

Abstract
Loop 5 (L5) is a conserved loop that projects from the α2-helix adjacent to the nucleotide site of all kinesin-family motors. L5 is critical to the function of the mitotic kinesin-5 family motors and is the binding site for several kinesin-5 inhibitors that are currently in clinical trials. Its conformational dynamics and its role in motor function are not fully understood. Our previous work using EPR spectroscopy suggested that L5 alters the nucleotide pocket conformation of the kinesin-5 motor Eg5 [1]. EPR spectra of a spin-labeled nucleotide analog bound at the nucleotide site of Eg5 display a highly immobilized component that is absent if L5 is shortened or if the inhibitor STLC is added [1], which X-ray structures suggest stabilizes a L5 conformation pointing away from the nucleotide site. These data, coupled with the proximity of L5 to the nucleotide site suggest L5 could interact with a bound nucleotide, modulating function. Here we use molecular dynamics (MD) simulations of Eg5 to explore the interaction of L5 with the nucleotide site in greater detail. We performed MD simulations in which the L5-domain of the Eg5•ADP X-ray structure was manually deformed via backbone bond rotations. The L5-domain of Eg5 was sufficiently lengthy that portions of L5 could be located in proximity to bound ADP. The MD simulations evolved to thermodynamically stable structures at 300K showing that L5 can interact directly with bound nucleotide with significant impingement on the ribose hydroxyls, consistent with the EPR spectroscopy results. Taken together, these data provide support for the hypothesis that L5 modulates Eg5 function via interaction with the nucleotide-binding site.
Keywords: kinesin-5, nucleotide-analog spin probes, EPR spectroscopy, molecular dynamics simulation
1. INTRODUCTION
Kinesin-family motors, G-proteins and myosin motors have evolved from a common ancestor protein and share the P-loop, Switch 1 and Switch 2 triphosphate binding domain motif at the nucleotide site [2–4]. All three families are α–β–α proteins with a core β-sheet platform flanked on both sides by α-helices. The Walker-A sequence [5], or P-loop, is a glycine-rich loop (L4) motif located between the core β3-strand and the flanking α2-helix and is diagnostic for ATPase proteins. The α2-helix adjacent to the P-loop in the kinesin-family motors is unique in the superfamily of proteins in that the α-helical repeat is broken two turns from the P-loop into the helix by a surface-exposed element, Loop5 (L5). The α2-helix then reforms immediately following L5 and extends along the entire surface of the protein. L5 is further of variable length. Homology modeling suggests L5 can be as short as three amino acids in kinesin-14 motors [6, 7], while it is six amino acids in length in the X-ray structure of ncd [8] and 8 amino acids in the structure of kinesin-1 [9]. Conversely, kinesin-5 has the longest known L5-domains, 17 amino acids in length in Eg5 [10].
The X-ray structures of Eg5 with and without bound mitotic inhibitors such as monastrol and S-trityl-L-cysteine (STLC) show that the inhibitors interact with L5 and thus imply that L5 can serve a regulatory function for Eg5 chemomechanics [10–14]. In the X-ray structures with inhibitor, the inhibitor is bound in a pocket formed by the protein surface and L5, with L5 bent away from the nucleotide site. Although X-ray structures with bound inhibitor show small shifts in structural elements, how these would integrate into the observed inhibition remains unresolved. This is in part due to the fact that the L5-domain is in approximately the same conformation in the X-ray structures in both the presence and the absence of inhibitors. Point mutations of the L5-domain, replacement of the Eg5 L5-domain with a kinesin-1 L5-domain, or deletion of 7 residues in the human Eg5 L5 all decrease the ATPase activity of Eg5 [12, 15, 16]. Replacement of the Eg5 L5 with the comparable domain from Neurospora eliminates STLC inhibition [17, 18]. All of these observations support a regulatory function for L5.
Most intriguingly, our previous work using nucleotide-analog electron paramagnetic resonance spectroscopy (EPR) probes at the nucleotide site of Eg5 shows two spectral components representing probes bound at the nucleotide site but having different probe mobilities [1]. When the experiments are repeated with the mutant Eg5-ΔL5 protein with a dramatically shortened L5-domain (amino acids 125–131 deleted), only the more mobile component is present in the spectrum. The immobilized component is no longer seen. A similar spectroscopic result is seen with the Eg5•SLADP protein in the presence of STLC. The data raise the possibility that one mechanism of L5 modulation of Eg5 function is via an interaction of L5 with the bound nucleotide, which with EPR probe mobility as the reporter signal, is manifested via a restriction of EPR probe mobility. This would of course require that the L5-domain be very flexible in order to reach over to the nucleotide site. Additional evidence supports this possibility. The x-ray structures of the kinesin-family proteins KIF2 (PDB ID 2GRY [19]), KIF2C (PDB ID 2HEH [6]), KIF22 (PDB ID 3BFN [20]) and kinesin13 (PDB ID 3EDL [21]) all show disordered components of L5, implying flexibility. The effects of L5 mutations on inhibitor binding likewise have been observed to support flexibility [16]. Spectrosocopic data measuring the rotational correlation time of a tryptophan residue within L5 have revealed a very high degree of structural flexibility in the absence of drug inhibitors [22], and EPR studies indicate high mobility for probes located within L5 [1].
Every experimental approach has strengths and weaknesses. X-ray crystallography provides high-resolution structures, but each structure is only a single snapshot of a multi-conformation process, so crucial states may still be missing from the X-ray database. In particular, X-ray structures of the microtubule (MT)-bound states have not been possible. The structure of the MT-bound states must instead be inferred from X-ray structures solved in the absence of MTs. Electron microscopy can resolve secondary structure in a MT-bound state, but not the MT-free states. EPR spectroscopy on the other hand provides lower resolution information. However in the present context, EPR spectroscopy has significant advantages in that it can be used in the presence and absence of MTs in experimental conditions mimicking physiological conditions. EPR spectroscopy can also very easily resolve multiple protein conformations in equilibrium as is the case noted above for Eg5. Computational simulation has proven to be a powerful tool for atomic-level examinations of different conformations of flexible kinesin-family structural elements in X-ray structures [15, 23–27] and their relationship to observations from other experimental techniques. Here we have used the human Eg5•ADP X-ray structure [10] as our starting point for molecular mechanics and molecular dynamics modeling of an Eg5 structure with a deformed L5 loop in order to study possible interactions of L5 with the nucleotide site. Our modeling suggests that the L5 loop in Eg5 is sufficiently long to interact with hydrophobic elements of the bound nucleotide and the adjacent nucleotide pocket, as well as to allow the formation of hydrogen bonds between the negative-charge rich loop and the nucleotide. The simulations imply that L5-nucleotide interaction could play a modulating role in Eg5 function.
2. METHODS
The X-ray structure of human kinesin-5 (Eg5) with bound ADP (PDB ID 1II6, ref [10]) served as the starting point for the simulations. Only the A-chain of the dimeric structure was retained. Structures were visualized and manipulated using the Chimera [28] graphics program. The bond between the backbone N and Cα atoms of amino acids 117 or 133, at either end of Loop 5, was removed. The loop was then sequentially maneuvered by φ - ψ backbone bond rotations of amino acids 117–133 so that Loop-5 was bent over the ADP moiety in the active site. Six separate starting structures were generated in this manner with Loop 5 coming straight down over the nucleotide or coming down with a twist in a figure-8 motif. After manipulations, the distance between the broken-bond atoms was less than 2Å with approximately the correct bond-angle orientation. The bond was then reestablished using the Chimera function “bond”. For the simulation of the Eg5•ADP/STLC complex, the starting structure was the X-ray structure of the complex (PDB ID 3KEN, ref. [14]). The structure was not modified prior to MD simulation.
Figure 8.
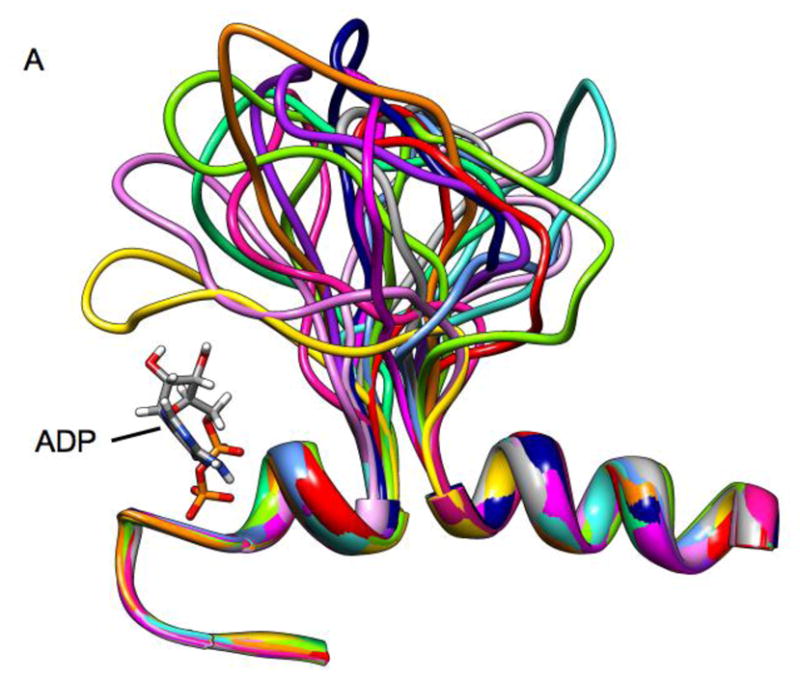
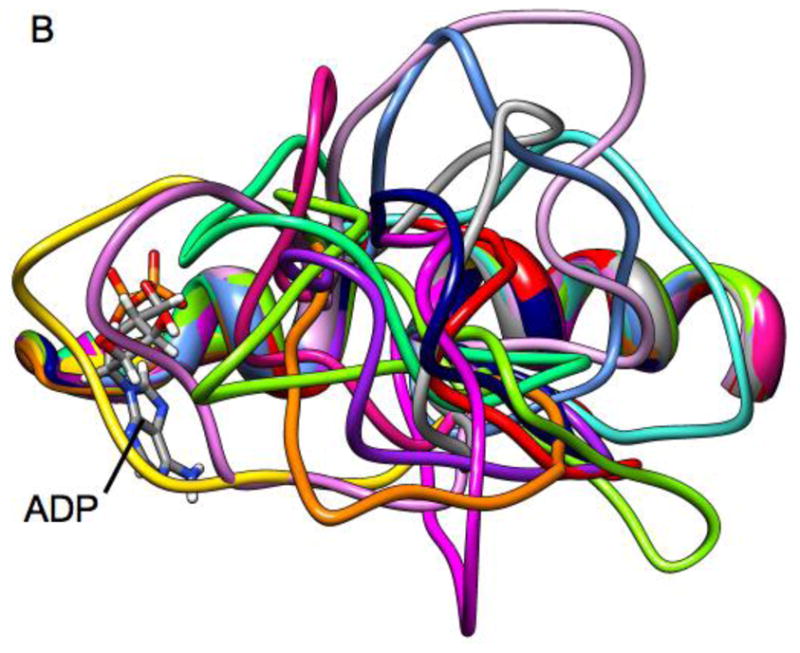
Ribbon diagrams of a superposition of Loop 5 conformations from high temperature molecular dynamics simulation. Individual frames are from a 10 ns simulation with frames separated by 600 ps.
Molecular dynamics simulations were performed using the Amber 10 [29] suite of codes. Charges for ADP and STLC were determined by first performing a single-point energy calculation at the Hartree-Fock level of theory using a 6-31G* basis set to obtain electrostatic charges using Gaussian03 [30]. These were then fit to the molecules using the RESP procedure [31]. Van der Waals parameters for Mg2+ were taken from [32] as modified in [26]. The tLeaP module of Amber 10 was used to add hydrogens to the structure. Histidines were protonated as appropriate for their local environment. Na+ counterions were then added to produce overall charge neutrality. A box of TIP3P water molecules [33] was used to solvate the system with a minimum 10Å border between the edge of the box and the closest point on the protein surface. Amber uses a geometric algorithm to place waters. The total size of all simulations after the addition of water molecules was in excess of 55,000 atoms.
The entire protein-water system was energy minimized using 1500 steps of the steepest descents algorithm and a subsequent 1500 steps of the conjugate gradient algorithm. The minimization quickly corrected any incorrect bond lengths and angles resulting from the editing of the structure to modify L5. The minimized system was then gradually heated and maintained at 298K using the Berendsen algorithm [34] for maintenance of temperature via coupling to an external bath. The particle-mesh Ewald procedure [35–37] with a non-bonded cutoff of 8Å was used to handle electrostatic interactions. Periodic boundary conditions were employed at constant pressure. MD simulations employed the SHAKE algorithm [38] and a 1fs time step. All simulations were done for 4ns. Protein Data Bank format files for all final structures shown in Figs 6–7 and in Figs. S2–S5 (supplementary online material) are available by contacting the authors.
Figure 6.

Ribbon representation of the interactions (ionic, hydrogen bonds, and hydrophobic) that result in the forces stabilizing the location of Loop5 (red) in one observed conformation from the MD simulations.
Figure 7.

Ribbon representation of the interactions (ionic, hydrogen bonds, and hydrophobic) that result in the forces stabilizing the location of Loop5 (red) in one observed conformation from the MD simulations.
To generate the Eg5-ΔL5 mutant, amino acids 125–131 were deleted from the PDB file of Eg5•ADP (PDB ID 1II6[10]). Backbone φ - ψ bonds in the remaining L5 domain were rotated to bring the amino and carboxyl termini of amino acids 124 and 132 within 2Å at approximately the correct bond angle, and the bond between them reestablished. The structure was then energy minimized as described above and the resulting structure used as the starting point for simulations with the Eg5-ΔL5 mutant. The shortened, mutant L5 was then modified as described above using φ - ψ backbone bond rotations to test whether it could interact with ADP at the active site.
High temperature simulations were used to further explore conformations available to L5. These used the generalized Born approach. In order to eliminate crystal forces, the starting structure for the high-temperature simulations was the MD-simulated structure of Eg5•ADP (PDB ID 1II6 [10]) following 5 ns of MD simulation using the particle mesh Ewald algorithm in a box of explicit TIP3P water molecules (described above). The water molecules were removed from the resulting Protein Data Bank format file. For the generalized Born simulation, the structure was then energy minimized using 1000 steps of steepest descent minimization followed by 1000 steps of conjugate gradient minimization, with all atoms except those in Loop5 (amino acids 117–133) restrained by a weak harmonic constraint (1.0 Kcal/mol/Å2). The evolution of the minimized structure was then simulated for 10 ns following gradual heating to 600K. All atoms except those in Loop5 were again restrained by the harmonic constraint. Backbone ω-bonds were constrained to remain in the trans-conformation.
3. RESULTS
Experimental motivation
Figure 1 shows spectra obtained from Eg5 in the presence of the nucleotide-analog spin probe, 2′,3′-SLADP. Similar spectra are also shown in [1]. The structure of 2′,3′-SLADP is in Fig. S1. The three central peaks, P2–P4, arise primarily from free probe tumbling rapidly in solution. When the EPR probe binds to Eg5, the adjacent protein surface restricts probe motion in the face of thermal agitation. This results in a broadening of the spectrum and the presence of the down-field and up-field components P1 and P5 in the spectra. However, as is clear from the spectrum for Eg5•2′,3′-SLADP (red), there is an additional low-field shoulder component denoted by P1* and a corresponding up-field P5*component that are missing in the spectra of Eg5-ΔL5•2′,3′-SLADP (blue) and Eg5•2′,3′-SLADP in the presence of STLC (cyan). The P1*–P5* component to the spectrum has even greater splitting than that of the P1–P5 components. Greater low-field to high-field splitting in the EPR spectrum implies more restricted mobility of the EPR probe [39]. Spectra similar to that of Eg5-ΔL5•2′,3′-SLADP, and missing the second more immobilized P1*–P5* component, have also been obtained with ncd and kinesin-1 [40]. The L5 domain of kinesin-1 is 8 amino acids in length [9]. For ncd it is six amino acids in length [8]. Additional and more complete details of the experimental observations are in [1].
Figure 1.

X-band EPR spectra of 2′,3′-SLADP bound to Eg5 (red), Eg5-ΔL5 (blue) and Eg5 in the presence of the inhibitor, STLC (cyan). The horizontal axis is magnetic field. The vertical axis is the derivative of absorbance. The spectra are 9.0 mT wide with center field at 350 mT. The figure is similar a previously published figure [1] and is present to motivate the computational modeling.
Fig. 2 shows a ribbon diagram of the X-ray structure of Eg5•ADP (PDB ID 1II6, ref. [10] overlaid with the structure of Eg5•ADP co-crystallized with bound STLC (PDB ID 2WOG, ref. [13]). L5 is colored green and magenta in Eg5•ADP. Magenta is the portion that is removed in the Eg5-ΔL5 construct. L5 is colored blue in the Eg5•ADP/STLC structure. As is clear, both L5-domains bend away from the nucleotide site containing bound ADP (red). The conformation of the L5-domain of Eg5•ADP/STLC is further stabilized by hydrophobic interactions with STLC (dark gray). The structure of Eg5•ADP co-crystalized with bound monastrol (PDB ID 1X88, ref [12]) shows a similar L5 orientation with monastrol in the same spatial location as STLC.
Figure 2.

An overlay of ribbon diagrams of the structures of Eg5•ADP [10] and Eg5•ADP with bound STLC [13]. Eg5•ADP is colored coral except for L5, which is colored green and magenta. The magenta segment is the portion of L5 that is removed in the Eg5-ΔL5 construct. Eg5•ADP with bound STLC is colored turquoise, except for L5, which is blue. STLC is gray. ADP is red.
Summarizing the experimental observations, the P1*–P5* component of the EPR spectrum is seen only for Eg5•2′,3′-SLADP. It is not seen for Eg5-ΔL5•2′,3′-SLADP, for K•2′,3′-SLADP or for ncd•2′,3′-SLADP. The latter all contain shortened L5-domains. It is further not seen in the spectrum of Eg5•2′,3′-SLADP/STLC. X-ray structures suggest that STLC results in a further stabilization of the L5-domain in a conformation pointing away from the nucleotide site [13, 14]. These observations all suggest that L5 could modulate wild-type Eg5 function via an interaction with the active site. We use MD simulation to investigate this possibility.
Molecular dynamics simulations of the Loop 5 domain
Initial (control) simulations were done with the unmodified x-ray structure of Eg5•ADP (PDB ID 1II6, ref [10]) to examine the inherent stability of the native L5 in the X-ray structure. Fig. 3A, B shows the result of the simulation of Eg5•ADP. Fig. 3A is a ribbon diagram side view of the region of the protein adjacent to L5. Fig. 3B is the view from the top of Fig. 3A. The original X-ray structure is in turquoise. The structure to which it evolves during the MD simulation is in coral. As is clear from the figures, L5 is displaced from ADP at the nucleotide site in the X-ray structure and remains displaced during the MD simulation. Fig. 4A shows the rms deviation of the Cα-Cα backbone atoms for the evolution of the X-ray structure of Eg5•ADP (red trace). The plot shows a leveling off to a new equilibrium in approximately 2 ns with a maximum rms displacement of slightly less than 3Å. Fig. 4B (red trace) shows the RMS deviation of L5 (Cα-Cα atoms, amino acids 117–133) during the same simulation, with a slightly lower RMS deviation, suggesting relatively more perturbation of the entire solvated protein from the X-ray structure. The maximum Cα displacement of L5 is 4.5Å at Tyr 125. Thus there is only a small perturbation of the protein and L5 during the simulation. Most importantly, L5 does not move adjacent to ADP during the simulation. It remains at a location where it does not interact with ADP, as seen in the X-ray structure. A similar simulation was also done using the X-ray structure of the Eg5•ADP/STLC complex as the initial structure. The MD simulation again showed that the X-ray structure was stable on the MD time scale, with L5 bent away from the nucleotide site and not interacting with ADP (data not shown).
Figure 3.


A. The X-ray structure of Eg5•ADP (turquoise), Eg5•ADP following 4ns of MD simulation (coral), the constructed structure of Eg5-ΔL5•ADP distorted to be as close as possible to ADP at the nucleotide site (red), and the structure of Eg5-ΔL5•ADP following 4 ns of MD simulation (purple) are shown as ribbon diagrams. The structures were superimposed using a least-squares distance minimization of the P-loop Cα atoms. ADP from the X-ray structure of Eg5•ADP is in dark gray. Only the regions of the proteins adjacent to the nucleotide site and L5 are shown. Fig. 3A is oriented from the side with the L5 at the top of the panel. B. Same a panel A, but looking down from the top of panel A.
Figure 4.


A. The RMS deviation of the MD-simulated structures as a function of time. Color coding: Eg5•ADP (red), Eg5-ΔL5•ADP (gray). The cyan, green, orange, blue and magenta plots are color-coded to the structures in Fig. 5. B. RMS deviation of only the Loop5 domain during the simulation. Color-coding is as in panel A.
An additional simulation was done using the Eg5-ΔL5 construct. Fig. 3A, B show the side and top views of our constructed (Methods) Eg5-ΔL5-domain modified via φ – ψ angle backbone rotations to bring it as close as possible to ADP (red trace). It is clear from the figure that the Eg5-ΔL5 construct is simply too short to interact significantly with the ADP moiety at the nucleotide site. The purple trace shows the evolved structure after 4 ns of MD simulation, which has moved away slightly farther from the nucleotide. Fig. 4A (gray trace) gives the rms deviation as a function of MD-simulated time. Again a new equilibrium is reached after approximately 2 ns, with a slightly greater rms value (5 Å) than for the wild-type L5-domain structure. The increased rms value appears to arise from random displacements of loops away from the nucleotide site, and not any components at the nucleotide site. Fig. 4B (gray trace) gives the rms deviation of only the L5 domain during the simulation, which is somewhat larger than observed for the wild-type protein (red trace). However, none of the values suggest extremely large-scale movements during the MD simulation. The maximum Cα-Cα displacement of the ΔL5-domain is 9.5Å at Pro 121. This is near the top apex of the loop, which points in different directions at the beginning (red) and end (purple) of the simulation. Three other attempts at modifying the L5-domain of the Eg5-ΔL5 construct via φ – ψ angle backbone rotations gave similar results in that the resulting structure did not interact with ADP at the nucleotide site (data not shown). From the molecular dynamics simulations, we conclude that the L5-domain of the Eg5-ΔL5 construct cannot significantly interact with ADP, consistent with the EPR spectra. Furthermore, the L5-domain of the MD simulation of the X-ray structure of Eg5 represents an equilibrium conformation that is compatible with the X-ray structure.
The question remains whether there are other L5 conformations that can interact with ADP, but have not yet been caught in the X-ray database? To investigate this question, L5 was deformed using computer graphical techniques to attempt to maximize any possible interaction with ADP (Methods). Six structures were generated and then analyzed using MD simulation. Fig. 5A, B and C give side, top and end-on (from nucleotide site) views of the L5 and the adjacent region following 4ns MD simulations for five of the deformed L5-domains and the X-ray structure (magenta with the ΔL5 deletion colored in black) again shown for comparison. Figs. 4A,B give the rms deviations (Cα – Cα) from the initial simulation structure as a function of time for the entire protein (Fig. 4A) and for Loop5 (Fig. 4B). Traces for structures for which the intact Loop5 domain has been deformed over the nucleotide-binding site are color-coded to the colors of the structures in Fig. 5. The important observation is that all structures, including the X-ray structure, level off to a new equilibrium structure with a rms deviation value 2–3Å after 2–3 ns of MD simulation. There is somewhat more total relative variability in the RMS deviations of the L5 domain alone. The initial deformations of the L5 domain were based solely on backbone bond rotations. The variability in the RMS deviations is related to the subsequent movements of the L5 domain early in the simulations as intra-protein interactions were forming. However, these are not extremely large-scale movements of L5. Most importantly, the perturbations of the protein in the MD simulations resulting from the modifications of L5 are not causing any significantly increased destabilization of the protein beyond the changes that are seen in the MD simulation using the Eg5•ADP X-ray structure (PDB ID 1II6) as the starting point of the simulation.
Figure 5.

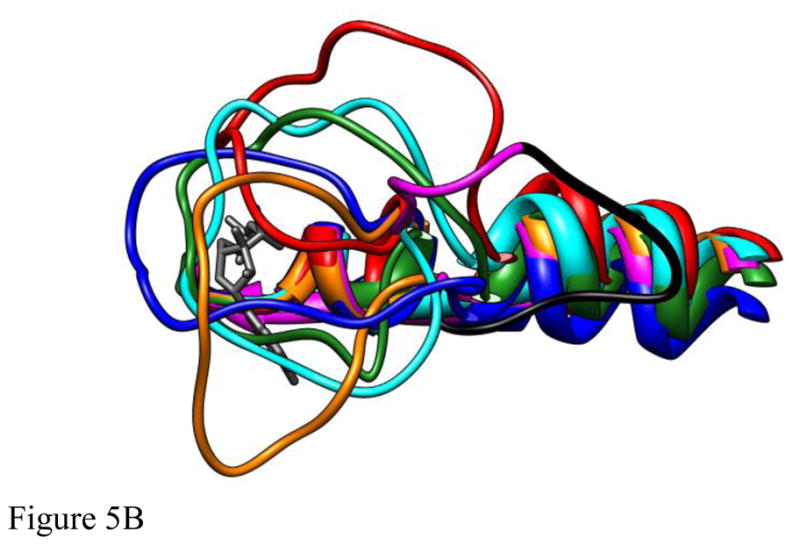
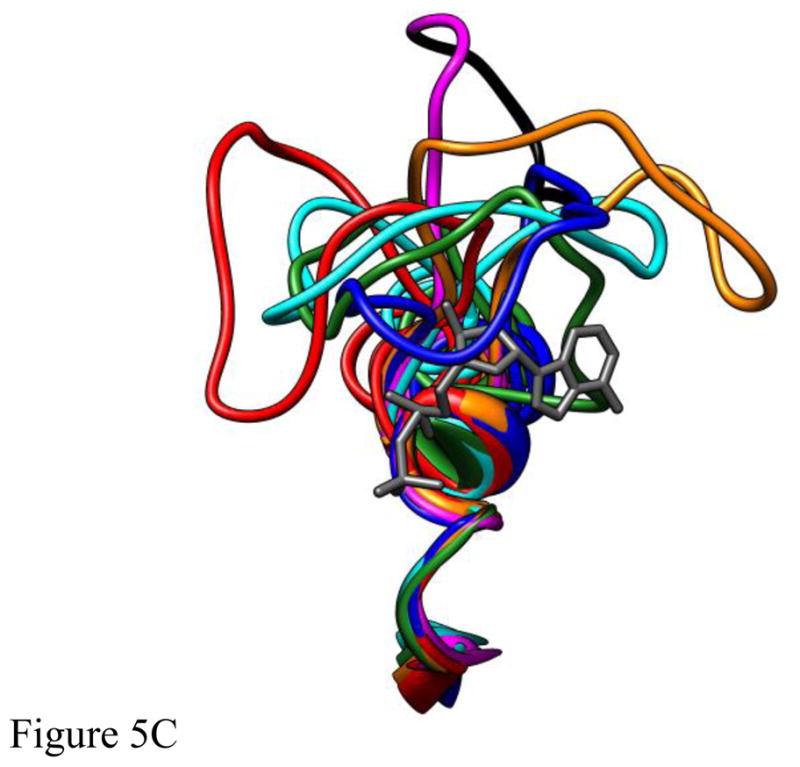
The structures of five different deformations of L5 in the Eg5•ADP structure (blue, cyan, red, green, orange) interacting with ADP bound at the nucleotide site (dark gray) following 4ns of MD simulation. The structures were superimposed using a least-squares distance minimization of the P-loop Cα atoms. The structure of Eg5•ADP (magenta with the domain deleted for Eg5-ΔL5 in black) is shown for comparison. Fig. 5A is oriented from the side with L5 at the top of the panel. Fig. 5B looks down from the top of panel A. Fig. 5C looks from a direction facing the bound ADP.
Fig. 6 shows higher resolution detail of representative stabilizing forces for the interactions of L5 with the nucleotide and the remainder of the protein for the simulations in which the conformation of L5 was modified. In Fig. 6, there are extensive intra-loop5 interactions in addition to interactions with the α2- (Thr112-Thr114) and α3-helices (Arg221). There is additional interaction with the short α-helix at the C-terminus of the β1-strand (Leu30-Arg33). This includes a hydrophobic stacking of the side-chain of Leu30, the side-chain ring of Trp127, the adenine ring of ADP and the phenyl ring of Phe113 (latter not shown for clarity). Fig. 7 shows an alternative conformation. In addition to intra-loop5 stabilizing interactions, there are interactions with the α1- (Lys77) and α2-helices (Thr112), the β3-strand (Arg24), with the side-chain ring of Trp127 located in a pocket formed by the hydrophobic portions of the side-chains of Asp130, Ala133, Arg119 and Thr112. The simulations in Figs. 6 and 7 were chosen randomly as representative. Other deformed conformations of L5 obtained from MD simulations with different initial deformations of L5 are shown in Figs. S2–S5 (Supplementary online material). In general, these conformations show interactions of the short α-helix at the C-terminus of the β1-strand (Leu30-Arg 33) and the α1-helix (Lys77) with L5. Interestingly, a recently solved X-ray crystal structure of Eg5 in the presence of AMPPNP (3HQD) shows some movement of these elements relative to their position in other structures (1II6, and 3KEN) [10, 41], indicating that some flexibility in these elements may accommodate different stable conformations of L5. As a further test of the stability of the deformed L5 conformations, the simulations were extended to 24 ns total simulation time. Compared to the L5 position after 4 ns (Fig. 4), the rms displacements of L5 (Cα – Cα) after 24 ns were 3.1Å (red), 2.2Å (orange), 2.3Å (green), 2.1Å (cyan), 3.5Å (blue), 2.8Å (magenta). All L5 domains remained projected over the nucleotide with little change in position during the additional 20 ns of simulation.
The above simulations demonstrate the MD time-scale stability of L5 conformations that impinge upon the nucleotide site. The L5 conformations were manually formed. A remaining question is whether L5 can actually deform into such impinging conformations in the face of thermal displacement. To address this question, we used the generalized Born approach for high-temperature MD simulations (details described in Methods). Higher temperature allows the simulation to more rapidly cross energy barriers than would be possible at physiological temperatures. Thus the feasible conformational space may thus be more rapidly sampled. Figure 8 shows the simulation approach applied to the Eg5•ADP X-ray structure (PDB ID 1II6, see Methods for details). Figure 8 shows the simulated location of L5 every 600ps during a 10 ns simulation run at 600K. Fig. 8A shows a “side” view with L5 at the top of the frame and ADP (standard atom colors) on the left-hand side of the frame. Fig. 8B shows the same structures from a point of view that looks down on L5 with the α2-helix underneath L5 and ADP partly obscured by L5 at the left-hand side. The EPR spectroscopy observation that raised the possibility of interaction between L5 and the nucleotide was from observations of 2′,3′-SLADP bound at the nucleotide site of Eg5 and the hinderance of the mobility the pyrrolidine spin-ring moiety. The spin ring is bridged by a carbonyl linkage to O2′- or the O3′-position the ribose ring of ADP. The ring plus leash is approximately 5.5Å in length and the pyrrolidine ring is 6.6Å across at its widest (spanning across the methyl groups). A diagram of SLADP with spin-ring moiety is in Fig. 1, ref. [40]. The important observation is that approximately half of the L5 orientations seen in Fig. 8 would provide additional restrictions on the motion of the attached spin probe beyond that of the native nucleotide site itself. This is again compatible with the experimental observation that the EPR probes bound to Eg5 see additional restriction of mobility.
4. DISCUSSION
Kinesin-family motors contain an unusual Loop 5 structure that breaks the α2-helix two-turns from the P-loop and projects out from the α2-helix into the aqueous environment. L5 is of variable length in different kinesin-family motors. L5 is surprisingly absent from kinesin’s evolutionary cousins, the myosin motors and the G-proteins [3], suggesting kinesin motor-specific function(s). The function of L5 remains unresolved. Interest in L5 function has been heightened by X-ray crystallography observations that L5 is involved in the binding of small-molecule, allosteric inhibitors of Eg5 [11, 12, 14, 42]. The specificity of the allosteric inhibitors to kinesin-5 motors and the obligatory involvement of Eg5 in mitosis offer the potential for the development of novel anti-cancer therapies.
In the x-ray structures of all kinesin-family motors with a nucleotide or a nucleotide analog at the active site, the ribose hydroxyls point out into the aqueous environment. This would place the spin moiety of 2′,3′-SLADP (Fig. S1) also projecting into the aqueous environment where it would be in a position to interact with a sufficiently long L5. The presence of a more immobilized component in the experimentally observed EPR spectrum of Eg5•2′,3′-SLADP, coupled with the absence of the more immobilized component in a species with a shortened L5-domain, or an L5 domain stabilized in a conformation away from the nucleotide site as indicated by the X-ray structure of Eg5•2′,3′-SLADP/STLC, initially suggested a direct interaction of L5 with the bound nucleotide. Supporting this idea, the MD simulations in this work demonstrate that the 18 amino-acid long L5-domain is sufficient in length to interact with the nucleotide. Furthermore we find that the shortened L5-domain of Eg5-ΔL5 does not interact significantly with the nucleotide in the MD simulations. Multiple attempts at deforming the ΔL5-domain into a configuration that would stably interact with the nucleotide failed. Interaction of L5 with the nucleotide site is further supported by studies suggesting that mutations of L5 perturb the Eg5 kinetic cycle [1, 12, 15–18, 22].
Surprisingly, the length of the L5-domain allowed for multiple conformations of L5 that formed new stable equilibrium conformations on the MD time scale (Figs 6–7 and Figs. S2–S5). Depending upon the specific simulation, inter-protein interactions were seen between L5 and the α1-, α2- and α3-helices, the β1-strand, and the short α-helix at the C-terminus of the β1-strand. To again emphasize the size of L5, we note that the latter short α-helix is located on the opposite side of the bound ADP adenine ring from the point of projection of L5 out of the α2-helix. Another unusual feature of L5 is the presence of the tryptophan residue, Trp127. In Figs. 6 and 7 it is involved in extensive hydrophobic interactions helping to stabilize the new conformation of L5. However, in other MD-simulated conformations, it is exposed to solvent. Although we are seeing multiple equilibrium conformations of L5 interacting with the nucleotide, we cannot rule out the possibility that given sufficient time, the intra-protein energetics would result in the multiple conformations evolving into a smaller subset, or a single preferred interacting conformation. Existing data from Maliga, et al. [22] indicate that W127 is highly dynamic in the absence of bound drug, and even exhibits some dynamics in the presence of drug. These may be short-lived conformations that nonetheless have a profound effect on Eg5 kinetic properties.
A significant observation is that stable interactions with the nucleotide site were formed for all six of our initial conditions with the intact L5 domain deformed over the nucleotide site. No simulation failed to interact with the nucleotide. The reason is that the L5 domain is sufficiently lengthy that such interactions are readily feasible and to be expected. However, the control MD simulations using the X-ray structure as the starting point showed an additional equilibrium position for the L5 domain with it pointing away from the nucleotide-site, not interacting with bound nucleotide, and stabilized primarily by W127 interacting with other hydrophobic parts of the protein. These observations are not in conflict. They provide further support for the hypothesis that there are multiple equilibrium configurations for L5, each representing a local free energy minimum on a global free energy surface, with X-ray structures to date having only captured one of the conformations of L5. These nucleotide-interacting conformations of L5 need not necessarily be long-lived; explaining why the MD/EPR spectroscopy approach used here has identified them while crystallography has yet to capture them. The simulations with Eg5-ΔL5 provide further support. Here unlike native Eg5, the deformed L5 of Eg5-ΔL5 that served as the initial condition for the simulation was too short to form any new intra-protein contacts to stabilize an interaction with the nucleotide. Thus the deformed conformations were not stable.
X-ray structures are fixed snapshots of a multi-conformation process. MD based on initial X-ray structures and EPR spectroscopy data has revealed functionally relevant conformations of kinesin motors in the past, many years before those conformations were revealed using experimental techniques. Initial X-ray structures from the year 1996 showed a single conformation of Switch 1, pulled away from the nucleotide opening the nucleotide pocket [8, 9, 43]. MD simulations performed in 2001 on the Switch 1 domain at the nucleotide-site of kinesin-family motors yielded multiple, stable, equilibrium conformations, just as we see here for the Eg5 L5. In particular, these simulations revealed a stable conformation with the Switch 1 domain adjacent to the nucleotide, closing the nucleotide-pocket [26]. Only very recently in 2010, an X-ray crystal structure was solved for a kinesin family member having a similar closed Switch 1 conformation [41].
Relationship to other work
A number of differing functions have been proposed for L5. In the X-ray structures of kinesin-family motors, the adenine ring is bound in a hydrophobic slot formed on one side by the adenine-binding loop frequently termed N4 [4]. This includes the ring of Pro27 and the propyl-group of Arg26 in Eg5. The “lid” of the pocket on the solvent side is conserved as a histidine, tyrosine or phenylalanine in kinesin-family motors (Phe113 in Eg5). Thus other than hydrophobic stacking, there is little to hold intact the adenine-binding pocket of kinesin-family motors. MD simulations originally showed this could result in a concurrent rotation of the amino-acid side-chain lid ring and adenine ring out of the binding pocket of ncd•ADP or kinesin•ADP. The rotation of the two stacked rings was stopped by steric interference from amino acid side chains at the base of L5. This led to our original suggestion that L5 served simply as a “bumper stop” to modulate nucleotide binding [26]. In myosins the lid is instead the hydrophobic portion of long, charged side chains, with the resultant salt-bridge providing additional stabilization. In G-proteins, there are additional ionic and hydrogen-bonding interactions to the bound guanine ring to help stabilize the bound nucleotide in the binding site. This may help to explain why L5 is missing in the latter two protein families.
A number of lines of evidence have suggested that an interaction between L5 and the adjacent α3-helix may likewise be involved in modulating motor function. A direct interaction was first observed in the X-ray structure of the non-motile kinesin-10 motor NOD [44], with the first 6 amino acids of L5 swinging away from the nucleotide site and packing against the α3-helix. EM reconstructions of microtubule-bound Drosophila kinesin-5 motor KLP61F likewise indicated that L5 undergoes a microtubule-binding dependent interaction with the α3-helix that was postulated to be involved in the transition between force-generating and diffusional modes of MT binding [45]. Spectroscopic and biochemical data have additionally linked L5 to the force-generating cycle through changes in the conformation of the neck-linker [1, 15, 46] that have been proposed to be modulated via interaction with the α3-helix and concomitant perturbations of Switch 1 by the α3-helix [15]. We observed only minimal interaction of L5 with the α3-helix in our simulations (a single interaction with Arg221 in two of the six simulations). However, our observations and the above observations are not in conflict. Our distortions of L5 to generate the initial conditions for the MD simulations were biased to emphasize interactions with the nucleotide site. L5 is clearly of sufficient length to generate interactions with the α3-helix that are even more extensive than are currently present in the X-ray and EM databases.
In summary, as previously noted, mutations of L5 have been implicated in perturbations of the Eg5 kinetic cycle [1, 12, 16–18, 22]. Here we have suggested another possible function for L5 in kinesin-5 motors, modulation of nucleotide binding via direct physical interaction with the nucleotide site. L5 is of surprisingly variable length over the breadth of the kinesin-family motors. There is likewise minimal sequence conservation. Considering the structural and functional results in toto, it seems plausible that L5 may play multiple functions that are dependent on the specific kinesin-family motor in question.
Supplementary Material
Highlights.
Loop5 in Eg5 projects from the protein adjacent to the nucleotide site.
Loop5 modulates Eg5 function by unresolved mechanisms.
Spectroscopy has suggested that loop5 may interact with the nucleotide site.
MD simulations are used to quantitatively test the interaction hypothesis.
The simulations show loop5 can interact extensively with the nucleotide site.
Acknowledgments
This work was supported by NIH grants GM077067 (E.P., N.N.), AR053720 (E.P.), AR042895 (R.C., N.N.) and GM072656 (S.R., A.L.). The computational portion of the research was performed using EMSL, a national scientific user facility sponsored by the Department of Energy’s Office of Biological and Environmental Research and located at Pacific Northwest National Laboratory.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Larson AG, Naber N, Cooke R, Pate E, Rice SE. The conserved L5 loop establishes the pre-powerstroke conformation of the Kinesin-5 motor, Eg5. Biophys J. 2010;98:2619–2627. doi: 10.1016/j.bpj.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rayment I, Smith C, Yount RG. The active site of myosin. Annu Rev Physiol. 1996;58:671–702. doi: 10.1146/annurev.ph.58.030196.003323. [DOI] [PubMed] [Google Scholar]
- 3.Kull FJ, Vale RD, Fletterick RJ. The case for a common ancestor: kinesin and myosin motor proteins and G proteins. J Muscle Res Cell Motil. 1998;19:877–886. doi: 10.1023/a:1005489907021. [DOI] [PubMed] [Google Scholar]
- 4.Vale RD. Switches, latches, and amplifiers: common themes of G proteins and molecular motors. J Cell Biol. 1996;135:291–302. doi: 10.1083/jcb.135.2.291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walker JE, Saraste M, Runswick MJ, Gay NJ. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold. Embo J. 1982;1:945–951. doi: 10.1002/j.1460-2075.1982.tb01276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J, Shen Y, Tempel W, Landry R, Arrowsmith CH, Edwards AM, Sundstrom M, Weigelt J, Bochkarev A, Park H. Crystal Structure of the KIF2C motor domain (CASP Target) 2006 http://www.rcsb.org/pdb/explore/explore.do?structureId=2HEH.
- 7.Greene L, Henikoff S. Kinesin home page. 1996 http://www.cellbio.duke.edu/kinesin/
- 8.Sablin EP, Kull FJ, Cooke R, Vale RD, Fletterick RJ. Crystal structure of the motor domain of the kinesin-related motor ncd. Nature. 1996;380:555–559. doi: 10.1038/380555a0. [DOI] [PubMed] [Google Scholar]
- 9.Kull FJ, Sablin EP, Lau R, Fletterick RJ, Vale RD. Crystal structure of the kinesin motor domain reveals a structural similarity to myosin. Nature. 1996;380:550–555. doi: 10.1038/380550a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Turner J, Anderson R, Guo J, Beraud C, Fletterick R, Sakowicz R. Crystal structure of the mitotic spindle kinesin Eg5 reveals a novel conformation of the neck-linker. J Biol Chem. 2001;276:25496–25502. doi: 10.1074/jbc.M100395200. [DOI] [PubMed] [Google Scholar]
- 11.Yan Y, Sardana V, Xu B, Homnick C, Halczenko W, Buser CA, Schaber M, Hartman GD, Huber HE, Kuo LC. Inhibition of a mitotic motor protein: where, how, and conformational consequences. J Mol Biol. 2004;335:547–554. doi: 10.1016/j.jmb.2003.10.074. [DOI] [PubMed] [Google Scholar]
- 12.Maliga Z, Mitchison TJ. Small-molecule and mutational analysis of allosteric Eg5 inhibition by monastrol. BMC Chem Biol. 2006;6:2. doi: 10.1186/1472-6769-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kaan HY, Ulaganathan V, Hackney DD, Kozielski F. An allosteric transition trapped in an intermediate state of a new kinesin-inhibitor complex. Biochem J. 2010;425:55–60. doi: 10.1042/BJ20091207. [DOI] [PubMed] [Google Scholar]
- 14.Kim ED, Buckley R, Learman S, Richard J, Parke C, Worthylake DK, Wojcik EJ, Walker RA, Kim S. Allosteric drug discrimination is coupled to mechanochemical changes in the kinesin-5 motor core. J Biol Chem. 2010;285:18650–18661. doi: 10.1074/jbc.M109.092072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Behnke-Parks WM, Vendome J, Honig B, Maliga Z, Moores C, Rosenfeld SS. Loop L5 acts as a conformational latch in the mitotic kinesin EG5. J Biol Chem. 2010;286:5242–5253. doi: 10.1074/jbc.M110.192930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brier S, Lemaire D, DeBonis S, Forest E, Kozielski F. Molecular dissection of the inhibitor binding pocket of mitotic kinesin Eg5 reveals mutants that confer resistance to antimitotic agents. J Mol Biol. 2006;360:360–76. doi: 10.1016/j.jmb.2006.04.062. [DOI] [PubMed] [Google Scholar]
- 17.Brier S, Lemaire D, Debonis S, Forest E, Kozielski F. Identification of the protein binding region of S-trityl-L-cysteine, a new potent inhibitor of the mitotic kinesin Eg5. Biochemistry. 2004;43:13072–82. doi: 10.1021/bi049264e. [DOI] [PubMed] [Google Scholar]
- 18.Brier S, Lemaire D, DeBonis S, Kozielski F, Forest E. Use of hydrogen/deuterium exchange mass spectrometry and mutagenesis as a tool to identify the binding region of inhibitors targeting the human mitotic kinesin Eg5. Rapid Commun Mass Spectrom. 2006;20:456–62. doi: 10.1002/rcm.2329. [DOI] [PubMed] [Google Scholar]
- 19.Wang J, Shen Y, Tempel W, Landry R, Arrowsmith CH, Edwards AM, Sundstrom M, Weigelt J, Bochkarev A, Park H. Crystal structure of the human KIF2 motor domain in complex with ADP. 2006 http://www.rcsb.org/pdb/explore/explore.do?structureId=2GRY.
- 20.Zhu H, Shen Y, Tempel W, Dobrovetsky E, MacKenzie F, Arrowsmith CH, Edwards AM, Weigelt J, Bochkarev A, Park H. Crystal structure of the motor domain of human kinesin family member. 2007;22 http://www.rcsb.org/pdb/explore/explore.do?structureId=3BFN.
- 21.Tan D, Rice WJ, Sosa H. Structure of the kinesin13-microtubule ring complex. Structure. 2008;16:1732–1739. doi: 10.1016/j.str.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maliga Z, Xing J, Cheung H, Juszczak LJ, Friedman JM, Rosenfeld SS. A pathway of structural changes produced by monastrol binding to Eg5. J Biol Chem. 2006;281:7977–7982. doi: 10.1074/jbc.M511955200. [DOI] [PubMed] [Google Scholar]
- 23.Hariharan V, Hancock WO. Insights into the mechanical properties of the kinesin neck linker domain from sequence analysis and molecular dynamics simulations. Cell Molec Bioeng. 2009;2:177–189. doi: 10.1007/s12195-009-0059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khalil AS, Appleyard DC, Labno AK, Georges A, Karplus M, Belcher AM, Hwang W, Lang MJ. Kinesin’s cover-neck bundle folds forward to generate force. Proc Natl Acad Sci U S A. 2008;105:19247–52. doi: 10.1073/pnas.0805147105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mennella V, Tan DY, Buster DW, Asenjo AB, Rath U, Ma A, Sosa HJ, Sharp DJ. Motor domain phosphorylation and regulation of the Drosophila kinesin 13, KLP10A. J Cell Biol. 2009;186:481–90. doi: 10.1083/jcb.200902113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Minehardt TJ, Cooke R, Pate E, Kollman PA. Molecular dynamics study of the energetic, mechanistic, and structural implications of a closed phosphate tube in ncd. Biophys J. 2001;80:1151–1168. doi: 10.1016/S0006-3495(01)76092-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wriggers W, Schulten K. Nucleotide-dependent movements of the kinesin motor domain predicted by simulated annealing. Biophys J. 1998;75:646–61. doi: 10.1016/S0006-3495(98)77555-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 29.Case DA, Darden TA, Cheatham TEI, Simmerling CL, Wang J, Duke RE, Luo R, Crowley M, Walker RC, Zhang W, Merz KM, Wang B, Hayik S, Roitberg A, Seabra G, Kolossváry I, Wong KF, Paesani F, Vanicek J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Yang L, Tan C, Mongan J, Hornak V, Cui G, Mathews DH, Seetin MG, Sagui C, Babin V, Kollman PA. AMBER10. University of California; San Francisco: 2008. [Google Scholar]
- 30.Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Montgomery JJA, Vreven T, Kudin KN, Burant JC, Millam JM, Iyengar SS, Tomasi J, Barone V, Mennucci B, Cossi M, Scalmani G, Rega N, Petersson GA, Nakatsuji H, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Klene M, Li X, Knox JE, Hratchian HP, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi R, Pomelli C, Ochterski JW, Ayala PY, Morokuma K, Voth GA, Salvador P, Dannenberg JJ, Zakrzewski VG, Dapprich S, Daniels AD, Strain MC, Farkas O, Malick DK, Rabuck AD, Raghavachari K, Foresman JB, Ortiz JV, Cui Q, Baboul AG, Clifford S, Cioslowski J, Stefanov BB, Liu G, Liashenko A, Piskorz P, Komaromi I, Martin RL, Fox DJ, Keith T, Al-Laham MA, Peng CY, Nanayakkara A, Challacombe M, Gill PMW, Johnson B, Chen W, Wong MW, Gonzalez C, Pople JA. Gaussian03. Gaussian, Inc; Wallingford, CT: 2004. [Google Scholar]
- 31.Bayly CI, Ciepak WD, Cornell WD, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J Phys Chem. 1993;97:10269–10280. [Google Scholar]
- 32.Aqvist J. Modelling of ion-ligand interactions in solutions and biomoleculaes. J Mol Struct (Theochem) 1992;256:135–152. [Google Scholar]
- 33.Jorgensen WL, Chandrasekar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Comp Phys. 1983;79:926–935. [Google Scholar]
- 34.Berendsen HJC, Postma JPM, Gunsteren WFv, DiNola A, Ghaak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;113:3684–3690. [Google Scholar]
- 35.Darden T, York D. Particle mesh Ewald - an Nlog(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. [Google Scholar]
- 36.Essman U, Preera M, Berkowitz T, Darden H, Pedersen G. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
- 37.Hawkins GD, Cramer CJ, Truhlar DG. Parameterized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J Chem Physics. 1996;100:19824–19839. [Google Scholar]
- 38.Ryckaert AJ, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comp Phys. 1977;23:327–341. [Google Scholar]
- 39.Griffith OH, Jost PC. Lipid spin labels in biological membranes. In: Berliner LJ, editor. Spin Labeling Theory and Applications. Academic Press; NY: 1976. pp. 454–523. [Google Scholar]
- 40.Naber N, Minehardt TJ, Rice S, Chen X, Grammer J, Matuska M, Vale RD, Kollman PA, Car R, Yount RG, Cooke R, Pate E. Closing of the nucleotide pocket of kinesin-family motors upon binding to microtubules. Science. 2003;300:798–801. doi: 10.1126/science.1082374. [DOI] [PubMed] [Google Scholar]
- 41.Parke CL, Wojcik EJ, Kim S, Worthylake DK. ATP hydrolysis in Eg5 kinesin involves a catalytic two-water mechanism. J Biol Chem. 2010;285:5859–5867. doi: 10.1074/jbc.M109.071233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kaan HY, Ulaganathan V, Rath O, Prokopcova H, Dallinger D, Kappe CO, Kozielski F. Structural basis for inhibition of Eg5 by dihydropyrimidines: stereoselectivity of antimitotic inhibitors enastron, dimethylenastron and fluorastrol. J Med Chem. 2010;53:5676–5683. doi: 10.1021/jm100421n. [DOI] [PubMed] [Google Scholar]
- 43.Kozielski F, Sack S, Marx A, Thormahlen M, Schonbrunn E, Biou V, Thompson A, Mandelkow EM, Mandelkow E. The crystal structure of dimeric kinesin and implications for microtubule-dependent motility. Cell. 1997;91:985–994. doi: 10.1016/s0092-8674(00)80489-4. [DOI] [PubMed] [Google Scholar]
- 44.Cochran JC, Sindelar CV, Mulko NK, Collins KA, Kong SE, Hawley RS, Kull FJ. ATPase cycle of the nonmotile kinesin NOD allows microtubule end tracking and drives chromosome movement. Cell. 2009;136:110–122. doi: 10.1016/j.cell.2008.11.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bodey AJ, Kikkawa M, Moores CA. 9-Angstrom structure of a microtubule-bound mitotic motor. J Mol Biol. 2009;388:218–224. doi: 10.1016/j.jmb.2009.03.008. [DOI] [PubMed] [Google Scholar]
- 46.Rosenfeld SS, Xing J, Jefferson GM, King PH. Docking and rolling, a model of how the mitotic motor Eg5 works. J Biol Chem. 2005;280:35684–35695. doi: 10.1074/jbc.M506561200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.