

Abstract
A novel anti-cancer agent was constructed by fusing a gene encoding the scFV that targets both glycosylated and unglycosylated forms of CD133 to a gene fragment encoding deimmunized PE38KDEL. The resulting fusion protein, dCD133KDEL, was studied to determine its ability to bind and kill tumor-initiating cells in vitro and in vivo. The anti-CD133 scFV selectively bound HEK293 cells transfected with the CD133 receptor gene. Time course viability studies showed that dCD133KDEL selectively inhibited NA-SCC and UMSCC-11B, two head and neck squamous cell carcinomas that contain a CD133 expressing subpopulation. Importantly, the drug did not inhibit the viability of hematopoietic lineages measured by long-term culture initiating cell and colony-forming assays from sorted human CD34+ progenitor cells. In addition to in vitro studies, in vivo tumor initiation experiments confirmed that CD133 sorted cells implanted into the flanks of nude mice grew faster and larger than unsorted cells. In contrast, cells that were pretreated with dCD133KDEL prior to implantation showed the slowest and lowest incidence of tumors. Furthermore, UMSCC-11B-luc tumors treated with multiple intratumoral injections of dCD133KDEL showed marked growth inhibition leading to complete degradation of the tumors, not observed with an irrelevant control targeted toxin. Experiments in immunocompetent mice showed that toxin deimmunization resulted in a 90% reduction in circulating anti-toxin levels. These studies show that dCD133KDEL is a novel anti-cancer agent effective at inhibiting cell proliferation, tumor initiation, and eliminating established tumors by targeting the CD133 subpopulation. This agent shows significant promise for potential development as a clinically useful therapy.
Keywords: pseudomonas exotoxin, xenograft model, CD133, head and neck cancer, cancer stem cells
Introduction
Evidence has mounted over the last decade that cancers contain a small subset of their own stem-like cells termed cancer stem cells (CSC). These CSC are known to enhance tumor inititation, self-renew, and also differentiate into phenotypically diverse cancer cells with more limited proliferative potential (1,2,3). A point of controversy regarding CSC is the existence of plasticity between stem cells and their more differentiated derivatives and some believe that more differentiated cancer cells can become reprogrammed and revert back to CSC (4). CSC are more resistant to current chemotherapy agents and have distinct cell surface markers (5,6). Because of this, it is important to develop new therapeutics to specifically target CSC. CD133 (prominin-1) is a 5-transmembrane glycoprotein whose function remains unknown. It is expressed on hematopoietic, endothelial, and neuronal stem cells (7,8,9) and has recently been identified as a CSC marker in a variety of carcinomas as well (6,10,11,12,13).
Targeted toxins (TT) are a class of biological drug consisting of a catalytic protein toxin chemically or genetically linked to a ligand recognizing a specific marker expressed on cancer cells (14). The catalytic destruction of the target cell is dependent on the internalization of the target receptor. A recent study has shown that antibodies targeting the CD133 receptor are efficiently internalized (15). Other studies have shown that CD133+ cells possess a very strong ability to initiate tumors, but CD133− cells do not (16,17). Thus, we reasoned that a targeted toxin directed against CSC via CD133 might prove highly disruptive to the dynamic process of tumor initiation and progression.
A novel targeted toxin called deimmunized CD133KDEL (dCD133KDEL) was synthesized using an anti-CD133 scFv reactive against loop two of the extracellular domain of CD133. This scFv was taken from the monoclonal antibody that has been shown to recognize both the glycosylated and unglycosylated forms of CD133 (18). This scFv was then cloned onto the same molecule containing a truncated form of pseudomonas exotoxin A (PE38) that has been successfully established as a clinically useful toxin (19). Studies show that fewer than 1000 molecules of PE38 delivered to the cytosol are sufficient to bring about cell death (20). In addition, we added a Lys-Asp-Glu-Leu (KDEL) C-terminus signal to our drug that provides enhanced tumor killing by preventing luminal ER protein secretion (21). Also, genetic engineering was used to address a major shortcoming of targeted toxins, their immunogenicity. To accomplish this, the PE toxin was mutated to remove immunogenic epitopes that were recently mapped (22) Studies show that three separate TT made with this deimmunized variant have highly reduced anti-toxin levels in mice despite multiple treatments with the drug compared to the non-mutated parental form (23, 24, 25).
In this paper, we show that dCD133KDEL specifically kills CD133+ tumor initiating cells and can arrest the proliferation of head and neck carcinoma cells in vitro and in vivo. dCD133KDEL is a novel deimmunized TT that target tumor initiating cells and is effective at preventing tumorigenesis and treating established tumors. This represents a significant and highly selective tool that can be utilized to study stem cell populations and as a possible clinical adjunct to chemotherapy.
Materials and Methods
Construction of dCD133KDEL
dCD133KDEL was synthesized from the fusion of DNA fragments encoding the scFV portion (anti-CD133scFV) of clone 7 (18) and a deimmunized, truncated form of pseudomonas exotoxin 38 (23, 24, 25). The assembled fusion gene contained (5' to 3') an NcoI restriction site, the ATG initiation codon, the gene for CD133 scFV, the DNA sequence encoding a seven amino-acid EASGGPE linker, the gene encoding for the first 362 amino acids of truncated deimmunized with the DNA sequence for KDEL replacing the REDLK at the c-terminus, followed by a NotI restriction site at the 3' end of the fusion gene. The resulting 1846 base pair gene was spliced into the pET28c bacterial expression vector containing an inducible isopropyl-b-D-thiogalactopyranoside T7 promoter and a kanamycin selection gene (Figure 1A). To verify that the dCD133KDEL gene had been cloned correctly and in frame, DNA sequence analysis was performed at the University of Minnesota BioMedical Genomics Center. The CD133scFV was separately cloned into the pET28c bacterial expression vector and produced to determine CD133 expression of various cell lines in flow cytometry studies.
Figure 1.

A) Plasmid map for dCD133KDEL shows the gene position. B) A large single peak of protein detected at an absorbance of 280 nm was collected and then analyzed by SDS-PAGE under non-reducing conditions. The gel lanes from left to right are: 1) PE38KDEL 7mut, 2) CD133 scFV, 3) dCD133KDEL, and 4) Molecular weight standard. C) Flow cytometry study showing that the anti-CD133 scFV recognizes the CD133 receptor. HEK cells were transfected with a gene encoding the CD133 receptor and then reacted with the anti-CD133 scFv labeled with FITC. As controls, cells were also reacted with RFB4 (anti-B cells)-FITC or a no treatment blank. Flow cytometry was performed using a FACS Caliber and data plotted as a histogram. D) CD133 expression was measured on various human carcinoma lines. The anti-CD133scFV was tagged with FITC and then reacted with the head and neck cancer cell lines NA-SCC and UMSCC-11B. The Caco-2 colorectal carcinoma was included as a positive control cell line since it known to overexpress CD133. Controls included cells reacted with anti-EpCAM-FITC, anti-CD45-FITC, and anti-CD19-FITC. Expression of CD45 and CD19 is mostly restricted to normal malignant hematopoietic cells and thus served as negative controls.
Purification of CD133scFV and dCD133KDEL
Purification of CD133 scFV and dCD133KDEL was performed as described previously (26). Briefly, each protein was expressed and purified from inclusion bodies using a Novagen pET expression system (Novagen, Madison WI) followed by a 2-step purification consisting of ion exchange fast protein liquid chromatography (Q sepharose Fast Flow, Sigma) and size exclusion chromatography (Hiload Superdex 200, Pharmacia). The purified protein was then analyzed by SDS-PAGE and stained with Commasie Brilliant Blue to determine purity.
Cell Lines and Culturing Technique
UMSCC-11B is a squamous cell carcinoma cell line that was derived from larynx tumor following chemotherapy (27). UMSCC11B-luc were transfected using a luciferase reporter construct, and were maintained under 10ug/ml of blastocidin. Cells were transfected using Invitrogen's Lipofectamine™ Reagent. NA-SCC is another squamous cell carcinoma line isolated from a tongue tumor (28). Both lines were obtained from Dr. Frank Ondrey (University of Minnesota) who originally obtained them from their originator, Dr. Thomas E. Carey, Dept. of Otolaryngology-Head and Neck Surgery, University of Michigan in 2009. NA and UMSCC cell lines were authenticated this year by STR testing performed by the Fragment Analysis Facility, John Hopkins University. Caco-2 (a colorectal carcinoma) and HEK293 (a human embryonic kidney cell line) were obtained from ATCC and have not been authenticated, but were positive for the appropriate markers. Only cells that were greater than 90% viable were used for experimentation.
Flow Cytometry and CD133+ Cell Enrichment
Flow cytometry was performed using a FACS Caliber at the University of Minnesota's Flow Cytometry Core Facility. FITC-labeled antibodies were used and results were analyzed using FLOWJO. Sorting was performed using a magnetic bead selection kit following manufacturers instructions (Stem Cell Technologies, Tukwila, WA). Briefly, cells were concentrated to 2 × 108cells/mL. FITC-labeled CD133scFV was added to cells at a concentration of 1.0ug/mL and incubated for 15 minutes. The EasySep FITC Selection Cocktail® was then added followed by the EasySep® Magnetic Nanoparticles after a 15 minute incubation. The cells were mixed and then placed within the magnet. Unbound cells were eluted and bound cells collected.
Time Course Viability Assays
Trypan blue viability assays were performed by plating 10,000 cells/well into 24 well plates. Toxin and media were replaced daily at 0.01nM for NA-SCC cells or 0.03nM for UMSCC-11B cells. Wells were harvested every other day using typsin digestion and counted on a hemoctyometer via trypan blue staining. Untreated wells typically became confluent around day 8. For the time course viability assays on CD133 negative sorted cells, UMSCC-11B cells were incubated with CD133 scFV-FITC and sorted using a FACS ARIA at the University of Minnesota's Flow Cytometry Core Facility. 20,000 cells/well were sorted into 24 well plates. The rest of the time course viability assay was performed as described above.
Hematopoietic Colony-Forming Unit Assays
Two types of assays were performed: short-term (2 week) hematopoietic colony-forming assays (CFC) and long-term (5 week) culture assays (LT-CIC) (29). For CFC assays, progenitor cells were sorted from human umbilical cord blood (UCB). CD34+ cells were sorted using magnetic selection, and collected in MethoCult GF+ H4435 (StemCell Technologies, Vancouver, BC, Canada; Cat. No. 04435) consisting of 1% methylcellulose, 30% FBS, 1% BSA, 50ng/ml stem cell factor, 20ng/ml granulocyte–macrophage colony-stimulating factor, 20ng/ml interleukin (IL)-3, 20ng/ml, IL-6, 20ng/ml granulocyte colony stimulating factor, and 3U/ml erythropoietin.. Targeted toxin was added at the desired dosages and cultures were incubated at 37°C, 5% CO2 for 2 weeks and then scored for colony-forming units according to standard criteria after 2 weeks. CFU-GEMM, CFU-GM, and BFU-E were counted individually. For LT-CIC assays, were cultured in commercial Myelocult media from StemCell Technologies with M2-10B4 stromal cells. Colonies were quantitated after 5 weeks.
Tumor Initiation Experiment
For the tumor initiation study, UMSCC-11B squamous carcinoma cells were injected into the right flank of nude mice at a concentration of 300,000 cells/mouse. Tumor size was measured using digital calipers and tumor volume (width × length × height) was calculated. Prior to injection cells were treated with 0.03nM of either dCD133KDEL or the non-specific toxin, CD22KDEL. A third group of cells was left untreated and unsorted. For the sorted group, cells were sorted for CD133 expression with a magnetic bead kit as previously described, plated overnight, harvested, and injected the following day.
Tumor Treatment Study and Imaging
For experiment 1, nude mice were injected with 3 million UMSCC11B-luc cells into the right flank. Tumors were treated with dCD133KDEL, CD3CD3KDEL, or PBS starting on day 8 when they had become palpable. A single course of treatment consisted of an injection of 20ug of drug given every other day (MWF) and mice were given a total of 8 injections. Controls were treated with PBS. Tumor volume was measured with calipers over time. For experiment 2, nude mice were injected with 6 million UMSCC-11B-luc cells in the right flank. Tumors were treated starting on day 10 post-inoculation. Treatment was done as in experiment 1, except mice received 16 injections. Also, an anti-B cell control, CD19KDEL, was used. Tumor volume was calculated prior to treatment. Since tumors were marked with a luciferase reporter gene, mice were imaged to determine their bioluminescent activity as described previously (23). Briefly, mice were injected with 100ul of 30mg/ml luciferin substrate 10 minutes prior to imaging and then anesthetized via inhalation of isoflurane gas. The mice were then imaged using the Xenogen Ivis 100 imaging system and analyzed with Living Image 2.5 software (Xenogen Corporation, Hopkington MA). Five-minute exposures were made and units for the regions of interest (ROI) were expressed as photons/sec/cm2/sr. For experiment 1, tumor treatment was stopped on day 24. For experiment 2, treatment was stopped on day 45.
Histology
Suspected tumor free survivors were sacrificed and for histology evaluation at the termination of the experiment. Liver, Kidney, and skin (from the tumor site) samples were taken. These samples were embedded in OCT compound (Miles, Elkhark, IN), snap frozen in liquid nitrogen, and stored at −80°C until sectioning. Sections of the tissues were cut, thawed, and mounted on glass slides. Slices were fixed for 5 minutes in acetone and then stained with Hematoxylin and Eosin (H&E).
Determining immunogenicity of deimmunized CD133KDEL
To determine whether mutated CD133KDEL elicited less of an anti-toxin response than non-mutated parental toxin, female Balb/c (n=5/group) were injected intraperitoneally once weekly with 0.25 μg of either deimmunized CD133KDEL or non-muted parental PE38KDEL toxin. Each week, five days after injection, the mice were bled via face vein. Serum was isolated using centrifugation, frozen, and the level of anti-PE38KDEL IgG in each serum sample was measured by ELISA. Briefly, PE38KDEL was bound 96-well microtiter plates at a concentration of 5ug/ml overnight at 4°C. Unbound protein was washed away with PBS-T and blocking was performed for 1 h with 5% milk/PBS-T. Serum samples were diluted appropriately and 100 μl of each was added to wells in triplicate. After 3 hours, each well was washed with PBS-T. Peroxidase-conjugated rabbit anti-mouse IgG (Sigma) was added to each well. After 2 hours samples were washed and o-Phenylenediamine dihydrochloride substrate was added. After 30 min, the absorbance at 490 nm was measured using a microplate reader. Quantification of actual anti-PE38KDEL IgG present in each sample was determined by comparing the absorbance values in each well to a standard curve prepared using M40-1 monoclonal anti-PE38KDEL antibody from Dr. Robert Kreitman (NIH, Bethesda, MD).
Results
Anti-CD133 scFv construct binds selectively
SDS-PAGE analysis showed that following ion exchange and size exclusion chromatography, dCD133KDEL was greater than 95% pure (Figure 1B).
To establish that the anti-CD133 scFv construct recognized the CD133 receptor, HEK293 cells were transfected with a gene encoding the CD133 receptor. Figure 1C is a histogram plot showing a high degree of reactivity of the anti-CD133-FITC with the transfected cells. Anti-CD22-FITC, however, did not bind the transfected cells. Interestingly, it appears that there is a high intensity and a low intensity peak indicating populations with high and low antigen densities. Figure 1D shows the expression of CD133 on various cell populations measured by flow cytometry. UMSCC-11B and NA-SCC cell lines were 4.0% and 5.9% CD133+, respectively. These cells were tested with anti-CD19-FITC as a negative control and were less than 1.2% CD19+. Caco-2, a human colorectal cell line, was tested as positive control and was 79% positive for CD133 expression. These numbers are from representative experiments that have been duplicated at least 3 times each. Together, these findings showed that the CD133 scFV is specific to the CD133 receptor that is selectively expressed on a small subpopulation of cancer cells.
Time Course Viability Assays Show Proliferation Inhibition
If CD133 is a marker for CSC, then selective killing of this subpopulation of cells should be sufficient to inhibit proliferation (3). To test this, dCD133KDEL was added to cells in a trypan blue viability time course assay to determine the number of viable cells. As seen in Figure 2A, the proliferation of UMSCC-11B cells was almost completely ablated, while a non-specific control toxin targeting CD22 did not inhibit cell proliferation. dCD133KDEL had the same effect on NA-SCC cells, another head and neck carcinoma line (Figure 2B). To determine whether dCD133KDEL was having off-target effects, we sorted UMSCC-11B cells for CD133− cells and performed a time course viability assay. As seen in Figure 2C, the targeted toxin showed no effect on the CD133− sorted cells. These plots show representative experiments that have each been reproduced.
Figure 2.

Trypan blue viability studies show that dCD133KDEL selectively inhibits head and neck cancer cells over time UMSCC-11B (A) or NA-SCC (B) cells were incubated with dCD133KDEL and viability was determined over time by directly counting cells growing in tissue culture wells using a trypan blue vital stain. Live cells exclude the dye and dead cells incorporate it. CD22KDEL, CD19KDEL (anti-B cell) and CD3CD3KDEL (anti-T cell) TT were included as negative controls. Media and toxin were replenished daily. In C, UMSCC-11B cells were sorted for CD133- cells and immediately incubated with dCD133KDEL to determine non-specific killing. dCD133KDEL did not inhibit CD133- UMSCC-11B cells treated immediately following sorting. Error bars were calculated for all data points, but are not visible in some instances since error margins were very slight.
Effect on Human Progenitor Colony Formation
CD34+ UCB cells are considered a rich source of normal hematopoietic progenitor cells. To determine the effect of dCD133KDEL on the development of hematopoietic cells derived from CD34+ progenitors, sorted CD34+ cells were treated with drug and evaluated in two standardized assays of hematopoietic stem/progenitor cell survival and function. Notably, the UCB CD34+ cells used for this study were first studied using 2-color flow cytometry and 67% coexpressed CD34 and CD133, twenty-four percent were CD34+CD133− and none of the cells were CD34−CD133+ (data not shown). dCD133KDEL drug concentrations were selected based on the results in Figure 2. Initially, studies of hematopoietic colony formation as a measure of hematopoietic progenitor cells were done by incubating the UCB CD34+ cells with dCD133KDEL Media and drug were renewed weekly in each well. After 2 weeks, hematopoietic colonies were counted. Figure 3 shows that only the cells cultured in the presence of intact DT toxin underwent significant reductions in colony formation. Even at concentrations of dCD133KDEL that greatly exceeded those used to inhibit cancer stem cells in the trypan blue viability assays, there was minimal effect on CFU-GM, CFU-GEMM, and BFU-E colony development. Additionally, we added dCD133KDEL to 5-week long-term culture initiating cell (LT-CIC) assays that better quantify putative hematopoietic stem/progenitor cells. As with the CFC study, there was minimal effect of dCD133KDEL on human LT-CICs after 5 weeks in culture. These experiments were reproduced with similar results (data not shown). Together, these results suggest that dCD133KDEL is not destructive to normal human hematopoietic stem/progenitor cells that may express CD133.
Figure 3.
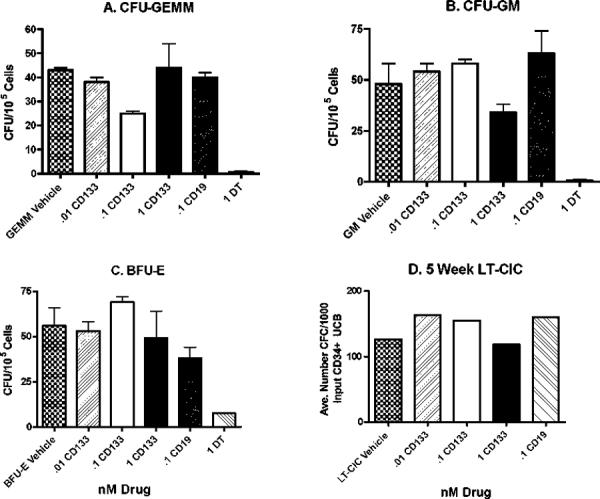
dCD133KDEL does not inhibit hematopoietic colony development. CD34+ sorted umbilical cord cells were cultured in the presence of dCD133KDEL under optimized colony growth conditions. Two types of assays were performed over the entire course of the culture period. The first was short-term (2 week) colony formation assay measuring (A) CFU-GEMM, (B) CFU-GM, and (C) BFU-E. The second was a long-term (5 week) LTC-IC Assay (D). Colonies were visually scored in a blinded study.
CD133+ Cells Efficiently Initiate Tumors
CD133+ CSC possess the unique ability to initiate tumors (16, 17). Thus, a tumor initiation study was performed to determine whether cells pretreated with dCD133KDEL would form tumors in vivo (Figure 4). UMSCC-11B cells were pretreated with 0.03nM of dCD133KDEL for 6 days prior to inoculation in the flanks of nude mice. Control mice were injected with either untreated cells or cells treated with a non-specific toxin targeting CD3. UMSCC-11B cells were also enriched for CD133+ cells using magnetic bead separation prior to inoculation. When cells were sorted using the magnetic bead separation and run using flow cytometry, the expression of CD133+ cells increased 4 fold (data not shown). As seen in Figure 4, cells treated with dCD133KDEL prior to injection had the lowest incidence of tumor formation (1 of 4). The single tumor that formed was markedly smaller than tumors formed by the control or CD133 enriched cells. CD133+ enriched cells formed tumors at the highest incidence, formed earlier and grew much larger than control tumors. These results further indicate that we are killing CSC and supports the fact that CD133 is a CSC marker in head and neck carcinomas.
Figure 4.

Study of the tumor initiation ability of CD133+ cells, a hallmark of tumor stem cells. Groups of nude mice (4–8/group) were injected in the flank with CD133-enriched cells. CD133+ cells were enriched from cultured UMSCC-11B cells using magnetic bead separation. These had the highest tumor incidence and rate of growth. Another group received cells treated with dCD133KDEL prior to implantation. These had the lowest incidence and lowest growth rate. The controls received unsorted tumor cells that were either untreated or pre-treated with an irrelevant control targeted toxin, CD3CD3KDEL. All groups received 300,000 cells. The plot shows the tumor growth rate over time and the table within the figure shows the tumor incidence. The mean tumor volume of the CD133+ sorted group is statistically different than the control group and the dCD133-depleted group using the student T tests (p< 0.0045 and 0.0012, respectively).
Tumor Treatment Studies Show Impressive Tumor Regression
In experiment 1, animals with palpable tumors were given intratumoral injections of 20ug/injection of dCD133KDEL. Visual measurement of tumor progression with calipers showed that tumors regressed and were gone by day 24 when treatment was stopped and conttol tumors progressed (Figure 5A). The same animals were imaged in real time and the images showed that control tumors given either PBS (M1–M3) or treated in an identical fashion with anti-T cell CD3CD3KDEL (M4, M5) did not regress (Figure 5B). No tumor was detected by day 24 in treated mice indicating that real time imaging correlated with caliper data. In experiment 2 (Figure 5C), animals were given twice the number of tumor cells, so tumors grew faster, and treatment was delayed until day 10. Although tumors grew more aggressively, all mice treated with dCD133KDEL responded to treatment. Mice treated with control CD19KDEL did not. Together, the combined experiments showed that by killing the CD133+ subpopulation of cells, the tumors were unable to generate more cells and eventually regressed.
Figure 5.

Tumor treatment studies show tumor regression. In A, tumor size was measured for Experiment 1 using digital calipers. A student's paired T test was performed on the average tumor size over the course of the experiment between the control and treated groups. The two curves are significantly different with a p value of 0.0001. Experiment 1 (B) and Experiment 2 (C) are shown with tumor bioluminescent and images showing that dCD133KDEL inhibits the progression of UMSCC-11B-luc nude mice flank tumors. For Experiment 1 (B), 3 million cells were injected and tumors were treated with dCD133KDEL (M6–M10) while controls were treated intratumorally with either PBS (M1–M3) or the anti-T CD3CD3KDEL (M4–M5) starting on day 8. This also shows that dCD133KDEL inhibits the progression of flank tumors and correlates with the caliper data. The arrows indicate where treatment (Tx) began and ended. For experiment 2 (C), tumors were induced by injection of 6 million cells. Intratumoral treatment was begun on day 10. Tumors were treated exactly as they were in experiment 1 except mice were given 16 injections instead of 8. Controls were untreated or treated with anti-B cell CD19KDEL. Animals were imaged weekly for both experiments. Bioluminescence intensity was measured. D) Histology photomicrograph shows skin tissue taken from the tumor injection site of a representative tumor-free mouse from Experiment 1. As a control, a photo is shown of skin tissue taken from a PBS-treated control mouse. Tumor is clearly visualized in controls, but absent in the drug treated mouse.
Histological analysis was performed on tumors from experiment 1. Figure 5D shows a skin section from a mouse, taken on day 59, in which an epithelial-like tumor is prominent. The tumor is robust and vascularized. Other sections revealed necrosis. Also shown is healthy skin from a mouse that was treated intratumorally with dCD133KDEL, which prevented tumor growth and eventually led to complete tumor elimination. The skin pathology looks mostly normal with intact hair follicles, although some evidence of inflammation is still present. The section is part of the histological analysis that confirms the animal was tumor free. The livers and kidneys of both treated and non-treated animals showed no evidence of tumor indicating that it did not metastasize to these organs.
Anti-toxin Levels Greatly Reduced in dCD133KDEL Immunized Mice
To ensure that we had effectively deimmunized dCD133KDEL, BALB/c mice were immunized with either mutated dCD133KDEL or parental non-mutated PE38KDEL. Mice (n=5/group) were immunized intraperitoneally once a week and were bled on day 63. Despite 9 weekly immunizations with drug (0.25 ug/injection), the dCD133KDEL group had significantly lower serum anti-toxin levels than the animals immunized with parental toxin (Figure 6). Generally, deimmunization resulted in an average 90% reduction in anti-toxin levels.
Figure 6.
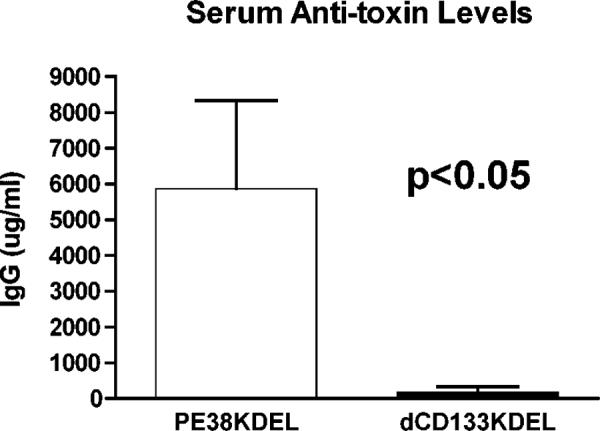
The immunogenicity of dCD133KDEL. The comparative ability of mutated dCD133KDEL and non-mutated parental toxin PE38KDEL to induce the production of anti-toxin antibodies in normal, immunocompetent mice was determined by measuring anti-PE38(KDEL) serum IgG levels on samples of mice immunized weekly with 0.25 μg of either drug (n=5 group). Animals were bled on day 63 after 9 weekly immunizations. Serum IgG anti-toxin levels were measured by indirect ELISA and quantification of antibodies was determined using a standard curve. Differences in the data sets between the PEKDEL-immunized and the dCD133KDEL groups were significant by Student T test (p<0.05).
Discussion
The major contributions of this manuscript are the development of a novel targeted toxin, dCD133KDEL, which targets CD133 tumor-initiating cells and its efficacy against two head and neck carcinoma lines. dCD133KDEL is a new and powerful reagent for three main reasons. The drug 1) targets only a small subpopulation of the total tumor, 2) has been successfully deimmunized bypassing a major clinical problem with targeted toxins, and 3) has a mechanism of action unlike typical chemotherapy agents. Targeted toxins function by binding to cell surface receptors, internalizing, and enzymatically inhibiting protein synthesis (30). CD133 is readily internalized rendering it an excellent marker for a targeted toxin (14). The fact that we observed heterogenous peaks in CD133 expression in transfected HEK cells in Figure 1C supports the argument that CD133 cycles and internalizes.
Bonnet and Dick reported the first CSC subpopulation in leukemia in 1997 (31). Since then investigators have validated the presence of CSC subpopulations not only in leukemia, but in many carcinomas as well. CD133 in particular has been identified as a CSC marker in these carcinomas (6, 10–13). Wei et al showed in their work that CD133 is a CSC marker for the head and neck laryngeal cancer line HEP-2. In their study, they showed that CD133+ sorted cells initiated tumors and uniquely possessed clonogenic capacity when compared with CD133− cells (16). We were able to confirm CD133 as a marker for tumor initiation cells in the current study with a different head and neck cell line UMSCC-11B.
Also unique to our study is the use of a new monoclonal antibody (clone 7) that binds all forms of the CD133 receptor (18). This is important because commercial antibodies currently available recognize an epitope that can be masked upon differentiation (32). In contrast, the scFV from clone 7 was used in our construction of dCD133KDEL recognizes only the CD133 peptide backbone, avoiding the issue of epitope masking or differential glycosylation. Specificity was determined by showing that our anti-CD133 scFV recognized cells transfected with DNA encoding the CD133 receptor. Furthermore, two head and neck cell lines contained subpopulations of CD133 expressing cells at similar levels to other head and neck cell lines described in the literature (16, 17, 33). When dCD133KDEL was tested in vitro, we discovered that it selectively inhibited cancer cell expansion in both cell lines. “We found that about 5% of the UMSCC-11B head and neck cancer cells used in the study were Annexin V positive following CD133KDEL treatment indicating apoptotic death. Since flow studies also showed that about 5% of the UMSCC-11B population were CD133+, this suggests an apoptotic death for CD133 cells. We also attempted dual staining to confirm that the same population that was Annexin positive was CD133 positive. However, these mechanistic studies proved complicated since earlier treatment with dCD133KDEL interfered with our ability to later recognize and quantitate CD133+ cells. We attributed this difficulty to either blocking interference or the high modulation rate of CD133.”
Since tumor initiation is a hallmark of CSC, UMSCC-11B tumor cells were pretreated with dCD133KDEL and transplanted into nude mice. These cells formed the lowest incidence of tumors when compared with controls, while cells enriched for CD133+ expression formed the largest tumors at the fastest rate. Furthermore, in two separate studies tumor cells were injected into the flanks of nude mice and when palpable were directly treated with dCD133KDEL. All treated tumors regressed over time, unlike control tumors and all treated animals were impressively tumor free after 79 days. We favor the explanation that dCD133KDEL acts to inhibit the self-renewing ability of CD133+ CSC. However, it is possible that dCD133KDEL has a bystander killing effect but this was not supported by our in vitro studies showing that dCD133KDEL did not kill CD133 negative cancer cells.
Several studies suggest the existence of another CSC population in some tumors that is CD133− (34, 35). For example, Chen et al discovered CD133− cells within glioblastoma primary tumors that had the capability to self-renew and support long-term growth in vitro and initiate tumors in vivo (35). dCD133KDEL may be a helpful biological tool in validating the existence of these cells and in helping to develop more inclusive therapies that may target all CSC and progenitors simultaneously.
The development of a novel TT that selectively targets CSC may have unique implications for the ubiquitous problem of carcinoma drug resistance and subsequent relapse. Several studies have shown that CSC are resistant to current chemotherapeutic agents (5, 6, 10, 36, 37). Since TT's work by a different mechanism and because it selectively kills CSC, dCD133KDEL may possess the unique ability to target the very cells responsible for drug resistance. Adding a TT as an adjunct to chemotherapy has been shown to be more effective than using either therapy alone (38, 39). Studies are underway to determine whether dCD133KDEL may be a useful adjunct to chemotherapy.
One of the major limitations of TT in clinical therapy is immunogenicity. Patients will start developing antibodies to the toxin and thus the number of treatments is limited. To address this problem, we mutated 7 immunogenic epitopes that account for the majority of antibodies produced against this form of PE toxin. We then used this mutated construct to create our fusion protein. As a result, we showed that even after 9 immunizations there is very little antibody produced against the toxin moiety of dCD133KDEL and 40% of the animals had no antibody response at all.
Drug safety issues regarding the affects of dCD133KDEL on normal human hematopoietic progenitor cells are important and need to be addressed prior to any therapeutic consideration. Therefore, we isolated CD34+ cells from UCB an established source of normal human hematopoietic stem cells, cultured them with dCD133KDEL and then determined their ability to form various hematopoietic colonies in established CFU (colony-forming unit) assays. To be sure, we changed media containing drug weekly and cultured the cells in long-term colony initiation assays for 5 weeks. Only toxin alone inhibited survival of hematopoietic progenitor cells. These results indicate that although the UCB CD34+ cells co-express CD133, these cells are resistant to treatment with dCD133KDEL These findings could be explained by multiple normal stem cell populations that are CD133− since 24% of the starting stem cells were CD34+CD133−. Rutella et al discovered a stem cell population in human cord blood that was CD133− and still capable of differentiating into multiple cell types (40). Also, Surronen et al showed that CD133+ cells can be generated from normal CD133− cells (41). Perhaps dCD133KDEL destroyed normal CD133+ cells and replacements emerged from the CD133− fraction. Alternatively, CD34+CD133+ cells may be metabolically quiescent enough or have other drug resistance mechanism to prevent significant killing by dCD133KDEL. Finally, the expression level of CD133 on tumor cells may be greater than the expression level on normal progenitors (42). This could allow for the observed selective killing of CSC over normal stem cells. Sehl et al, used mathematical modeling to conclude that in order for a treatment to be safe it must be highly selective and able to target quiescent cancer stem cells (43) and since dCD133KDEL is selective and because cells do not have to be actively cycling for targeted toxins to be taken up and effectively cause apoptosis, dCD133KDEL warrants further investigation.
Several cell populations in the body express CD133 and therefore may be potential targets for the toxin, causing either short-term or long-term health effects. Thus, extensive toxicity studies will be necessary to validate a CD133 targeted drug for clinical use. These studies will not be trivial. CD133 expression on normal cells has proven somewhat complicated since CD133 has been shown to undergo post-translational modulation such that mRNA and internal protein expression does not correlate with the surface expression (44). Also, the commercial antibodies such as AC133 only bind certain forms of the receptor (18,32). Furthermore, studies in an “ontarget” model closer to humans than rodents may be necessary.
In summary, we developed a novel deimmunized TT that selectively binds the cancer stem cell marker CD133. dCD133KDEL impressively inhibited cell proliferation in vitro, decreased tumor initiation, does not kill normal human hematopoietic progenitor cells, and caused complete tumor regression in a an accepted model of human head and neck cancer which is an xenotransplant flank model. This work represents the development of a new cancer therapeutic that function by selectively targeting the minority cancer stem cell subpopulation within the tumor. We believe dCD133KDEL warrants further study as a possible solution for drug resistant relapse in human carcinoma.
Acknowledgements
This work was supported in part by the US Public Health Service Grants RO1-CA36725, R01 HL077923 awarded by the NCI and the NIAID, DHHS, the Randy Shaver Foundation, the Lion's Children's Cancer Fund, and the William Lawrence and Blanche Hughes Fund.
Financial Support: This work was supported in part by the US Public Health Service Grants RO1-CA36725, R01 HL077923 awarded by the NCI and the NIAID, DHHS, the Randy Shaver Foundation, the Lion's Children's Cancer Fund, and the William Lawrence and Blanche Hughes Fund.
Abbreviations
- dCD133KDEL
deimmunized pseudomonas exotoxin fused to anti-CD133 scFv with a KDEL terminus
- aa
amino acid
- Ab
antibody
- CD3
cluster determinant 3
- CD19
cluster determinant 19
- CD22
cluster determinant 22
- DT390
truncated diphtheria toxin
- ER
endoplasmic reticulum
- FITC
fluorescein isothiocyanate
- KDEL
Lys-Asp-Glu-Leu
- TT
Targeted toxin
- KDEL
amino acid sequence Lys-Asp-Glu-Leu
- mAb
monoclonal antibody
- PE
pseudomonas exotoxin
- photons/sec/cm2/sr
photons per second per square centimeter per steradian
- scFv
recombinant single chain VH and VL domain
Footnotes
Conflict of Interest: None
References
- 1.Deonarain MP, Kousparou CA, Epenetos AA. Antibodies targeting cancer stem cells: a new paradigm in imunotherapy? MAbs. 2009;1:12–25. doi: 10.4161/mabs.1.1.7347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Al-Hajj M, Clarke MF. Self-renewal and solid tumor stem cells. Oncogene. 2004;23:7274–82. doi: 10.1038/sj.onc.1207947. [DOI] [PubMed] [Google Scholar]
- 3.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 4.Gupta PB, Chaffer CL, Weinberg RA. Nat Med. 2009;15:1010–2. doi: 10.1038/nm0909-1010. [DOI] [PubMed] [Google Scholar]
- 5.Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J of Clin Onc. 2008;26:2839–2845. doi: 10.1200/JCO.2007.15.1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Boman BM, Wicha MS. Cancer Stem Cells: A Step Toward the Cure. J Clin Oncol. 2008;26:2795–9. doi: 10.1200/JCO.2008.17.7436. [DOI] [PubMed] [Google Scholar]
- 7.Yin AH, Miraglia S, Zanjani ED, Almeida-Porada G, Ogawa M, Leary AG, et al. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997;90:5002–12. [PubMed] [Google Scholar]
- 8.Peichev M, Naiyer AJ, Pereira D, Zhu Z, Lane WJ, Williams M, et al. Expression of VEGFR-2 and AC133 by circulating human CD34(+) cells identifies a population of functional endothelial precursors. Blood. 2000;95:952–8. [PubMed] [Google Scholar]
- 9.Uchida N, Buck DW, He D, Reitsma MJ, Masek M, Phan TV, et al. Direct isolation of human central nervous system stem cells. Proc Natl Acad Sci U S A. 2000;97:14720–5. doi: 10.1073/pnas.97.26.14720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fabrizi E, di Martino S, Pelacchi F, Ricci-Vitiani L. Therapeutic implications of colon cancer stem cells. World J Gastroenterol. 2010;16:3871–7. doi: 10.3748/wjg.v16.i31.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harper LJ, Piper K, Common J, Fortune F, Mackenzie IC. Stem cell patterns in cell lines derived from head and neck squamous cell carcinoma. J Oral Pathol Med. 2007;36:594–603. doi: 10.1111/j.1600-0714.2007.00617.x. [DOI] [PubMed] [Google Scholar]
- 12.Wright MH, Calcagno AM, Salcido CD, Carlson MD, Ambudkar SV, Varticovski L. Brca1 breast tumors contain distinct CD44+/CD24− and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 2008;10:R10. doi: 10.1186/bcr1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Du Z, Qin R, Wei C, Wang M, Shi C, Tian R, et al. Pancreatic cancer cell resistant to chemoradiotherapy rich in “stem-cell-like” tumor cells. Dig Dis Sci. 2011;56:741–50. doi: 10.1007/s10620-010-1340-0. [DOI] [PubMed] [Google Scholar]
- 14.Fitzgerald D, Pastan I. Targeted toxin therapy for the treatment of cancer. J Natl Cancer Inst. 1989;81:1455–63. doi: 10.1093/jnci/81.19.1455. [DOI] [PubMed] [Google Scholar]
- 15.Pavon LF, Gamarra LF, Marti LC, Amaro Junior E, Moreira-Filho CA, Camargo-Mathias MI, et al. Ultrastructural characterization of CD133+ stem cells bound to superparamagnetic nanoparticles: possible biotechnological applications. Cell Reprogram. 2010;12:391–403. doi: 10.1111/j.1365-2818.2008.02049.x. [DOI] [PubMed] [Google Scholar]
- 16.Wei XD, Zhou L, Cheng L, Tian J, Jiang JJ, Maccallum J. In Vivo investigation of CD133 as a putative marker of cancer stem cells in HEP-2 cell line. Head Neck. 2009;31:94–101. doi: 10.1002/hed.20935. [DOI] [PubMed] [Google Scholar]
- 17.Friedman S, Min L, Schultz A, Thomas D, Lin RY. CD133+ Anaplastic thyroid cancer cells initiate tumors in immunodeficient mice and are regulated by thyrotropin. PLoS One. 2009;4:e5395. doi: 10.1371/journal.pone.0005395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swaminathan SK, Olin MR, Forster CL, Cruz KS, Panyam J, Ohlfest JR. Identification of a novel monoclonal antivody recognizing CD133. J Immunol Methods. 2010;361:110–5. doi: 10.1016/j.jim.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 19.Kreitman RJ, Stetler-Stevenson M, Margulies I, Noel P, Fitzgerald DJ, Wilson WH, et al. Phase II trial of recombinant immunotoxin RFB4(dsFv)-PE38 (BL22) in patients with hairy cell leukemia. J Clin Oncol. 2009;27:2983–90. doi: 10.1200/JCO.2008.20.2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kreitman RJ, Pastan I. Accumulation of a recombinant immunotoxin in a tumor in vivo: fewer than 1000 molecules per cell are sufficient for complete responses. Cancer Res. 1998;58:968–75. [PubMed] [Google Scholar]
- 21.Kreitman RJ, Pastan I. Importance of the glutamate residue of KDEL in increasing the cytotoxicity of Pseudomonas exotoxin derivatives and for increased binding to the KDEL receptor. Biochem J. 1995;307:29–37. doi: 10.1042/bj3070029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Onda M, Beers R, Xiang L, Nagata S, Wang QC, Pastan I. An immunotoxin with greatly reduced immunogenicity by identification and removal of B-cell epitopes. Proc Natl Acad Sci U S A. 2008;105:11311–6. doi: 10.1073/pnas.0804851105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stish BJ, Oh S, Chen H, Dudek AZ, Kratzke RA, Vallera DA. Design and modification of EGF4KDEL 7mut, a novel bispecific ligand-directed toxin, with decreased immunogenicity and potent anti-mesothelioma activity. Br J Cancer. 2009;101:1114–23. doi: 10.1038/sj.bjc.6605297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsai AK, Oh S, Chen H, Shu Y, Ohlfest JR, Vallera DA. A novel bispecific ligand-directed toxin designed to simultaneously target EGFR on human glioblastoma cells and uPAR on tumor neovasculature. J Neurooncol. 2011;103:255–66. doi: 10.1007/s11060-010-0392-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vallera DA, Oh S, Chen H, Shu Y, Frankel AE. Bioengineering a unique deimmunized bispecific targeted toxin that simultaneously recognizes human CD22 and CD19 receptors in a mouse model of B-cell metastases. Mol Cancer Ther. 2010;9:1872–83. doi: 10.1158/1535-7163.MCT-10-0203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vallera DA, Chen H, Sicheneder AR, Panoskaltsis-Mortari A, Taras EP. Genetic alteration of a bispecific ligand directed toxin targeting human CD19 and CD22 receptors resulting in improved efficacy against systemic. Leuk Res. 2009;33:1233–42. doi: 10.1016/j.leukres.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Worsham MJ, Chen KM, Meduri V, Nygren AO, Errami A, Schouten JP, et al. Epigenetic events of disease progression in head and neck squamous cell carcinoma. Arch Otolaryngol Head Neck Surg. 2006;132:668–77. doi: 10.1001/archotol.132.6.668. [DOI] [PubMed] [Google Scholar]
- 28.Abu-Ali S, Fotovati A, Shirasuna K. Tyrosine-kinase inhibition results in EGFR clustering at focal adhesions and consequent exocytosis in uPAR down-regulated cells of head and neck cancers. Mol Cancer. 2008;7:47. doi: 10.1186/1476-4598-7-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tian X, Kaufman DS. Hematopoietic development of human embryonic stem cells in culture. Methods Mol Biol. 2008;430:119–33. doi: 10.1007/978-1-59745-182-6_8. [DOI] [PubMed] [Google Scholar]
- 30.Kreitman RJ. Recombinant immunotoxins containing truncated bacterial toxins for the treatment of hematologic malignancies. BioDrugs. 2009;23:1–13. doi: 10.2165/00063030-200923010-00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonnet D, Dick J. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3:730–7. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 32.Kemper K, Sprick MR, de Bree M, Scopelliti A, Vermeulen L, Hoek M, et al. The AC133 epitope, but not the CD133 protein, is lost upon cancer stem cell differentiation. Cancer Res. 2010;70:719–29. doi: 10.1158/0008-5472.CAN-09-1820. [DOI] [PubMed] [Google Scholar]
- 33.Zhu W, Hai T, Ye L, Cote GJ. Medullary thyroid carcinoma cell lines contain a self-renewing CD133+ population that is dependent on ret proto-oncogene activity. J Clin Endocrinol Metab. 2010;95:439–44. doi: 10.1210/jc.2009-1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beier F, Beier CP, Aschenbrenner I, Hildebrandt GC, Brümmendorf TH, Beier D. Identification of CD133-/telomerase low progenitor in glioblastoma-derived cancer stem cell lines. Cell Mol Neurobiol. 2011;31:337–43. doi: 10.1007/s10571-010-9627-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen R, Nishimura MC, Bumbaca SM, Kharbanda S, Forrest WF, Kasman IM, et al. A hierarchy of self-renewing tumor-initiating cell types in glio-blastoma. Cancer Cell. 2010;17:362–375. doi: 10.1016/j.ccr.2009.12.049. [DOI] [PubMed] [Google Scholar]
- 36.Dylla SJ, Beviglia L, Park IK, Chartier C, Raval J, Ngan L, et al. Colorectal cancer stem cells are enriched in xenogeneic tumors following chemotherapy. PLoS One. 2008;3:e2428. doi: 10.1371/journal.pone.0002428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dallas NA, Xia L, Fan F, Gray MJ, Gaur P, van Buren G, 2nd, et al. Chemoresistant colorectal cancer cells, the cancer stem cell phenotype, and increased sensitivity to insulin-like growth factor-I receptor inhibition. Cancer Res. 2009;69:1951–7. doi: 10.1158/0008-5472.CAN-08-2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hassan R, Broaddus VC, Wilson S, Liewehr DJ, Zhang J. Anti–Mesothelin Immunotoxin SS1P in Combination with gemcitabine results in increased activity against mesothelin-expressing tumor xenografts. Clin Cancer Res. 2007;13:7166–71. doi: 10.1158/1078-0432.CCR-07-1592. [DOI] [PubMed] [Google Scholar]
- 39.Pearson JW, Sivam G, Manger R, Wiltrout RH, Morgan AC, Jr, Longo DL. Enhanced therapeutic efficacy of an immunotoxin in combination with chemotherapy against an intraperitoneal human tumor xenograft in athymic mice. Cancer Res. 1989;49:4990–5. [PubMed] [Google Scholar]
- 40.Rutella S, Bonanno G, Marone M, De Ritis D, Mariotti A, Voso MT, et al. Identification of a novel subpopulation of human cord blood CD34- CD133- CD7-CD45+ lineage-cells capable of lymphoid/NK cell differentiation after in vitro exposure to IL-15. J Immunol. 2003;171:2977–88. doi: 10.4049/jimmunol.171.6.2977. [DOI] [PubMed] [Google Scholar]
- 41.Suuronen EJ, Wong S, Kapila V, Waghray G, Whitman SC, Mesana TG, et al. Generation of CD133+ cells from CD133- peripheral blood mononuclear cells and their properties. Cardiovasc Res. 2006;70:126–35. doi: 10.1016/j.cardiores.2006.01.014. [DOI] [PubMed] [Google Scholar]
- 42.Smith LM, Nesterova A, Ryan MC, Duniho S, Jonas M, Anderson M, et al. CD133/prominin-1 is a potential therapeutic target for antibody-drug conjugates in hepatocellular and gastric cancers. Br J Cancer. 2008;99:100–9. doi: 10.1038/sj.bjc.6604437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sehl ME, Sinsheimer JS, Zhou H, Lange KL. Differential destruction of stem cells: implications for targeted cancer stem cell therapy. Cancer Res. 2009;69:9481–9. doi: 10.1158/0008-5472.CAN-09-2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sgambato A, Puglisi MA, Errico F, Rafanelli F, Boninsegna A, Rettino A, et al. Post-translational modulation of CD133 expression during sodium butyrate-induced differentiation of HT29 human colon cancer cells: implications for its detection. J Cell Physiol. 2010;224:234–41. doi: 10.1002/jcp.22124. [DOI] [PubMed] [Google Scholar]