

Abstract
FADD (Fas-associated protein with death domain) is a cytosolic adapter protein essential for mediating death receptor-induced apoptosis. It has also been implicated in a number of non-apoptotic activities including embryogenesis, cell-cycle progression, cell proliferation, and tumorigenesis. Our recent studies have demonstrated that high levels of phosphorylated FADD in tumor cells correlates with increased activation of the anti-apoptotic transcription factor NF-κB and is a biomarker for aggressive disease and poor clinical outcome. These findings suggest that inhibition of FADD phosphorylation is a viable target for cancer therapy. A high throughput screen using a cell-based assay for monitoring FADD-kinase activity identified NSC 47147 as a small molecule inhibitor of FADD phosphorylation. The compound was evaluated in live cells and mouse tumors for its efficacy as an inhibitor of FADD-kinase activity through the inhibition of CK1α. NSC 47147 was shown to decrease levels of phosphorylated FADD and NF-κB activity such that combination therapy lead to greater induction of apoptosis and enhanced tumor control as compared to either agent alone. The studies described here demonstrate the utility of bioluminescent cell based assays for the identification of active compounds and the validation of drug target interaction in a living subject. In addition, the presented results provide proof of principle studies as to the validity of targeting FADD-kinase activity as a novel cancer therapy strategy.
Keywords: FADD, phosphorylation, non-invasive molecular imaging, NF-κB, chemotherapy
Introduction
FADD (Fas-associated protein with death domain) was first identified as a cytosolic adapter protein essential for mediating death receptor-induced apoptosis (1–3). The FADD protein links to the cytoplasmic tail of active death receptors (DR), such as Fas, DR4, and DR5, where it binds procaspases-8 and -10, leading to the formation of the death-inducing signaling complex (DISC) at the cytosolic-side of the cell membrane. DISC formation initiates intracellular processing and activation of procaspases, which, in turn, initiates cleavage of the downstream targets, caspase-3, -6, and -7, and subsequently, apoptosis (4–6). Besides its role in regulating death receptor-induced apoptosis, FADD is also implicated in a number of death receptor-induced non-apoptotic activities including, embryogenesis, cell-cycle progression, cell proliferation, and tumorigenesis (6–10). Many of these non-apoptotic activities are determined by the level of phosphorylation of a specific C-terminal serine (Ser194) in a region distinct from the proapoptotic function related to the death domain (11–12).
Recent studies have led to a better understanding of the FADD gene and its location on chromosome 11q13.3 a hot spot for chromosomal amplification in a number of human cancers including breast, bladder, esophagus, lung, and head and neck carcinomas (13, 14). Our recent studies provide evidence of overexpression of FADD mRNA and protein in human lung adenocarcinoma and its correlation to NF-κB activation. We have also demonstrated that high-levels of phosphorylated FADD (pFADD) predominantly localized to the nucleus in lung tumor cells, is a biomarker for aggressive disease as well as for poor clinical outcome (13). The molecular basis for this correlation stems from the role of pFADD as a potent mediator of the non-apoptotic transcription factor NF-κB (13, 15), a known regulator of cell fate decisions such as, resistance to programmed cell death and lack of proliferation control (16).
Phosphorylation of FADD at serine 194 has been shown to be mediated by casein kinase Iα (CK1α) (4) and FIST-HIPK3 (FADD-interacting serine-threonine kinase/homeodomain-interacting protein kinase 3) (17) but the exact regulation and role of pFADD in cancer are not well understood. In this research, we utilize a bioluminescent cell-based assay to characterize NSC 47147 as a potent inhibitor of FADD phosphorylation and to evaluate its potential as a therapeutic agent in cancer treatment.
Materials and Methods
Antibodies and reagents
NSC 47147 was a generous gift from Ricardo Pérez-Tomás, University of Barcelona. SP600125 was purchased from Calbiochem (EMD Chemicals, San Diego, CA), CKI7 from Toronto Research Chemicals (New York On., Canada) and bortezomib from Sigma Aldrich (St. Louis, MO). The NCI Diversity Set was acquired from the NCI/DTP Open Chemical Repository (http://dtp.cancer.gov). Cisplatin was obtained from the University of Michigan pharmacy and D-luciferin was purchased from Promega (Madison, WI). Rabbit polyclonal antibodies to phospho-FADD (Ser194), phospho-c-Jun (Ser63), phospho-β-Catenin (Ser45), phospho-I-κBα (Ser32/36), β-catenin, I-κBα, caspase-3, cleaved caspase-3 (Asp175), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and mouse monoclonal antibody to c-Jun were purchased from Cell Signaling Technology (Danvers, MA). Goat polyclonal antibody to casein kinase Iα was purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and mouse monoclonal antibody to FADD from BD Pharmingen.
Cell culture
A549 (lung epithelial carcinoma), Jurkat (T lymphocyte) and SW620 (colorectal adenocarcinoma) cells were purchased from the American Type Culture Collection (ATCC). Cell cultures were maintained in a humidified incubator at 37°C and 5% CO2. A549 and Jurkat cells were grown in RPMI 1640, SW620 cells in Leibovitz’s L-15 medium. Each supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen, Carlsbad, CA) and 100 units/ml penicillin. ATCC cell lines were tested routinely for mycoplasma and purity. All ATCC lines were expanded immediately upon receipt and multiple vials of low passage cells were maintained in liquid N2. No vial of cells was cultured for more than 1–2 months. A549-FKR and SW620-BGCR cells have been previously described (18–19). A549-FKR findings were validated using freshly obtained A549 cultures from the ATCC. Cultures were maintained in a humidified incubator at 37°C and 5% CO2 and all cell culture experiments were done in serum-containing media. For in vitro and in vivo experiments, cells were removed from tissue culture dishes using 0.05% trypsin containing EDTA. Cell cultures were between 70% and 90% confluent at the time of harvest.
Western analysis
A549 and Jurkat cells were seeded at the appropriate density in six-well plates 24 hours before compound treatment. A549 cells were treated, washed twice with ice-cold PBS and lysed with extraction buffer [(1% NP40, 150 mM NaCl, 25 mM Tris (pH 8.0) supplemented with complete phosphatase and protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany)]. Cell lysates were rocked at 4°C for 30 minutes. Particulate material was removed by centrifugation at 13,000 rpm for 15 minutes at 4°C. The supernatants were collected and protein content estimated by a detergent compatible protein assay kit from Bio-Rad (Hercules, CA). Whole cell lysates containing equal amounts of protein (10–20 μg) were separated by 12% Bis-Tris polyacrylamide gels (Invitrogen, Carlsbad, CA), and transferred to PVDF membranes. The membranes were probed against specific primary antibodies followed by HRP-conjugated secondary antibodies and visualized using the Enhanced Chemiluminescence Plus Western Blotting System (GE Healthcare, Piscataway, NJ).
Bioluminescent FADD-Kinase reporter assay
The bioluminescent FADD-kinase reporter assay was executed as previously described (18). Briefly, A549 expressing FKR cells were seeded (1×105 cells/well) in opaque 96-well plates, 24-hours prior to assaying. Compound stocks were prepared in DMSO and diluted 1:100 in phosphate buffered saline. Intermediate stocks (10 μl) were added to the assay plates using the Beckman Biomek NXP Laboratory Automation Workstation (Beckman Coulter, Fullerton CA). Unless otherwise noted, cells were incubated with test compound at 37°C, 5% CO2 for 1 hour (CKI7) and 6 hours (SP600125 and NSC 47147) at the indicated concentrations. Live-cell luminescent imaging was read with an EnVision Xcite Multi-label Reader (PerkinElmer, Shelton, CT) 10 minutes after addition of D-luciferin (100 μg/ml final concentration) to the assay medium. Percent change in FKR activity was calculated as Acontrol/Asample × 100.
CK1α inhibition assays
CK1α enzymatic activity was evaluated using Lance Ultra CK2α1/β Kinase Assay (PerkinElmer, Shelton, CT) according to manufacturer’s instructions. Recombinant CK1α was purchased from ProQinase (Freiburg, Germany). Serial dilutions of NSC 47147 (1 to 100μM) and CKI7 (1 to 300 μM), were incubated with 25 nM CK1α enzyme, 50nM ULight-Topo-IIα (Thr1342) Peptide and 1 μM ATP, final concentrations of inhibitors were in 2% DMSO. Kinase reactions were terminated after 30 minutes by the addition of EDTA. Experiments were performed in triplicate and the data was derived from a minimum of three independent experiments. Percent inhibition was calculated as % inhibition = (rate no inhibitor) − (rate with inhibitor)/(rate no inhibitor) × 100.
A bioluminescent reporter cell line expressing a construct designed to monitor changes in CK1α activity recently described by our lab (SW620 BGCR), was used to provide additional evidence that NSC 47147 inhibits FADD phosphorylation through inhibition of CK1α. (19). In brief, SW620-BGCR cells were plated in 12-well plates and treated with 250 μM CKI7 and 3 μM NSC 47147 for 3 hours. Bioluminescence was acquired on an IVIS 200 imaging platform (Caliper Life Science, Hopkinton, MA, USA) after adding 100 μg/ml d-luciferin (Xenogen, Alameda, CA, USA). Region of interest (ROI) values were calculated for each exposure and analyzed. Bioluminescence measurements were performed in triplicate.
Cell viability assay
A549 cells were monitored for viability based on ATP levels 24-hours post NSC 47147 treatment. NSC 47147/Bortezomib, cisplatin sensitization experiments were preincubated with cisplatin for 24 hours followed by the addition of various doses of NSC 47147 or Bortezomib for 24 hours. Percent viability was measured using Cell Titer Glo Luminescent Cell Viability Assay (Promega, Madison, WI) and measured using the EnVision Xcite Multilabel Reader (PerkinElmer, Shelton, CT). Percent viability was calculated as Asample/Acontrol × 100.
Phospho-IκB functional assay
Phospho-IκB levels were monitored in whole cell lysates using AlphaScreen SureFire p-IκB (S32/36) assay kit, (PerkinElmer, Shelton, CT), a homogenous bead-based assay designed to measure the phosphorylation of endogenous p-IκB (S32/36) in cell lysates, according to the manufacturer’s protocol. TNFα mediated I-κBα phosphorylation was measured in cells treated with 10 ng/ml TNFα (Invitrogen, Carlsbad, CA) for 0, 5, 15 and 30 minutes following 6-hour pretreatment with 6 μM NSC 47147.
Flow Cytometry
A549 cells were pretreated with cisplatin (10 μM) or control vehicle for 18 hours, followed by treatment with 6 μM NSC 47147 for 6 hours after which cells were trypsinized, counted and pelleted at 1 × 106 cells/sample. The pellet was resuspended in phosphate buffered saline, fixed by drop wise addition of an equivalent volume of ice-cold 100%ethanol. The samples were placed on ice for 20-minutes and stored at 4°C until day of analysis. On day of analysis, the cells were pelleted, ethanol decanted, and pellet resuspended in propidium iodide/RNAase (50 μg/ml/100 μg/ml) phosphate buffered saline solution and analyzed at the Flow Cytometry Core Facility at the University of Michigan Cancer Center.
In vivo studies
Tumor xenografts expressing FKR reporter were established by implanting 2 × 106 stably transfected A549- FKR cells onto both flanks of nu/nu CD-1 male nude mice (Charles River Labs, MA). When tumors reached a volume of approximately 100–150 mm3, treatment was initiated. All mouse experiments were approved by the University Committee on the Use and Care of Animals of the University of Michigan.
In vivo bioluminescence imaging and tumor growth studies
For bioluminescence imaging, mice bearing A549-FKR xenograft were given a single intraperitoneal (i.p.) injection of 0.5 mg/kg NSC 47147 or vehicle control (DMSO). Following treatment, the mice were anesthetized with 2% isofluorane/air mixture and given a single i.p. injection of 150 mg/kg D-luciferin in phosphate buffered saline. Bioluminescent images were acquired beginning 5-minutes after luciferin injection and at designated times post-treatment. Relative luminescence was calculated as the ratio of bioluminescence at each time point over bioluminescence pretreatment.
For the in vivo tumor growth studies, tumor-bearing mice were randomized into four groups: vehicle (DMSO), cisplatin (2 mg/kg), NSC 47147 (3 mg/kg), and a combination of cisplatin and NSC 47147 (2 and 3 mg/kg, respectively). Cisplatin was dissolved in water and NSC 47147 in 20% DMSO/water. Both agents were administered via i.p. injection. NSC 47147 was given daily for eight days either alone or co-administered in combination with cisplatin. Cisplatin was given on days 1 and 7 in all cisplatin-containing regimens. Tumor volume was calculated according to the equation for a prolate spheroid: tumor volume = (p/6) × (L ×W2), where L and W represent the longer and shorter dimension of the tumor, respectively. Data are expressed as the ratio of tumor volume at various times post-treatment compared with tumor volume on day one. Mice were monitored daily and tumor size and animal weight measured every 2 or 3 days until day 13.
Immunohistochemistry
A549-FKR tumor bearing mice were treated each day for 4 days with vehicle, cisplatin, NSC 47147 or cisplatin with NSC 47147. On the final day, mice were sacrificed 2 hours after injection, and tumors were excised and fixed in 10% neutral-buffered formalin for 48 hours (n = 6 tumors for each of the 4 groups). Tumor slices (5μm) were prepared according to standard procedures and stained with hematoxylin-eosin. Level of apoptosis was detected with an ApopTag Peroxidase In Situ Apoptosis Detection Kit (Chemicon, Inc.) which stains for apoptosis by labeling and detecting DNA strand breaks using the TUNEL method. Representative images were taken on an Olympus Bx-51 microscope with 20X magnification.
Statistical analysis
All data were expressed as the mean ± standard error of mean (SEM) from at least three independent experiments. Graph Pad Prism v.5 (GraphPad Prism version 5.01, GraphPad Software) nonlinear regression analysis, sigmoidal dose-response (variable slope) was used to generate the IC50 values. For tumor growth analysis, ANOVA was done on the proportional change of volume from baseline at each time point. If there was a significant difference, all pairwise comparisons were run adjusting for the multiple comparisons using Tukey’s HSD (honest significant differences). Differences between groups were considered significant when the P value was ≤ 0.05.
Results
Identification of a FADD-kinase inhibitor, NSC 47147
Using the A549-FKR reporter assay to screen an NCI compound diversity set, we identified NSC 47147, a tripyrrole alkaloid compound that inhibits FADD phosphorylation (Fig. 1). The compound increases FKR reporter activity in a concentration-dependent manner with an IC50 of 2.0 μM (Fig. 2A). Comparatively, the JNK inhibitor (SP600125), known to downregulate FIST-HIPK3 activity, and the CK1α inhibitor (CKI7) also demonstrate a concentration-dependent increase in FKR bioluminescence with IC50 values of 22 μM and 143 μM, respectively (Fig. 2A). These results are supported by a concomitant decrease in phosphorylated FADD (pFADD) expression following treatment with SP600125, and CKI7 (Fig. 2B). Changes in expression levels were not due to differences in protein loading as demonstrated by expression of GAPDH. It is important to note that in response to inhibition of FADD phosphorylation, corresponding levels of total FADD also show a decrease. We have previously demonstrated that inhibition of FADD phosphorylation results in its ubiquitin-dependent degradation. Using MG132, a proteosome inhibitor, have confirmed that a decrease in total FADD levels in response to FADD dephosphorylation (i.e., in the presence of a FADD kinase inhibitor) can be reversed by inhibiting proteosomal degradation (18).
Figure 1.
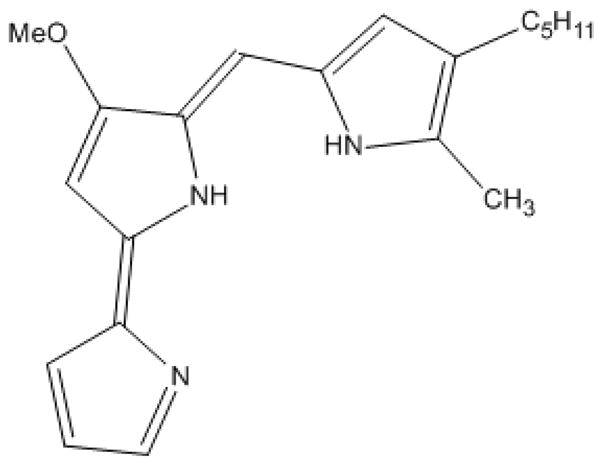
Compound structure for NSC 47147
Figure 2. Identification of NSC 47147 as a FADD-kinase inhibitor.

(A) A549-FKR cells were treated with NSC 47147 and SP600125 for 6 hours and CKI7 for 1 hour, at the indicated concentrations followed by bioluminescent measurement. NSC 47147 blocks phosphorylation of FADD with greater potency than FADD-kinase inhibitors, SP600125 and CKI7. (B) A549-FKR cells were treated as described for FKR reporter assay and lysates analyzed with specific antibodies by western blot. Treatment with FADD-kinase inhibitors, CKI7 and SP600125 show dose dependent decrease in phosphorylated FADD protein that correlates with FKR activity. Total and phosphorylated β-catenin and cJun, respective targets of CKI7 and SP600125, have been shown for comparison. (C) A549 and Jurkat cells were treated for 6 hours with NSC 47147. Western blot analysis reveals NSC 47147 leads to a decrease in pFADD and p-β-catenin levels.
To delineate possible FADD-kinase targets of NSC 47147, we examined whether the compound inhibited phosphorylation of β-catenin, a downstream target of CK1α. Western analysis of A549 and Jurkat cells following 6-hour treatment with NSC 47147 demonstrate a decrease in phosphorylated β-catenin (p-β-catenin) protein (Fig. 2C). The data shows a trend similar to the inhibition of p-β-catenin expression following treatment with CKI7 (Fig 2B). Three hour treatment of 250 μM CKI7 and 3 μM NSC 47147 of our recently described CK1α reporter cell line (SW620-BGCR) resulted in 10-fold induction in bioluminescence activity indicative of CKIα inhibition (Fig 3A). In this assay, CKI7 resulted in 7.5 fold increase in activity. To examine further the effect of NSC 47147 on CK1α inhibition, we directly evaluated CK1α enzymatic activity in response to the compound (Fig. 3B). It is worth mentioning that maximal inhibition by NSC 47147 was not observed due to its limited aqueous solubility. However, the resulting IC50 for NSC 47147, 6 μM, supports the ability of NSC 47147 to inhibit CK1α. Taken together; these results substantiate our finding that NSC 47147 inhibits phosphorylation of FADD through inhibition of FADD-kinase CK1α.
Figure 3. NSC 47147 inhibits CK1α.

(A) SW620 CK1α reporter cells were treated with 3 μM NSC 47147 and 250 μM CKI7 for 3 hours. Data shows 10-fold increase in bioluminescence indicative of CK1α inhibition in response to NSC 47147 treatment. CKI7, a CK1α inhibitor, is shown as a positive control. (B) NSC 47147 was evaluated using a TR-FRET biochemical assay for its direct effect on CK1α enzymatic activity. The in vitro assay reveals direct inhibition of CK1α activity by NSC 47147.
Effect of NSC 47147 on cell viability
To examine the correlation of NSC 47147 activation of the FKR reporter and corresponding decrease in pFADD on cell viability, A549 lung carcinoma cells were treated with increasing concentrations of NSC 47147 and ATP levels were measured at 24-hours post-treatment. The increase in FKR reporter activity directly correlates with a decrease in percent cell viability. Results demonstrate similar IC50s (2 μM) for both inhibition of pFADD, as indicated by increased reporter activity, and corresponding decrease in cell viability (Fig. 4).
Figure 4. Inhibition of FADD phosphorylation correlates with decreased cell viability.

Concentration dependent inhibition by NSC 47147 in A549-FKR cell based assay shown plotted against percent cell viability. Cells were treated with NSC 47147 for 6 hours in the FKR assay and 24 hours to assess viability. ATP levels were quantified following treatment using Promega Cell Titer Glo. Identical IC50 values for each analysis suggest a correlation between inhibition of FADD phosphorylation and decreased cell viability.
NSC 47147 inhibits NF-κB activation in A549 lung carcinoma cells
Our previous studies have demonstrated a close correlation between levels of pFADD and NF-κB activation (13). Based on this evidence, we sought to examine the effect of NSC 47147 on the inhibition of both endogenous and TNFα-induced levels of phosphorylated IκBα, critical to transcriptional activity of NF-κB (16). Treatment of A549 cells with 1, 3, and 6 μM NSC 47147 resulted in 31, 63, and 76 percent decrease in pIκBα activity, respectively, along with a corresponding decrease in protein expression of both pIκBα and total IκBα (Fig. 5A and 5B). A549 cells stimulated with TNFα following pretreatment with NSC 47147, demonstrate that the inhibitor also blocks TNFα induced IκBα phosphorylation (Fig. 5C). Western blot analysis demonstrates that NSC 47147 inhibits TNFα-induced expression of pIκBα at 5-minutes post-stimulation (Fig. 5D) as well as inhibiting the rapid IKK-dependent phosphorylation of IκBα before its degradation and the continued phosphorylation and turnover of newly synthesized IκBα 30-minutes after TNFα stimulation (Fig. 5D) (20).
Figure 5. NSC 47147 attenuation of NF-κB activity.

(A) The phosphorylation status of I-κBα was evaluated using AlphaScreen pIκB assay and by (B) western blot analysis in A549 cells following 6-hour treatment with 0, 1, 3, and 6 μM NSC 47147. Results show a decrease in luminescence indicative of pIκB inhibition. Western blot confirms decreasing levels of pIκB proteins and total I-κBα proteins in response to NSC 47147. (C) TNFα induced pIκB levels after 6-hour treatment with and without NSC 47147. A549 cells were stimulated with 10 ng/ml TNFα following pre-treatment with 6 μM NSC 47147. Cellular lysates were subjected to analysis by pIκB AlphaScreen and western blot.
NSC 47147 as a chemosensitizing agent
We hypothesized that pFADD mediated NF-κB inhibition by NSC 47147 would sensitize tumor cells to chemotherapeutic agents and enhance tumor cell death. To test this hypothesis, A549 cells were treated with NSC 47147 or Bortezomib, a small molecule NF-κB inhibitor, in combination with cisplatin, a chemotherapeutic agent known to induce apoptosis. The cells were pretreated with cisplatin (10 μM) for 18 hours followed by 24-hour treatment with NSC 47147 or Bortezomib following which, ATP levels were measured. As shown in figure 6A, NSC 47147 treatment in combination with cisplatin leads to an increase in cell death (* P value ≤ 0.05) as compared to NSC 47147 treatment alone. As shown in figure 6B, treatment of A549 cells with bortezomib and cisplatin also resulted in a marked (* P value ≤ 0.05) decrease in cell viability. These data suggest that the enhanced sensitivity to cisplatin in response to NSC 47147 or bortezomib treatment is due to inhibition of NF-κB.
Figure 6. NSC 47147 chemosensitizes A549 lung cancer cells to cisplatin induced apoptosis.

(A, B) A549 cells were preincubated with 10 μM cisplatin for 24 hours followed by increasing concentrations of NSC 47147 or Bortezomib for 24 hours. ATP levels following treatment were quantified using Promega Cell Titer Glo. Data shows inhibition of NF-κB by NSC 47147 or bortezomib in combination with cisplatin leads to greater cell death as compared to NSC 47147 or bortezomib alone (* P ≤ 0.05). (C) A549 lung cancer cells treated with NSC 47147 in the absence of presence of cisplatin were subjected to cell cycle analysis. Data shows more cells in sub-G1 with NSC 47147/cisplatin combination therapy (12.3%) as compared to control (1.8%), NSC 47147 (1.6%) and cisplatin alone (5.2%). (D) A549 cells treated with NSC 47147, cisplatin or both agents were evaluated for inhibition of pFADD levels and induction of apoptosis by assessing the levels of cleaved caspase-3.
To further confirm the cooperative effects of cisplatin and NSC 47147, A549 cells pretreated with cisplatin (10 μM) for 18 hours followed by 6-hour treatment with NSC 47147 (6 μM) were subjected to cell cycle analysis. Figure 6C shows 12.3% of cells in sub-G1 phase in response to NSC 47147/cisplatin combination treatment as compared to vehicle control (1.8%), NSC 47147 (1.6%) and cisplatin (5.2%) alone. Western analysis on similarly treated cells, resulted in an increase in caspase-3 cleavage with cisplatin, no effect with NSC 47147 alone, and a marked increase in cleaved caspase-3 levels (17 and 19 kDa bands) with combined NSC 47147 and cisplatin treatment (Fig. 6D), thus supporting the capacity of NSC 47147 to augment cisplatin-induced apoptosis.
Effect of NSC 47147 in tumor xenograft models
Having established the efficacy of NSC 47147 toward suppression of FADD phosphorylation and subsequent NF-κB inhibition in cells, we next sought to investigate the effects of the compound in a mouse tumor model. To establish tumors, we implanted A549-FKR cells into the flanks of nude mice. When tumors reached a volume between 100–150 mm3, we monitored bioluminescence over time in mice treated with NSC 47147 or vehicle control. Bioluminescence imaging shows an increase in FKR reporter activity following treatment with NSC 47147, maximum reporter activity was reached within 6-hours followed by sustained, although lower, activity up to 36-hours (Fig. 7A). Representative images of NSC 47147 treated mice are shown in Figure 7B.
Figure 7. NSC 47147 inhibits FADD phosphorylation in an A549-FKR xenograft model and sensitizes tumors to an apoptotic stimulus.

(A) Athymic nude mice bearing A549-FKR expressing xenografts were treated with vehicle control (DMSO) or NSC 47147 (0.5 mg/kg) by i.p. injection. Mice were imaged for bioluminescence at the indicated times. Relative luminescence was calculated as the ratio of bioluminescence at each time point to the basal bioluminescence prior to treatment. Data points represent mean relative luminescence ± SEM. (B) Representative bioluminescence images of tumor-bearing mice prior to treatment (basal) and 3, 6 and 18 hours post–treatment with NSC 47147 (0.5 mg/kg) . (C) Tumor-bearing mice were treated with 3 mg/kg NSC 47147 once daily for 8 days and/or 2 mg/kg cisplatin on days 1 and 7 by i.p injection and tumor volume calculated through day 13. Data are plotted as mean relative tumor volume ± SEM. (D) Immunohistochemical staining of A549-FKR xenografts after once-daily treatment for 4-days with 3 mg/kg NSC 47147 and/or 2 mg/kg cisplatin. Tumors were harvested and fixed on day 4; sections were stained for apoptosis as described in materials and methods. Figure shows duplicate images of representative fields.
The chemosensitizing effects of NSC 47147 on tumor progression were evaluated in A549-FKR mouse xenografts treated with NSC 47147 (3mg/kg) once daily for 8 days with or without cisplatin (2mg/kg) i.p. injections on days 1 and 7. Significant reduction (P ≤ 0.05) in tumor volume was observed at day 13 for the NSC 47147/cisplatin combination therapy as compared to control or either chemical agent alone (Fig. 7C). Notably, mice treated with the combination therapy had no increase in tumor volume through the duration of the study, whereas mice treated with cisplatin or NSC 47147 alone resumed tumor growth at day 13 and day 7, respectively.
Based on these results, we evaluated the degree of apoptosis in the tumor xenografts following treatment with NSC 47147 alone or in combination with cisplatin. Immunohistological staining for apoptosis demonstrate areas of cell death within tumors of cisplatin treated mice, a small amount of apoptosis in tumors of NSC 47147 treated mice, and a pronounced increase in apoptosis throughout the tumors of mice receiving the combination of cisplatin and NSC 47147 (Fig. 7D).
Discussion
Although FADD was originally identified as a key mediator of apoptosis, it has recently been identified as a modulator of a number of death receptor dependent and independent non-apoptotic activities including, embryogenesis, cell-cycle progression, cell proliferation, and tumorigenesis (6, 8, 9). Previous research from our lab has revealed phosphorylated FADD (pFADD) as a biomarker for poor clinical outcome in human lung adenocarcinoma. Our studies have demonstrated a strong correlation between high levels of phosphorylated FADD and elevated NF-κB activity, the anti-apoptotic actions of which lead to the formation of aggressive phenotypes, resistance to chemotherapeutic agents, and poor clinical outcome (13, 15, 21). In this study, we provide evidence that abrogation of NF-κB signaling through small molecule inhibition of FADD phosphorylation is a novel and viable approach for cancer therapy.
Comprehensive studies recently conducted in A549 and Jurkat cell lines have provided insight into the mechanistic basis for FADD and its involvement in NF-κB activation and tumorigenesis (13, 22, 23). Additionally, it was in Jurkat cells that CK1α was identified as the kinase that phosphorylates FADD (4). Research from our lab has shown these lines to express high levels of phosphorylated FADD protein, thereby providing the impetus toward selecting these models for the identification and evaluation of a potential FADD-kinase inhibitor. Using A549 lung cancer cells expressing our recently described FADD-Kinase Reporter, FKR (18), we performed a high-throughput screen with an NCI diversity compound collection and identified NSC 47147 as a potent inhibitor of FADD phosphorylation. NSC 47147 is a prodigiosin, a family of natural red pigments synthesized by a variety of microorganisms. Prodigiosins have been shown to possess antineoplastic properties, demonstrating an ability to initiate cell-cycle arrest and apoptosis (24, 25).
Treatment of A549-FKR cells with NSC 47147 showed a concentration-dependent increase in reporter activity with one to two logs greater potency than previously described FADD-kinase inhibitors, SP600125 and CKI7 (IC50 = 2, 22 and 150 μM, respectively). The increase in reporter bioluminescence following treatment with inhibitors of FADD-kinases, FIST-HIPK3 and CK1α, suggests that the mechanism of action of NSC 47147 could be, in part, due to its inhibition of either of these kinases. Western analysis of A549 and Jurkat cells treated with NSC 47147 show a dose dependent depletion of pFADD protein with a concomitant decrease in phospho-β-catenin, also a CK1α substrate (26). No decrease in c-Jun activation was apparent, indicating that the compound obtrudes CK1α activity, but does not influence FIST-HIPK3 (data not shown). The dose dependent decrease in pFADD and p-β-catenin proteins in both cell lines suggest that NSC 47147 may affect pFADD levels by either direct inhibition of CK1α or by impinging on an upstream signaling event. The results presented here reveal inhibitory activity in a CK1α cell based assay as well as direct inhibition in a CK1α biochemical assay, thereby providing mechanistic evidence that NSC 47147 is a CK1α inhibitor. CK1α regulates multiple oncogenic pathways, in addition to its proposed involvement in pFADD mediated NF-κB regulation, namely the β-catenin/WNT signaling axis (26, 27) making it a rational target for drug therapy.
A decrease in pFADD levels as detected by an increase in FADD-kinase reporter activity directly correlated with a reduction in cell viability suggesting that the inhibition of FADD phosphorylation and increase in cytotoxicity are a consequence of the same biological event. It has been proposed that the molecular basis for the correlation between pFADD levels and cell death stems from its role as a potent activator of NF-κB, an anti-apoptotic transcription factor (13). Previous research has shown that overexpression of FADD stimulates NF-κB promoter activity (15, 28)and that FADD-deficient cells are more susceptible to viral infection due to defects in NF-κB activation (29). Furthermore, FADD plays an integral role in the recently described TRADDosome complex, a central mediator of NF-κB signaling (30). Although it was previously unappreciated, our published work has shown that increased levels of phosphorylated FADD result in elevated NF-κB activation (13). The data presented in this paper provide further evidence that the phosphorylated form of FADD rather than FADD is a key component of the NF- κB activating complex.
The role of NF-κB in tumorigenesis has been well established and is based on its action as a regulator of cell fate decisions. It is often dysregulated in cancer cells leading to uncontrolled cell proliferation and resistance to therapeutic intervention (16). These data show that A549 lung carcinoma cells treated with NSC 47147, yield a decrease in NF-κB activation as measured by the phosphorylation status of I-κBα. Phosphorylation of I-κBα by IKK (I kappa kinase) requires recruitment of the TRADDosome complex wherein IKK plays an essential role (30). These results confirm previous reports that TNFα-induced NF-κB activation is dependent on levels of FADD expression (15, 31, 32) and provide further evidence of NSC 47147 as an inhibitor of FADD phosphorylation.
Tumor cells with constitutively active NF-κB are known to be resistant to chemotherapeutic agents, a resistance that can be alleviated by inhibition of NF-κB signaling (16). We hypothesized that pretreatment with NSC 47147 would chemosensitize A549 lung cancer cells toward an apoptotic stimulus leading to enhanced cell death. Results from our cell cycle analysis show a higher percentage of cells in the sub G1 population with a parallel increase in caspase-3 activity, thereby providing evidence that NSC 47147 sensitizes cells to cisplatin leading to decreased cell viability. We believe the mechanistic basis for the proposed synergy is that NSC 47147, by inhibiting FADD phosphorylation, abates TRADDosome mediated NF-κB activation. In the absence of NF-κB signaling, tumor cells become sensitive to apoptotic stimuli (i.e. in response to cisplatin treatment). In this regard, our data shows that bortezomib induced NF-κB inhibition in combination with cisplatin also results in a decrease in cell viability. The ability of NSC 47147 to exhibit a similar outcome when used in combination with cisplatin provides additional mechanistic evidence that the cytotoxic effects of NSC 47147 are at least in part due to its inhibition of NF-κB.
Studies from our laboratory, as well as others, have also demonstrated a role of pFADD in G2/M progression (23, 33). Therefore, we expected NSC 47147 to arrest cells at the G2/M phase of the cell cycle consistent with its proposed mechanism as an inhibitor of pFADD levels. As expected, the fraction of cells in G2/M following NSC 47147 treatment was 19.1% compared to 11.4% for control cells thereby providing additional support that NSC 47147 inhibits FADD phosphorylation leading to cell cycle arrest and cell death.
Having demonstrated the in vitro effect of NSC 47147 alone and in combination with cisplatin, we next examined the compounds efficacy as a chemotherapeutic agent in a mouse tumor model. Mouse tumor xenografts expressing the FKR reporter showed an increase in bioluminescence following treatment with NSC 47147 demonstrating the ability of the compound to inhibit activation of FADD in an animal model. The combined NSC 47147 and cisplatin therapy resulted in a decrease in tumor volume greater than the effect of either compound alone. Consistent with the reduction in tumor volume, TUNEL staining revealed enhanced apoptotic activity in mouse tumors treated with combination drug therapy compared to either agent alone. Treatment of mice with 3 mg/kg NSC 47147 did not result in overt toxicity in our treatment groups as well as by other research groups (34, 35). This, combined with the capacity of NSC 47147 to control tumor burden when used in combination with a known chemotherapy agent, provides validation for the investigation of more efficacious and biologically available inhibitors of FADD phosphorylation. The identification of compounds which, when co-administered with chemotherapeutic drugs, increase their efficacy is of significant clinical importance.
In summary, our previous research has identified phosphorylated FADD as a prognostic biomarker for poor clinical outcome. The mechanistic basis for this biomarker is the ability of pFADD to promote the anti-apoptotic actions of NF-κB. The results presented here identify a small molecule inhibitor of FADD phosphorylation that induces cell death through abrogation of NF-κB activity. We have provided proof of principle studies showing inhibition of FADD phosphorylation through CK1α may be a viable target for anticancer therapy either as a single agent, but more interestingly, in combination with clinically approved chemotherapeutic agents.
Acknowledgments
This work was supported by the US National Institutes of Health research grants R01CA129623 (AR), R21CA131859 (AR), U24CA083099 (BDR), P50CA093990 (BDR) and by an RSNA Resident Seed Grant (T.W.). Grant support for NSC 47147 synthesis was provided by the Ministerio de Sanidad, Spain and the European Union (FIS PI061226). We would also like to thank the member of the Center for Molecular Imaging for their technical help and constructive criticisms.
Abbreviations list
- FADD
Fas associated death domain
- PBS
phosphate buffered saline
- i.p
intraperitoneal
- ANOVA
analysis of variance
Footnotes
No potential conflicts of interest.
References
- 1.Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81:505–12. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- 2.Zhang J, Winoto A. A mouse Fas-associated protein with homology to the human Mort1/FADD protein is essential for Fas-induced apoptosis. Mol Cell Biol. 1996;16:2756–63. doi: 10.1128/mcb.16.6.2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boldin MP, Goncharov TM, Goltsev YV, Wallach D. Involvement of MACH, a novel MORT1/FADD-interacting protease, in Fas/APO-1-and TNF receptor-induced cell death. Cell. 1996;85:803–15. doi: 10.1016/s0092-8674(00)81265-9. [DOI] [PubMed] [Google Scholar]
- 4.Alappat EC, Feig C, Boyerinas B, Volkland J, Samuels M, Murmann AE, et al. Phosphorylation of FADD at serine 194 by CKIalpha regulates its nonapoptotic activities. Mol Cell. 2005;19:321–32. doi: 10.1016/j.molcel.2005.06.024. [DOI] [PubMed] [Google Scholar]
- 5.Elrod HA, Sun SY. Modulation of death receptors by cancer therapeutic agents. Cancer Biol Ther. 2008;7:163–73. doi: 10.4161/cbt.7.2.5335. [DOI] [PubMed] [Google Scholar]
- 6.Tourneur L, Chiocchia G. FADD: a regulator of life and death. Trends Immunol. 2010;31:260–9. doi: 10.1016/j.it.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 7.Kabra NH, Kang C, Hsing LC, Zhang J, Winoto A. T cell-specific FADD-deficient mice: FADD is required for early T cell development. Proc Natl Acad Sci U S A. 2001;98:6307–12. doi: 10.1073/pnas.111158698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Imtiyaz HZ, Zhou X, Zhang H, Chen D, Hu T, Zhang J. The death domain of FADD is essential for embryogenesis, lymphocyte development, and proliferation. J Biol Chem. 2009;284:9917–26. doi: 10.1074/jbc.M900249200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang J, Kabra NH, Cado D, Kang C, Winoto A. FADD-deficient T cells exhibit a disaccord in regulation of the cell cycle machinery. J Biol Chem. 2001;276:29815–8. doi: 10.1074/jbc.M103838200. [DOI] [PubMed] [Google Scholar]
- 10.Hua ZC, Sohn SJ, Kang C, Cado D, Winoto A. A function of Fas-associated death domain protein in cell cycle progression localized to a single amino acid at its C-terminal region. Immunity. 2003;18:513–21. doi: 10.1016/s1074-7613(03)00083-9. [DOI] [PubMed] [Google Scholar]
- 11.Tourneur L, Buzyn A, Chiocchia G. FADD adaptor in cancer. Med Immunol. 2005;4:1. doi: 10.1186/1476-9433-4-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Werner MH, Wu C, Walsh CM. Emerging roles for the death adaptor FADD in death receptor avidity and cell cycle regulation. Cell Cycle. 2006;5:2332–8. doi: 10.4161/cc.5.20.3385. [DOI] [PubMed] [Google Scholar]
- 13.Chen G, Bhojani MS, Heaford AC, Chang DC, Laxman B, Thomas DG, et al. Phosphorylated FADD induces NF-kappaB, perturbs cell cycle, and is associated with poor outcome in lung adenocarcinomas. Proc Natl Acad Sci U S A. 2005;102:12507–12. doi: 10.1073/pnas.0500397102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gibcus JH, Menkema L, Mastik MF, Hermsen MA, de Bock GH, van Velthuysen ML, et al. Amplicon mapping and expression profiling identify the Fas-associated death domain gene as a new driver in the 11q13.3 amplicon in laryngeal/pharyngeal cancer. Clin Cancer Res. 2007;13:6257–66. doi: 10.1158/1078-0432.CCR-07-1247. [DOI] [PubMed] [Google Scholar]
- 15.Hu WH, Johnson H, Shu HB. Activation of NF-kappaB by FADD, Casper, and caspase-8. J Biol Chem. 2000;275:10838–44. doi: 10.1074/jbc.275.15.10838. [DOI] [PubMed] [Google Scholar]
- 16.Karin M, Cao Y, Greten FR, Li ZW. NF-kappaB in cancer: from innocent bystander to major culprit. Nat Rev Cancer. 2002;2:301–10. doi: 10.1038/nrc780. [DOI] [PubMed] [Google Scholar]
- 17.Rochat-Steiner V, Becker K, Micheau O, Schneider P, Burns K, Tschopp J. FIST/HIPK3: a Fas/FADD-interacting serine/threonine kinase that induces FADD phosphorylation and inhibits fas-mediated Jun NH(2)-terminal kinase activation. J Exp Med. 2000;192:1165–74. doi: 10.1084/jem.192.8.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khan AP, Schinske KA, Nyati S, Bhojani MS, Ross BD, Rehemtulla A. High-throughput molecular imaging for the identification of FADD kinase inhibitors. J Biomol Screen. 2010;15:1063–70. doi: 10.1177/1087057110380570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nyati S, Ranga R, Ross BD, Rehemtulla A, Bhojani MS. Molecular imaging of glycogen synthase kinase-3beta and casein kinase-1alpha kinases. Anal Biochem. 2010;405:246–54. doi: 10.1016/j.ab.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nasuhara Y, Adcock IM, Catley M, Barnes PJ, Newton R. Differential IkappaB kinase activation and IkappaBalpha degradation by interleukin-1beta and tumor necrosis factor-alpha in human U937 monocytic cells. Evidence for additional regulatory steps in kappaB-dependent transcription. J Biol Chem. 1999;274:19965–72. doi: 10.1074/jbc.274.28.19965. [DOI] [PubMed] [Google Scholar]
- 21.Monks NR, Biswas DK, Pardee AB. Blocking anti-apoptosis as a strategy for cancer chemotherapy: NF-kappaB as a target. J Cell Biochem. 2004;92:646–50. doi: 10.1002/jcb.20080. [DOI] [PubMed] [Google Scholar]
- 22.Wajant H, Haas E, Schwenzer R, Muhlenbeck F, Kreuz S, Schubert G, et al. Inhibition of death receptor-mediated gene induction by a cycloheximide-sensitive factor occurs at the level of or upstream of Fas-associated death domain protein (FADD) J Biol Chem. 2000;275:24357–66. doi: 10.1074/jbc.M000811200. [DOI] [PubMed] [Google Scholar]
- 23.Bhojani MS, Chen G, Ross BD, Beer DG, Rehemtulla A. Nuclear localized phosphorylated FADD induces cell proliferation and is associated with aggressive lung cancer. Cell Cycle. 2005;4:1478–81. doi: 10.4161/cc.4.11.2188. [DOI] [PubMed] [Google Scholar]
- 24.Perez-Tomas R, Vinas M. New insights on the antitumoral properties of prodiginines. Curr Med Chem. 2010;17:2222–31. doi: 10.2174/092986710791331103. [DOI] [PubMed] [Google Scholar]
- 25.Montaner B, Perez-Tomas R. The prodigiosins: a new family of anticancer drugs. Curr Cancer Drug Targets. 2003;3:57–65. doi: 10.2174/1568009033333772. [DOI] [PubMed] [Google Scholar]
- 26.Knippschild U, Wolff S, Giamas G, Brockschmidt C, Wittau M, Wurl PU, et al. The role of the casein kinase 1 (CK1) family in different signaling pathways linked to cancer development. Onkologie. 2005;28:508–14. doi: 10.1159/000087137. [DOI] [PubMed] [Google Scholar]
- 27.Knippschild U, Gocht A, Wolff S, Huber N, Lohler J, Stoter M. The casein kinase 1 family: participation in multiple cellular processes in eukaryotes. Cell Signal. 2005;17:675–89. doi: 10.1016/j.cellsig.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 28.Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, et al. IPS-1, an adaptor triggering RIG-I-and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–8. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- 29.Balachandran S, Thomas E, Barber GN. A FADD-dependent innate immune mechanism in mammalian cells. Nature. 2004;432:401–5. doi: 10.1038/nature03124. [DOI] [PubMed] [Google Scholar]
- 30.Michallet MC, Meylan E, Ermolaeva MA, Vazquez J, Rebsamen M, Curran J, et al. TRADD protein is an essential component of the RIG-like helicase antiviral pathway. Immunity. 2008;28:651–61. doi: 10.1016/j.immuni.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 31.Ermolaeva MA, Michallet MC, Papadopoulou N, Utermohlen O, Kranidioti K, Kollias G, et al. Function of TRADD in tumor necrosis factor receptor 1 signaling and in TRIF-dependent inflammatory responses. Nat Immunol. 2008;9:1037–46. doi: 10.1038/ni.1638. [DOI] [PubMed] [Google Scholar]
- 32.Hsu H, Shu HB, Pan MG, Goeddel DV. TRADD-TRAF2 and TRADD-FADD interactions define two distinct TNF receptor 1 signal transduction pathways. Cell. 1996;84:299–308. doi: 10.1016/s0092-8674(00)80984-8. [DOI] [PubMed] [Google Scholar]
- 33.Scaffidi C, Volkland J, Blomberg I, Hoffmann I, Krammer PH, Peter ME. Phosphorylation of FADD/MORT1 at serine 194 and association with a 70-kDa cell cycle-regulated protein kinase. J Immunol. 2000;164:1236–42. doi: 10.4049/jimmunol.164.3.1236. [DOI] [PubMed] [Google Scholar]
- 34.Yamamoto C, Takemoto H, Kuno K, Yamamoto D, Tsubura A, Kamata K, et al. Cycloprodigiosin hydrochloride, a newH(+)/Cl(−) symporter, induces apoptosis in human and rat hepatocellular cancer cell lines in vitro and inhibits the growth of hepatocellular carcinoma xenografts in nude mice. Hepatology. 1999;30:894–902. doi: 10.1002/hep.510300417. [DOI] [PubMed] [Google Scholar]
- 35.Zhang J, Shen Y, Liu J, Wei D. Antimetastatic effect of prodigiosin through inhibition of tumor invasion. Biochem Pharmacol. 2005;69:407–14. doi: 10.1016/j.bcp.2004.08.037. [DOI] [PubMed] [Google Scholar]