

Abstract
Most of the currently used cancer chemotherapies are based on compounds that inhibit general cellular mechanisms, such as DNA replication or tubulin function, and lack specificity in relation to features of the cancer cell. Recent advances in genomic studies have increased our knowledge of tumor cell biology, and a panoply of new targets have been postulated. This has provided an opportunity to develop and validate drugs that specifically target cancer cells through their unique genetic characteristics. Identification of MUC16/CA125 both as a marker and a driver of transformation led us to design a target-based high-content screen to identify and classify compounds that exhibit differential effect on MUC16-expressing cells. We developed a co-culture assay in 384-well plate containing isogenic ovarian cancer cells that are positive or negative for the MUC16 protein. High-throughput screening of our small-molecule pilot library led to the identification of compounds preferentially cytotoxic to MUC16-positive or -negative cells using a Preferential Score analysis. We compared screening results in both A2780 and SK-OV-3 ovarian cancer cells in single and co-culture settings. We also identified compounds that were cytotoxic for both types of ovarian cancer cells regardless of the MUC16 status. Compounds that were preferentially targeting MUC16 cells were subsequently confirmed by caspase-induction assays. The isogenic, dual-color fluorescence strategy is an innovative approach that can effectively identify novel drug candidates selectively targeting cancer cells that have unique molecular properties.
Keywords: Ovarian cancer, MUC16/CA125, high-throughput drug screening, dual-fluorescence screens, isogenic paired cell lines, compound screening
INTRODUCTION
Treatments for human malignancies are evolving from classic cytotoxic chemotherapy agents to more sophisticated targeted therapy (1, 2). Since uncontrolled proliferation has been a hallmark of malignant behavior, many of the cytotoxic compounds in current use have been directed toward DNA synthesis, damage, and repair processes. More recently, knowledge about molecular biology of cancer cells has afforded new opportunities to address other cancer-specific traits and decrease the host toxicity from cancer therapeutics. The Cancer Genome Atlas (TCGA) promises to provide a new catalog of cancer targets based on mutation, allelic loss, gene amplification, altered gene expression, and other cancer-specific changes to the DNA, RNA, and protein profile of the cancer cells (3). However, the high-throughput technologies of the TCGA focus on nucleic acids will need to be complemented by biologic studies confirming the value of the target abnormalities identified. Since epithelial cancers can show dozens or hundreds of changes in genetic expression, there is an urgent need to validate targets nominated because of altered gene expression and select cancer-specific targets for therapeutic development.
Ovarian cancer is the most common cause of death from gynecologic malignancy in the developed world (4, 5). The tethered mucin MUC16/CA125 is elevated in about 80% of ovarian cancers, but is not expressed significantly outside the female reproductive track (6, 7). This selectivity has been utilized for both disease monitoring and therapeutic intervention (8, 9). The regulation of MUC16 expression in cancer cells is not well understood (7).
CA125 has been shown to be an independent adverse prognostic factor in clinical studies. In results from the TCGA, MUC16 amplification or mutations are present in more than 10% of all serous ovarian tumors, suggesting that either transcriptional or post transcriptional regulation changes must be common in ovarian cancer (10, 11). Targeting MUC16/CA125 may be an effective strategy for ovarian cancer-specific therapy but thus far, no clinical approach to MUC16/CA125 targeting has been successful. We have also established that expression of even small portions of the MUC16 gene transforms 3T3 cells and confers an invasive, more aggressive phenotype (manuscript submitted). These characteristics imply that MUC16 confers novel biological properties to cancer cells that are both selective and functionally important.
By using isogenic cell lines of different backgrounds (e.g., A2780 and SK-OV-3), each differing only in the expression of physiologic levels of MUC16, we can readily isolate MUC16-specific effects for investigation of biology and therapy (6). High-content screening offers several advantages because one can measure simultaneously different cell features and exclude compounds that have nonspecific, off-target effects on cellular viability, morphology, and behavior. The goal of this study was to introduce and implement a dual-fluorescence isogenic screening strategy to identify and classify small molecules that specifically target cells differing from the parental line by expression of a single protein (12). In this pilot study, we establish the feasibility of an isogenic dual-fluorescence screening application for identification of MUC16-specific compounds for development in the treatment of ovarian cancer.
MATERIEALS AND METHODS
Cell lines
A2780 cells were obtained as a gift from Dr. Thomas Hamilton (Fox Chase Cancer Center) and cultured in RPMI-1640 with 10 mM HEPES, 10% fetal bovine serum (FBS, Hyclone, Logan, UT), 2mM L-glutamine and 0.2 U/ml insulin (Eli Lilly & Co, Indianapolis, IL), penicillin 100 U/ml, and streptomycin 100 μg/ml. The SK-OV-3, SK0V8, CAOV3 and OVCAR3 cells were obtained through the American Type Culture Collection (ATCC, Manassas, VA) and propagated according to those instructions. The T80 immortalized human ovarian epithelial cells were the generous of Dr. Robert Bast. MUC16 fragments were created using pBK-CMV-MUC16-B53 DNA as a template and introduced into EcoRV and NotI multiple cloning sites of the phrGFP II-C vector (Stratagene, LaJolla, CA) (6, 13). These constructs, containing elements of MUC16 (MUC16c678–GFP and MUC16c344–GFP), were transfected into CA125-negative ovarian cell lines A2780 and SK-OV-3, respectively. In the SKOV8 cells, an siRNA to CA125 was introduced in a pSilencer vector to reduce the MUC16 expression with a scrambled siRNA which had no effect (Awtrey C, personal communication). We also introduced the red fluorescent mCherry vector (Clontech, Mountainview, CA) into the cell lines expressing MUC16 to differentiate vector-only cells from MUC16-expressing cells. Stable transfectants were selected with G418 and enriched by fluorescence activated cell sorting (FACS) for either green fluorescent protein (GFP) alone or GFP and mCherry (MUC16-positive cells). The identity of the created cell lines were confirmed by FACS analysis with antibodies to GFP and human VK8 and OC125, which recognize the MUC16 external domain. The culture supernatants of these transfectants were tested periodically for MUC16 expression by ELISA (ADVIA Centaur, Siemens Healthcare Diagnostics, Deerfield, IL) and Western Blot (Bio-Rad Laboratories, Hercules, CA).
Cell growth in single or co-culture in 384-well plate
A2780-GFP and A2780-MUC16/mCherry cells were plated separately or simultaneously in 384-well plates at different final cell density: 250, 500, 1000, and 1500 cells per well. When A2780-GFP and A2780-MUC16/mCherry cells were co-cultured; a ratio of 1:1 was used. Plates were fixed and stained as described above at 48h, 72h, and 96h after plating and analyzed on the IN Cell 1000. Twenty-four hours before each time point, 5μL of Alamar Blue (AbD Serotec, Oxford, UK) were added and conversion of resazurin was assessed using the LEADSeeker (GE Healthcare, Piscataway, NJ). The data from growth were curve experiments analyzed on the log scale. Effect of time was estimated using linear regression.
Compound libraries
The library used for the pilot screen combines 3,119 chemicals obtained commercially from Prestwick (Washington, DC) and MicroSource (Gaylordsville, CT). The Prestwick Chemical Library is a unique collection of 1,119 high purity chemical compounds, all off patent and carefully selected for structural diversity and broad spectrum, covering several therapeutic areas from neuropsychiatry to cardiology, immunology, rheumatology, anesthesia, with known safety and bioavailability in humans. The library contains 90% of marketed drugs and 10% bioactive alkaloids or related substances. The MicroSource Library contains 2,000 biologically active and structurally diverse compounds from known drugs, experimental bioactives, and pure natural products. The library includes a reference collection of 160 synthetic and natural toxic substances (inhibitors of DNA/RNA synthesis, protein synthesis, cellular respiration, and membrane integrity), a collection of 80 compounds representing classical and experimental pesticides, herbicides, and endocrine disruptors, and a unique collection of 720 natural products and their derivatives. The collection includes simple and complex oxygen heterocycles, alkaloids, sesquiterpenes, diterpenes, pentacyclic triterpenes, sterols, and many other diverse representatives(14–16). Compound library stocks were formulated in 10% dimethyl sulfoxide (DMSO) at 100 μM, to be added at 10% of final assay volume for a 10 μM compound concentration in 1 % DMSO (15)
High-throughput screening
The pilot screening process involved testing 3,119 compounds at 10μM concentration with 1% DMSO in duplicates on our automated platform (CRS F3 Robot System, Thermo CRS). Compounds were plated in 384-well format plate (Corning: 3985) at 100μM in 10% DMSO in a 5 μL volume using the TPS-384, sealed and stored in −20°C until use. The day of the screen, plates were thawed and centrifuged at 1000 rpm for 1min. High controls (1% DMSO final) and Low controls (10μM Killer Mix containing a mixture of cytotoxic compounds including staurosporine) were plated in a 5μL volume in column 13 and 14, respectively. A2780 and SK-OV-3 cells were trypsinized with 0.25% Trypsin/EDTA and diluted in their respective culture media with P/S and 10% FBS. A2780 and SK-OV-3 cells were plated into the Corning 3985 microtiter plates at a final density of 1500 and 1000 cells per well, respectively, in 45 μL using Multidrop 384 (Thermo Electron Corporation, Waltham, MA). After dispensing, all plates were placed in a Stericult incubator at 37°C and 5% CO2. After 72h incubation, media was aspirated using the Biotek Washer (Biotek Instruments, Winooski, VT) and 50 μL of 4% paraformaldehyde in Phosphate-Buffered Saline (PBS, Invitrogen, Carlsbad, CA) was added by Multidrop 384. After 20min incubation, the fixation solution was removed and 50 μL of staining solution containing 0.05% Triton X-100 and 10μM of Hoechst 33342 (Invitrogen, Carlsbad, CA) in PBS was added. The plates were allowed to incubate for 10 min in the dark and then washed twice with PBS. 50μL of PBS was finally added, and plates were sealed and stored in 4 °C. Plates were imaged using the automated widefield microscope IN Cell 1000 (GE Healthcare, Piscataway, NJ) to detect GFP, mCherry, and Hoechst staining. The fully automated linear track robotic platform CRS F3 (Thermo Electron Corporation, Waltham, MA) was used to image-barcoded plates on the IN Cell 1000. 4 fields per well were imaged with a Plan Apo 10X objective and 1×1 binning. The dichroic cube 61002bs (Chroma) was used to separate the GFP channel (475–535), mCherry channel (565–620), and the Hoechst channel (360-Open), and exposure times of 150ms for each channel was used. Screening data files from the IN Cell 1000 were loaded into the custom-built data management system, ORIS (Oncology Research Informatics System) powered by ChemAxon Cheminformatics tools (ChemAxon, Newark, DE) that links compound identification, plating information, and data analysis. For dose response studies, hit compounds were tested in a 12-point serial dilution and run according to the method described above. After image acquisition and analysis, GFP and mCherry count were plotted in Sigma Plot to calculate IC50.
Statistical analysis of the high-throughput screening
For each plate, the data were transformed into percent inhibition using medians of the plate’s positive and negative controls, and then normalized by median polish algorithm in order to eliminate systematic row or column differences (17). For each well, the preferential effect of mCherry over GFP was measured by T= (Percent Inhibition mCherry - Percent Inhibition GFP)/(2- Percent Inhibition mCherry). If mCherry and GFP have 100% and 0% inhibition, respectively, then T=1. The statistic T is decreasing if the percent inhibition of mCherry and GFP get closer to each other or percent inhibition of mCherry is decreasing. We tested the null hypothesis that mCherry and GFP have equal percent inhibition. The distribution of statistic T under this hypothesis can be estimated using values of T for positive and negative control wells. For each compound we calculate Preferential Score (PS) as its value of T normalized by mean and standard deviation of the estimated null distribution. High values of PS (>3) present evidence against null hypothesis and are consistent with preferential effect of mCherry. Compounds with high PS values in both the A2780 and the SK-OV-3 duplicates were selected for validation
Image analysis
Images were analyzed using the developer Toolbox Software (GE Healthcare, Piscataway, NJ) where a custom-built algorithm was designed to segment GFP and mCherry cells based on the Hoechst segmentation. The count of GFP, mCherry, and Hoechst objects per well was used as the measurement. Batch analysis was performed to process all plates.
Caspase-3 activity assay
A Caspase-3 Activity Assay kit was purchased from Roche Diagnostics GmbH, Germany and used according to manufacturer’s instructions. Assays were performed in quadruplicate, and means were evaluated using Student’s t test between MUC16-negative and MUC16-positive cells. The assays were repeated at least 3 separate times.
RESULTS
Co-culture of fluorescence-labeled cells
To identify small molecules that target preferentially the MUC16 protein, we co-cultured in the same well two isogenic cell lines that differ only by expression of the MUC16 protein. The protein encoded by MUC16 is a large glycoprotein that consists of a small cytoplasmic C-terminal, a potential external cleavage site and a tandem repeat domain that covers most of the external part of the protein (Fig. 1a). We expressed the carboxy terminus of the molecule, using the last 344–678 amino acids that include one or more of the tandem repeat sequences recognized as the CA125 antigen. Using both parental A2780 and SK-OV-3 human ovarian cancer cells, we engineered A2780-GFP and SK-OV-3-GFP cells that expressed GFP fluorophore alone. A2780-mCherry and SK-OV-3-mCherry cell lines were engineered that expressed the combination of mCherry fluorophore and the carboxy terminal region of the MUC16 protein. Markers expression was confirmed by FACS analysis. Expression of the transgene was not completely uniform. About 80% of A2780-GFP cells were found GFP-positive, whereas about 71% of A2780-mCherry were mCherry-positive (Fig. 1b). This difference in GFP and mCherry proportions could be explained by the different imaging settings used for the FACS and microscopy techniques. Low GFP and mCherry signals captured by the microscope were excluded to avoid any measurement of background noise in the analysis.
Figure 1.

Cellular growth of MUC16-expressing and control A2780 cells in single and co-culture.
(a) Structure of the MUC16 transmembrane protein. (b) FACS analysis of A2780-GFP (top) and A2780-mCherry cells (bottom) showing percent of cells expressing each fluorophore. (c) Representative fluorescent images of the co-culture of A2780-GFP and A2780-mCherry cells at 20X. (d) Plot of A2780-GFP (and A2780-MUC16-mCherry growth as a function of seeding density. (e) GFP based counts in A2780-GFP monoculture and with A2780-MUC16-mCherry co-culture.(f) mCherry-based counts in single-A2780-MUC16-mCherry monoculture and with A2780-GFP co-culture.
We had previously established that the growth characteristics of the cell lines in vitro were identical. For the high-content screen, we assessed the cell growth for the A2780-GFP and mCherry-labeled cell lines cultured separately in 384-well plates from 48–96h of incubation (Fig. 1c). Total nuclei count was measured by the Hoechst staining. Both cell lines exhibited statistically similar growth over time at different initial cell-seeding densities. Cell doubling time was calculated based on the exponential phase of the curve for the cell density of 1500 cells per well. The doubling time for A2780-GFP was estimated at 25.9h, whereas A2780-mCherry doubling time was around 24.9h and was not statistically different from the GFP-vector control. Similar results were obtained when measuring the cellular proliferation by metabolic activity measurement using the Alamar Blue assay (Suppl. Fig. 1a). Co-culture of A2780-GFP and A2780-mCherry cells was also examined. The cells were seeded into 384-well plate at 1:1 ratio and cell growth was determined as described above. The growth curves from the same initial cell-seeding density (500–1500 cells per well) were generated and compared with those from the single-cell-line cultures. A2780-GFP or A2780-mCherry grown separately or in co-culture grew at the same rate (Fig. 1d–f; Suppl. Fig. 1b). A final seeding density of 1500 cells/well and a 72h growth period were selected as final test conditions. Similar studies were done for the SK-OV-3 isogenic pair studies, and 1500 cells per well was chosen.
Target-based high-content screen
The dual-color fluorescence cellular assay was developed to perform in a miniaturized 384-well microplate format. In the initial study, the paired A2780 cell-line constructs were seeded into 384-well microplates containing 10μM of test compounds in 1% DMSO (v/v) at a final density of 1500 cells/well. For controls, 1% DMSO (v/v) was used as a High control and 10μM of Killer Mix in 1% DMSO (v/v) was used as a Low control. The cells were incubated with compounds for 72 hours, followed by fixation in 4% paraformaldehyde and staining with Hoechst to identify nuclei. Finally, the plates were imaged on the In Cell Analyzer 1000 (GE Healthcare, Piscataway, NJ), and cytotoxicity was assessed by custom analysis module using Developer configured for fluorescence object count. We performed a control run to assess the inter- and intra-plate variability using High control (1% DMSO) and Low Control (10μM Killer Mix) plated in 3 different plates each (Suppl. Fig. 2a&b). The control plates with 1% DMSO and 10μM Killer Mix gave a Z′ of 0.50 and 0.54 for GFP and mCherry, which indicated that the assay was both robust and amenable to high-throughput screening.
Using the pilot compound library (15) and the screening conditions described, we performed three separate assessments, one with the A2780-GFP control cells, one with the A2780-mCherry-MUC16+ cells, and a third with co-culture (Fig. 2). Each of these analyses was performed in duplicate (noted as set 1 and set2) and the results are presented in Fig. 3. In panel 3a, the correspondence of the two GFP replicates is presented as the size and the color of the dot for each compound. The consistency of the replicate analyses is illustrated through the consistency of the size and the dot for each compound. The position on the X–Y grid represents the correspondence between two separate replicates of the GFP analysis in co-culture of the GFP/mCherry cells. It can be seen that the dots fall in a consistent area, roughly along the 45-degree line of identity. A similar analysis of mCherry results in the A2780 cell line is presented in Fig. 3c. Two nominated “hits” from the mCherry analysis, quinacrine and rutilantinone, are highlighted.
Figure 2.

High-content screening workflow. Compounds were plated at 10μM final concentration in 384-well plates before dual cell seeding. Cells were added and after 72h incubation, cells were fixed and stained with Hoechst 33342. GFP, mCherry, and Hoechst fluorescence were detected using the IN Cell 1000 widefield microscope with a 10X PlanFluor objective, and 4 fields per well were acquired. Based on the nuclear mask, GFP and mCherry count was measured through automated analysis with developer. Statistical analysis and in-house calculations were performed to assess the effect of compounds on cell viability.
Figure 3.

Consistency of the target-based high-content screen. Each compound was tested in 4 separate replicates, and each test is represented by a single dot. In each panel, the PS for the first replicate of the single cell culture condition is illustrated by dot size (small to large), while the second replicate is illustrated by the intensity of the blue color of the dot. The position on the X–Y axes represents the PS in the first co-culture replicate (X axis) and the PS of the second replicate (Y axis). Figure 3a represents the GFP [MUC16(−)] data for A2780 cells, while Figure 3b represents the GFP [MUC16(−)] data for the SK-OV-3 cell line. Similarly, Figure 3c illustrates the screening PS for the mCherry [MUC16(+)] cells, and Figure 3d represents the PS for all the compounds in the SK-OV-3[MUC16(+)] cells. While the a and b panels identify no highly specific compounds for MUC16-negative cell lines, the lower panels identify candidate MUC16(+) selective compounds. Quinacrine and rutilantinone are identified as examples.
To confirm the MUC16 specificity of our A2780 results, we developed a second syngeneic dual-fluorescence screen with an independent ovarian cell line, SK-OV-3. The parent SK-OV-3-GFP cell had no MUC16 expression, while paired SK-OV-3-mCherry-MUC16+ cells display MUC16/CA125 on the cell surface. Following method development analogous to the A2780 development, we were able to perform a similar dual-fluorescence SK-OV-3-based screen with the same 3,119 compound library. In that analysis, a second set of three screens was performed in duplicate; these are illustrated in Figures 3b&d. As with the A2780 results, the consistency of the replicates of single SK-OV-3 GFP (Fig. 3b) or SK-OV-3 mCherry (Fig. 3d) screen is represented by the size and blue intensity of the individual dots, each representing a single compound. The position on the X-Y axis represents the homology between the two dual culture replicates, while the distance from the origin represents the compound activity against the specified cell line. A two dimensional analysis of the replication data, presented as nuclear counts, is presented in Supplemental Figure 3.
The correspondence between the SK-OV-3 and A2780 cell isogenic screens is shown in Fig. 4. In panel 4a, the mean GFP PS for the SK-OV-3 single culture system is represented by the size of the dot, while the GFP PS for the A2780 cell pair is indicated by the intensity of the blue color. From this analysis, it becomes clear that there are no outstanding, high PS “hits” for the two parental lines when compared with the MUC16+ lines. In contrast, panel 4b indicates that for the MUC16 + mCherry co-culture systems, several highly selective compounds were identified with consistently high PS acrose both cell types. The quinacrine and rutilantinone dots are identified for convenience in this figure.
Figure 4.

Comparison of high-content results with the two ovarian cell lines, A2780 and SK-OV-3. Each compound was tested is represented by a single dot. In each figure, the mean PS for the SK-OV-3 single-cell-line culture condition is illustrated by dot size (small to large), while the mean PS for A2780 single-cell-line culture is illustrated by the intensity of the blue color of the dot. The position on the X–Y axes represents the mean PS in the SK-OV-3 co-culture (X axis), and the mean PS of the A2780 co-culture condition (Y axis). Figure 4a represents the GFP [MUC16(−)] data for A2780 and SK-OV-3 cells while Figure 4b illustrates the screening PS for the mCherry [MUC16(+)] cells from both backgrounds. While the a panel identify no highly specific compounds for MUC16 negative cell lines, the b panel identify candidate MUC16(+) selective compounds. Quinacrine and rutilantinone are again identified as examples. Other potential “hits” are identified in Table 1a.
The most positive results for the A2780 and SK-OV-3 analyses are shown in Table 1a. We initially identified thirteen relatively selective MUC16 candidate compounds, as shown in Table 1a, including carboplatin, quinacrine, andrographolide, oxyphenbutazone, acriflavinium, and rutilantinone, which appeared to be selectively more toxic for the MUC16-expressing A2780-mCherry cells, based on the selection criteria of individual PS ≥3 for both the individual analyses and the dual-fluorescence study. This gave an initial screening value of 0.416% tested compounds. The A2780 dual screening co-culture system was highly representative of the individual results, both in selectivity and magnitude of effect (Table 1a, columns 3–8). This consistency will decrease the complexity of the screening in the future and provide a cheaper, more efficient and specific strategy for high-content screening of the MUC16 target lesion.
Table 1a. Selective effects of candidate compounds.
Growth inhibition effects are listed for both individual culture and co-culture conditions. The relative performance scores for the individual culture and the co-culture conditions are also listed for both the A2780 primary screen and the SK-OV-3-based confirmatory screen.
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| No | Compound | A2780 Ind AVG PS | A2780 Ind AVG GEP | A2780 Ind AVG mCH | A2780 Dual AVG PS | A2780 Dual AVG GEP | A2780 Dual AVG mCH | SK-OV-3 Ind AVG PS | SK-OV-3 Ind AVG GEP | SK-OV-3 Ind AVG mCH | SK-OV-3 Dual AVG PS | SK-OV-3 Dual AVG GEP | SK-OV-3 Dual AVG mCH |
| 1 | Andrographolide | 4 | 42% | 81% | 5 | 53% | 68% | 0 | 92% | 89% | 0 | 91% | 85% |
| 2 | 4′-Hydroxychalcone | 3 | 39% | 73% | 3 | 52% | 77% | 2 | 72% | 79% | 1 | 69% | 80% |
| 3 | Cycloheximide | 6 | 36% | 90% | 5 | 56% | 80% | 0 | 76% | 74% | 1 | 71% | 77% |
| 4 | Doxazosin mesylate | 4 | 33% | 72% | 3 | 45% | 63% | 0 | 22% | 18% | 0 | 19% | 15% |
| 5 | 7-Desoxypurpurogalli n-7-carboxylic acid, methylester, trimethlyester | 4 | 33% | 77% | 3 | 38% | 63% | 1 | 2% | 5% | 1 | 7% | 15% |
| 6 | Carboplatin | 4 | 32% | 75% | 3 | 41% | 61% | 0 | 8% | 6% | 0 | 17% | 12% |
| 7 | Gitoxin | 3 | 12% | 49% | 6 | 3% | 57% | 0 | 57% | 54% | 0 | 63% | 56% |
| 8 | Acetylglucosamine | 4 | 47% | 90% | 3 | 42% | 69% | 1 | 0% | 2% | 0 | 0% | 2% |
| 9 | Oxyphenbutazone | 8 | 11% | 90% | 7 | 10% | 69% | 3 | 79% | 88% | 0 | 92% | 90% |
| 10 | Quinacrine | 3 | 19% | 60% | 5 | 11% | 52% | 2 | 45% | 53% | 7 | 0% | 64% |
| 11 | Ellipticine | 3 | 39% | 64% | 9 | 0% | 69% | 1 | 56% | 57% | 2 | 46% | 62% |
| 12 | Acriflavinium hydrochloride | 5 | 0% | 60% | 7 | 0% | 61% | 1 | 72% | 75% | 3 | 52% | 76% |
| 13 | Rutilantinone | 4 | 34% | 75% | 9 | 42% | 97% | 0 | 79% | 76% | 2 | 53% | 68% |
The confirmatory screens for the same compounds with a second dual-fluorescence isogenic ovarian cancer cell pair (SK-OV-3) are shown in Table 1a, from columns 9–14. As can be seen, only one compound, quinacrine, had a confirmed specificity for MUC16+ cells over the parental MUC16(−) cell line. The structures are shown in Table 1b. Based on the dual selection with the two independent cell-based assays, only quinacrine was consistently more toxic to MUC16-expressing cells in both the A2780 and the SK-OV-3 dual-fluorescence screens. Additional comparisons for these 4 compounds were done for 2 additional isogenic cell line pairs (T80 and SKOV8) and two MUC16 expressing lines CAOV3 and OVCAR3. The results shown in Table 1c, confirm that, for the most part, the expression of MUC16 conferred a modest sensitivity to each agent compared to matched the MUC16 negative isogenic cell line.
Table 1b. Structures of Candidate Compounds.
The structures for the 13 candidate compounds are shown.
| No | Compound | Structure |
|---|---|---|
| 1 | Andrographolide |
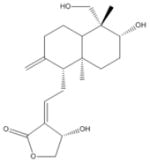
|
| 2 | 4′-Hydroxychalcone |
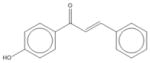
|
| 3 | Cycloheximide |
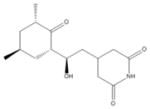
|
| 4 | Doxazosin mesylate |
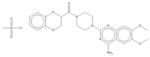
|
| 5 | 7-Desoxypurpurogalli n-7-carboxylic acid, methylester, trimethlyester |
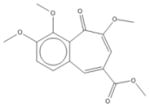
|
| 6 | Carboplatin |
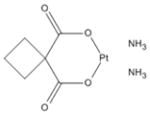
|
| 7 | Gitoxin |
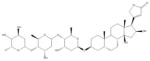
|
| 8 | Acetylglucosamine |
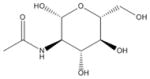
|
| 9 | Oxyphenbutazone |
|
| 10 | Quinacrine |
|
| 11 | Ellipticine |
|
| 12 | Acriflavinium hydrochloride |
|
| 13 | Rutilantinone |
|
Table 1c. Additional Confirmation of Candidates.
To test the MUC16 selectivity of the nominated compounds following the A2780 isogenic screens additional matched, isogenic MUC16 positive/negative pairs were examined. The IC50’s, obtained graphically following 96 well plate Alamar Blue assays are shown. While 3 of 4 compounds were confirmed as more selective for MUC16 expressing cells in the SKOV3 pair, 4 of four were confirmed in the T80 paired cell lines and 3 of 4 (except Quinacrine) were more toxic for the MUC16 expressing SKOv8 cells than for the stable siRNA MUC16 knockdowns which show less than 10% of the parental levels of MUC16.
| Compounds | SKOV3 | T80 | SKOV8 | CAOV3 | OVCAR3 | |||
|---|---|---|---|---|---|---|---|---|
| No MUC16 | MUC16+ve | No MUC16 | MUC16 +ve | No MUC16 | MUC16 +ve | MUC16 +ve | MUC16 +ve | |
| Knock Down | Scramble | WT | WT | |||||
| Ellipticine | 4.5 | 4 | 0.3 | 0.25 | 0.2 | 0.11 | 0.6 | 0.7 |
| Quinacrine dihydrochloride dihydrate | 4.125 | 3.5 | 4.5 | 3.5 | 0.5 | 1.1 | 8 | 7 |
| Acriflavinium hydrochloride | 4.25 | 5.5 | 1.8 | 1.4 | 0.5 | 0.25 | 0.7 | 1 |
| Rutilantinone | 50 | 47.5 | 10.8 | 9.5 | 19 | 15 | 9 | 10 |
In order to confirm these findings in a different functional assay, induction of apoptosis was used to validate the screening results in both A2780 and SKOV3 matched cell lines. We examined the effect of quinacrine and three other A2780 screen-nominated compounds (ellipticine, acriflavinium hydrochloride, and rutilantinone) (Fig. 5) that met the selection criteria for MUC16 targeting in the A2780 pair but not in the SK-OV-3 assay. No compound was found to be MUC16 selective in SK-OV-3 cell screen that was not also identified as selective in the A2780 cell pair.
Figure 5.

Validation of quinacrine and other candidate compounds by caspase-3 activity. (a). Quinacrine structure and images of a representative field for GFP, mCherry, Hoechst, and a merged image. (b and c) Candidate compounds and the results of caspase-3 activation studies (n=3). Only quinacrine showed statistically selective caspase activation in MUC16 + cells compared with the isogenic parental cell line.
Four compounds (highlighted in yellow in Table 1a) were tested for the induction of caspase-3 activity (a marker of apoptosis) in both of the test cell-line pairs (Fig. 5b&c). Only quinacrine statistically increased caspase-3 activation in the A2780-MUC16-mCherry-positive cells compared with the A2780-phrGFP cells (p<0.0001) (Fig. 5a&b). In SKOV3-MUC16-mCherry transfectants, caspase-3 activation was significantly elevated compared with the vector control for quinacrine but not for rutilantinone, ellipticine, and acriflavinium. Thus, in both cell-line pairs, quinacrine is confirmed as relatively selective for MUC16-bearing ovarian cancer cells compared with the parental line. Some of the additional compounds had modest selection advantages for MUC16-based targeting in A2780, but did not appear to have a selective effect for MUC16-expressing cells in another ovarian cancer cell background. As shown in Fig. 5c, Ellipticine, Acriflavinium and Rutilantinone each increase caspase 3 activity over untreated controls, none of these three are differentially active in MUC16(+) A2780 or SK-OV-3 cells. While some of these compounds might have activity in selected cell backgrounds, the two-stage screening system allowed the early selection of candidate compounds that were MUC16-specific before the slower, labor-intensive confirmatory screening was performed by hand.
DISCUSSION
As The Cancer Genome Atlas is completed for specific tumors, the biology of various target genetic alterations will need to be confirmed, and the opportunities for targeting specific genetic alterations will increase very rapidly. Efficient strategies for lead compound generation will be needed to accelerate development and credentialing of individual cancer-specific targets. In this work, we describe the implementation of strategy for high-throughput screening of a single genetic alteration, using a syngeneic, dual-fluorescence strategy. For some genes, it is possible to quickly select candidate compounds for drug development. In this co-culture method, the cost of sequential screening can be avoided, and variation between the screening steps is reduced. Within each well, the variations in dispensing, handling, location/edge effects have been excluded and the procedure is highly economical, based on the small volume of each test well. The co-culture strategy is clearly dependent on a similar growth curve between the two isogenic pairs.
In this study, we demonstrate that use of two different cell pairs, each isogeneic except for MUC16 expression, provided a highly selective screen and allowed a high probability of true “hits” that are independent of the background cell biology of the different tumor cell lines. This redundancy in different cellular backgrounds was able to rapidly eliminate inconsistent effects that depend more on cellular context than on the specific genetic alteration under study. By stringently defining hits as positive wells in two independent systems, the high-throughput approach described herein led to a very low rate of unconfirmed candidacy, and in a full screening enterprise, would facilitate selection of candidates for further drug development.
The co-culture system was also validated in the comparison with the individual culture results. The co-culture results were highly consistent with the individual culture data and were relatively consistent across the two screening systems. In this study, only quinacrine was actually selected as a positive, MUC16 selective “hit” after the confirmatory screen. A simpler, co-culture analysis of the two test systems would have called the same single compound positive, and no false positives were identified in the 3,119 compounds that completed the entire double selection testing. The independent confirmation by caspase-3 activation represented a separate mechanism that did not depend on growth inhibition alone and gives further confirmation regarding the validity and specificity of the high-content-screening results. While our selection rule is highly stringent, it greatly increases the specificity of the MUC16 targeting and has the advantage of a “hit rate” below 0.1% which will hopefully yield a tractable number of hits for secondary screening when several hundred thousand compounds are tested.
The applications of this isogenic, dual-fluorescence strategy are potentially broad. Screening of a much larger library is likely to be successful in identification of MUC16+ hits. Our pilot data suggest that this strategy may yield fewer than 1 hit/thousand candidates from a larger library, quickly reducing the number of potential therapeutic agents into a manageable number for more extensive confirmation. Alternatively, a third isogenic pair could be used to further validate the selective nature of the selected compounds, if a large, unmanageable number were identified from the 2 cell-line panels. The technique could also be readily adapted to screening libraries of either siRNAs or shRNAs. The isogenic character of the paired cell lines would readily facilitate the selection of targets that could be subsequently confirmed by additional siRNA candidates.
Certain limitations are also notable. The studies would be much more complex if the MUC16+/− cell lines were not identical in their growth characteristics over the days of evaluation. Any paracrine or endocrine interactions between the cancer and the host would be much more difficult to model in this system, and the advantages of high-content screening would be lost. Finally the selective expression of MUC16 in mullerian tissue is an important advantage (6). If the expression of the target is not essentially confined to tumor tissue, the selectivity in vitro might be insufficient to nominate candidates for additional testing. Nonetheless, this is the first practical implementation of an isogenic, dual-fluorescence high-content screen for cancer drug development. The screen is now being applied to a library of over 350,000 compounds for additional testing.
Supplementary Material
Acknowledgments
The authors acknowledge the help and support of MSKCC core facilities, especially the High Throughput Drug Screening Facility, DNA Sequencing, Clinical Chemistry Laboratory, Flow Cytometry and Biostatistics Divisions.
This work is supported by NIH Grant # PO1-CA52477-17 and by Mr. William H. and Mrs. Alice Goodwin and the “Commonwealth Foundation for Cancer Research” and “The Experimental Therapeutics Center of Memorial Sloan-Kettering Cancer Center.”
References
- 1.Aggarwal S. Targeted cancer therapies. Nat Rev Drug Discov. 2010;9:427–8. doi: 10.1038/nrd3186. [DOI] [PubMed] [Google Scholar]
- 2.McClellan M, Benner J, Schilsky R, Epstein D, Woosley R, Friend S, et al. An accelerated pathway for targeted cancer therapies. Nat Rev Drug Discov. 2011;10:79–80. doi: 10.1038/nrd3360. [DOI] [PubMed] [Google Scholar]
- 3.Heng HH. Cancer genome sequencing: the challenges ahead. BioEssays: news and reviews in molecular, cellular and developmental biology. 2007;29:783–94. doi: 10.1002/bies.20610. [DOI] [PubMed] [Google Scholar]
- 4.Cannistra SA. Is there a “best” choice of second-line agent in the treatment of recurrent, potentially platinum-sensitive ovarian cancer? J Clin Oncol. 2002;20:1158–60. doi: 10.1200/JCO.2002.20.5.1158. [DOI] [PubMed] [Google Scholar]
- 5.Cannistra SA. Cancer of the ovary. N EnglJ Med. 2004;351:2519–29. doi: 10.1056/NEJMra041842. [DOI] [PubMed] [Google Scholar]
- 6.Dharma Rao T, Park KJ, Smith-Jones P, Iasonos A, Linkov I, Soslow RA, et al. Novel Monoclonal Antibodies Against the Proximal (Carboxy-Terminal) Portions of MUC16. Appl Immunohistochem Mol Morphol. 2010 doi: 10.1097/PAI.0b013e3181dbfcd2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bast RC, Jr, Spriggs DR. More than a biomarker: CA125 may contribute to ovarian cancer pathogenesis. Gynecol Oncol. 2011;121:429–30. doi: 10.1016/j.ygyno.2011.04.032. [DOI] [PubMed] [Google Scholar]
- 8.Anderson NS, Bermudez Y, Badgwell D, Chen R, Nicosia SV, Bast RC, Jr, et al. Urinary levels of Bcl-2 are elevated in ovarian cancer patients. Gynecol Oncol. 2009;112:60–7. doi: 10.1016/j.ygyno.2008.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Reinartz S, Kohler S, Schlebusch H, Krista K, Giffels P, Renke K, et al. Vaccination of patients with advanced ovarian carcinoma with the anti-idiotype ACA125: immunological response and survival (phase Ib/II) Clinical cancer research: an official journal of the American Association for Cancer Research. 2004;10:1580–7. doi: 10.1158/1078-0432.ccr-03-0056. [DOI] [PubMed] [Google Scholar]
- 10.Zorn KK, Tian C, McGuire WP, Hoskins WJ, Markman M, Muggia FM, et al. The prognostic value of pretreatment CA 125 in patients with advanced ovarian carcinoma: a Gynecologic Oncology Group study. Cancer. 2009;115:1028–35. doi: 10.1002/cncr.24084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Torrance CJ, Agrawal V, Vogelstein B, Kinzler KW. Use of isogenic human cancer cells for high throughput screening and drug discovery. Nature Biotechnology. 2001:19. doi: 10.1038/nbt1001-940. [DOI] [PubMed] [Google Scholar]
- 13.Yin BW, Lloyd KO. Molecular cloning of the CA125 ovarian cancer antigen: identification as a new mucin, MUC16. J Biol Chem. 2001;276:27371–5. doi: 10.1074/jbc.M103554200. [DOI] [PubMed] [Google Scholar]
- 14.Shelton CC, Tian Y, Shum D, Radu C, Djaballah H, Li YM. A miniaturized 1536-well format gamma-secretase assay. Assay Drug Dev Technol. 2009;7:461–70. doi: 10.1089/adt.2009.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Seideman JH, Shum D, Djaballah H, Scheinberg DA. A high-throughput screen for alpha particle radiation protectants. Assay Drug Dev Technol. 2010;8:602–14. doi: 10.1089/adt.2010.0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shum D, Smith JL, Hirsch AJ, Bhinder B, Radu C, Stein DA, et al. High-content assay to identify inhibitors of dengue virus infection. Assay Drug Dev Technol. 2010;8:553–70. doi: 10.1089/adt.2010.0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tukey JW. Some thoughts on clinical trials, especially problems of multiplicity. Science. 1977;198:679–84. doi: 10.1126/science.333584. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.