

Abstract
This study tested the hypothesis that sensitivity to the Ca2+-induced loss of mitochondrial membrane potential (ΔΨm) and the sensitivity of the loss of ΔΨ to mitochondrial permeability transition pore (PTP) inhibitors are different for neurons and astrocytes. Primary cultures of rat cortical neurons and astrocytes were exposed to the Ca2+ ionophore 4-Br-A23187 and ΔΨm was monitored with the fluorescent probe tetramethylrhodamine methyl ester (TMRM). Ca2+ ionophore caused a decline in ΔΨm in both cell types that was partially inhibited by cyclosporin A (CsA) in astrocytes but not in neurons. Another PTP inhibitor, 2-aminoethoxy-diphenylborate, was ineffective at protecting against mitochondrial depolarization but depolarization was inhibited by FK506, an immunosuppressant drug similar to CsA that does not inhibit the PTP. CsA and FK506 both significantly reduced the ionophore-induced rise in [Ca2+]i in both neurons and astrocytes. We conclude that the protective effects of CsA and FK506 against Ca2+ ionophore-induced mitochondrial membrane depolarization in intact astrocytes is not due to PTP inhibition but is possibly a consequence of inhibiting the rise in [Ca2+]i.
Keywords: Permeability transition pore, immunophilin, TMRM, calcium homeostasis, mitochondria, 4-Br-A23187
Introduction
Cell death induced by a variety of insults is often preceded by an abnormal increase in [Ca2+]i. During cellular Ca2+ overload, mitochondria accumulate Ca2+, which under some circumstances triggers opening of the inner membrane permeability transition pore (PTP). Opening of this non-selective pore results in the loss of mitochondrial membrane potential (ΔΨm), loss of Ca2+ and other mitochondrial solutes, including metabolic cofactors of <1500 Da, and osmotic swelling (Crompton, 1999; Di Lisa et al., 2001; Petronilli et al., 2001; Zoratti and Szabo, 1995). The PTP is activated by Ca2+ and oxidative stress and inhibited by Mg2+, adenine nucleotides and the immunosuppressant drug cyclosporin A (CsA) (Fournier et al., 1987; Gunter and Pfeiffer, 1990; Halestrap et al., 1997a; Halestrap et al., 1997b; Kristal and Dubinsky, 1997). Brain mitochondria display heterogeneity in their response to Ca2+-induced MPT, with neuronal mitochondria being relatively resistant to inhibition by CsA, compared to astrocyte mitochondria (Bambrick et al., 2006; Brustovetsky and Dubinsky, 2000; Kristian et al., 2002). Considering the importance of mitochondrial dysfunction in neural cell death, the cellular basis for the heterogeneous responses of brain mitochondria to large Ca2+ loads could provide insight into the mechanisms responsible for selective cell vulnerability and cell-selective targets of neuroprotective drugs, e.g., CsA (Li et al., 2000; Uchino et al., 1998; Yoshimoto and Siesjo, 1999; Sullivan et al., 2011). Previous experiments performed with digitonin-permeabilized primary cultures of cerebellar granule neurons and cortical astrocytes demonstrated that mitochondrial Ca2+ uptake, and resistance to Ca2+-induced bioenergetic dysfunction, was potentiated by CsA in astrocytes but not neurons (Bambrick et al., 2006). The much greater complexity of intact cells compared to isolated mitochondria or permeabilized cells can influence mitochondrial activities and their response to stress. The goal of this study was to test the hypothesis that mitochondria within intact neurons and astrocytes display differential sensitivity to inhibition of Ca2+-induced PTP opening by CsA, as evaluated by measuring the response of ΔΨm following exposure of cells to a Ca2+ ionophore. Comparisons of the effects of PTP inhibitors on ionophore-induced rise in [Ca2+]i uncovered an additional effect of immunosuppressant drugs that influences the interpretation of their effects on mitochondria and the PTP.
MATERIALS AND METHODS
Cell culture
Cerebral cortex astrocytes and neurons were isolated from 17 day in utero Sprague-Dawley rat fetuses. All animal procedures were carried out according to the National Institutes of Health and the University of Maryland, Baltimore, Guidelines for the Care and Use of Laboratory Animals. Cortical neurons were grown on 25 mm coverslips for 10–17 days in vitro, at a density of ~50,000 cell/coverslip, in Neurobasal medium with glutamine, penicillin, streptomycin and B27 supplement in 95% air/5% CO2 at 37°C. Glial proliferation was prevented by adding cytosine-arabinofuranoside (5 μM) 24 hr after plating. Immunocytochemical measurements of glial fibrillary acid protein (GFAP) confirmed that cultures contained <1% glia. Cortical astrocytes were cultured in DMEM/F12 (1/1) 10% FBS with penicillin and streptomycin in 95% air/5% CO2 at 37°C at a density of 20,000 cells/coverslip. For astrocytes, the culture medium was changed every 3 days and the cells were used at 14–17 days in vitro within 48 hr after changing the medium. Cultures were >95% astrocytes by GFAP immunocytochemistry.
Fluorescence microscopy
Imaging of Intracellular Mitochondrial Membrane Potential
Mitochondrial membrane potential (ΔΨm) was followed in intact cortical neurons and astrocytes by epifluorescence microscopy. Cells were loaded with 50 nM tetramethylrhodamine methyl ester perchlorate (TMRM, Molecular Probes Eugene, OR) in DMSO (0.1% final concentration) for 20 min at 37°C. The coverslips were mounted in the chamber of a Nikon Eclipse TE2000-S inverted microscope (Objectives: SFluor 20×0.75 N.A. and 40×1.2 N.A.) in recording solution containing 120 mM NaCl, 3.5 mM KCl, 20 mM HEPES, 1.3 mM CaCl2, 5 mM NaHCO3, 1.0 mM MgCl2, 15 mM glucose, and 5 nM TMRM, pH 7.4. Experiments were performed at room temperature. Single cell fluorescence of TMRM was imaged by excitation at 547 nm (Polychrome IV, Till, Munich, Germany), and emission at 620 nm. Image sequences (50 msec exposure time, 2×2 binning, 10 s timelapse) were acquired by an ORCA-ER CCD camera (Hamamatsu Photonics, Hamamatsu, Germany). Data acquisition and analysis were conducted using Metafluor 5.0 (Universal Imaging, West Chester, PA) imaging software.
Ca2+ influx into the cell was stimulated by the addition of the calcium ionophore 4-bromo-A23187 (5 μM). The mitochondrial uncoupler FCCP (5 μM) was applied at the end of experiments to indicate the level of fluorescence of TMRM that reflected maximal dissipation of ΔΨm. Data are presented as TMRM fluorescence at time t= [ Ft-FFCCP/F0] x 100%.
Imaging of Intracellular Ca2+
Cells were loaded with 2 μM of Fura FF (Molecular Probes, Eugene, OR) at room temperature in culture medium. Neurons were loaded for 20 min and astrocytes were loaded for 40 min followed by a 15 min dye de-esterification period. Cells were illuminated alternately with 340 and 380 nm and fluorescence was monitored at 510 nm with the same imaging system used for TMRM. Ratios were calculated after subtraction of a background image from each wavelength (340 and 380). [Ca2+]i levels were expressed as the ratio of emitted fluorescence on excitation at 340 and 380 nm. Cells were pre-incubated with treatment drugs; 2 μM of CsA, 1 μM of FK506 and 100 μM of 2-aminoethoxydiphenyl borate (2-APB) for 30 min. Drugs were also present in all solutions during the entire experiment.
Statistical Analysis
All data are expressed as means ± S.E.M. of n cells. Data from individual cultures were pooled to calculate group means. Data presented represent 4–8 coverslips from 2–4 independent cell preparations. Statistical significance was assessed by one-way ANOVA test followed by the Tukey test for multiple comparisons. For data that were not normally distributed, the Mann-Whitney test or Kruskal-Wallis non-parametric ANOVA with Dunn’s post hoc test for multiple comparisons of unbalanced data was used. p<0.05 was considered to be statistically significant.
Materials
FK-506 was from LC Laboratories (Woburn, MA, USA). All cell culture reagents were from GIBCO-BRL. All other chemical reagents were from Sigma USA.
RESULTS
Ca2+ ionophore-induced mitochondrial depolarization in cortical astrocytes and neurons
Baseline TMRM fluorescence intensity in both astrocytes and neurons was stable, declining less than 10% over 30 min, and was not affected by the presence of PTP inhibitors. Exposure of cells to 5 μM of 4-Br-A23187 resulted in a rapid decline in TMRM fluorescence, as shown in Figure 1. Fluorescence changes were measured at 2, 5, and 10 min after the addition of the ionophore and came to completion by 10 min. Figure 2 compares these changes for astrocytes and neurons at 10 min after exposure to 4-Br-A23187. With ionophore alone, the decline in fluorescence intensity was approximately 80% for astrocytes and 75% for neurons. Preincubation of cells with CsA for 30 min attenuated the decrease in the astrocyte TMRM signal but actually increased the fluorescence loss in neurons (Figures 1 and 2). In addition to being a well-characterized inhibitor of the mitochondrial PTP, CsA is also a potent inhibitor of specific protein phosphatases, e.g., calcineurin. The effect of FK506 on ionophore-induced mitochondrial depolarization was tested as a control since it also inhibits these phosphatases but does not inhibit the PTP (Connern and Halestrap, 1994). Surprisingly, FK506 also protected against the loss of TMRM fluorescence in astrocytes. As with CsA, FK506 also slightly promoted the drop in TMRM signal in neurons. Since the positive affect of FK506 with astrocytes suggests that PTP inhibition is not necessary for the inhibition of mitochondrial depolarization by CsA, another PTP inhibitor without known effects on protein phosphorylation was tested. 2-aminoethoxy-diphenyl borate (2-APB) was shown previously to inhibit PTP opening in brain mitochondria under conditions where CsA is ineffective (Chinopoulos et al., 2003). This agent displayed no significant effect on the fluorescent decline with either astrocytes or neurons (Figure 2).
Figure 1. Ca2+-induced loss of TMRM fluorescence in astrocytes and neurons.
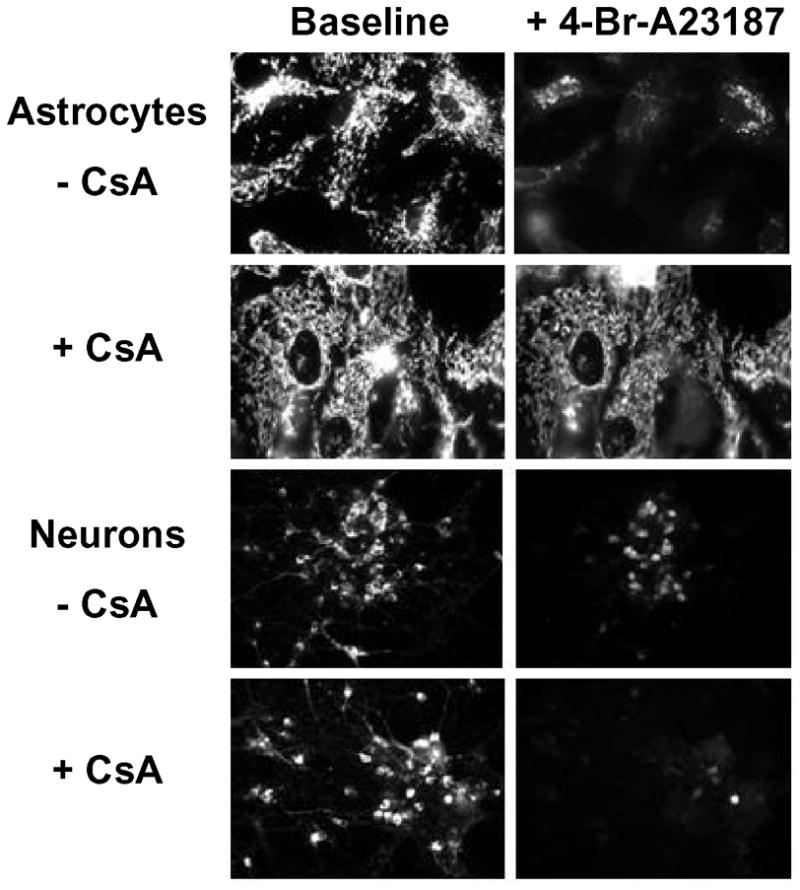
Mitochondrial membrane potential (ΔΨm) was followed in intact cortical neurons and astrocytes by epifluorescence microscopy with the fluorophore TMRM. Images are representative of those obtained for rat cortical astrocytes and neurons before and 10 minutes after the addition of the Ca2+ ionophore 4-Br-A23187 (5 μM) in the absence or presence of 45 min pretreatment and continued exposure to the PTP inhibitor cyclosporin A (CsA; 2 μM).
Figure 2. Changes in mitochondrial membrane potential (ΔΨm) following exposure of astrocytes and neurons to Ca2+ ionophore.
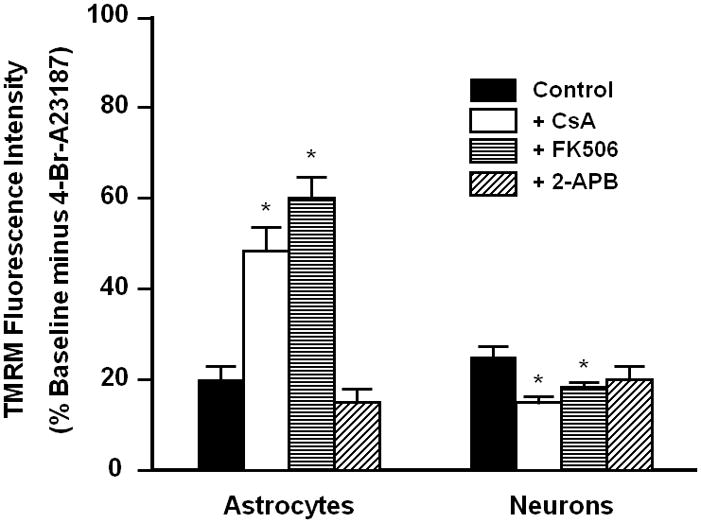
Images such as those in Fig. 1 were analyzed for TMRM fluorescence intensity within the cell bodies of astrocytes and neurons before and 10 min after the addition of the Ca2+ ionophore 4-Br-A23187 (5 μM) in the absence (Control) or presence of a 45 min pretreatment and continued exposure to cyclosporine A (CsA, 2 μM), FK506 (1 μM), or 2-aminoethoxy diphenyl borate (100 μM). Data are expressed as percent of initial fluorescence before addition of ionophore, with 0% defined as the minimal fluorescence obtained 2 min after the addition of the mitochondrial uncoupler FCCP (5 μM) at 10 min after addition of ionophore. * p<0.05 when compared to control cells
Effects of Ca2+ ionophore, CsA, and FK506 on astrocyte and neuronal [Ca2+]i
Additional experiments were performed to determine how CsA could protect against loss of ΔΨm in astrocytes by a PTP independent mechanism. The low affinity Ca2+-sensitive dye Fura FF AM was used to evaluate the large increases in [Ca2+]i induced by exposure of astrocytes and neurons to 4-Br-A23187. Cortical astrocytes were heterogeneous in their response to 4-Br-A23187 with at least two distinct populations evident: those exhibiting a high initial peak and plateau (high responders) and those presenting a low transient initial peak followed by a delayed slight increase in [Ca2+]i (low responders) (Figure 3A). Pre-incubation of astrocytes with CsA totally abolished the high responder response (Figure 3B). Incubation with FK506 also reduced the number of cells that responded to the ionophore with a high initial [Ca2+]i peak (Figure 3C). Addition of the mitochondrial respiratory uncoupler FCCP at the end of the experiments where the initial rise in [Ca2+]i was suppressed by CsA or FK506 resulted in an abrupt increase in [Ca2+]i (Figure 3B,C).
Figure 3. Ca2+ionophore-induced rise in [Ca2+]i in cortical astrocytes.

Changes in Fura FF ratios (340/380) induced by 5 μM 4-Br-A23187 were heterogeneous among individual cortical astrocytes (light gray lines, n=100), with two distinct populations evident, those with high initial peak and plateau and those with low transient initial peak followed by delayed slight increase in [Ca2+]i (A). In contrast, the [Ca2+]i increase after 4-Br-A23187 was uniformly low in all cells preincubated with CsA (n=134) (B). Incubation with FK506 also reduced the number of cells with high initial [Ca2+]i peak (n=148) (C). Mean [Ca2+]i values (solid lines with SEM (dark gray) in A,B,C) of CsA and FK506-incubated cells were significantly lower than control cells. 5 μM of FCCP application 10 min after exposure to 4-Br- A23187 induced a further increase in treated cells (B,C).
Exposure of cortical neurons to the Ca2+ ionophore induced a uniform rise in [Ca2+]i that was more rapid than that observed in astrocytes (Figure 4A). The presence of either CsA or FK506 slightly inhibited the rate and extent to which [Ca2+]i rose in response to ionophore (Figure 4B.C), but not to the extent observed for astrocytes (Figure 3). The addition of FCCP resulted in a relatively very small additional rise in [Ca2+]i under all conditions. While there was no significant difference in the initial Fura FF ratio for astrocytes and neurons, the ratios observed after addition of ionophore in the absence or presence of CsA or FK506 were significantly greater for neurons than astrocytes (compare 3A and 4A, p<0.001).
Figure 4. Ca2+ionophore-induced rise in [Ca2+]i in cortical neurons.

Changes in Fura FF ratio (340/380) following exposure of cortical neurons to 5 μM of 4-Br-A23187 and 5 μM of FCCP. 4-Br- A23187 caused a large rapid increase in [Ca2+]i in both drug-treated (B,C) and untreated (A) cells (n= 211). Panel D provides a direct comparison between the mean increases (solid lines) shown in panels A,B, and C), demonstrating that the elevation observed in control cells was significantly higher than that of cells preincubated with 2 μM CsA (B) (n=286) or with 1 μM of FK506 (C) (n=166).
Discussion
The original aim of this study was to determine if PTP opening in response to elevated [Ca2+]i is different for astrocytes and neurons and whether the sensitivity to pharmacologic inhibition is different between these two major types of brain cells. Although CsA inhibited the drop in ΔΨm induced by Ca2+ ionophore in astrocytes, inhibition was also observed with FK506, an immunophilin binding drug that does not inhibit the PTP (Fig. 2). Moreover, inhibition was not observed with 2-APB, a PTP inhibitor that is a non-immunophilin-binding drug (Fig. 2). Measurements of intracellular Ca2+ in response to Ca2+ ionophore demonstrated that both CsA and FK506 substantially dampened the rise in cytosolic Ca2+ caused by 4-Br-A23187 in astrocytes, indicating that this effect is most likely responsible for their protection against mitochondrial depolarization. In contrast to the effects of CsA and FK506 on astrocytes, these agents slightly promoted rather than inhibited neuronal mitochondrial depolarization in response to Ca2+ ionophore. It is also possible that these agents affected the TMRM measurements by influencing the membrane potential of the plasma membrane rather than that of the mitochondria; however, such effects would necessarily be different for the two cell types. Similar to the effects of these drugs on astrocyte [Ca2+]i, they slightly reduced the ionophore-induced rise in [Ca2+]i in neurons.
These results fail to provide evidence for involvement of a CsA-sensitive PTP in mitochondrial depolarization in both astrocytes and neurons, albeit for different reasons. In astrocytes, the CsA inhibition of depolarization is most likely due to dampening the rise in intracellular Ca2+ since both effects are also observed with FK506. It is possible, however, that despite their effects on intracellular Ca2+, these agents both inhibit mitochondrial depolarization that is independent of PTP or of changes in Ca2+. These results do not exclude the possibility of a CsA-sensitive PTP but do reveal an additional mechanism of action for both CsA and FK506 that may obscure the direct effect of CsA on mitochondria. In neurons, neither CsA or FK506 protect against depolarization even though they reduce the rise in [Ca2+]i. It is possible, however, that CsA protection against loss of mitochondrial membrane potential in neurons would be observed at lower concentration of Ca2+ ionophore since both brain and liver mitochondria lose their sensitivity to protection against PTP opening by CsA at very high Ca2+ loads (Chinopoulos et al., 2003). Also, it is possible that at high levels of Ca2+ ionophore, the cycling of Ca2+ in and out of the mitochondrial membrane could dominate over flux through the PTP, thus obscuring inhibition by PTP inhibitors. Moreover, our experiments only utilized a rise in intracellular Ca2+ to induce mitochondrial membrane depolarization and the sensitivity to CsA could be different if oxidative stress plus Ca2+ were used as PTP inducers.
The ability of both CsA and FK506 to lower the large rise in [Ca2+]i elicited by ionophore could be very important for interpreting the cytoprotective effects of these drugs in paradigms known to be associated with pathologic elevations of [Ca2+]i. Thus neuroprotection by both CsA and FK506 has been reported in animal models of stroke and traumatic brain injury, where large elevations of intracellular Ca2+ are known to occur in both astrocytes and neurons (Alessandri et al., 2002; Friberg and Wieloch, 2002; Kuroda et al., 1999; Singleton et al., 2001; Sullivan et al., 2011). Inhibition of the PTP or regulation of protein phosphorylation-dependent apoptotic pathways are typically invoked as explanations for cytoprotection by immunophilin-based drugs. Our results indicate that their effects on intracellular Ca2+ should also be considered.
There are several possible mechanisms for the ability of these drugs to inhibit a rise in intracellular Ca2+. They may relieve inhibition of noncapacitative Ca2+ channels (Mignen et al., 2003). CsA has also been reported to stimulate uptake of Ca2+ into the endoplasmic reticulum through the endoplasmic reticulum Ca2+-ATPase (SERCA) (Smaili et al., 2001). Although direct entry of Ca2+ into cells via 4-Br-A23187 is primarily responsible the rise in [Ca2+]I in our experiments, additional mechanisms including those that both add to, and buffer the rise in Ca2+ can affect both the rate and extent of Ca2+ elevation. It is also possible that CsA and FK506 could lower the effective concentration of the Ca2+ ionophore by stimulated its active efflux through the multidrug resistance transporter (Morjani and Madoulet, 2010); however, this mechanism is unlikely due to the high concentration of ionophore that was used and its strong affinity to cell membranes.
Acknowledgments
Grant Support: Supported by DAMD 17-03-1-0745 to LLB, NIH NS07375 to SK, and NIH NS34152 and HD016596 to G.F.
Reference List
- Alessandri B, Rice AC, Levasseur J, DeFord M, Hamm RJ, Bullock MR. Cyclosporin A improves brain tissue oxygen consumption and learning/memory performance after lateral fluid percussion injury in rats. J Neurotrauma. 2002;19:829–841. doi: 10.1089/08977150260190429. [DOI] [PubMed] [Google Scholar]
- Bambrick LL, Chandrasekaran K, Mehrabian Z, Wright C, Krueger BK, Fiskum G. Cyclosporin A increases mitochondrial calcium uptake capacity in cortical astrocytes but not cerebellar granule neurons. J Bioenerg Biomembr. 2006;38:43–47. doi: 10.1007/s10863-006-9004-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brustovetsky N, Dubinsky JM. Dual responses of CNS mitochondria to elevated calcium. J Neurosci. 2000;20:103–113. doi: 10.1523/JNEUROSCI.20-01-00103.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinopoulos C, Starkov AA, Fiskum G. Cyclosporin A-insensitive permeability transition in brain mitochondria: Inhibition by 2-aminoethoxydiphenyl borate. J Biol Chem. 2003;278:27382–27389. doi: 10.1074/jbc.M303808200. [DOI] [PubMed] [Google Scholar]
- Connern CP, Halestrap AP. Recruitment of mitochondrial cyclophilin to the mitochondrial inner membrane under conditions of oxidative stress that enhance the opening of a calcium-sensitive non-specific channel. Biochem J. 1994;302:321–324. doi: 10.1042/bj3020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crompton M. The mitochondrial permeability transition pore and its role in cell death. Biochem J. 1999;341:233–249. [PMC free article] [PubMed] [Google Scholar]
- Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P. Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart. J Biol Chem. 2001;276:2571–2575. doi: 10.1074/jbc.M006825200. [DOI] [PubMed] [Google Scholar]
- Fournier N, Ducet G, Crevat A. Action of cyclosporine on mitochondrial calcium fluxes. J Bioenerg Biomembr. 1987;19:297–303. doi: 10.1007/BF00762419. [DOI] [PubMed] [Google Scholar]
- Friberg H, Wieloch T. Mitochondrial permeability transition in acute neurodegeneration. Biochimie. 2002;84:241–250. doi: 10.1016/s0300-9084(02)01381-0. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258:C755–C786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- Halestrap AP, Connern CP, Griffiths EJ, Kerr PM. Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol Cell Biochem. 1997a;174:167–172. [PubMed] [Google Scholar]
- Halestrap AP, Woodfield KY, Connern CP. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J Biol Chem. 1997b;272:3346–3354. doi: 10.1074/jbc.272.6.3346. [DOI] [PubMed] [Google Scholar]
- Kristal BS, Dubinsky JM. Mitochondrial permeability transition in the central nervous system: induction by calcium cycling-dependent and -independent pathways. J Neurochem. 1997;69:524–538. doi: 10.1046/j.1471-4159.1997.69020524.x. [DOI] [PubMed] [Google Scholar]
- Kristian T, Weatherby TM, Bates TE, Fiskum G. Heterogeneity of the calcium-induced permeability transition in isolated non-synaptic brain mitochondria. J Neurochem. 2002;83:1297–1308. doi: 10.1046/j.1471-4159.2002.01238.x. [DOI] [PubMed] [Google Scholar]
- Kuroda S, Janelidze S, Siesjo BK. The immunosuppressants cyclosporin A and FK506 equally ameliorate brain damage due to 30-min middle cerebral artery occlusion in hyperglycemic rats. Brain Res. 1999;835:148–153. doi: 10.1016/s0006-8993(99)01535-8. [DOI] [PubMed] [Google Scholar]
- Li PA, Kristian T, He QP, Siesjo BK. Cyclosporin A enhances survival, ameliorates brain damage, and prevents secondary mitochondrial dysfunction after a 30-minute period of transient cerebral ischemia. Exp Neurol. 2000;165:153–163. doi: 10.1006/exnr.2000.7459. [DOI] [PubMed] [Google Scholar]
- Mignen O, Thompson JL, Shuttleworth TJ. Calcineurin directs the reciprocal regulation of calcium entry pathways in nonexcitable cells. J Biol Chem. 2003;278:40088–40096. doi: 10.1074/jbc.M306365200. [DOI] [PubMed] [Google Scholar]
- Morjani H, Madoulet C. Immunosuppressors as multidrug resistance reversal agents. Methods Mol Biol. 2010;596:433–446. doi: 10.1007/978-1-60761-416-6_19. [DOI] [PubMed] [Google Scholar]
- Petronilli V, Penzo D, Scorrano L, Bernardi P, Di LF. The mitochondrial permeability transition, release of cytochrome c and cell death. Correlation with the duration of pore openings in situ. J Biol Chem. 2001;276:12030–12034. doi: 10.1074/jbc.M010604200. [DOI] [PubMed] [Google Scholar]
- Singleton RH, Stone JR, Okonkwo DO, Pellicane AJ, Povlishock JT. The immunophilin ligand FK506 attenuates axonal injury in an impact-acceleration model of traumatic brain injury. J Neurotrauma. 2001;18:607–614. doi: 10.1089/089771501750291846. [DOI] [PubMed] [Google Scholar]
- Smaili SS, Stellato KA, Burnett P, Thomas AP, Gaspers LD. Cyclosporin A inhibits inositol 1,4,5-trisphosphate-dependent Ca2+ signals by enhancing Ca2+ uptake into the endoplasmic reticulum and mitochondria. Journal of Biological Chemistry. 2001;276(26):23329–40. doi: 10.1074/jbc.M100989200. [DOI] [PubMed] [Google Scholar]
- Sullivan PG, Sebastian AH, Hall ED. Therapeutic window analysis of the neuroprotective effects of cyclosporine a after traumatic brain injury. J Neurotrauma. 2011;28:311–318. doi: 10.1089/neu.2010.1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchino H, Elmer E, Uchino K, Li PA, He QP, Smith ML, Siesjo BK. Amelioration by cyclosporin A of brain damage in transient forebrain ischemia in the rat. Brain Res. 1998;812:216–226. doi: 10.1016/s0006-8993(98)00902-0. [DOI] [PubMed] [Google Scholar]
- Yoshimoto T, Siesjo BK. Posttreatment with the immunosuppressant cyclosporin A in transient focal ischemia. Brain Res. 1999;839:283–291. doi: 10.1016/s0006-8993(99)01733-3. [DOI] [PubMed] [Google Scholar]
- Zoratti M, Szabo I. The mitochondrial permeability transition. Biochim Biophys Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]