

Abstract
We investigated the role of a Ca2+ channel and intracellular calcium concentration ([Ca2+]i) in osmotic stress-induced JNK activation and tight junction disruption in Caco-2 cell monolayers. Osmotic stress-induced tight junction disruption was attenuated by 1,2-bis(2-aminophenoxyl)ethane-N,N,N′,N′-tetraacetic acid (BAPTA)-mediated intracellular Ca2+ depletion. Depletion of extracellular Ca2+ at the apical surface, but not basolateral surface, also prevented tight junction disruption. Similarly, thapsigargin-mediated endoplasmic reticulum (ER) Ca2+ depletion attenuated tight junction disruption. Thapsigargin or extracellular Ca2+ depletion partially reduced osmotic stress-induced rise in [Ca2+]i, whereas thapsigargin and extracellular Ca2+ depletion together resulted in almost complete loss of rise in [Ca2+]i. L-type Ca2+ channel blockers (isradipine and diltiazem) or knockdown of the CaV1.3 channel abrogated [Ca2+]i rise and disruption of tight junction. Osmotic stress-induced JNK2 activation was abolished by BAPTA and isradipine, and partially reduced by extracellular Ca2+ depletion, thapsigargin, or CaV1.3 knockdown. Osmotic stress rapidly induced c-Src activation, which was significantly attenuated by BAPTA, isradipine, or extracellular Ca2+ depletion. Tight junction disruption by osmotic stress was blocked by tyrosine kinase inhibitors (genistein and PP2) or siRNA-mediated knockdown of c-Src. Osmotic stress induced a robust increase in tyrosine phosphorylation of occludin, which was attenuated by BAPTA, SP600125 (JNK inhibitor), or PP2. These results demonstrate that CaV1.3 and rise in [Ca2+]i play a role in the mechanism of osmotic stress-induced tight junction disruption in an intestinal epithelial monolayer. [Ca2+]i mediate osmotic stress-induced JNK activation and subsequent c-Src activation and tyrosine phosphorylation of tight junction proteins. Additionally, inositol 1,4,5-trisphosphate receptor-mediated release of ER Ca2+ also contributes to osmotic stress-induced tight junction disruption.
Keywords: Calcium Channels, Cell junctions, Intestine, Phosphotyrosine, Src
Introduction
Epithelial tight junctions provide the barrier function that prevents the diffusion of toxins, allergens, and pathogens from external environment into the tissues and systemic circulation. Preserving the tight junction integrity, therefore, is essential for the maintenance of normal epithelial homeostasis. Various types of stress are known to disrupt the barrier function in the gastrointestinal mucosa. Stress-induced epithelial barrier disruption plays an important role in the pathogenesis of numerous gastrointestinal diseases (1). Osmotic stress is one type of stress that the intestine faces under physiological (2) and pathophysiological conditions (3–5). Our recent study showed that osmotic stress disrupts intestinal epithelial tight junctions leading to barrier dysfunction by a JNK2 (c-Jun N-terminal kinase 2)-dependent mechanism (6). Understanding the biochemical mechanisms that underlie stress-induced cellular responses leading to disruption of epithelial tight junction and barrier function is important in developing therapeutics for the treatment of stress-related diseases.
Tight junctions are multiprotein complexes that serve as a permeability barrier, regulating the passage of ions and small molecules through a paracellular pathway. They are organized by the interactions of transmembrane proteins such as occludin, tricellulin, claudins, and junctional adhesion molecules with cytoplasmic scaffold proteins such as ZO-1, ZO-2, and ZO-3 (7). ZO-1 anchors the transmembrane proteins to the actin cytoskeleton and recruits an array of signaling proteins such as protein kinases, protein phosphatases, G proteins, etc. (8). Tight junction proteins are phosphorylated on tyrosine, serine, and threonine residues, which regulate protein-protein interactions and the integrity of tight junctions (9–12).
Calcium (Ca2+) is a highly versatile intracellular signaling element that can regulate numerous cellular functions and is required for the maintenance of integrity of intercellular junctional complexes. It is well established that extracellular Ca2+ is essential for the maintenance of intercellular junctions, including adherens junctions and tight junctions. Although it is not required for protein-protein interactions at tight junctions, extracellular Ca2+ is required for the maintenance of tight junction integrity. Depletion of extracellular Ca2+ leads to disruption of adherens junctions and tight junctions and increase in paracellular permeability in Caco-2, Madin-Darby canine kidney cells, and pulmonary and endothelial cell monolayers (13–16). Studies also indicated that the basal level of intracellular Ca2+ is required for the assembly of tight junctions and maintenance of its integrity (17, 18). Interestingly, a few studies showed that an increase in intracellular Ca2+ concentration ([Ca2+]i) leads to disruption of tight junctions in endothelial monolayers (14). Therefore, the potential role of a rise in [Ca2+]i and subsequent mechanisms associated with tight junction regulation are rather confusing at this point and warrants further investigation (19).
In the present study, we demonstrate that an elevation in [Ca2+]i plays a crucial role in the mechanism of osmotic stress-induced disruption of tight junctions. We show, for the first time, that both CaV1.3-mediated influx of extracellular Ca2+ and the release of ER Ca2+ through IP32-gated Ca2+ release channels disrupt tight junctions in an intestinal epithelial monolayer. The present study also provides evidence to indicate that [Ca2+]i-induced tight junction disruption in osmotic stress-treated cell monolayers is mediated by the activation of JNK2 and c-Src-mediated tyrosine phosphorylation of tight junction proteins.
EXPERIMENTAL PROCEDURES
Chemicals
Cell culture medium (DMEM), fetal bovine serum (FBS), and antibiotics were procured from Cellgrow® (Manassas, VA). DMEM without Ca2+ was purchased from Invitrogen. Transfection reagents Opti-MEM®, LipofectamineTM 2000, OligofectamineTM, and PlusTM reagents were purchased from Invitrogen. SP600125 (anthra(1,9-cd)pyrazol-6(2H)-one-1,9-pyrazoloanthron), thapsigargin, and PP2 (4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine) were purchased from Calbiochem, EMD Chemicals (San Diego, CA). BAPTA-AM, genistein, isradipine, diltiazem, and FITC-inulin were purchased from Sigma. Fura-2AM (acetoxymethyl ester) and pluronic acid were from Invitrogen. Xestospongin-C was purchased from Cayman Chemical (Ann Arbor, MI). Streptavidin-agarose was obtained from Pierce Biotechnology, Inc. and other chemicals were of analytical grade purchased from either Sigma or Fisher Scientific.
Antibodies
Rabbit polyclonal anti-JNK1/2(pTpY183/185) (p-JNK) antibody was purchased from BIOSOURCE/Invitrogen. Rabbit polyclonal anti-JNK/SAPK was purchased from Upstate Biotech Inc. (Charlottesville, VA). HRP-conjugated anti-mouse IgG, HRP-conjugated anti-rabbit IgG, and anti-β-actin antibodies were obtained from Sigma. Rabbit polyclonal anti-ZO-1, rabbit polyclonal anti-occludin, and HRP-conjugated mouse monoclonal anti-occludin antibodies were purchased from Zymed Laboratories Inc.. Alexa Fluor 488-conjugated anti-mouse IgG, Cy3-conjugated anti-rabbit IgG, and Alexa Fluor 488-conjugated phalloidin were purchased from Molecular Probes (Eugene, OR). Mouse anti-GFP antibody was purchased from Clontech (Mountain view, CA). Mouse monoclonal CaV1.3 antibodies were obtained from Abcam (Cambridge, MA). Goat anti-CaV1.2 antibody was purchased from Santa Cruz Biotechnology Inc. (Santa Cruz, CA). Biotin-conjugated anti-Tyr(P) antibodies were purchased from BD Transduction Laboratories (San Jose, CA).
Antisense Oligonucleotides and siRNA
Antisense oligonucleotides for JNK1 (AS-Jnk1) and JNK2 (AS-Jnk2) and the scrambled sequence for AS-Jnk1 (MS-Oligo) were designed as described previously (6) and custom synthesized in phosphorothioate form by Sigma Genosys. Human c-SRC and c-YES-specific siRNA and the corresponding control RNA were purchased from Dharmacon (Lafayette, CO).
shRNA for CaV1.3
A vector-based short hairpin RNA (shRNA) method was used to silence CaV1.3 gene expression in Caco-2 cells. Two targeting sequences were chosen against the nucleotide sequence of human CaV1.3 (Gene ID 776 CACNA1D; NM_000720.2) using Dharmacon siDesigner software (Target 1, CCGAATAGCTCCAAGCAAA (sequence position 290–308); Target 2, GGAAGACCCAGAGATACA (sequence position 5697–5715)). The sequences were further verified by BLAST search by human genome databases, and no matches were found other than CaV1.3, confirming the uniqueness of these sequences. To construct shRNA vectors, two pairs of oligonucleotides containing the antisense sequence, hairpin loop region (TTGATATCCG), and sense sequence with cohesive BamHI and HindIII sites were synthesized (Integrated DNA Technologies Inc., Coralville, IA) as follows: top strand 1, 5′-GATCCCGCCGAATAGCTCCAAGCAAATTGATATCCGTTTGCTTGGAGCTATTCGGTTTTTTCCAAA-3′ and bottom strand 1, 5′-AGCTTTTGGAAAAAACCGAATAGCTCCAAGCAAACGGATATCAATTTGCTTGGAGCTATTCGGCGG-3′, top strand 2, 5-GATCCCGGGAAGACCCAGAGATACATTGATATCCGTGTATCTCTGGGTCTTCCCTTTTTTCCAAA-3′ and bottom strand 2, 5′-AGCTTTTGGAAAAAAGGGAAGACCCAGAGATACACGGATATCAATGTATCTCTGGGTCTTCCCGG-3′. The corresponding forward and reverse strands were annealed and inserted at BamHI and HindIII sites into pRNATin-H1.2/Neo (GenScript Corp., Piscataway, NJ) vector, which induces expression of shRNA by the H1.2 promoter and cGFP reporter gene by the cytomegalovirus promoter (Promega, WI). The constructs were transformed into DH5α Escherichia coli competent cells for preparation of plasmid DNA. Plasmid DNA were isolated and purified using the Qiagen Plasmid miniprep kit (Valencia, CA). Insertion of the shRNA sequence was confirmed by releasing it by digestion with BamHI and HindIII.
Cell Culture
Caco-2 cells purchased from ATCC (Manassas, VA) were grown under standard cell culture conditions as described (20). In brief, Caco-2 cells were cultured in DMEM containing 10% (v/v) fetal bovine serum (FBS), high glucose, l-glutamine, pyruvate, and antibiotics (penicillin, streptomycin, and gentamicin). The cells were grown as monolayers in 100-mm Petri dishes or T-75 flasks. Experiments were conducted on cells grown in polycarbonate membrane transwell inserts (Costar, MA) of varying diameters (6.5, 12, and 24 mm). Studies were done on 9–11 (6.5 mm), 12–14 (12 mm), and 16–19 days (24 mm).
Transfection
Caco-2 cells grown to about 75% confluence were transfected using 1 ml of antibiotic and serum-free Opti-MEM® containing 150 nm oligonucleotides (MS-oligo, AS-Jnk1, or AS-Jnk2), c-Src siRNA, or nonspecific siRNA using 3.15 μl of Oligofectamine reagent as described previously (6). After 24 h, the cell monolayers were trypsinized and seeded onto Transwell inserts of 6.5-, 12-, or 24-mm diameters and used for experiments on days 3 or 4 post-seeding. Transfection of shRNA for CaV1.3 was performed using LipofectamineTM 2000 reagent according to the manufacturer's protocol. Briefly, cells were transfected with 1 μg of plasmid DNA (shRNA1, shRNA2, or empty vector) using Lipofectamine 2000 and Plus reagent in 50 μl of Opti-MEM, and incubated for 30 min at room temperature. The transfection mixture was then replaced with normal DMEM containing serum and antibiotics. After 24 h, cell monolayers were trypsinized, seeded onto Transwell inserts from Costar (Cambridge, MA), and used for experiments on days 3 or 4 after seeding.
Osmotic Stress and Treatment with Inhibitors
Cell monolayers in transwells were incubated in 600 mosM DMEM (osmolarity adjusted with mannitol) in apical or basal compartment to induce osmotic stress. Barrier disruption was evaluated by measuring TER and inulin flux. SP600125 (1 μm) or genistein (100 μm) were administered 50 min prior to osmotic stress and PP2 (3 μm) was administered 30 min prior to osmotic stress. For extracellular Ca2+ depletion, Ca2+-free medium (CFM) was used to prepare the hyperosmolar medium and administered either to apical (CFM-A) or basal (CFM-B) compartments. Intracellular Ca2+ depletion was achieved by preloading cells with 10 μm BAPTA-AM. Ca2+ channel blockers, thapsigargin (1 μm), isradipine (1 μm), or diltiazem (100 μm) were administered 30 min prior to osmotic stress and 1 μm xestospongin-C was added 15 min before osmotic stress.
Measurement of TER
TER was measured as described previously (20) using a Millicell-ERS Electrical Resistance System (Millipore, Bedford, MA). TER was calculated as Ω·cm2 by multiplying it with the surface area of the monolayer. The TER of the polycarbonate membrane in Transwells (∼30 Ω·cm2) was subtracted from all readings.
Unidirectional Flux of Inulin
Transwells with the cell monolayers were incubated under varying experimental conditions in the presence of FITC-inulin (0.5 mg/ml) in the basal well. At varying times of treatment, 50 μl of apical medium was withdrawn and fluorescence was measured using a plate reader (BioTEK Instruments, Winooski, VT). The flux into the apical well was calculated as the percent of total fluorescence administered into the basal well per hour per cm2 surface area.
Immunofluorescence Microscopy
Cell monolayers were fixed in 3% paraformaldehyde in PBS. Following permeabilization in 0.2% Triton X-100, cell monolayers were blocked in 4% nonfat milk in TBST (20 mm Tris, pH 7.2, and 150 mm NaCl). They were incubated for 1 h with primary antibodies (rabbit polyclonal anti-ZO-1 and mouse monoclonal anti-occludin), followed by a 1-h incubation with secondary antibodies, Alexa Fluor 488-conjugated anti-mouse IgG and Cy3-conjugated anti-rabbit IgG antibodies. The fluorescence was examined under a Zeiss LSM 5 laser scanning confocal microscope, and images from x-y sections (1 μm) were collected using LSM 5 Pascal software. Images were stacked using the software, Image J (NIH), and processed by Adobe Photoshop (Adobe Systems Inc., San Jose, CA).
Intracellular Ca2+ Measurements
Caco-2 cell monolayers were grown on 20-mm diameter glass-bottom microwell dishes of (MatTek, Ashland, MA) for 5 days. Cells were serum starved for 12 h and washed with HBSS prior to incubation in HEPES-buffered solution (134 mm NaCl, 6 mm KCl, 2 mm CaCl2, 1 mm MgCl2, 10 mm HEPES, and 10 mm glucose, pH 7.4) containing the fluorescent Ca2+ probe, Fura-2 AM (10 μm), and 0.5% pluronic F-127 for 30 min, followed by a 15-min wash. Fura-2-loaded Caco-2 cells were alternately excited at 340 or 380 nm using a PC driven hyperswitch (Ionoptix, MA). Ratios were collected every second at 510 nm using a Dage MTI iCCD camera and Ionwizard software (Ionoptix). Intracellular Ca2+ concentrations were calculated using Equation 1,
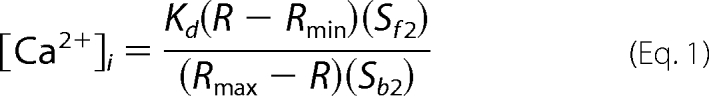 |
where R is the 340/380 nm ratio, Rmin and Rmax are the minimum and maximum ratios determined in Ca2+-free and saturating Ca2+ solutions, respectively. Sf2/Sb2 is the Ca2+ free/Ca2+ replete-ratio of emissions at 380 nm excitation, and Kd is the dissociation constant for Fura-2. Rmin, Rmax, Sf2, and Sb2 were determined by increasing the Ca2+ permeability of Caco-2 cells with ionomycin (10 mm), and perfusing cells with a high Ca2+ (10 mm) or Ca2+-free (10 mm EGTA) solution. The in situ apparent dissociation constant (Kd) for Fura-2 used in this study was 224 nm. Eight to 10 cells in each monolayer were analyzed simultaneously, and experiments were repeated in 3–5 monolayers.
Immunoprecipitation
Caco-2 cell monolayers were washed with ice-cold PBS and proteins were extracted in hot lysis buffer D (0.3% SDS in 10 mm Tris buffer, pH 7.4, containing 1 mm sodium orthovanadate, 10 mm sodium fluoride, 1 mm PMSF, and 10 μl/ml of protease inhibitors mixture). Homogenate was heated at 100 °C for 10 min, suspended with a 25-gauge syringe 5 times, and centrifuged to collect the clear supernatant. Protein content was measured by the BCA method (Pierce). Phospho-Tyr was immunoprecipitated as described before (21) using biotin-conjugated anti-Tyr(P) antibody. Immunocomplexes were isolated by precipitation using streptavidin-agarose and immunoblotted for ZO-1 and occludin using mouse monoclonal anti-ZO-1 and HRP-conjugated mouse monoclonal anti-occludin antibodies.
Cell Lysate and Immunoblot Analysis
Proteins from the cells were extracted in heated lysis buffer D as described above (21). An aliquot was taken for protein estimation, and the lysate was mixed with equal volume of Laemmli's sample buffer (two times concentrated), heated at 100 °C for 10 min. Proteins were separated in a 7% gel using SDS-polyacrylamide gel electrophoresis and transferred to PVDF membrane. The membranes were probed for p-JNK, JNK 1/2, p-Src, CaV1.2, CaV1.3, c-Src, ZO-1, occludin, or β-actin by using a combination of the specific primary antibodies with corresponding HRP-conjugated anti-mouse IgG or HRP-conjugated anti-rabbit IgG secondary antibodies. The blot was developed using the chemiluminescence (ECL) system (Amersham Biosciences/GE Healthcare). Densitometric analysis of specific bands was done using the Image J software.
Statistical Analysis
The observed data in the two different groups was compared using Student's t tests for grouped data. Significance in all tests was set at 95% or greater confidence level.
RESULTS
Intracellular Ca2+ Is Required for Osmotic Stress-induced Tight Junction Disruption
A recent study demonstrated that osmotic stress disrupts tight junctions and induces barrier dysfunction in Caco-2 cell monolayers and mouse ileum (6). In the present study, we evaluated the effect of [Ca2+]i on osmotic stress-induced tight junction disruption. Osmotic stress reduced TER (Fig. 1A) and increased inulin permeability (Fig. 1B) in a time-dependent manner. Pretreatment of cell monolayers with BAPTA-AM (a cell permeable Ca2+ chelator) significantly attenuated a osmotic stress-induced decrease in TER and increase in inulin permeability. BAPTA-AM pretreatment also attenuated osmotic stress-induced redistribution of occludin and ZO-1 from the intercellular junctions into the intracellular compartment (Fig. 1C). BAPTA treatment, by itself, produced no significant effect on TER, inulin permeability, or distribution of occludin and ZO-1.
FIGURE 1.

Intracellular calcium is required for osmotic stress-induced tight junction disruption. A and B, Caco-2 cell monolayers were pretreated with (□, ■) or without (○, ●) BAPTA-AM for 20 min and osmotic stress (●, ■) was induced as described under “Experimental Procedures.” TER (A) and unidirectional inulin flux (B) were measured at varying times. Values are mean ± S.E. (n = 6). Asterisks indicate the values that are significantly (p < 0.05) different from corresponding values for control group, and # indicates the values that are significantly (p < 0.05) different from corresponding values for osmotic stress without BAPTA. C, cell monolayers in different groups at 1 h after OS were fixed and stained for occludin and ZO-1 by the immunofluorescence method. Preloading cells with BAPTA did not cause a significant change in TER or inulin permeability.
Ca2+ Influx and ER Ca2+ Release Are Involved in Osmotic Stress-induced Tight Junction Disruption
The effect of osmotic stress on [Ca2+]i was evaluated in Fura-2-loaded Caco-2 cell monolayers. Osmotic stress rapidly increased [Ca2+]i reaching a maximum by 15 min (Fig. 2A). Pretreatment of cell monolayers with thapsigargin (an ER Ca2+-ATPase inhibitor that depletes ER Ca2+) partially reduced the osmotic stress-induced increase in [Ca2+]i. Similarly, removal of extracellular Ca2+ also partially reduced osmotic stress-induced [Ca2+]i. Osmotic stress-induced increase in [Ca2+]i was almost abolished when cells were simultaneously treated with thapsigargin and Ca2+-free medium (Fig. 2, A and B).
FIGURE 2.

Extracellular calcium influx and intracellular store-operated calcium release are necessary for osmotic stress-induced tight junction disruption. A and B, Fura-2-loaded Caco-2 cell monolayers were exposed to OS in the presence or absence of thapsigargin (TG) and/or CFM. Real time (A) change in the levels of intracellular calcium was measured for up to 45 min as described under “Experimental Procedures.” Steady state calcium levels are averaged (B); 8–10 cells in each of 3 monolayers per group were analyzed. Values are mean ± S.E. Asterisks indicate the values that are significantly (p < 0.05) different from corresponding baseline values, and # indicates the values that are significantly (p < 0.05) different from value for OS group. C and D, Caco-2 cell monolayers were pretreated with thapsigargin or incubated with calcium-free medium in the apical compartment (CFM-A) or basal compartment (CFM-B). Osmotic stress was induced by administration of hyperosmotic medium as described under “Experimental Procedures.” Control groups, with or without TG or CFM, received no osmotic stress. TER (C) and unidirectional inulin flux (D) were measured after 1 h. Values are mean ± S.E. (n = 6). Asterisks indicate the values that are significantly (p < 0.05) different from corresponding values for the control group, and # indicates the values that are significantly (p < 0.05) different from corresponding values for OS without TG or CFM treatment. E, cell monolayers exposed to OS with or without TG and CFM were fixed at 1 h after OS and stained for occludin and ZO-1 by immunofluorescence method.
Removal of extracellular Ca2+ or pretreatment of cells with thapsigargin also attenuated a osmotic stress-induced decrease in TER (Fig. 2C) and increase in inulin permeability (Fig. 2D). This effect on osmotic stress-induced changes in TER and inulin permeability was significantly greater when Ca2+ depletion and thapsigargin treatment were applied simultaneously. Interestingly, depletion of Ca2+ from the apical buffer, but not from the basal buffer, was effective in preventing the osmotic stress effect on barrier function. Ca2+ depletion in the basolateral buffer rather exacerbated osmotic stress-induced inulin permeability (Fig. 2D). Thapsigargin treatment or removal of Ca2+ from apical or basal buffers did not alter TER or inulin flux in the absence of osmotic stress. Osmotic stress-induced redistribution of occludin and ZO-1 was also attenuated by thapsigargin or Ca2+-free medium on the apical surface (Fig. 2E). Removal of Ca2+ from the basal buffer failed to prevent osmotic stress-induced redistribution of occludin and ZO-1.
CaV1.3 Contributes to Osmotic Stress-induced Ca2+ Influx and Tight Junction Disruption
The requirement of extracellular Ca2+ in the apical compartment indicated that Ca2+ influx through Ca2+ channels on the apical membrane is involved in osmotic stress-induced barrier dysfunction. Some of the well characterized Ca2+ channels on the apical membrane of intestinal epithelial cells include L-type voltage-gated channels, such as Cav1.3 (19). Therefore, we evaluated the effect of diltiazem and isradipine (L-type Ca2+ channel blockers) on the osmotic stress-induced increase in [Ca2+]i and disruption of barrier function. Both diltiazem and isradipine partially reduced osmotic stress-induced [Ca2+]i (Fig. 3A), and significantly attenuated the osmotic stress-induced increase in inulin permeability (Fig. 3B) and redistribution of occludin and ZO-1 from the intercellular junctions (Fig. 3C).
FIGURE 3.

CaV1.3 mediates osmotic stress-induced calcium influx and tight junction disruption. A, Fura-2-loaded Caco-2 cell monolayers were exposed to OS in the presence or absence of isradipine (Isra). Real time (A) change in the levels of [Ca2+]i was measured as described under “Experimental Procedures.” Steady state [Ca2+]i was evaluated. Asterisks indicate the values that are significantly (p < 0.05) different from baseline values, and # indicates the values that are significantly (p < 0.05) different from corresponding values for the OS group. B, Caco-2 cell monolayers were pretreated with or without diltiazem or isradipine for 40 min prior to incubation with or without OS. Inulin permeability was measured at 1 h after OS. Values are mean ± S.E. (n = 6). Asterisks indicate the values that are significantly (p < 0.05) different from the corresponding values for control group, and # indicates the values that are significantly (p < 0.05) different from corresponding values for the “None” OS group. C, cell monolayers in different groups at 1 h after osmotic stress were fixed and stained for occludin and ZO-1 by the immunofluorescence method. D, protein extracts from Caco-2 cells transfected with the empty vector or CaV1.3-shRNAs were immunoblotted for CaV1.3 and CaV1.2. Band densities were evaluated and the values are mean ± S.E. (n = 3). Asterisks indicate the values that are significantly (p < 0.05) different from the corresponding value for Vector group. E, inulin permeability was measured during osmotic stress in monolayers of cells transfected with empty vector or CaV1.3-shRNA. Asterisks indicate the values that are significantly (p < 0.05) different from corresponding values for vector-transfected cells. F, cell monolayers in different groups at 1 h after osmotic stress were fixed and stained for ZO-1 and GFP by the immunofluorescence method. G and H, intracellular calcium was measured during oxidative stress in vector or CaV1.3-shRNA-transfected cell monolayers. Steady state values were evaluated and the values are mean ± S.E. (n = 3). Asterisks indicate the values that are significantly (p < 0.05) different from corresponding baseline values, and # indicates the values that are significantly different (p < 0.05) from values for vector group.
Evidence indicates that Cav1.3 contributes to Ca2+ absorption from the apical surface (19). Therefore, we evaluated the effect of Cav1.3 gene silencing on osmotic stress-induced tight junction disruption. Cav1.3 shRNA reduced the Cav1.3 protein in Caco-2 cells compared with that in vector-transfected cells (Fig. 3D). In contrast, shRNA did not alter the Cav1.2 protein, indicating the specificity of shRNA. Knockdown of CaV1.3 did not alter the basal level of [Ca2+]i. Knockdown of CaV1.3 effectively blocked the osmotic stress-induced increase in inulin permeability (Fig. 3E) and attenuated redistribution of ZO-1 from intercellular junctions (Fig. 3F). CaV1.3 knockdown by shRNA markedly reduced the osmotic stress-induced increase in [Ca2+]i (Fig. 3, G and H).
IP3 Receptor-mediated ER Ca2+ Release Is Involved in Osmotic Stress-induced Tight Junction Disruption
A significant reduction of the osmotic stress-induced increase in [Ca2+]i by thapsigargin indicated that ER Ca2+ release is a partial contributor to elevated [Ca2+]i. One of the mechanisms associated with ER Ca2+ release involves IP3 receptor activation. Therefore, we evaluated the effect of xestospongin C (IP3 receptor antagonist) on the osmotic stress effect. Pretreatment of cell monolayers with xestospongin C significantly attenuated the osmotic stress-induced decrease in TER (Fig. 4A) and increase in inulin permeability (Fig. 4B). Xestospongin C also prevented osmotic stress-induced redistribution of occludin and ZO-1 from intercellular junctions (Fig. 4C). Xestospongin C, by itself, showed a slight but significant increase in inulin permeability in the absence of osmotic stress.
FIGURE 4.

IP3 receptor-mediated intracellular calcium release is involved in osmotic stress-induced tight junction disruption. Caco-2 cell monolayers were pretreated with or without xestospongin-C (Xe-C) for 30 min and OS was induced as described under “Experimental Procedures.” TER (A) and unidirectional inulin flux (B) were measured at 1 h after osmotic stress. Values are mean ± S.E. (n = 6). Asterisks indicate the values that are significantly (p < 0.05) different from the corresponding control values, and # indicates the values that are different from values for OS without Xe-C. C, cell monolayers in different groups at 1 h after osmotic stress were fixed and stained for occludin and ZO-1 by the immunofluorescence method.
Intracellular Ca2+ Mediates Osmotic Stress-induced JNK Activation
A recent study demonstrated that osmotic stress rapidly activates JNK and that osmotic stress-induced tight junction disruption is mediated by JNK2 activity (6). Therefore, we examined the potential role of [Ca2+]i in osmotic stress-induced JNK activation. Results show that the osmotic stress-induced increase in p-JNK2 was abrogated by pretreatment of cell monolayers with BAPTA-AM (Fig. 5A). The osmotic stress-induced increase in p-JNK2 was also significantly attenuated by pretreatment of cells with thapsigargin or isradipine or by the removal of extracellular Ca2+ from the apical buffer (Fig. 5, C and D). The osmotic stress-induced increase in the level of p-JNK was also attenuated by shRNA-mediated knockdown of CaV1.3 (Fig. 5, E and F).
FIGURE 5.

Intracellular calcium mediates osmotic stress-induced JNK2 activation. A and B, Caco-2 cell monolayers were pretreated with or without BAPTA-AM for 30 min prior to osmotic stress. At varying times protein extracts were immunoblotted for phospho-JNK (p-JNK). Band densities were evaluated and the values are mean ± S.E. (n = 3). Asterisks indicate the values that are significantly (p < 0.05) different from the corresponding value for the control group (None). C and D, osmotic stress was induced in cell monolayers pretreated with or without thapsigargin (TG) or isradipine (Isra) or incubated with CFM. Protein extracts at varying times were immunoblotted for p-JNK and total JNK. Band densities were evaluated and the values are mean ± S.E. (n = 3). Asterisks indicate the values that are significantly (p < 0.05) different from the corresponding value for the control group (0 min), and # indicates that values that are significantly different from the corresponding value for the OS group. E and F, total and p-JNK were analyzed by immunoblot analysis in protein extracts from vector or shRNA-transfected cell monolayers in the presence or absence of osmotic stress. Asterisks indicate the values that are significantly (p < 0.05) different from the corresponding value for the control group, and # indicates the values that are significantly different from corresponding value for the vector group.
JNK-mediated c-Src Activation Is Involved in Osmotic Stress-induced Tight Junction Disruption
Previous studies indicated that tyrosine kinases, especially c-Src, play an important role in tight junction disruption by hydrogen peroxide (22) or acetaldehyde (23). In the present study, we evaluated the effect of genistein (a broad range tyrosine kinase inhibitor) and PP2 (Src kinase selective inhibitor) on osmotic stress-induced barrier dysfunction. Both genistein (Fig. 6A) and PP2 (Fig. 6B) significantly attenuated the osmotic stress-induced increase in inulin permeability. Genistein (Fig. 6C) and PP2 (Fig. 6D) also prevented osmotic stress-induced redistribution of occludin and ZO-1 from the intercellular junctions. Genistein or PP2 by themselves produced no significant effect on inulin permeability or distribution of tight junction proteins.
FIGURE 6.

Osmotic stress disrupts tight junctions by a tyrosine kinase-dependent mechanism. A and B, Caco-2 cell monolayers were pretreated with or without genistein (A) or PP2 (B) for 30 min and osmotic stress was induced as described under “Experimental Procedures.” Inulin flux was measured at 1 h after osmotic stress. Values are mean ± S.E. (n = 6). Asterisks indicate the values that are significantly (p < 0.05) different from the corresponding control values, and # indicates the values that are different (p < 0.05) from the corresponding values for OS without the inhibitor group. C and D, cell monolayers pretreated with genistein (C) or PP2 (D) were fixed at 1 h after OS and stained for occludin and ZO-1 by the immunofluorescence method.
c-Src is well established to play a role in tyrosine phosphorylation of tight junction proteins and disruption of tight junctions (22). In the present study, we evaluated the role of c-Src activation in osmotic stress-induced tight junction disruption. Osmotic stress induced a transient increase in the level of c-Src(pY418), the activated c-Src (Fig. 7A). Osmotic stress-induced elevation of c-Src(pY418) was attenuated by SP600125 (JNK inhibitor) and PP2 (Src kinase inhibitor). Osmotic stress-induced elevation of c-Src(pY418) was also attenuated by removal of extracellular Ca2+ or pretreatment of cells with BAPTA-AM or isradipine (Fig. 7B). Transfection of Caco-2 cells with c-Src-specific siRNA reduced the level of c-Src (Fig. 7C) and attenuated the osmotic stress-induced increase in inulin permeability (Fig. 7D). Knockdown of c-SRC also prevented osmotic stress-induced redistribution of occludin and ZO-1 from the intercellular junctions (Fig. 7E). Transfection of Caco-2 cells with c-Yes-specific siRNA reduced the level of c-Yes without altering the c-Src level (Fig. 7F), but failed to influence the osmotic stress-induced increase in inulin permeability (Fig. 7G).
FIGURE 7.

Osmotic stress activates c-Src by an intracellular calcium and JNK-dependent mechanism. A, Caco-2 cell monolayers were pretreated with or without SP600125 or PP2 for 30 min prior to osmotic stress. At varying times protein extracts were immunoblotted for c-Src(pY418) and total c-Src. B, OS was induced in cell monolayers pretreated with or without BAPTA-AM or isradipine (Isra) or incubated with CFM. Protein extracts at 30 min after osmotic stress were immunoblotted for Src(pY418) and total c-Src. Asterisks indicate the band density values that are significantly (p < 0.05) different from the corresponding value for the control group, and # indicates that values that are significantly different from the corresponding value for “None” in the OS group. C, proteins extracted from Caco-2 cells transfected with nonspecific RNA (NS-RNA) or c-Src siRNA were immunoblotted for c-Src and β-actin. Asterisks indicate the band density value that is significantly (p < 0.05) different from the corresponding value for the NS-RNA group (gray bar). D, Caco-2 cell monolayers transfected with NS-RNA or c-Src siRNA were exposed to osmotic stress for 1 h. Inulin permeability was measured. Values are mean ± S.E. (n = 6). Asterisks indicate the values that are significantly (p < 0.05) different from the corresponding control values. # indicates values that are significantly different from the corresponding value for the NS-RNA group. E, untreated and osmotic stress-treated cell monolayers were fixed and stained for occludin and ZO-1. F, proteins extracted from Caco-2 cells transfected with nonspecific RNA or c-Yes siRNA were immunoblotted for c-Src and β-actin. G, Caco-2 cell monolayers transfected with NS-RNA or c-Yes siRNA were exposed to osmotic stress for 1 h. Inulin permeability was measured. Values are mean ± S.E. (n = 6).
Osmotic Stress Induces Tyrosine Phosphorylation of Occludin and ZO-1 by Ca2+, JNK, and Src Kinase-dependent Mechanism
Previous studies showed that tyrosine phosphorylation of tight junction proteins is a key mechanism involved in oxidative stress-induced tight junction disruption and barrier dysfunction (10, 11). Therefore, we examined the effect of osmotic stress on tyrosine phosphorylation of occludin and ZO-1. Osmotic stress induced a robust increase in the level of tyrosine-phosphorylated occludin (Fig. 8, A and C). Osmotic stress-induced occludin phosphorylation was abolished by pretreatment of cells with SP600125. A similar effect of osmotic stress on tyrosine phosphorylation of ZO-1 was observed, however, ZO-1 phosphorylation was weak. Our recent study indicated that JNK2 activity is high in underdifferentiated 4-day-old cell monolayers and knockdown of JNK by specific antisense oligonucleotides (AS-Jnk1 and AS-Jnk2) significantly enhanced tight junction integrity in Caco-2 cell monolayers (6). The present study shows that high levels of tyrosine-phosphorylated occludin and ZO-1 were detected in 4-day-old missense oligo-transfected cells (Fig. 8, B and D). The levels of tyrosine-phosphorylated occludin and ZO-1 were significantly low in cells transfected with AS-Jnk2 and AS-Jnk1. AS-Jnk2 was more effective than AS-Jnk1 in reducing tyrosine phosphorylation of occludin and ZO-1. Furthermore, osmotic stress-induced tyrosine phosphorylation of occludin was attenuated also by pretreatment of cells with BAPTA-AM (Fig. 8E) or PP2 (Fig. 8F).
FIGURE 8.

Osmotic stress induces tyrosine phosphorylation of occludin and ZO-1 by a calcium-, JNK-, and c-Src-dependent mechanism. A, Caco-2 cell monolayers (14 days old, differentiated) were incubated with or without OS for 1 h in the presence or absence of SP600125. Phosphotyrosine was immunoprecipitated from proteins extracted under denaturing conditions and the immunocomplexes were immunoblotted for occludin and ZO-1. B, phosphotyrosine immunoprecipitated from cells transfected with different oligos (on day 4 post transfection) were immunoblotted for occludin and ZO-1. C and D, densitometric analysis of bands in blots related to experiments described in panels A (C) and B (D), respectively. Values are mean ± S.E. (n = 3). Asterisks indicate the values that are significantly (p < 0.05) different from the corresponding control values or values for MS oligo-transfected cells. # indicates the values that are significantly (p < 0.05) different from the corresponding values for the OS group without SP600125. E and F, phosphotyrosine was immunoprecipitated from proteins extracted from cell monolayers with osmotic stress in the presence or absence of BAPTA (E) or PP2 (F) and immunoblotted for occludin.
DISCUSSION
A significant body of evidence indicates that stress is associated with the pathogenesis of many diseases. One of the major targets of stress-induced tissue injury is the gastrointestinal tract. Our recent study indicated that osmotic stress disrupts the intestinal epithelial tight junctions and induces barrier dysfunction by a JNK2-dependent mechanism. The present study shows that an initial signal triggered by osmotic stress is a rise in [Ca2+]i. The study provides evidence that osmotic stress-induced elevation of [Ca2+]i involves both CaV1.3-mediated Ca2+ influx and IP3 receptor-gated release of store-operated Ca2+. The evidence also demonstrates that [Ca2+]i mediates stress-induced JNK2 activation and that JNK-mediated c-Src activation and tyrosine phosphorylation of tight junction proteins are the downstream mechanisms to JNK activation leading to tight junction disruption and barrier dysfunction.
A rapid increase in [Ca2+]i, abrogation of osmotic stress-induced tight junction disruption, and barrier dysfunction by BAPTA demonstrate that intracellular [Ca2+]i plays a crucial role in the osmotic stress-induced tight junction disruption. Although the role of extracellular calcium in formation of adherens junction and maintenance of tight junction integrity is a well established fact, information on the role of [Ca2+]i in tight junction regulation is sparse. A few studies have indicated that [Ca2+]i is required for the assembly of tight junctions (14) and for EGF-mediated preservation of tight junctions in acetaldehyde-treated cell monolayers (24). On the other hand, activation of basolateral TRPV4 channel leads to disruption of barrier function in a mammary epithelial cell line (25), and ER calcium release is associated with barrier disruption by ethanol in brain endothelial cells (26) and bacterial infection in Caco-2 cells (27).
A partial reduction of osmotic stress-induced increase in [Ca2+]i by removal of extracellular calcium indicates that calcium influx from the extracellular source is involved in this response. Removal of extracellular calcium also attenuates osmotic stress-induced tight junction disruption and barrier dysfunction. Interestingly, removal of extracellular calcium from only the apical buffer was sufficient to attenuate osmotic stress-induced tight junction disruption. This observation indicates that calcium influx via an apical calcium channel is involved in osmotic stress-induced tight junction disruption. Removal of calcium from the basal medium exacerbated the effect of osmotic stress on tight junction integrity, which is likely mediated by destabilization of adherens junction that is located just basal to tight junctions.
The observation that Ca2+ influx from the apical medium is involved in osmotic stress-induced tight junction disruption raised the question about what specific Ca2+ channel is involved in osmotic stress-induced Ca2+ influx and tight junction disruption. Attenuation of a osmotic stress-induced increase in Ca2+ and tight junction disruption by diltiazem and isradipine indicated that the L-type voltage-gated Ca2+ channel is involved in osmotic stress-induced tight junction disruption. Several types of voltage-gated Ca2+ channels have been described in the apical membrane of intestinal epithelial cells (19). CaV1.3, is a major voltage-gated Ca2+ channel in the intestinal epithelium. CaV1.3 is involved not only in Ca2+ absorption (19), but also in Ca2+ signaling leading to myosin light chain kinase activation (28). Our present study demonstrates that knockdown of CaV1.3 significantly reduces the osmotic stress-induced increase in Ca2+ and abrogates osmotic stress-induced tight junction disruption and barrier dysfunction. These results demonstrate that CaV1.3 plays a crucial role in osmotic stress-induced Ca2+ influx and tight junction disruption in the intestinal epithelium.
A partial reduction of osmotic stress-induced Ca2+ increase by thapsigargin indicates that ER Ca2+ release also contributes to a osmotic stress-induced increase in [Ca2+]i. A significant attenuation of osmotic stress-induced tight junction disruption and barrier dysfunction by thapsigargin demonstrates that ER Ca2+ release is required for osmotic stress-induced tight junction disruption. IP3 receptor activation is a major mechanism involved in ER Ca2+ release (29). A significant attenuation of osmotic stress-induced tight junction disruption by xestospongin-C, a selective antagonist of IP3 receptor, suggests that IP3 receptors are involved in a osmotic stress-induced rise in [Ca2+]i and disruption of tight junctions. Hence, both extracellular Ca2+ influx and ER Ca2+ release are involved in osmotic stress-induced increase in [Ca2+]i and tight junction disruption (Fig. 9). These findings raise a question of whether the two sources of Ca2+ contribute to the same pool of [Ca2+]i in osmotic stress-treated cells or form two different [Ca2+]i pools leading to activation of different downstream signaling pathways. The answer to this question is not clear at this time. It is possible that a high level of Ca2+ achieved by multiple sources of Ca2+ is required to reach a threshold that trigger downstream signals leading to JNK activation and tight junction disruption.
FIGURE 9.

Schematic outline of mechanisms associated with osmotic stress-induced tight junction disruption. Results of this study are summarized. The initial signal appears to be an increase in intracellular calcium caused by both influx through the apical calcium channel, CaV1.3, and intracellular release of ER calcium by IP3R-mediated mechanism. Rise in intracellular calcium mediates activation of JNK, which in turn activates c-Src leading to tyrosine phosphorylation of tight junction proteins and disruption of tight junctions.
JNK, originally known as stress-activated protein kinase is activated in cells by various types of stress (30). Our recent study demonstrated that osmotic stress-induced tight junction disruption and barrier dysfunction in the intestinal epithelium is mediated by JNK2 (6). Knockdown of JNK enhances tight junction integrity and attenuates osmotic stress-induced tight junction disruption in Caco-2 cell monolayers (6). Osmotic stress-induced tight junction disruption was absent in ileum of JNK2 knock-out mice. Additionally, cold-restraint stress-induced tight junction disruption in rat ileum was attenuated by the administration of a JNK-selective inhibitor, SP600125. The observation that [Ca2+]i plays a role in osmotic stress-induced tight junction disruption raised the question whether JNK activation and rise in [Ca2+]i are interdependent. The time course studies indicated that the increase in [Ca2+]i reached a peak within 5 min after administration of the hyperosmotic mannitol. On the other hand, JNK activation required about 15 min with the peak level achieved at 30 min. Therefore, we reasoned that [Ca2+]i mediates osmotic stress-induced JNK activation. Osmotic stress-induced JNK activation was indeed attenuated by extracellular Ca2+ depletion or treatment of cells with thapsigargin, diltiazem, or isradipine, indicating that both Ca2+ influx and ER Ca2+ are required for osmotic stress-induced JNK activation. This also provides supporting evidence to the speculation that the two sources of Ca2+ are required to maintain [Ca2+]i at a concentration high enough to induce JNK activation. The mechanism associated with Ca2+-induced JNK activation is unknown. It is likely that [Ca2+]i activates MKK7, which in turn activates JNK2.
A significant body of evidence indicates that c-Src activity causes tight junction disruption in different epithelia including Caco-2 cell monolayers (22). c-Src regulates phosphorylation of occludin and other tight junction and adherens junction proteins leading to dissociation of protein-protein interactions and disruption of tight junctions (22). In the present study, we examined the potential role of the Src kinase in osmotic stress-induced tight junction disruption. Attenuation of osmotic stress-induced tight junction disruption and barrier dysfunction by genistein and PP2 displayed a clear indication of the role of Src kinase in osmotic stress-induced tight junction disruption. The present study shows that osmotic stress causes a rapid and transient increase in the level of c-Src(pY418) indicating the activation of c-Src by osmotic stress. Attenuation of osmotic stress-induced c-Src activation by SP600125 indicated that Src activation is downstream to JNK activation. Activation of c-Src by osmotic stress was also attenuated by BAPTA, extracellular Ca2+ depletion, or isradipine, demonstrating that Ca2+-mediated JNK activation is responsible for c-Src activation. The role of c-Src in osmotic stress-induced tight junction disruption was confirmed by knockdown of c-SRC using specific siRNA. Osmotic stress-induced increase in inulin permeability and redistribution of occludin and ZO-1 were effectively attenuated by c-SRC knockdown. Interestingly, specific knockdown of c-YES did not influence osmotic stress-induced paracellular permeability.
Previous studies demonstrated that c-Src activation leads to phosphorylation of many tight junction and adherens junction proteins. Phosphorylation of occludin on specific tyrosine residues results in loss of its interaction with ZO-1 and enhanced susceptibility to hydrogen peroxide-induced tight junction disruption in Caco-2 and Madin-Darby canine kidney cell monolayers (10, 11). Similarly, Src-mediated tyrosine phosphorylation of β-catenin on specific tyrosine residues attenuates its interaction with E-cadherin (31). The present study examined the effect of osmotic stress on tyrosine phosphorylation of occludin and ZO-1. Tyrosine phosphorylation of ZO-1 was weak, whereas osmotic stress induced a robust increase in tyrosine phosphorylation of occludin. Osmotic stress-induced tyrosine phosphorylation of occludin was abrogated by SP600125, indicating the role of JNK activity in osmotic stress-induced occludin tyrosine phosphorylation. The role of JNK in occludin phosphorylation was further supported by a reduced level of basal occludin phosphorylation in cells transfected with JNK-specific antisense oligonucleotides. Osmotic stress-induced occludin tyrosine phosphorylation was also attenuated by pretreatment of cell monolayers with BAPTA or PP2. These studies indicate that tyrosine phosphorylation of occludin is mediated by Ca2+-induced JNK activation and subsequent Src activation. It is likely that tyrosine phosphorylation of occludin results in loss of interaction between occludin and ZO-1. The mechanism of JNK-mediated c-Src activation is unclear at this time. But, evidence indicates that c-Src activity can be regulated by Ser/Thr phosphorylation (32), allowing us to speculate that JNK may directly phosphorylate c-Src.
Our previous study demonstrated that occludin is phosphorylated by c-Src (10). Both mass spectrometric analysis and point mutation studies indicated that only Tyr-398 and Tyr-402 are phosphorylated by c-Src. In the present study, the data show that osmotic stress-induced occludin Tyr phosphorylation is completely attenuated by Src kinase inhibitor, suggesting that Tyr-398 and Tyr-402 are phosphorylated during OS-induced TJ disruption.
A question that arises in this study is whether CaV1.3-mediated Ca2+ influx regulates the stored calcium level, and therefore ER release of Ca2+. An answer to this question is unclear at this point. Our data show that removal of extracellular calcium or depletion of stored calcium by thapsigargin only partially prevents a OS-induced rise in [Ca2+]i. However, the voltage-gated Ca2+ channel blocker, CaV1.3 knockdown, and xestospongin-C all effectively attenuate OS-induced TJ disruption. Based on these data we speculate that a threshold level of [Ca2+]i is required to be achieved to induce TJ disruption. Therefore, reduction of the OS-induced rise in [Ca2+]i to half either by calcium-free medium or thapsigargin attenuates OS-induced TJ disruption.
In summary, this study demonstrates that osmotic stress induces a rapid increase in [Ca2+]i via CaV1.3-mediated extracellular Ca2+ influx and IP3 receptor-mediated ER Ca2+ release. This increase in [Ca2+]i caused a rapid activation of JNK2 and disruption of tight junction. Furthermore, c-Src activation and tyrosine phosphorylation of occludin mediates the JNK2 effect on tight junction integrity.
This work was supported, in whole or in part, by National Institutes of Health Grants R01-DK55532 (to R. K. R.), R01-AA12307 (to R. K. R.), HL067061 (to J. H. J.), and HL094378 (to J. H. J.).
- IP3
- inositol 1,4,5-trisphophate
- JNK
- N-terminal c-Jun kinase
- AS-Jnk
- antisense oligonucleotide for JNK
- SP600125
- anthra(1,9-cd)pyrazol-6(2H)-one-1,9-pyrazoloanthron
- TER
- transepithelial electrical resistance
- ZO
- zona occludens
- BAPTA-AM
- 1,2-bis-(o-aminophenoxy)-ethane-N,N,N′,N′-tetraacetic acid-acetoxymethyl ester
- OS
- osmotic stress
- PP2
- 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine
- CFM
- Ca2+-free medium
- TRPV4
- transient receptor potential vanilloid family Ca2+ channel 4
- CaV1.2
- CaV1.3, voltage-dependent calcium channels 1.2 and 1.3
- ER
- endoplasmic reticulum.
REFERENCES
- 1. Mawdsley J. E., Rampton D. S. (2005) Gut 54, 1481–1491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Madara J. L. (1983) J. Cell Biol. 97, 125–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Cheromcha D. P., Hyman P. E. (1988) Dig. Dis. Sci. 33, 78S–84S [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schilli R., Breuer R. I., Klein F., Dunn K., Gnaedinger A., Bernstein J., Paige M., Kaufman M. (1982) Gut 23, 326–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Vernia P., Gnaedinger A., Hauck W., Breuer R. I. (1988) Dig. Dis. Sci. 33, 1353–1358 [DOI] [PubMed] [Google Scholar]
- 6. Samak G., Suzuki T., Bhargava A., Rao R. K. (2010) Am. J. Physiol. Gastrointest. Liver Physiol. 299, G572–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Van Itallie C. M., Anderson J. M. (2004) Physiology 19, 331–338 [DOI] [PubMed] [Google Scholar]
- 8. Rao R. (2008) Front. Biosci. 13, 7210–7226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Balda M. S., Matter K. (2008) J. Cell Sci. 121, 3677–3682 [DOI] [PubMed] [Google Scholar]
- 10. Elias B. C., Suzuki T., Seth A., Giorgianni F., Kale G., Shen L., Turner J. R., Naren A., Desiderio D. M., Rao R. (2009) J. Biol. Chem. 284, 1559–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kale G., Naren A. P., Sheth P., Rao R. K. (2003) Biochem. Biophys. Res. Commun. 302, 324–329 [DOI] [PubMed] [Google Scholar]
- 12. Seth A., Sheth P., Elias B. C., Rao R. (2007) J. Biol. Chem. 282, 11487–11498 [DOI] [PubMed] [Google Scholar]
- 13. Balda M. S., González-Mariscal L., Contreras R. G., Macias-Silva M., Torres-Marquez M. E., García-Sáinz J. A., Cereijido M. (1991) J. Membr. Biol. 122, 193–202 [DOI] [PubMed] [Google Scholar]
- 14. Brown R. C., Davis T. P. (2002) Stroke 33, 1706–1711 [DOI] [PubMed] [Google Scholar]
- 15. Ma T. Y., Tran D., Hoa N., Nguyen D., Merryfield M., Tarnawski A. (2000) Microsc. Res. Tech. 51, 156–168 [DOI] [PubMed] [Google Scholar]
- 16. Mandel L. J., Bacallao R., Zampighi G. (1993) Nature 361, 552–555 [DOI] [PubMed] [Google Scholar]
- 17. Ye J., Tsukamoto T., Sun A., Nigam S. K. (1999) Am. J. Physiol. Renal Physiol. 277, F524–F532 [DOI] [PubMed] [Google Scholar]
- 18. Stuart R. O., Sun A., Bush K. T., Nigam S. K. (1996) J. Biol. Chem. 271, 13636–13641 [DOI] [PubMed] [Google Scholar]
- 19. Nakkrasae L. I., Thongon N., Thongbunchoo J., Krishnamra N., Charoenphandhu N. (2010) J. Physiol. Sci. 60, 9–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Suzuki T., Elias B. C., Seth A., Shen L., Turner J. R., Giorgianni F., Desiderio D., Guntaka R., Rao R. (2009) Proc. Natl. Acad. Sci. U.S.A. 106, 61–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rao R. K., Basuroy S., Rao V. U., Karnaky K. J., Jr., Gupta A. (2002) Biochem. J. 368, 471–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Basuroy S., Sheth P., Kuppuswamy D., Balasubramanian S., Ray R. M., Rao R. K. (2003) J. Biol. Chem. 278, 11916–11924 [DOI] [PubMed] [Google Scholar]
- 23. Atkinson K. J., Rao R. K. (2001) Am. J. Physiol. Gastrointest. Liver Physiol. 280, G1280–G1288 [DOI] [PubMed] [Google Scholar]
- 24. Suzuki T., Seth A., Rao R. (2008) J. Biol. Chem. 283, 3574–3583 [DOI] [PubMed] [Google Scholar]
- 25. Reiter B., Kraft R., Günzel D., Zeissig S., Schulzke J. D., Fromm M., Harteneck C. (2006) FASEB J. 20, 1802–1812 [DOI] [PubMed] [Google Scholar]
- 26. Haorah J., Knipe B., Gorantla S., Zheng J., Persidsky Y. (2007) J. Neurochem. 100, 324–336 [DOI] [PubMed] [Google Scholar]
- 27. Vikström E., Bui L., Konradsson P., Magnusson K. E. (2010) Eur. J. Cell Biol. 89, 584–597 [DOI] [PubMed] [Google Scholar]
- 28. Mace O. J., Morgan E. L., Affleck J. A., Lister N., Kellett G. L. (2007) J. Physiol. 580, 605–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Berridge M. J., Bootman M. D., Roderick H. L. (2003) Nat. Rev. Mol. Cell Biol. 4, 517–529 [DOI] [PubMed] [Google Scholar]
- 30. Lin A. (2003) Bioessays 25, 17–24 [DOI] [PubMed] [Google Scholar]
- 31. Sheth P., Seth A., Atkinson K. J., Gheyi T., Kale G., Giorgianni F., Desiderio D. M., Li C., Naren A., Rao R. (2007) Biochem. J. 402, 291–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chackalaparampil I., Shalloway D. (1988) Cell 52, 801–810 [DOI] [PubMed] [Google Scholar]