5 knockdown samples, suggests that Mgat5-generated N-glycans are necessary for cell surface localization of VEGF-R2 and galectin-3 (Fig. 2E, panels 2 and 3). Analysis of whole cell lysates indicates that the reduction of cell surface localization of VEGF-R2 is not due to reduced expression levels (Fig. 2E, panel 4) and that galectin-3 expression was unaffected by disruption of the Mgat5 gene (Fig. 2E, panel 5).
To test the hypothesis that galectin-3 facilitates the retention of VEGF-R2 on the plasma membrane directly, we conducted the cell surface biotinylation and cross-linking experiments as described above using galectin-3 knockdown HUVE cells. Galectin-3 knockdown cells were generated by transfection with lentiviral particles expressing shRNA directed against galectin-3, resulting in 97% reduction of expression at the mRNA level (Fig. 2F) and ∼70% reduction at the protein level (F, inset). The plasma membrane fractions of control and galectin-3 knockdown cells were subjected to Western blot analysis using anti-VEGF-R2 antibodies. A reduced level of VEGF-R2 was detected in the galectin-3 knockdown sample relative to the control sample, indicating that less VEGF-R2 cell surface localization occurs in these cells (Fig. 2G, panel 1). As expected, galectin-3 was not detected on the cell surface of galectin-3 knockdown cells (Fig. 2G, panel 2). Total expression of VEGF-R2 was comparable in galectin-3 knockdown and control cells, indicating that the decreased level of cell surface VEGF-R2 in galectin-3 knockdown cells was not due to a reduction in the total expression of VEGF-R2 (Fig. 2G, panel 3). These findings are consistent with our hypothesis that galectin-3 retains VEGF-R2 on the plasma membrane.
Because we hypothesize that galectin-3 retains VEGF-R2 on the plasma membrane, galectin-3 must therefore interact with VEGF-R2 prior to VEGF-A binding, as this interaction induces rapid receptor internalization. To test if galectin-3 binds unphosphorylated VEGF-R2, we isolated VEGF-R2 from cells that had been unstimulated or stimulated with VEGF-A for 5 min prior to lysis. The cell lysates were incubated with galectin-3-conjugated Sepharose beads, and the bound proteins were analyzed by Western blot analysis with antibodies directed against unphosphorylated and phosphorylated (Tyr-1175) VEGF-R2. As expected, the receptor is phosphorylated only in VEGF-A stimulated cells (Fig. 2H, panels 1 and 3). The presence of an anti-VEGF-R2-reactive component in the galectin-3-bound fractions of both the stimulated and unstimulated cells shows that VEGF-R2 binding to galectin-3 is independent of its phosphorylation state (Fig. 2H, panel 2). These data support the prediction that galectin-3 facilitates VEGF-R2 plasma membrane retention. Whole cell lysates probed with antibodies against VEGF-R2 indicate that an equal amount of receptor was present in both samples (Fig. 2H, panel 4).
Galectin-3 Retains VEGF-R2 on the Plasma Membrane
To better characterize the apparent retention of VEGF-R2 on the plasma membrane by galectin-3, the cellular localization of the receptor was examined by confocal microscopy. Previous studies have shown that upon VEGF-A stimulation, VEGF-R2 is rapidly internalized by clathrin-dependent, early endosomal antigen 1 (EEA1)-positive early endosomes rather than in caveolae-dependent endosomes (25). Thus, the degree of internalization of VEGF-R2 was assessed with respect to the EEA1 marker. The distribution of VEGF-R2 in control and both galectin-3 and Mgat5 knockdown cells was evaluated after stimulation with VEGF-A to initialize VEGF-R2 internalization. Mgat5 and galectin-3 knockdown cells displayed significantly greater colocalization of VEGF-R2 and EEA1 as compared with control cells as evaluated by Costes analysis (correlation coefficient of control shRNA, 0.176 ± 0.082; galectin-3 shRNA, 0.350 ± 0.132; Mgat5 shRNA, 0.564 ± 0.239) consistent with the prediction that galectin-3 retains VEGF-R2 on the plasma membrane in a carbohydrate-dependent manner (Fig. 3, A and B).
FIGURE 3.

VEGF-R2 internalization is increased in Mgat5 and galectin-3 knockdown cells. To determine whether cells lacking Mgat5 or galectin-3 have increased levels of internalized VEGF-R2, knockdown (shGal3, shMgat5) and shControl endothelial cells were stimulated with VEGF, fixed, permeabilized, probed with anti-VEGF-R2 and anti-EEA1 monoclonal antibodies, and analyzed by confocal microscopy. A, representative images of EEA1- and VEGF-R2-stained HUVE cells. Scale bar = 30 μm; inset scale bar = 10 μm. The arrows indicate areas of VEGF-R2 colocalized with EEA1. The results are representative of two independent experiments. B, Costes correlation coefficient normalized to total EEA1 staining. Data are expressed as mean ± S.E. *, p < 0.01; **, p < 0.05 as compared with shControl cells. The results are representative of two independent experiments (n = at least 5/group). C, to assess VEGF-R2 endocytosis, shControl and knockdown (shMgat5 and shGal3) cells were also biotinylated to label extracellular proteins and treated with VEGF-A to stimulate VEGF-R2 internalization for the indicated time periods. Cell surface bound biotin was then removed by a reducing media wash, and the cells were lysed. The internalized biotinylated proteins, along with whole cell lysates, were separated by SDS-PAGE and analyzed by Western blot analysis for VEGF-R2 and β-actin, respectively. The results are representative of two independent experiments. D, quantification of internalized biotin labeling. Data are expressed mean ± S.E. AU, arbitrary units.
To further evaluate the role of galectin-3 in VEGF-R2 internalization, biotin-based endocytosis assays were performed. Cell surface proteins of galectin-3 and Mgat5 knockdown endothelial cells were biotinylated, and internalization was initiated by stimulation with VEGF-A. Endocytosis was stopped, and cell-surface bound biotin was removed by L-glutathione-containing media. Internalized VEGF-R2 was then detected in the intracellular fraction by Western blot analysis of the components isolated by streptavidin pull-down. Consistent with the results obtained by confocal microscopy, a significantly greater amount of VEGF-R2 was internalized in galectin-3 and Mgat5 knockdown cells relative to non-targeting shRNA control cells (Fig. 3, C and D). Taken together, these findings indicate that galectin-3 reduces VEGF-R2 internalization by binding the receptor's carbohydrate antennae, thus retaining a greater portion of the receptor on the plasma membrane upon VEGF-A stimulation.
Knockdown of Galectin-3 and Mgat5 Reduces VEGF-mediated Angiogenesis in Vitro
Recent studies in our laboratory demonstrated that galectin-3 and Mgat5 influence aspects of the angiogenic process, specifically migration and differentiation, when endothelial cells are stimulated with VEGF-A (12). To better characterize the role of galectin-3 and Mgat5 with respect to VEGF-A-mediated angiogenesis, an in vitro angiogenesis assay was employed to measure the formation of capillary sprouts in a three-dimensional matrix, as described by Korff and Augustin (26). Control, galectin-3, and Mgat5 knockdown HUVE cell spheroids were embedded in a collagen matrix and overlaid with media containing VEGF-A. After 24 h, the cumulative length of the sprouts, indicative of angiogenic potential, was evaluated. Stimulation with VEGF-A resulted in a robust angiogenic response in control cells (7.59 ± 1.73-fold change over cells in media alone), which was substantially reduced in galectin-3 (1.98 ± 0.31-fold change over cells in media alone) and Mgat5 (2.69 ± 0.84-fold change over cells in media alone) knockdown cells (Fig. 4). These findings are consistent with the hypothesis that galectin-3 directly influences VEGF-A-mediated angiogenic signaling.
FIGURE 4.

Galectin-3 and Mgat5 knockdown cells display reduced angiogenesis in vitro. To determine the role of galectin-3 and Mgat5 in angiogenesis, an in vitro HUVE cell sprouting assay was used. Endothelial cell spheroids were embedded in a collagen matrix and stimulated with media containing 50 ng/ml VEGF-A. After 24 h, the cumulative length of the sprouts for each spheroid was quantified by ImageJ (top panel). Data are expressed as mean fold change over control-treated cells incubated in media alone ± S.E. *, p < 0.05 as compared with shControl cells. The results are representative of three independent experiments (n = at least 4/group). Also shown are representative images of sprouting spheroids (bottom panel). Scale bar = 100 μm.
Galectin-3 and Mgat5 Knockout Mice Show Reduced Corneal Neovascularization
Because VEGF-A and VEGF-R2 are the primary mediators of angiogenesis in the cornea (27, 28), the consequences of galectin-3 disruption on VEGF-A-mediated angiogenesis in vivo were evaluated by employing a clinically relevant suture model of in vivo corneal neovascularization using Gal3−/− and Gal3+/+ mice. As a control for N-glycan-dependent corneal neovascularization, the experiments were repeated in Mgat5−/− and Mgat5+/+ mice. In this model, corneal neovascularization was induced from the limbic vein toward a suture located in the normally avascular cornea (29). A suture with two stromal incursions covering approximately 90° of the corneal circumference was placed in Gal3−/−, Mgat5−/−, and wild-type mice, and the extent of neovascularization was evaluated 7 days post-surgery by labeling angiogenic vasculature with the endothelial cell marker FITC-conjugated Bandeiraea simplicifolia (BS1) lectin. A 42.2% reduction (± 4.2%) of corneal neovascularization was observed in Gal3−/− mice as compared with wild-type controls (Fig. 5A). Similarly, Mgat5−/− animals displayed a 47.3% reduction (± 8.4%) of corneal neovascularization relative to wild-type control mice (Fig. 5B). These findings suggest that galectin-3 and the N-glycan branching generated by Mgat5 play a proangiogenic role in the cornea.
FIGURE 5.

Galectin-3 and Mgat5 knockout mice demonstrate reduced corneal neovascularization. To determine the effect of galectin-3 and Mgat5 on corneal neovascularization, the corneal suture model was used. A, galectin-3 null mice have reduced suture-induced neovascularization. A single suture was placed ∼2 mm above the limbic vessel in the cornea of wild-type (Gal-3+/+) or galectin-3 knockout mice (Gal-3−/−). After 7 days, the mice were perfused intracardially by injection of FITC-BSI and analyzed by fluorescence microscopy. i, the area of fluorescent labeled vessels was measured by ImageJ. Data are expressed as mean ± S.E. (n = 4/group). *, p < 0.05 as compared with Gal-3+/+ mice. The results are representative of two independent experiments. ii, representative fluorescence images of corneas. The arrowheads indicate sutures. Scale bar = 100 μm. B, Mgat5 knockout mice demonstrate reduced corneal neovascularization. i, corneal neovascularization in wild-type (Mgat5+/+) and Mgat knock-out mice (Mgat5−/−) was evaluated using the corneal suture model. Data are expressed as mean ± S.E. (at least n = 4/group). *, p < 0.05 as compared with Mgat5+/+ mice. The results are representative of two independent experiments. ii, representative fluorescence images of corneas. The arrowheads indicate sutures. Scale bar = 200 μm. BS1, Bandeira simplicifolia.
DISCUSSION
This study was designed to test the hypothesis that galectin-3 modulates VEGF-A-mediated signaling by retaining VEGF-R2 on the plasma membrane. Here, we demonstrate that galectin-3 interacts with the N-glycans of VEGF-R2 on the plasma membrane in an Mgat5-dependent manner. Using confocal microscopy and biotin-based endocytosis assays, we also found that galectin-3 retains VEGF-R2 on the plasma membrane and observed that VEGF-A-induced receptor internalization was markedly increased in both galectin-3 and Mgat5 knockdown endothelial cells as compared with control cells. Angiogenesis experiments revealed that knockdown of galectin-3 and Mgat5 reduced VEGF-A-mediated angiogenesis in vitro. Using the suture model of corneal neovascularization, the extent of neovascularization was substantially less in the corneas of Gal3−/− and Mgat5−/− animals compared with control animals. These data strongly suggest that galectin-3 mediates VEGF-R2 plasma membrane localization and VEGF-A-induced angiogenesis.
VEGF-R2 is activated by binding to VEGF-A, which results in autophosphorylation of the receptor, initiates intracellular signaling cascades, and triggers the rapid endocytic internalization of the receptor (25). Although recent studies have demonstrated that VEGF-R2 signaling is not immediately attenuated and may, in fact, be enhanced upon internalization into the endosomal compartment (25, 30), initial binding and activation of VEGF-R2 on the plasma membrane is a critical preliminary step for signaling (31).
In this study, we found that the addition of exogenous galectin-3 induces time- and dose-dependent phosphorylation of VEGF-R2 in endothelial cells. One mechanism by which galectin-3 might mediate this effect is by increasing the density of receptor on the cell surface accessible to low levels of endogenous VEGF-A. Indeed, receptor density on the plasma membrane has been shown to be a major determinant of receptor-ligand complex formation and, thus, intracellular signaling. For example, diminished plasma membrane localization of the EGF receptor and TGF-β receptors I and II markedly reduce activation of ERK1/2 and Smad2/3, respectively (15). Consistent with this model, we found that galectin-3 binds both phosphorylated and unphosphorylated VEGF-R2, indicating that binding of galectin-3 to the receptor is independent of activation by VEGF-A. Because receptor cell surface density is regulated by the rate of endocytosis and exocytosis, factors that affect the endocytic uptake of receptors directly impact signaling as well. For example, because of an enhanced rate of constitutive endocytosis of receptors, reduced basic fibroblast growth factor and epidermal growth factor mediated ERK1/2 activation has been observed in mammary epithelial tumor cells isolated from Mgat5−/− animals (15). Alternatively, galectin-3 might activate VEGF-R2 by positioning the receptor in proximity to plasma membrane-restricted mediators. Activation of VEGF-R2 might be induced by increasing the number of interactions between receptors, thus promoting receptor kinase activity, or by facilitating interactions between VEGF-R2 and other cell surface signaling factors, such as integrins. That exogenous galectin-3 activates VEGF-R2 is noteworthy, as increased expression of galectin-3 has been associated with the invasiveness and metastatic potential of tumors of the head, neck, thyroid, liver, gastric tract, and colon (32–36). Because angiogenesis is required for tumor growth and metastasis (37), it is possible that increased expression of galectin-3 enhances tumorigenesis by activating VEGF-R2 signaling pathways, thus sensitizing endothelial cells to VEGF-A stimuli and promoting angiogenesis.
In its oligomeric form, galectin-3 is predicted to bring receptors into proximity that are otherwise spatially separated, and by promoting these interactions, galectin-3 is thought to significantly impact the function of plasma membrane glycoproteins (16, 39, 40). Our findings fit well into the overall theme of galectin-glycoprotein lattice formation, as recent literature has shown that other galectin family members regulate plasma membrane residency of glycoproteins. In neuroblastoma cells for example, galectin-1 binds and retains the receptor protein-tyrosine phosphatase β (RPTPβ) on the plasma membrane, facilitating cell-cell adhesion and migration (41). Galectin-9 has been shown to retain the glucose transporter 2 (GLUT2) on the surface of pancreatic β cells, which is necessary for glucose-stimulated insulin secretion. Disruption of this interaction leads to redistribution of the transporter into endosomes and lysosomes, resulting in impaired insulin secretion and metabolic dysfunction (42). In enterocyte-like HT-29 cells, galectin-4 is necessary for the localization of glycoproteins into detergent-resistant membranes on the cell surface. Its disruption causes accumulation of these glycoproteins inside the cell (43). Accordingly, we propose that galectin-3 oligomers cross-link VEGF receptors into a lattice formation on the cell surface and thereby delay their removal by endocytosis. The increased receptor density on the cell surface enhances VEGF-A-mediated signal transduction and angiogenesis (Fig. 6).
FIGURE 6.

Proposed model. Galectin-3 promotes clustering of VEGF-R2 by binding to Mgat5-synthesized β1,6GlcNAc-branched N-glycans. In the absence of galectin-3, VEGF receptors accumulate in endosomal vesicles (left panel). In the presence of galectin-3, VEGF-R2 is retained on the plasma membrane and angiogenic signaling is enhanced (right panel).
In summary, we found that galectin-3 binds the N-glycans of VEGF-R2, facilitating its plasma membrane retention and phosphorylation. We also show reduced in vitro angiogenesis in both galectin-3 and Mgat5 knockdown HUVE cells and reduced in vivo corneal neovascularization in Gal3−/− and Mgat5−/− mice. The data presented here suggest that by regulating VEGF-R2 internalization and phosphorylation, galectin-3 makes a significant contribution to VEGF-A-mediated angiogenesis and could therefore be a useful target for the development of antiangiogenic therapeutics.
Acknowledgments
We thank Jarel Gandhi for help with confocal imaging and Drs. Parastoo Sadrai and Reza Dana (Schepens Eye Research Institute) for help with the suture model. We also thank Dr. Zhiyi Cao for helpful comments and Drs. Rosana Meyer and Nader Rahimi (Boston University School of Medicine) for help with the sprouting assay. We thank Dr. Hakon Leffler (Lund University, Sweden) for the galectin-3 construct.
This work was supported by the National Eye Institute Grant EY07088 and by National Institutes of Health Grant P30 N5047243 for confocal microscopy performed at the Tufts Center for Neuroscience Research. This work was also supported, in part, by The New England Corneal Transplant Fund, the Massachusetts Lions Eye Research Fund, and an unrestricted grant from Research to Prevent Blindness to the New England Eye Center.
- VEGF-R2
- VEGF receptor 2
- Mgat5
- β1,6-N-acetylglucosaminyl transferase 5
- HUVE
- human umbilical vein endothelial
- qPCR
- quantitative PCR
- L-PHA
- leukoagglutinin of Phaseolus vulgaris
- ConA
- concanavalin A
- DTSSP
- 3,3′-dithiobis-sulfosuccinimidylpropionate
- EEA
- early endosomal antigen.
REFERENCES
- 1. Folkman J. (2006) Annu. Rev. Med. 57, 1–18 [DOI] [PubMed] [Google Scholar]
- 2. Folkman J. (1995) Nat. Med. 1, 27–31 [DOI] [PubMed] [Google Scholar]
- 3. Pluda J. M. (1997) Semin. Oncol. 24, 203–218 [PubMed] [Google Scholar]
- 4. Olsson A. K., Dimberg A., Kreuger J., Claesson-Welsh L. (2006) Nat. Rev. Mol. Cell Biol. 7, 359–371 [DOI] [PubMed] [Google Scholar]
- 5. Scott A., Mellor H. (2009) Biochem. Soc. Trans. 37, 1184–1188 [DOI] [PubMed] [Google Scholar]
- 6. Ferrara N., Gerber H. P., LeCouter J. (2003) Nat. Med. 9, 669–676 [DOI] [PubMed] [Google Scholar]
- 7. Gerber H. P., Hillan K. J., Ryan A. M., Kowalski J., Keller G. A., Rangell L., Wright B. D., Radtke F., Aguet M., Ferrara N. (1999) Development. 126, 1149–1159 [DOI] [PubMed] [Google Scholar]
- 8. Joœko J., GwóŸdŸ B., Jedrzejowska-Szypułka H., Hendryk S. (2000) Med. Sci. Monit. 6, 1047–1052 [PubMed] [Google Scholar]
- 9. Inoue M., Hager J. H., Ferrara N., Gerber H. P., Hanahan D. (2002) Cancer Cell 1, 193–202 [DOI] [PubMed] [Google Scholar]
- 10. Takahashi T., Yamaguchi S., Chida K., Shibuya M. (2001) EMBO J. 20, 2768–2778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nangia-Makker P., Honjo Y., Sarvis R., Akahani S., Hogan V., Pienta K. J., Raz A. (2000) Am. J. Pathol. 156, 899–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Markowska A. I., Liu F. T., Panjwani N. (2010) J. Exp. Med. 207, 1981–1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dumic J., Dabelic S., Flögel M. (2006) Biochim. Biophys. Acta 1760, 616–635 [DOI] [PubMed] [Google Scholar]
- 14. Ahmad N., Gabius H. J., André S., Kaltner H., Sabesan S., Roy R., Liu B., Macaluso F., Brewer C. F. (2004) J. Biol. Chem. 279, 10841–10847 [DOI] [PubMed] [Google Scholar]
- 15. Dennis J. W., Pawling J., Cheung P., Partridge E., Demetriou M. (2002) Biochim. Biophys. Acta 1573, 414–422 [DOI] [PubMed] [Google Scholar]
- 16. Partridge E. A., Le Roy C., Di Guglielmo G. M., Pawling J., Cheung P., Granovsky M., Nabi I. R., Wrana J. L., Dennis J. W. (2004) Science. 306, 120–124 [DOI] [PubMed] [Google Scholar]
- 17. Granovsky M., Fata J., Pawling J., Muller W. J., Khokha R., Dennis J. W. (2000) Nat. Med. 6, 306–312 [DOI] [PubMed] [Google Scholar]
- 18. Altin J. G., Pagler E. B. (1995) Anal. Biochem. 224, 382–389 [DOI] [PubMed] [Google Scholar]
- 19. Fabbri M., Fumagalli L., Bossi G., Bianchi E., Bender J. R., Pardi R. (1999) EMBO J. 18, 4915–4925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Meyer R. D., Latz C., Rahimi N. (2003) J. Biol. Chem. 278, 16347–16355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Streilein J. W., Bradley D., Sano Y., Sonoda Y. (1996) Invest. Ophthalmol. Vis. Sci. 37, 413–424 [PubMed] [Google Scholar]
- 22. Pola R., Ling L. E., Silver M., Corbley M. J., Kearney M., Blake Pepinsky R., Shapiro R., Taylor F. R., Baker D. P., Asahara T., Isner J. M. (2001) Nat. Med. 7, 706–711 [DOI] [PubMed] [Google Scholar]
- 23. Vaisman N., Gospodarowicz D., Neufeld G. (1990) J. Biol. Chem. 265, 19461–19466 [PubMed] [Google Scholar]
- 24. Cummings R. D., Kornfeld S. (1982) J. Biol. Chem. 257, 11230–11234 [PubMed] [Google Scholar]
- 25. Lampugnani M. G., Orsenigo F., Gagliani M. C., Tacchetti C., Dejana E. (2006) J. Cell Biol. 174, 593–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Korff T., Augustin H. G. (1999) J. Cell Sci. 112, 3249–3258 [DOI] [PubMed] [Google Scholar]
- 27. Gan L., Fagerholm P., Palmblad J. (2004) Acta Ophthalmol. Scand. 82, 557–563 [DOI] [PubMed] [Google Scholar]
- 28. Hosseini H., Nejabat M. (2007) Med. Hypotheses 68, 799–801 [DOI] [PubMed] [Google Scholar]
- 29. Cursiefen C., Chen L., Borges L. P., Jackson D., Cao J., Radziejewski C., D'Amore P. A., Dana M. R., Wiegand S. J., Streilein J. W. (2004) J. Clin. Invest. 113, 1040–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sawamiphak S., Seidel S., Essmann C. L., Wilkinson G. A., Pitulescu M. E., Acker T., Acker-Palmer A. (2010) Nature. 465, 487–491 [DOI] [PubMed] [Google Scholar]
- 31. Ewan L. C., Jopling H. M., Jia H., Mittar S., Bagherzadeh A., Howell G. J., Walker J. H., Zachary I. C., Ponnambalam S. (2006) Traffic 7, 1270–1282 [DOI] [PubMed] [Google Scholar]
- 32. Gillenwater A., Xu X. C., el-Naggar A. K., Clayman G. L., Lotan R. (1996) Head Neck 18, 422–432 [DOI] [PubMed] [Google Scholar]
- 33. Xu X. C., el-Naggar A. K., Lotan R. (1995) Am. J. Pathol. 147, 815–822 [PMC free article] [PubMed] [Google Scholar]
- 34. Hsu D. K., Dowling C. A., Jeng K. C., Chen J. T., Yang R. Y., Liu F. T. (1999) Int. J. Cancer 81, 519–526 [DOI] [PubMed] [Google Scholar]
- 35. Irimura T., Matsushita Y., Sutton R. C., Carralero D., Ohannesian D. W., Cleary K. R., Ota D. M., Nicolson G. L., Lotan R. (1991) Cancer Res. 51, 387–393 [PubMed] [Google Scholar]
- 36. Schoeppner H. L., Raz A., Ho S. B., Bresalier R. S. (1995) Cancer 75, 2818–2826 [DOI] [PubMed] [Google Scholar]
- 37. Carmeliet P., Jain R. K. (2000) Nature 407, 249–257 [DOI] [PubMed] [Google Scholar]
- 38. Deleted in proof. [Google Scholar]
- 39. Goetz J. G., Joshi B., Lajoie P., Strugnell S. S., Scudamore T., Kojic L. D., Nabi I. R. (2008) J. Cell Biol. 180, 1261–1275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lagana A., Goetz J. G., Cheung P., Raz A., Dennis J. W., Nabi I. R. (2006) Mol. Cell. Biol. 26, 3181–3193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Abbott K. L., Matthews R. T., Pierce M. (2008) J. Biol. Chem. 283, 33026–33035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ohtsubo K., Takamatsu S., Minowa M. T., Yoshida A., Takeuchi M., Marth J. D. (2005) Cell 123, 1307–1321 [DOI] [PubMed] [Google Scholar]
- 43. Stechly L., Morelle W., Dessein A. F., André S., Grard G., Trinel D., Dejonghe M. J., Leteurtre E., Drobecq H., Trugnan G., Gabius H. J., Huet G. (2009) Traffic 10, 438–450 [DOI] [PubMed] [Google Scholar]