

Abstract
T cell receptor (TCR) down-modulation after antigen presentation is a fundamental process that regulates TCR signal transduction. Current understanding of this process is that intrinsic TCR/CD28 signal transduction leads to TCR down-modulation. Here, we show that the interaction between programmed cell death 1 ligand 1 (PD-L1) on dendritic cells (DCs) and programmed death 1 (PD-1) on CD8 T cells contributes to ligand-induced TCR down-modulation. We provide evidence that this occurs via Casitas B-lymphoma (Cbl)-b E3 ubiquitin ligase up-regulation in CD8 T cells. Interference with PD-L1/PD-1 signalling markedly inhibits TCR down-modulation leading to hyper-activated, proliferative CD8 T cells as assessed in vitro and in vivo in an arthritis model. PD-L1 silencing accelerates anti-tumour immune responses and strongly potentiates DC anti-tumour capacities, when combined with mitogen-activated kinase (MAPK) modulators that promote DC activation.
Keywords: Cbl, dendritic cells, PD-1, PD-L1, TCR
INTRODUCTION
The immune system must protect the organism against infectious diseases and cancer without provoking autoimmunity. T lymphocytes play a key role in the induction of protective and long-lasting immunity, but, if uncontrolled, can cause autoreactive disease. For this reason, T cell activation is regulated at multiple levels, particularly during antigen presentation. Understanding these mechanisms is essential for designing effective therapies for the treatment of cancer, infectious diseases and autoimmune disorders. T cells recognize specific peptides in association with major histocompatibility complex (MHC) molecules that are expressed on the surface of antigen presenting cells (APCs) through binding of their respective T cell receptor (TCRs). However, MHC-peptide recognition is not sufficient for full T cell activation, and a range of co-stimulatory ligand–receptor interactions is also required that can provide either positive or negative signals. For instance the CD80–CD28 interaction is stimulatory, while others such as programmed cell death 1 ligand 1 (PD-L1)–programmed death 1 (PD-1) association are inhibitory. The relative contribution of co-stimulatory/co-inhibitory signals determines the activation state of T cells, leading to T cell proliferation and acquisition of effector activities or differentiation into anergic or regulatory T cells. Thus, the overall integration of positive and negative signals during co-stimulation provides a checkpoint at which T cell responses are modulated (Nurieva et al, 2006).
PD-L1 is a member of the B7 family of co-stimulatory/inhibitory molecules, which is expressed in a wide range of cell types, including T cells and dendritic cells (DCs; Latchman et al, 2004; Sharpe et al, 2007). PD-1 is transiently up-regulated in activated T cells during antigen presentation, and its ligation to PD-L1 recruits src homology 2 domain-containing tyrosine phosphatases 1 and 2 (SHP 1 and 2) to its intracellular switch motif. SHPs dephosphorylate effector molecules associated with the TCR leading to termination of TCR signal transduction (Chemnitz et al, 2004; Sheppard et al, 2004).
Ligand-induced TCR down-modulation is another regulatory process of T cell activation at the level of antigen presentation. TCRs are removed from the T cell surface shortly after activation, limiting signal transduction and avoiding excessive responses (Holst et al, 2008; Naramura et al, 2002; San Jose et al, 2000; Schonrich et al, 1991; Shamim et al, 2007). The current view of ligand-induced TCR down-modulation is that intrinsic TCR signalling following antigen recognition is sufficient for TCR down-modulation. However, the exact mechanism by which this occurs is still under extensive research. Ligand-induced TCR down-modulation is a complex, multi-mechanistic process (Lauritsen et al, 1998). It is well established that ligand-engaged TCR complexes are quickly internalized (Cai et al, 1997; Dietrich et al, 1998; Huppa et al, 2010; Lauritsen et al, 1998; Valitutti et al, 1995). Afterwards, other TCR complexes including non-engaged ones, are down-regulated following signal transduction from the triggered TCRs (San Jose et al, 2000). Nevertheless, up-regulation of E3 ubiquitin ligases of the Casitas B-lymphoma (Cbl) family in T cells contributes to ligand-induced TCR down-modulation as demonstrated by studies with Cbl knock-out (KO) mice (Naramura et al, 2002; Shamim et al, 2007). Cbl KO T cells have reduced TCR down-modulation following antigen presentation, leading to sustained signalling and hyper-activation (Chiang et al, 2000; Naramura et al, 2002; Shamim et al, 2007). Interestingly, to date, no additional extrinsic signals provided by DCs in the immunological synapse, which are critical for ligand-induced TCR down-modulation, have been identified.
Counteracting negative signals transmitted to effector T cells is a promising approach to achieve therapeutic efficacy for cancer and infectious diseases. Thus, the disruption of PD-L1/PD-1 signalling pathway, mainly by systemic administration of blocking antibodies, is receiving increasing interest in immunotherapy (Curran et al, 2010; Hirano et al, 2005; Hobo et al, 2010; Zhou et al, 2010). In addition, abrogation of Cbl ubiquitin ligases in effector T cells significantly enhances their anti-tumour/anti-viral activities (Bachmaier et al, 2000; Chiang et al, 2000; Naramura et al, 2002; Paolino et al, 2011), making them attractive therapeutic targets.
RESULTS
PD-L1 silencing in DCs inhibits CD8 TCR down-modulation
In this study, we investigated the consequences and therapeutic outcomes of silencing the co-stimulatory ligand PD-L1 in DCs during antigen presentation to T cells. Thus, we delivered a PD-L1-specific short hairpin (sh)RNA (termed p5) using a reported lentiviral platform (Arce et al, 2011). Transduction of bone marrow derived DCs (BM-DCs) resulted in the specific silencing of PD-L1 surface expression with no down-regulation of other classical DC maturation markers (Fig 1 of Supporting Information).
To test the effects of PD-L1 silencing on MHC class I and II antigen presentation, we co-delivered the shRNA p5 with IiOVA, a well-defined model antigen containing both MHC class I and class II epitopes (Fig 1A; Arce et al, 2011; Escors et al, 2008). Thus, BM-DCs were transduced with control lentivectors expressing GFP alone, IiOVA-GFP or IiOVA-p5-GFP. Inclusion of the shRNA did not alter gene expression from the lentivector (Fig 1A). BM-DCs transduced with IiOVA-GFP or IiOVA-p5-GFP activated proliferation of OVA-specific CD4 and CD8 T cells purified from transgenic OT-II and OT-I mouse strains, respectively (results not shown). However, while approximately 40% of CD8 T cells clustered around IiOVA-expressing DCs after 16 h of culture as assessed by microscopy, around 90% of CD8 T cells clustered around BM-DCs expressing IiOVA-p5 (Fig 1B). By comparison, no differences were observed for DC-CD4 OT-II cell association, which was around 10% (results not shown). Therefore, in agreement with other studies (Fife et al, 2009), these results suggested that the DC-CD8 T cell association was enhanced when PD-L1 was silenced.
Figure 1. PD-L1 silencing in antigen presenting DCs inhibits TCR down-modulation in CD8+ T cells.
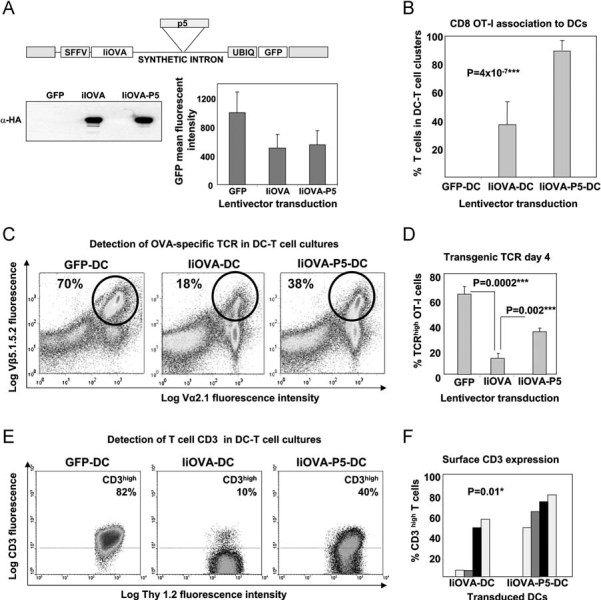
- Lentivector co-delivery platform of IiOVA with shRNA-p5 (top). IiOVA expression (immunoblot, lower left) and GFP expression (graph, right).
- Association between lentivector transduced DCs and antigen specific OT-I cells as indicated. Associated DC-T cells were quantified by microscopy.
- Surface expression of transgenic OVA TCR chains in OT-I cells co-cultured for 4 days with DCs transduced with the indicated lentivectors (top). Transgenic OT-I cells are Vβ5.1.5.2high and Vα2.1high (circles), the their percentages shown within the gate.
- As in (C), but plotting transgenic OVA-specific TCR surface expression in OT-I CD8+ T cells co-cultured with the indicated transduced DCs (bottom), as a bar graph with error bars (standard deviations). ***, highly significant differences.
- As in (C), with CD3 surface expression in Thy1.2-gated T cells. Percentages refer to CD3high T cells within the upper gate, delimited with a horizontal dotted line. Thy1.2 remains unchanged.
- Bar graph as in (E) plotting results from four independent experiments. Each of the experiments is represented as a differently shaded pair of bars. *, significant differences.
To investigate why increased DC-T cell clustering was observed upon down-regulation of PD-L1, we examined several potential factors. Interestingly, we found that TCR down-modulation was significantly inhibited when antigen was presented in the absence of PD-L1 (Fig 1C and D). Equivalent results were obtained by surface CD3 staining, the TCR component mediating signal transduction (Fig 1E and F), and by staining with OVA-specific class I pentamer (results not shown). Surface levels of the T cell lineage marker Thy1.2 remained unchanged, confirming the specificity of the TCR down-modulation (Fig 1E). It is worth noting that TCR down-modulation reached a maximum on day 3–4, and TCR levels gradually recovered afterwards (Fig 2 of Supporting Information). PD-L1 silencing in DCs significantly delayed TCR down-modulation but did not completely abrogate it (Fig 2 of Supporting Information). Persistent TCR signal transduction was demonstrated by intracellular detection of phosphorylated extracellular signal-regulated kinase (ERK) in TCRhigh CD8+ T cells (results not shown).
To confirm that a reduction in DC PD-L1 co-stimulation also reduced TCR down-modulation on activated T cells in vivo, we immunized Thy1.1 mice with BM-DCs transduced with the different lentivectors. Then, congenic Thy1.2 OT-I CD8+ T cells were adoptively transferred the following day and analysed 7 days later. OVA-specific OT-I cells primed in vivo by DCs with silenced PD-L1 down-regulated TCR expression to a lesser extent than T cells primed in the presence of PD-L1 (Fig 2). Collectively, our results demonstrate that DC-derived PD-L1 signalling contributes to ligand-induced TCR down-modulation after antigen presentation.
Figure 2. Vaccination with a lentivector co-delivering antigen and a PD-L1-specific shRNA expands antigen-specific TCRhigh CD8+ T cells.
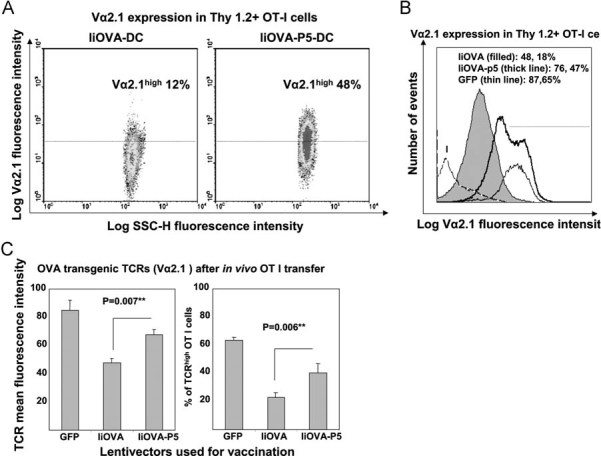
- Flow cytometry of OVA-specific transgenic TCR (Vα2.1 chain) surface expression in transferred Thy1.2+ CD8+ T cells in mice vaccinated with DCs transduced with the indicated lentivectors on top of the graph. Percentages of high Vα2.1 OT-I cells are shown within the graphs.
- As in (A), but in the form of a histogram comparing vaccinations with transduced DCs, including the GFP-DC vaccination control. Very low numbers of Thy1.2+ cells were found in GFP-DC vaccinated mice due to lack of proliferation. Lentivectors used for DC transductions are shown within the legend. Numbers refer to Vα2.1 mean fluorescent intensities, and the percentage to Vα2.1-positive OT-I cells, as defined by those cells within the 95% isotype gate (horizontal dotted line). The horizontal dotted line delimits the gate in which 95% of isotype-stained CD8+ T cells are excluded. I, Isotype staining control.
- OVA-specific TCR surface expression as in (B) in OT-I adoptively transferred into mice vaccinated with DCs transduced with the indicated lentivectors (bottom). **, indicate very significant differences.
PD-L1 silencing in DCs blocks expression of Cbl E3 ubiquitin ligases in CD8+ T cells
Recent evidence demonstrates that expression of Cbl E3 ubiquitin ligases significantly contributes to ligand-induced TCR down-modulation and limits TCR signal transduction (Bachmaier et al, 2000; Chiang et al, 2000; Naramura et al, 2002). Cbl KO T cells exhibit a reduced TCR down-modulation after antigen presentation, exactly to the same extent as observed in our experiments (Fig 1; Shamim et al, 2007). Therefore, we tested whether PD-L1 silencing in antigen-presenting DCs inhibited up-regulation of the two major Cbl proteins, c-Cbl and Cbl-b, in CD8+ T cells (Fig 3A and B). Our data show that, while antigen presentation by IiOVA-DCs strongly up-regulated both Cbl proteins, PD-L1 silencing in IiOVA-p5-DCs did not result in significant expression of Cbl-b or, to a lesser extent, c-Cbl (Fig 3B). We found that lack of Cbl-b expression rather than c-Cbl was consistent with the inhibition of CD3 internalization (Fig 3C and D).
Figure 3. Interference with PD-L1/PD-1 signalling inhibits TCR down-modulation by blocking Cbl-b up-regulation.
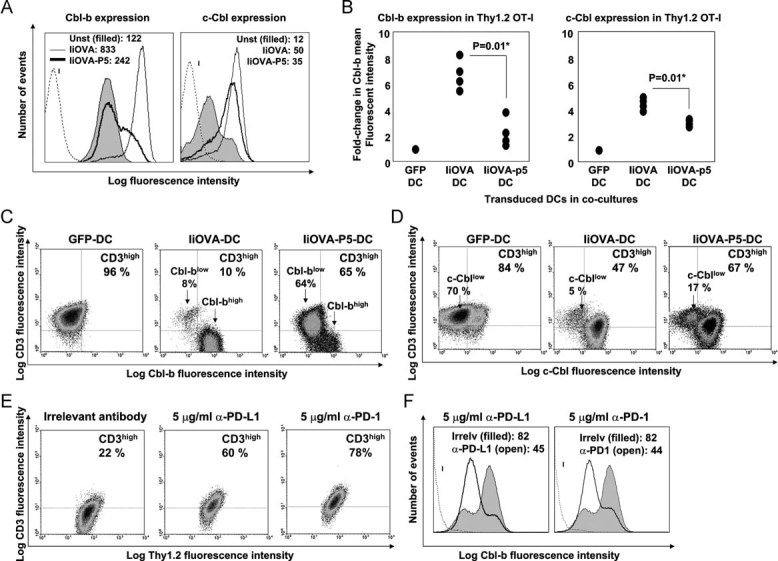
- Intracellular expression of Cbl-b and c-Cbl in OT-I cells co-cultured with lentivector-transduced DCs. Lentivectors used for transduction are indicated in the legend within the histograms. Numbers indicate mean fluorescent intensities. I, isotype control.
- Same as (A), as a scatter plot representing the fold-change in Cbl mean fluorescent intenisty compared to T cells co-cultured with GFP-DCs. Four independent experiments are plotted and analysed with the Mann–Whitney U-test. *, significant differences.
- Cbl-b and surface CD3 expression in OT-I cells co-cultured with lentivector-transduced DCs as indicated on top of the density plots. Percentages of CD3high T cells are shown, as well as populations with different Cbl levels (arrows).
- Same as in (C), but detecting c-Cbl.
- Surface CD3 and Thy1.2 expression in OT-I cells co-cultured with DCs expressing IiOVA, in the presence of the indicated concentrations of PD-L1 or PD-1 blocking antibodies (top of each graph). Percentages of CD3high T cells are indicated within each density plot.
- As in (E) but intracellular Cbl-b expression, represented as a histogram and gating in Thy1.2+ T cells. Histograms in this figure came from a single experiment out of two independent experiments. Mean fluorescent intensities are shown. I, isotype control.
To confirm that PD-L1/PD-1 co-stimulation was a regulator of TCR down-modulation and also exclude off-target shRNA-mediated mechanisms, we repeated our DC-T cell co-cultures in the presence of well-described PD-L1 or PD-1 blocking antibodies. Blockade of either the ligand or receptor reproducibly replicated our findings using PD-L1 silencing in DCs, including significant inhibition of TCR down-modulation and Cbl-b expression (Fig 3E and F and data not shown). In fact, CD3 internalization and Cbl-b expression were reduced in a dose-dependent manner (data not shown). A nearly complete abrogation of CD3 internalization was achieved with the highest concentration of PD-1 blocking antibody, while surface levels of Thy1.2 remained unchanged (Fig 3E). Collectively, these data demonstrate that the PD-L1/PD-1 interaction contributes to ligand-induced TCR down-modulation and that it depends on PD-1 expression in T cells after activation in our experimental system.
To further demonstrate the requirement for PD-1 expression in T cells, we silenced PD-1 in human T cells by delivering shRNAs using lentiviral vectors. We chose human cells due to their susceptibility to transduction in the absence of activation. Thus, PD-1-specific shRNAs and GFP were co-delivered under the control of the U6 and phosphoglycerate kinase (PGK) promoters. To overcome the necessity of specific antigen presentation, we stimulated human CD4+ T cells with monocytes and staphylococcus enterotoxin-B (SEB) superantigen, following a published protocol (Horgan et al, 1990). Accordingly, CD3 surface down-modulation was significantly inhibited following PD-1 silencing in stimulated GFP+ CD4+ T cells, although not completely abrogated (results not shown). We reproduced these results with shRNAs targeted to three different PD-1 regions.
PD-L1 silencing in DCs leads to hyperactivated pro-inflammatory TCRhigh CD8+ T cells
To investigate whether PD-L1-induced dysregulation of TCR internalization altered the function of primed T cells, we initially compared proliferation in T cells incubated with BM-DCs transduced with IiOVA or IiOVA-p5 lentivectors. As expected, an inverse correlation between surface expression of TCR chains and proliferation was observed (Fig 4A–C). In contrast, T cells stimulated in the presence of reduced PD-L1 signalling proliferated more extensively in vitro, even though they had significantly increased TCR levels (Fig 4B and C). These T cells produced increased levels of IFN-γ and IL-17, suggesting an enhanced pro-inflammatory phenotype (Fig 4D and E). Detection of annexin V in DC-T cell co-cultures indicated that apoptosis was not increased in T cells primed in the absence of PD-L1 (data not shown). Therefore, these data suggested that activation of CD8+ T cells without PD-L1 co-stimulation results in the expansion of a hyperactivated population of cells. This phenotype was very similar to that of hyperactivated TCRhigh T cells from Cbl KO mice (Chiang et al, 2000; Naramura et al, 2002), which spontaneously develop lethal autoimmune disorders (Naramura et al, 2002). Therefore, we tested whether T cells primed in the absence of PD-L1 co-stimulation could cause immune pathology in a well-established inflammatory arthritis model using OVA as a surrogate autoantigen (Arce et al, 2011; Fig 4F). Transfer of T cells primed by DCs transduced with IiOVA-p5 resulted in severe knee inflammation after intra-articular challenge with OVA compared to mice that had received T cells primed with DCs expressing IiOVA, which induced much less severe inflammation. Therefore, these results show that T cell priming without PD-L1 co-stimulation leads to the development of hyperactivated T cells, which cause significant immune pathology in vivo if directed towards an autoantigen (Fig 4F).
Figure 4. PD-L1 silencing in antigen presenting DCs results in hyperactivated pro-inflammatory TCRhigh CD8+ T cells.
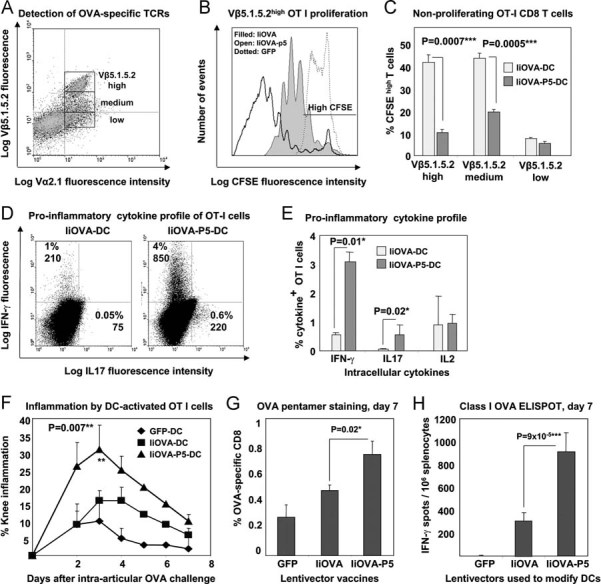
- The gating of OT-I cells according to Vβ5.1.5.2 expression is shown. T cell populations were separated into high, medium and low surface expression for proliferation analyses by CFSE dilution in TCRhigh OT-I co-cultured with lentivector-transduced DCs.
- Flow cytometry analyses of CFSE dilution in Vβ5.1.5.2high OT-I cells co-cultured for 4 days with DCs transduced with the indicated lentivectors (shown within the graph). The horizontal line within the graph delimits CFSE high T cells (95% of unstimulated OT-I).
- Same as B, showing the mean percentages of CFSE high OT-I cells with error bars are plotted (three experiments) according to Vβ5.1.5.2 surface expression (bottom). ***, highly significant differences.
- IFN-γ and IL-17 expression in OT-I cells co-cultured with lentivector-transduced DCs (top). Percentage and mean fluorescent intensities are shown within the gates.
- As in (D), plotting mean percentages and error bars from three experiments, including IL-2 expression. *, significant differences.
- Knee inflammation after intra-articular challenge with OVA in groups of five mice. One day before challenge, OT-I cells were co-cultured with lentivector-transduced DCs (legends within the graph) and intravenously transferred. Means are plotted with error bars (standard deviations). **, very significant differences.
- Percentage of OVA-specific endogenous CD8+ T cells in draining lymph nodes stained with class I OVA-pentamers, on day 7 after vaccination with the indicated lentivectors used for DC transduction (bottom). Means and error bars from three experiments are plotted.
- CD8+ responses by IFN-γ ELISPOT on day 7 after vaccination with lentivector-transduced DCs (bottom).
Our conclusions were further reinforced by studying the endogenous CD8+ T cell expansion after subcutaneous vaccination with lentivector-modified DCs. Effector CD8+ T cell expansion was significantly increased 7 days after immunization when PD-L1 was silenced in DCs as observed by pentamer-staining in draining lymph nodes (Fig 4G) and IFN-γ ELISPOT in splenocytes (Fig 4H). Numbers of pentamer-positive CD8+ T cells after vaccination with PD-L1-silenced IiOVA-DCs remained higher than in non-silenced vaccination controls up to 3 weeks after vaccination, with high pentamer fluorescent intensities that probably reflect high TCR surface levels (Fig 5). These results suggested an accelerated or uncontrolled CD8+ T cell activation/expansion in agreement with our in vitro studies using OT-I cells.
Figure 5. PD-L1 silencing in DCs accelerates expansion of endogenous antigen-specific CD8+ T cells.
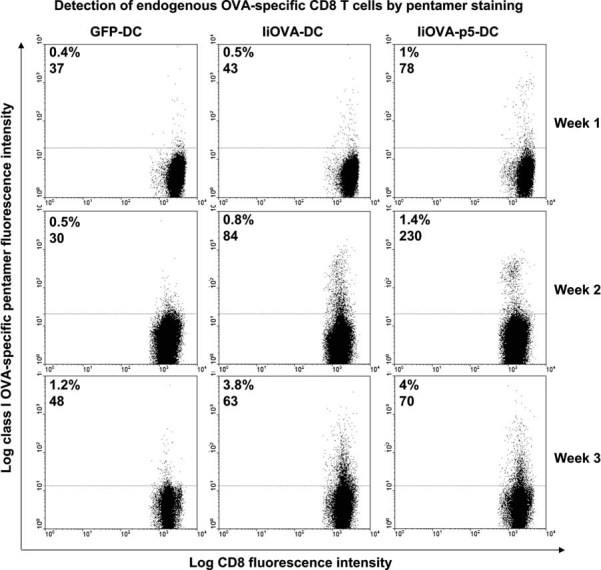
As in Fig 4G, flow cytometry analyses of endogenous OVA-specific CD8+ T cells expanded after vaccination with the indicated transduced DCs (top of the graphs). T cells from draining inguinal lymph nodes were stained with a class I-specific OVA pentamer, and percentages of pentamer-specific OT-I cells, with their corresponding mean fluorescent intensities are shown in each graph. Analyses were performed 1 week after vaccination (top row, indicated on the right of the graphs), 2 weeks (middle row) and 3 weeks (bottom row). Vaccination groups consisted of between 3 and 5 mice. Plots correspond to individual mice.
PD-L1 silencing in DCs accelerates anti-tumour immune responses
Our observation that lentivector delivery of antigen with PD-L1 silencing induced potent inflammatory T cells suggested that these T cells could be a useful means of enhancing immunity in the context of anti-tumour therapy. Thus, we tested the therapeutic efficacy in an EG7 mouse lymphoma model (EL4 cells expressing OVA as a surrogate tumour antigen). Subcutaneous administration of EG7 cells results in aggressive solid lymphomas, which can be effectively treated by vaccination with IiOVA-expressing DCs (Escors et al, 2008). DCs were transduced with lentivectors expressing GFP alone, IiOVA-GFP or IiOVA-p5-GFP and were injected subcutaneously when tumours were detectable (on day 4 or 5; Fig 6A). The therapeutic immunization with as few as 105 IiOVA-p5-GFP transduced DCs significantly reduced tumour growth compared to non-silenced controls (Fig 6B). As a consequence, a significant increase in lifespan of tumour-bearing mice was observed (Fig 6C). Surprisingly, overall survival after 1 month was similar when compared to non-silenced DC counterparts (Fig 6D). Thus, our data strongly suggested that PD-L1 silencing inhibited tumour growth and prolonged survival, but did not increase cure rates. All surviving tumour-free mice pooled from three independent experiments were resistant to re-challenge with EG7 cells demonstrating effective recall responses (Fig 6E).
Figure 6. PD-L1 silencing in antigen presenting DCs accelerates anti-tumour activities.
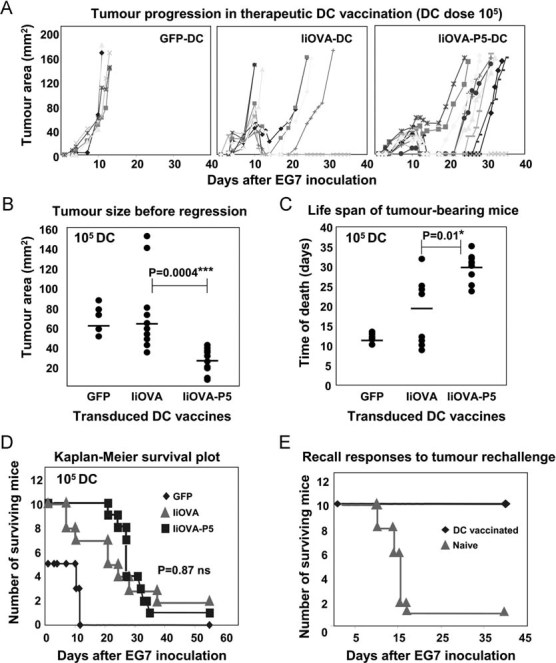
- Tumour progression in mice vaccinated with lentivector-transduced DCs. Data from individual mice are represented as independent lines.
- Same as (A) but on day 10 (peak of tumour size). Each mouse represented as a dot. Means from each group are indicated within the graph with horizontal lines. Comparisons were performed with the Mann–Whitney U-test. ***, highly significant differences.
- Time of death from tumour bearing mice. Vaccination groups indicated on the bottom. Means from each group are indicated within the graph with horizontal lines. Comparisons were performed with the Mann–Whitney U-test. *, significant differences.
- As in (A), representing the Kaplan–Meier survival plot. Statistical significance was tested with the log-rank test. Ns, no significant differences.
- Kaplan–Meier survival plot-in surviving tumour-free mice from experiments as in (D), re-challenged with EG7 lymphoma cells. A control group with naive mice was used.
PD-L1 silencing potentiates the adjuvant capacities of molecular DC activators
To test whether anti-tumour immunity could be further improved, we combined PD-L1 silencing with the expression of selected DC molecular activators (Arce et al, 2011; Escors et al, 2008). PD-L1 shRNA p5 was co-delivered with IiOVA and a constitutive activator of mitogen-activated kinase (MAPK) p38 (MKK6EE) or the ERK inhibitor MEK1 ΔNES AA (Fig 7A). We chose these MAPK modulators because we have previously demonstrated that p38 activation and ERK inhibition using these modulators increase expression of co-stimulatory and adhesion molecules in DCs such as CD80, CD40 and ICAM I (Arce et al, 2011; Escors et al, 2008). Vaccination with a low dose of transduced DCs (104/mouse) showed a synergistic increase in CD8+ responses by IFN-γ ELISPOT when p5 was co-delivered with MAPK modulators (Fig 7A). Immunization with as few as 104 PD-L1-silenced DCs activated with MAPK modulators resulted in effective control of tumour growth compared to mice receiving DC with IiOVA alone (Fig 7B). The combination of ERK inhibition with PD-L1 silencing was particularly effective in reducing tumour growth (Fig 7B). Interestingly, PD-L1 silencing with either ERK inhibition or p38 activation significantly increased tumour-free survival (Fig 7C). This suggests that whilst PD-L1 inhibition can generate a T-cell response superior at controlling tumour growth, additional co-stimulatory signals (via DC activation) are required to optimize T-cell functionality for efficient tumour killing (Fig 7C). These data demonstrate that in the absence of PD-L1 silencing, as few as 104 activated DCs are sufficient to prime potent CD8+ T cell responses, which can control tumour growth. We further show that by improving the potency of DCs, in conjunction with removal of PD-L1-based regulation, we can generate significantly enhanced T cell immunity, capable of controlling tumour growth in a therapeutic setting.
Figure 7. PD-L1 silencing increases adjuvant capacities of selected DC activators.
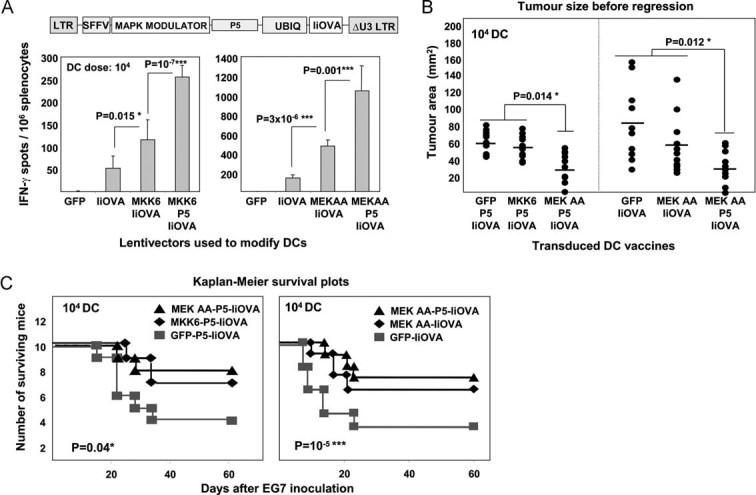
- Top, lentivector structure used for the experiments. Bottom, graphs represent CD8+ T cell responses by IFN-γ ELISPOT-In splenocytes 7 days after vaccination with the indicated lentivector-transduced DCs. MKK6, the p38 activator MKK6EE mutant; MEK AA, the ERK inhibitor MEK1 ΔNES AA.
- Tumour size on day 8 (peak of tumour size) in mice vaccinated with the indicated lentivector-transduced DCs. Group combinations from two different experiments have been combined in a single graph, and are separated with a vertical dotted line. Means from each group are indicated within the graph with horizontal lines. Statistical comparisons were performed using the Kruskall–Wallis non-parametric test. *, significant differences.
- As in (B), but representing the Kaplan–Meier survival plots. Vaccination groups are indicated within the plots. Statistical significance was tested with the log-rank test. *, significant differences; ***, highly significant differences.
DISCUSSION
We have provided evidence that PD-L1 in antigen presenting DCs contributes to ligand-induced TCR down-modulation. Ligand-induced TCR down-modulation is a complicated process in which more than one mechanism is involved (Lauritsen et al, 1998). Firstly, serially ligand-engaged TCRs internalize quickly (from seconds to hours) after ligand–MHC binding (Huppa et al, 2010; Valitutti et al, 1995). Secondly, non-engaged TCR complexes can also be down-regulated following signal transduction from previously engaged (and endocytosed) TCR complexes (San Jose et al, 2000). It is still under debate whether TCR down-modulation is caused by internalization followed by degradation or by preventing TCR recycling back to the membrane (Dietrich et al, 1998; San Jose et al, 2000; Valitutti et al, 1995). Interestingly, we have observed that TCR down-modulation progressively continues up to 3 days after initial antigen presentation by DCs. Subsequently, TCR surface levels recover again. This TCR down-modulation may be caused by either direct internalization or just a shift in recycling towards intracellular retention of TCR complexes (San Jose et al, 2000; Valitutti et al, 1995). Our results implicate PD-L1/PD-1 co-stimulation in the second, slower phase and they are in agreement with the following working model for regulation of T cell activation during antigen presentation by DCs (Fig 8): Firstly, antigen recognition (with additional co-stimulatory interactions) leads to TCR signal transduction. Following T cell activation, PD-1 is exposed to the T cell surface where it engages with PD-L1 on the surface of the APC. PD-1 signal transduction recruits SHP phosphatases and, importantly, leads to Cbl-b expression by a yet undefined pathway. Cbl-b ubiquitinates and inactivates key TCR signal transduction mediators, while removing TCRs from the T cell surface. Whether this removal is triggered by active internalization or a change in the dynamics of TCR recycling is yet unclear. Our data supports PD-1 as an early brake that fine-tunes T cell activation during antigen presentation after TCR signal transduction. In any case, our model can provide an explanation for results reported in several unrelated studies. Firstly, TCR/CD28 signal transduction per se is not required for full ligand-induced TCR down-modulation, as observed in Cbl-b KO CD8+ T cells (Shamim et al, 2007). Secondly, PD-1 blocking antibodies inhibit Cbl-b transcriptional up-regulation, amongst other negative regulators of T cell activation as shown by polymerase chain reaction (after reverse transcription) (RT-PCR; Nurieva et al, 2006). Thirdly, human T cells activated in vitro with anti-CD3/anti-CD28 antibodies maintain high surface TCR levels when compared to antigen presentation by APCs (Yang et al, 2010). And fourthly, old human CD8+ T cells, which maintain low surface TCR levels, significantly increase their surface expression after in vitro activation with anti-CD3/anti-CD28 antibodies only in the presence of PD-L1 blocking antibodies (Mirza et al, 2010). This last observation would implicate PD-L1/PD-1 homotypic T cell interactions (T cell–T cell) either in trans or in cis as suggested by others (Fooksman et al, 2010). The interpretation of all these results is, therefore, straightforward if PD-L1/PD-1 binding contributes to ligand-induced TCR down-modulation through Cbl-b expression.
Figure 8. PD-1-dependent early brake model of T cell activation.
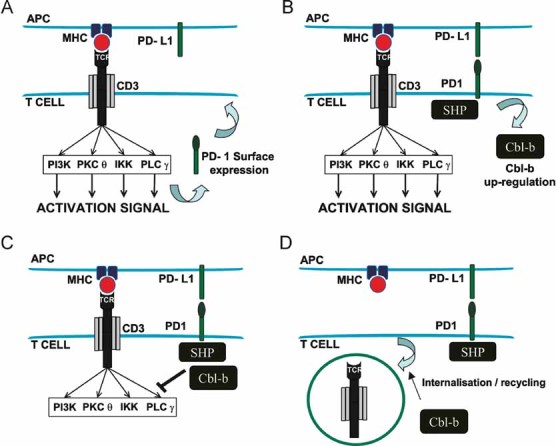
- Antigen presenting cell (APC; top) presents antigen in the context of MHC. T cell TCR recognizes the antigen (together with CD80–CD28 binding, not shown for simplification purposes) and leads to activating signal transduction. PD-1 surface expression is up-regulated.
- PD-1 on the surface engages with PD-L1 on the APC's surface, leading to recruitment of SHP phosphatases and up-regulating Cbl-b expression.
- SHP and Cbl-b de-phosphorylate and inactivate by ubiquitination, respectively, key components of the TCR signal transduction machinery.
- Cbl-b either triggers TCR down-modulation, or shifts the recycling equilibrium towards retention of TCR complexes.
Unexpectedly, we did not observe significant ligand-induced TCR down-modulation after 3–4 days co-culture of OVA-specific CD4+ T cells with IiOVA-expressing BM-DCs, whether PD-L1 was silenced or not. Accordingly, we were unable to test in the mouse model if PD-L1/PD-1 co-stimulation was involved in CD4+ TCR down-modulation. It is worth noting that unlike CD8+ T cells, only a minor proportion of OVA-specific OT-II T cells strongly associated to IiOVA-expressing DCs. This is in agreement with other reports in which naive CD4+ T cells detach quickly from APCs, but they get fully activated later by multiple, short-lived contacts (Celli et al, 2005). However, it is likely that PD-L1/PD-1 interaction may also be involved in CD4 TCR down-modulation as we did observe inhibition of TCR down-modulation in PD-1-silenced human CD4 T cells, but in the context of superantigen stimulation.
PD-L1/PD-1 interaction is receiving an increasing interest as a therapeutic target. While most of the studies use systemic administration of blocking antibodies, a minor proportion have silenced B7 inhibitory molecules in DCs (Breton et al, 2009; Hobo et al, 2010). In any case, therapeutic outcomes vary considerably and are quite dependent on the specific experimental system. In many instances, combination with other strategies has to be applied to achieve effective therapy (Breton et al, 2009; Curran et al, 2010; Hobo et al, 2010; Pilon-Thomas et al, 2010). In our experimental system, PD-L1 silencing in DCs alone did not improve overall cure rates compared to non-silenced DCs. PD-L1 silencing had to be combined with DC molecular activators to achieve increased therapeutic activities. Curiously, this phenomenon is observed in many other published studies and may account for the relative lack of efficacy of PD-L1/PD-1 blocking antibodies unless they are given in combination therapies. Thus, the anti-tumour efficacy of PD-L1/PD-1 blocking antibodies is enhanced by lentivector vaccination (Sierro et al, 2011; Zhou et al, 2010). In these two last reports, an increased recruitment of tumour infiltrating CD8+ T cells was observed after systemic administration of PD-L1/PD-1 antibodies. In agreement with this, we also observed a tendency to increased intra-tumour CD8+ T cell numbers in mice vaccinated with IiOVA-expressing PD-L1-silenced DCs. However, we could not demonstrate significance in relative numbers or TCR surface levels compared to vaccination with non-silenced DCs. In contrast to systemic administration of blocking antibodies, PD-L1 silencing in our experimental model is taking place in a limited number of DCs. Secondly, we could not differentiate a specific recruitment of effector CD8+ T cells from just simply an overall increase in CD8+ T cell numbers (Figs 4 and 5). However, in light to previous reports, it is likely that also in our case, there is an enhanced, active intra-tumoural recruitment of CD8+ T cells.
Summarizing, our results demonstrate that PD-L1 silencing in DCs may speed-up T cell expansion and cytokine responses (which is important for tumour immunotherapy) but other factors also contribute to the effectiveness of the immune response, for instance the functionality of activated T-cells which is a direct consequence of a range of co-stimulatory signals during antigen presentation. Further co-stimulation could enhance the expression of cytotoxic molecules in effector CD8+ T cells such as granzyme B and perforin, which could be responsible for the observed increase in therapeutic activity.
Our model highlights the contribution of PD-L1/PD-1 co-stimulation to ligand-induced TCR down-modulation during ‘physiological’ antigen presentation by DCs. This is a very complex process that is regulated at multiple levels, and there are other pathways that inhibit effector T cell activities without modifying TCR levels (Barber et al, 2006; Choi & Schwartz, 2007; Wherry, 2011). Defining the mechanism by which PD-1 engagement leads to Cbl-b up-regulation and modulates TCR levels is a key question in T cell physiology and a potential target for therapeutic intervention.
The paper explained
PROBLEM
Our study addresses a fundamental immunological mechanism for the control of T cell activation during antigen presentation, TCR down-modulation. This process limits TCR signal transduction and prevents T cell hyperactivation after antigen-presentation by dendritic cells. Current understanding of this process is that intrinsic TCR/CD28 signal transduction leads to TCR internalization. However, to date, no additional extrinsic signals have been identified which contribute to TCR down-modulation.
RESULTS
We show here that binding of PD-L1 on dendritic cells to PD-1 on T cells is implicated in ligand-induced TCR down-modulation, by promoting Cbl-b E3 ubiquitin ligase expression. This finding expands the role of PD-L1 in immune regulation to a critical step in T cell activation.
We have further analysed the consequences of preventing TCR down-modulation, in vitro and in vivo in two very different disease models; antigen-specific inflammatory arthritis and a T cell-derived lymphoma. By combining PD-L1 silencing with modulators of selected signalling pathways (MAPKs) in dendritic cells, we have increased their anti-tumour activities, obtaining therapeutic effects with doses 100-to-1000-fold lower than those currently used in experimental cancer models and in human clinical trials.
IMPACT
Our study will be of interest to a broad range of basic researchers in cell biology, immunology, rheumatology as well as clinicians. Our findings will be of particular importance for clinical application in autoimmune disorders and cancer, as they highlight TCR trafficking and its interference as a potential key therapeutic target.
MATERIALS AND METHODS
Cells and mice
293T, EG7 cells and BM-DCs were grown as described (Escors et al, 2008). Approval for the animal studies was obtained from the University College London Animal Ethics Committee. The following mouse strains were used: C57BL/6 mice, OT-I and OT-II mice. The last two strains express a transgenic TCR specific for MHC- class I and class II OVA epitopes, respectively. DC-T cell co-cultures were performed as described (Arce et al, 2011). T cells were added to DCs 3 days after transduction at a ratio of 10:1, and co-cultured for a minimum of 2 days and a maximum of 1 week. When indicated, isotype antibody control, purified anti-mouse PD-L1 clone MIH5 or anti-mouse PD-1 antibody clone RMP1-14 were added to DCs after lentivector transduction, at the concentrations indicated in the text. These last two antibodies are well-characterized blocking antibodies (Freeman et al, 2000; Kanai et al, 2003). Intracellular cytokine detection by flow cytometry was performed as described (Arce et al, 2011). Immunoblots using HA-specific antibodies were performed as described (Escors et al, 2008).
Plasmids, lentivector production, titration and cell transduction
Dual lentivectors co-expressing MAPK modulators along with GFP or HA-tagged IiOVA were used (Arce et al, 2011; Escors et al, 2008). The sequence of the PD-L1-specific shRNA used in the present experiments (designated p5) was 5′-CTCGAGAAGGTATATTGCTGTTGACAGTGAGCGAGCGTTGAAGATACAAGCTCAATAGTGAAGCCACAGATGTATTGAGCTTGTATCTTCAACGCCTGCCTACTGCCTCGGAATTC-3′. The sequences of the PD-1-specific shRNAs are: PD-1A 5′-GTGCTAAACTGGTACCGCATTTCAAGAGAATGCGGTACCAGTTTAGCATTTTTTACGCGT′-3′, PD-1B 5′-GCCACCATTGTCTTTCCTAGTTCAAGAGACTAGGAAAGACAATGGTGGTTTTTTACGCGT-3′ and PD-1C 5′-GCCAACACATCGGAGAGCTTTTCAAGAGAAAGCTCTCCGATGTGTTGGTTTTTTACGCGT-3′. For cloning and expression of PD-1-specific shRNAs, a lentivector was constructed based on pDUAL containing a U6 promoter replacing the SFFV promoter, and a PGK promoter replacing the ubiquitin promoter. Lentivectors were produced and titrated as described (Kochan et al, 2008; Selden et al, 2007). BMDCs were transduced using previously described protocols (Escors et al, 2008). Human CD4 T cells were transduced as described (Frecha et al, 2008).
Cell staining and flow cytometry
Surface and intracellular staining were performed as described previously (Escors et al, 2008). The following anti-mouse antibodies were purchased from eBioscience, with isotype controls: Biotinylated antibodies specific for Thy1.2, CD80, CD40, PD-L1, PD-L2, phycoerythrin (PE)-conjugated anti CD25, anti-CD8, anti-ICAM I, anti-MHC I, anti-MHC II, anti-CD3, PE-Cy7-conjugated anti-CD4, allophycocyanin (APC)-conjugated anti-Foxp3, anti-IFN-γ, anti-CD3, fluorescein isothiocyanate (FITC)-conjugated anti-IL17, anti IL-2 and anti-PD-1 clone J43. The following antibodies were purchased from BD Bioscience Pharmingen: Biotinylated anti-mouse Vβ 5.1 5.2 TCR, Alexa Fluor 647-conjugated anti-mouse phosphoERK 1/2. FITC- and PE-conjugated anti-Vα2 TCR were purchased from Caltag laboratories. Cbl-b goat polyclonal IgG and Alexa fluor 647-conjugated c-Cbl antibodies were purchased from Santa Cruz Biotechnology. Alexa Fluor 488-conjugated donkey anti-goat IgG was purchased from Invitrogen. Anexin V staining kit following the instructions of the manufacturer (BD Biosciences). Pentamer staining was performed as described (Arce et al, 2011).
Vaccination, T cell responses and tumour experiments
All vaccination experiments were repeated independently at least three times, using groups of five mice, as described (Breckpot et al, 2010; Escors et al, 2008; Goold et al, 2011). IFN-γ ELISPOTs were performed as described (Escors et al, 2008).
Therapeutic tumour experiments were performed as described before, using 10 mice per vaccination groups (Escors et al, 2008). Lentivector-transduced DCs at the indicated doses were injected subcutaneously when tumours were detectable (on day 4 or 5). Tumour growth was monitored daily and mice were sacrificed when tumour surface was higher than 140–150 mm2.
T cell purification, CFSE labelling, adoptive transfer and antigen-induced arthritis
Untouched CD4+ T or CD8+ T cells were purified from mouse spleens using the Dynal Mouse CD4 or CD8 Negative Isolation Kit (Invitrogen). T cells were labelled with CFSE as described (Arce et al, 2011). Purified OT-I cells were adoptively transferred intravenously (between 106 and 108 cells/mouse) into groups of five mice, when indicated. After transfer, antigen-induced arthritis was triggered by intra-articular injection of 100 µg of purified OVA (Sigma) in 10 µl of PBS. Knee inflammation was monitored daily. The experiment was repeated twice.
Statistical analysis
ELISPOT data was analysed as described (Arce et al, 2011; Escors et al, 2008). Normally, a minimum of three independent experiments was performed with five mice per group. For tumour experiments, 10 mice per group per experiment were used. Mean fluorescence intensities from surface or intracellular staining were analysed as described (Arce et al, 2011; Escors et al, 2008). Survival data from tumour experiments was compared using the log-rank test as described before (Escors et al, 2008). CD3 surface expression levels, tumour size between groups and lifespan of tumour-bearing mice were compared using the non-parametric Mann–Whitney U-test or Kruskall–Wallis test for multicomparisons.
Acknowledgments
We acknowledge Drs Anthony Antoniou, David Guilliano and Sergio Quezada for critical reading of the manuscript and helpful discussions. We also acknowledge Dr Els Verhoeyen and Prof François-Löic Cosset for providing the measles virus mutant envelope proteins for lentivector pseudotyping. Katarzyna Karwacz's work was supported by a Medical Research Council UK studentship. Frederick Arce was supported by a University College London Hospitals/University College London Comprehensive Biomedical Research Centre Award. David Escors is funded by an Arthritis Research UK Career Development Fellowship (18433).
Supporting information is available at EMBO Molecular Medicine online.
The authors declare that they have no conflict of interest.
Author contributions
All authors were involved in drafting the article or revising it critically for important intellectual content. KK, MC, CLB and DE responsible for study conception and design. KK, CB, DMD, FA and DE performed data acquisition and interpretation.
Supplementary material
Detailed facts of importance to specialist readers are published as ”Supporting Information”. Such documents are peer-reviewed, but not copy-edited or typeset. They are made available as submitted by the authors.
References
- Arce F, Breckpot K, Stephenson H, Karwacz K, Ehrenstein MR, Collins M, Escors D. Selective ERK activation differentiates mouse and human tolerogenic dendritic cells, expands antigen-specific regulatory T cells, and suppresses experimental inflammatory arthritis. Arthritis Rheum. 2011;63:84–95. doi: 10.1002/art.30099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmaier K, Krawczyk C, Kozieradzki I, Kong YY, Sasaki T, Oliveira-dos-Santos A, Mariathasan S, Bouchard D, Wakeham A, Itie A, et al. Negative regulation of lymphocyte activation and autoimmunity by the molecular adaptor Cbl-b. Nature. 2000;403:211–216. doi: 10.1038/35003228. [DOI] [PubMed] [Google Scholar]
- Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, Freeman GJ, Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- Breckpot K, Escors D, Arce F, Lopes L, Karwacz K, Van Lint S, Keyaerts M, Collins M. HIV-1 lentiviral vector immunogenicity is mediated by Toll-like receptor 3 (TLR3) and TLR7. J Virol. 2010;84:5627–5636. doi: 10.1128/JVI.00014-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton G, Yassine-Diab B, Cohn L, Boulassel MR, Routy JP, Sekaly RP, Steinman RM. siRNA knockdown of PD-L1 and PD-L2 in monocyte-derived dendritic cells only modestly improves proliferative responses to Gag by CD8(+) T cells from HIV-1-infected individuals. J Clin Immunol. 2009;29:637–645. doi: 10.1007/s10875-009-9313-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Kishimoto H, Brunmark A, Jackson MR, Peterson PA, Sprent J. Requirements for peptide-induced T cell receptor downregulation on naive CD8+ T cells. J Exp Med. 1997;185:641–651. doi: 10.1084/jem.185.4.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celli S, Garcia Z, Bousso P. CD4 T cells integrate signals delivered during successive DC encounters in vivo. J Exp Med. 2005;202:1271–1278. doi: 10.1084/jem.20051018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 associate with immunoreceptor tyrosine-based switch motif of programmed death 1 upon primary human T cell stimulation, but only receptor ligation prevents T cell activation. J Immunol. 2004;173:945–954. doi: 10.4049/jimmunol.173.2.945. [DOI] [PubMed] [Google Scholar]
- Chiang YJ, Kole HK, Brown K, Naramura M, Fukuhara S, Hu RJ, Jang IK, Gutkind JS, Shevach E, Gu H. Cbl-b regulates the CD28 dependence of T-cell activation. Nature. 2000;403:216–220. doi: 10.1038/35003235. [DOI] [PubMed] [Google Scholar]
- Choi S, Schwartz RH. Molecular mechanisms for adaptive tolerance and other T cell anergy models. Semin Immunol. 2007;19:140–152. doi: 10.1016/j.smim.2007.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curran MA, Montalvo W, Yagita H, Allison JP. PD-1 and CTLA-4 combination blockade expands infiltrating T cells and reduces regulatory T and myeloid cells within B16 melanoma tumors. Proc Natl Acad Sci USA. 2010;107:4275–4280. doi: 10.1073/pnas.0915174107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietrich J, Backstrom T, Lauritsen JP, Kastrup J, Christensen MD, von Bulow F, Palmer E, Geisler C. The phosphorylation state of CD3gamma influences T cell responsiveness and controls T cell receptor cycling. J Biol Chem. 1998;273:24232–24238. doi: 10.1074/jbc.273.37.24232. [DOI] [PubMed] [Google Scholar]
- Escors D, Lopes L, Lin R, Hiscott J, Akira S, Davis RJ, Collins MK. Targeting dendritic cell signalling to regulate the response to immunisation. Blood. 2008;111:3050–3061. doi: 10.1182/blood-2007-11-122408. [DOI] [PubMed] [Google Scholar]
- Fife BT, Pauken KE, Eagar TN, Obu T, Wu J, Tang Q, Azuma M, Krummel MF, Bluestone JA. Interactions between PD-1 and PD-L1 promote tolerance by blocking the TCR-induced stop signal. Nat Immunol. 2009;10:1185–1192. doi: 10.1038/ni.1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fooksman DR, Vardhana S, Vasiliver-Shamis G, Liese J, Blair DA, Waite J, Sacristan C, Victora GD, Zanin-Zhorov A, Dustin ML. Functional anatomy of T cell activation and synapse formation. Annu Rev Immunol. 2010;28:79–105. doi: 10.1146/annurev-immunol-030409-101308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frecha C, Costa C, Negre D, Gauthier E, Russell SJ, Cosset FL, Verhoeyen E. Stable transduction of quiescent T cells without induction of cycle progression by a novel lentiviral vector pseudotyped with measles virus glycoproteins. Blood. 2008;112:4843–4852. doi: 10.1182/blood-2008-05-155945. [DOI] [PubMed] [Google Scholar]
- Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, Fitz LJ, Malenkovich N, Okazaki T, Byrne MC, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–1034. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold HD, Escors D, Conlan TJ, Chakraverty R, Bennett CL. Conventional DC are required for the activation of helper-dependent CD8 T cell responses after cutaneous vaccination with lentiviral vectors. J Immunol. 2011;186:4565–4572. doi: 10.4049/jimmunol.1002529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, Rietz C, Flies DB, Lau JS, Zhu G, et al. Blockade of B7-H1 and PD-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res. 2005;65:1089–1096. [PubMed] [Google Scholar]
- Hobo W, Maas F, Adisty N, de Witte T, Schaap N, van der Voort R, Dolstra H. siRNA silencing of PD-L1 and PD-L2 on dendritic cells augments expansion and function of minor histocompatibility antigen-specific CD8+ T cells. Blood. 2010;116:4501–4511. doi: 10.1182/blood-2010-04-278739. [DOI] [PubMed] [Google Scholar]
- Holst J, Wang H, Eder KD, Workman CJ, Boyd KL, Baquet Z, Singh H, Forbes K, Chruscinski A, Smeyne R, et al. Scalable signaling mediated by T cell antigen receptor-CD3 ITAMs ensures effective negative selection and prevents autoimmunity. Nat Immunol. 2008;9:658–666. doi: 10.1038/ni.1611. [DOI] [PubMed] [Google Scholar]
- Horgan KJ, Van Seventer GA, Shimizu Y, Shaw S. Hyporesponsiveness of “naive” (CD45RA+) human T cells to multiple receptor-mediated stimuli but augmentation of responses by co-stimuli. Eur J Immunol. 1990;20:1111–1118. doi: 10.1002/eji.1830200525. [DOI] [PubMed] [Google Scholar]
- Huppa JB, Axmann M, Mortelmaier MA, Lillemeier BF, Newell EW, Brameshuber M, Klein LO, Schutz GJ, Davis MM. TCR-peptide-MHC interactions in situ show accelerated kinetics and increased affinity. Nature. 2010;463:963–967. doi: 10.1038/nature08746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai T, Totsuka T, Uraushihara K, Makita S, Nakamura T, Koganei K, Fukushima T, Akiba H, Yagita H, Okumura K, et al. Blockade of B7-H1 suppresses the development of chronic intestinal inflammation. J Immunol. 2003;171:4156–4163. doi: 10.4049/jimmunol.171.8.4156. [DOI] [PubMed] [Google Scholar]
- Kochan G, Escors D, Gonzalez JM, Casasnovas JM, Esteban M. Membrane cell fusion activity of the vaccinia virus A17–A27 protein complex. Cell Microbiol. 2008;10:149–164. doi: 10.1111/j.1462-5822.2007.01026.x. [DOI] [PubMed] [Google Scholar]
- Latchman YE, Liang SC, Wu Y, Chernova T, Sobel RA, Klemm M, Kuchroo VK, Freeman GJ, Sharpe AH. PD-L1-deficient mice show that PD-L1 on T cells, antigen-presenting cells, and host tissues negatively regulates T cells. Proc Natl Acad Sci USA. 2004;101:10691–10696. doi: 10.1073/pnas.0307252101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauritsen JP, Christensen MD, Dietrich J, Kastrup J, Odum N, Geisler C. Two distinct pathways exist for down-regulation of the TCR. J Immunol. 1998;161:260–267. [PubMed] [Google Scholar]
- Mirza N, Duque MA, Dominguez AL, Schrum AG, Dong H, Lustgarten J. B7-H1 expression on old CD8+ T cells negatively regulates the activation of immune responses in aged animals. J Immunol. 2010;184:5466–5474. doi: 10.4049/jimmunol.0903561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naramura M, Jang IK, Kole H, Huang F, Haines D, Gu H. c-Cbl and Cbl-b regulate T cell responsiveness by promoting ligand-induced TCR down-modulation. Nat Immunol. 2002;3:1192–1199. doi: 10.1038/ni855. [DOI] [PubMed] [Google Scholar]
- Nurieva R, Thomas S, Nguyen T, Martin-Orozco N, Wang Y, Kaja MK, Yu XZ, Dong C. T-cell tolerance or function is determined by combinatorial costimulatory signals. EMBO J. 2006;25:2623–2633. doi: 10.1038/sj.emboj.7601146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolino M, Thien CB, Gruber T, Hinterleitner R, Baier G, Langdon WY, Penninger JM. Essential role of E3 ubiquitin ligase activity in Cbl-b-regulated T cell functions. J Immunol. 2011;186:2138–2147. doi: 10.4049/jimmunol.1003390. [DOI] [PubMed] [Google Scholar]
- Pilon-Thomas S, Mackay A, Vohra N, Mule JJ. Blockade of programmed death ligand 1 enhances the therapeutic efficacy of combination immunotherapy against melanoma. J Immunol. 2010;184:3442–3449. doi: 10.4049/jimmunol.0904114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Jose E, Borroto A, Niedergang F, Alcover A, Alarcon B. Triggering the TCR complex causes the downregulation of nonengaged receptors by a signal transduction-dependent mechanism. Immunity. 2000;12:161–170. doi: 10.1016/s1074-7613(00)80169-7. [DOI] [PubMed] [Google Scholar]
- Schonrich G, Kalinke U, Momburg F, Malissen M, Schmitt-Verhulst AM, Malissen B, Hammerling GJ, Arnold B. Down-regulation of T cell receptors on self-reactive T cells as a novel mechanism for extrathymic tolerance induction. Cell. 1991;65:293–304. doi: 10.1016/0092-8674(91)90163-s. [DOI] [PubMed] [Google Scholar]
- Selden C, Mellor N, Rees M, Laurson J, Kirwan M, Escors D, Collins M, Hodgson H. Growth factors improve gene expression after lentiviral transduction in human adult and fetal hepatocytes. J Gene Med. 2007;9:67–76. doi: 10.1002/jgm.1000. [DOI] [PubMed] [Google Scholar]
- Shamim M, Nanjappa SG, Singh A, Plisch EH, LeBlanc SE, Walent J, Svaren J, Seroogy C, Suresh M. Cbl-b regulates antigen-induced TCR down-regulation and IFN-gamma production by effector CD8 T cells without affecting functional avidity. J Immunol. 2007;179:7233–7243. doi: 10.4049/jimmunol.179.11.7233. [DOI] [PubMed] [Google Scholar]
- Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8:239–245. doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, Qiu Y, Jussif JM, Carter LL, Wood CR, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004;574:37–41. doi: 10.1016/j.febslet.2004.07.083. [DOI] [PubMed] [Google Scholar]
- Sierro SR, Donda A, Perret R, Guillaume P, Yagita H, Levy F, Romero P. Combination of lentivector immunization and low-dose chemotherapy or PD-1/PD-L1 blocking primes self-reactive T cells and induces anti-tumor immunity. Eur J Immunol. 2011 doi: 10.1002/eji.201041235. DOI: 10.1002/eji.201041235. [DOI] [PubMed] [Google Scholar]
- Valitutti S, Muller S, Cella M, Padovan E, Lanzavecchia A. Serial triggering of many T-cell receptors by a few peptide–MHC complexes. Nature. 1995;375:148–151. doi: 10.1038/375148a0. [DOI] [PubMed] [Google Scholar]
- Wherry EJ. T cell exhaustion. Nat Immunol. 2011;131:492–499. doi: 10.1038/ni.2035. [DOI] [PubMed] [Google Scholar]
- Yang S, Dudley ME, Rosenberg SA, Morgan RA. A simplified method for the clinical-scale generation of central memory-like CD8+ T cells after transduction with lentiviral vectors encoding antitumor antigen T-cell receptors. J Immunother. 2010;33:648–658. doi: 10.1097/CJI.0b013e3181e311cb. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Xiao H, Liu Y, Peng Y, Hong Y, Yagita H, Chandler P, Munn DH, Mellor A, Fu N, et al. Blockade of programmed death-1 pathway rescues the effector function of tumor-infiltrating T cells and enhances the antitumor efficacy of lentivector immunization. J Immunol. 2010;185:5082–5092. doi: 10.4049/jimmunol.1001821. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.