

Abstract
In patients with HIV/AIDS, neuropathic pain is a common neurological complication. Infection with the HIV itself may lead to neuropathic pain, and painful symptoms are enhanced when patients are treated with nucleoside reverse transcriptase inhibitors (NRTI). The mechanisms by which NRTIs contribute to the development of neuropathic pain are not known. In the current studies, we tested the role of TNFα in antiretroviral drug-induced neuropathic pain. We administered 2′,3′-dideoxycytidine (ddC, one of the NRTIs) systemically to induce mechanical allodynia. We found that ddC induced overexpression of both mRNA and proteins of GFAP and TNFα in the spinal dorsal horn. TNFα was colocalized with GFAP in the spinal dorsal horn and with NeuN in the DRG. Knockdown of TNFα with siRNA blocked the mechanical allodynia induced by ddC. Intrathecal administration of glial inhibitor or recombinant TNF soluble receptor, reversed mechanical allodynia induced by ddC. These results suggest that TNFα is involved in NRTI-induced neuropathic pain.
Keywords: neuropathic pain, HIV, ddC, TNFα
1. Introduction
Highly active antiretroviral therapy (HAART) has markedly increased survival of patients with HIV/AIDS from the introduction since 1996 (Sacktor, 2002) (Beck et al., 1999). HAART contains nucleoside reverse transcriptase inhibitors (NRTIs) as an active component. NRTIs decrease plasma viral load, and can result in improvements in immune function (e.g. CD4 lymphocyte count recovery) in patients with HIV. However, drug toxicity of NRTIs limits the successful treatment in many individuals (see review) (Haas et al., 2006). Patients receiving NRTIs therapy develop a distal symmetric small fiber ‘dying back’ axonal neuropathy with pain (Dalakas, 2001; Dubinsky et al., 1989; Pardo et al., 2001; Reliquet et al., 2001; Simpson, 2002). Neuropathic pain associated with the use of NRTIs is clinically quite common (Berger et al., 1993; Luciano et al., 2003), though the mechanisms underlying this phenomenon are yet to be determined. The antiviral agent 2′,3′-dideoxycytidine (ddC, one of the NRTIs) to patients with HIV, was approved in 1992 as an antiretroviral drug for people with HIV infection. The ddC was used particularly for patients who are intolerant of or ineligible for AZT/ddI. The ddC is still widely used in clinics in Africa, Europe and Asia. Clinical studies show that ddC increases the risk of occurrence of neuropathic pain in HIV-1 infected patients (Arenas-Pinto et al., 2008; Dalakas et al., 2001; Lopez et al., 2004; McCarthy et al., 2000).
The cellular effects of NRTIs that could potentially contribute to the development of neuropathic pain, include cellular toxicity following the inhibition of mitochondrial DNA polymerase γ (Dalakas et al., 2001; Kakuda, 2000) and upregulation of CXCR4 receptor, SDF-1 and CCL2 (Bhangoo et al., 2007; Wallace et al., 2007). However, the detailed mechanism by which these patients with HIV/AIDS experienced pain remains unknown. Tumor necrosis factor alpha (TNFα) is expressed on activated macrophages and lymphocytes as well as other cell types (e.g. glia in the CNS). In the chronic pain state, previous studies indicate that TNFα is increased by spinal cord injury (Peng et al., 2006), spinal nerve ligation (Cui et al., 2008; Hao et al., 2007b), and formalin injection into the paw in rats (Choi et al., 2008; Zhou et al., 2008). We hypothesize that ddC may induce neuropathic pain through overexpression of TNFα in the DRG and/or spinal cord. In the present study, we found that TNFα played an important role in the neuropathic pain induced by ddC.
2. Methods
2.1. Animal experiments
Male Sprague-Dawley rats weighing 225–250 g were housed, one to four per cage approximately 7 days prior to the beginning of the study. Free access to food and water, and maintained on a 12:12, light:dark schedule at 21 °C and 60% humidity. All housing conditions and experimental procedures were approved by the University Animal Care and Use Committee, and were conducted in accordance with the ethical guidelines of the International Association for the Study of Pain(Zimmermann, 1983).
2.2. Intrathecal catheter implantation
For intrathecal administration, intrathecal catheters were implanted under isoflurane anesthesia (Hao et al., 2009). A polyethylene (PE-10) catheter filled with 0.9% saline was advanced 8 cm caudally through an incision in the atlanto-occipital membrane, to position its tip at the level of the lumbar enlargement. The rostral tip of the catheter was passed subcutaneously, externalized on top of the skull, and sealed with a stainless-steel plug. Animals showing neurological deficits after implantation were excluded. Animals were used within 5 days after implantation of the catheter.
2.3. Systemic ddC model
The ddC (Sigma, St Louis, MO) was freshly prepared in saline on the day of the experiment. The ddC- and vehicle-treated groups were given a one-time intraperitoneal (IP) injection of ddC (1 ml, 50 mg/kg) or saline (1 ml, vehicle), respectively (Joseph et al., 2004). Systemic ddC induced a significant decrease in the mechanical threshold.
2.4. Mechanical threshold
Animals were placed in transparent plastic cubicles on a mesh floor for an acclimatization period of at least 30 min on the morning of the test day. Mechanical threshold induced by systemic ddC was determined by assessing the rate response of paw withdrawal to von Frey hairs of graded tensile strength. Rats were placed in individual plastic cages with wire mesh bottoms, and after 1 h the mechanical threshold was measured. A series of 10 calibrated fine von Frey filaments (0.4, 0.7, 1.2, 1.5, 2.0, 3.6, 5.5, 8.5, 11.8, and 15.1 g) were presented serially to the left hind paw in ascending order of strength, with sufficient force to cause slight bending against the paw and held for 6 s. A positive response was defined as a rapid withdrawal and/or licking of the paw immediately upon application of the stimulus. Whenever a positive response to a stimulus occurred, the next smaller von Frey hair was applied, and whenever a negative response occurred, the next higher force was applied. In the absence of a response at a pressure of 15.1 g, animals were assigned to this cut-off value. Measurements were taken 1 day before and 1, 2, 3, 3.5, 4, 5, 6, and 7 weeks after surgery. According to the method described, a tactile stimulus producing a 50% likelihood of withdrawal was determined using the up-down method (Chaplan et al., 1994; Dixon, 1980). Behavioral testing was performed during the day portion of the circadian cycle (8:00 AM to 5:00 PM).
2.5. Reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA from the lumbosacral dorsal spinal cord tissue was isolated using TRIzol reagent (Invitrogen). The RNA sample was treated with DNase I at room temperature for 15 min to remove genomic DNA, and was then heated for 15 min at 65°C to inactivate the DNase I. First-strand cDNA was produced with Omniscript Reverse Transcriptase (Qiagen), as described (Sun et al., 2011). A similar reaction mixture without reverse transcriptase was prepared and run in parallel as a control. cDNA was amplified using the following primer sets: β-actin-forward (5′-CAG TTC GCC ATG GAT GAC GAT ATC-3′) and β-actin-reverse (5′-CAC GCT CGG TCA GGA TCT TCA TG-3′), GFAP- GFAP forward (5′-CTAATGACTATCGCCGCCAACT) and GFAP reverse (5′-GTCTTTACCACGATGTTCCTCTTG), TNFα-forward (5′-TGCCTCAGCCTCTTCTCATTCC 3-′) and TNFα-reverse (5′-GGGCAGCCTTGTCCCTTGAA-3′). The resulting RT-PCR products were separated by gel electrophoresis on 1.5% agarose gel. For densitometric analyses, amplification product bands were quantified with Quantity One analysis software (Bio-Rad, Hercules, CA, USA). The relative level of mRNA expression was shown as the ratio of the mRNA of TNFα and GFAP to that of β-actin.
2.6. Western Blots
The tissues were homogenized in protein lysis buffer (150 mM sodium chloride, 1.0% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris, pH 8.0) containing protease inhibitors and phosphatase inhibitors (Phosphatase Inhibitor Cockail 1/2) as previously described (Zheng et al., 2011). The homogenate was centrifuged at 18,000 g for 20 min at 4°C. The supernatant was collected and assayed for protein concentration using the DC protein assay kit (Bio-Rad, Hercules, CA, USA). Aliquots containing 30 μg of protein were dissolved in Laemmli buffer and boiled at 95 °C for 5 min, the proteins were separated by 10% Tris-glycine SDS-PAGE gel and transferred to a PVDF membrane. The membranes were blocked with 5% nonfat dry milk in PBS buffer, and then incubated with primary antibodies for 1h at room temperature, including rabbit anti-TNFα (1:500, Millipore, Billerica, MA, USA), mouse anti-GFAP (1:5000, Sigma, St. Louis, MO, USA), and mouse anti-β-actin (1:8000, Santa Cruz Biotechnology, Santa Cruz, CA, USA). The blots were incubated with a secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), developed in a chemiluminescence solution (Pierce Biotechnology, Rockford, IL, USA). Quantification of Western blots was done from the obtained chemiluminescence values (Bio-Rad ChemiDoc, Hercules, CA, USA). Targeted protein bands were normalized using the amount of β-actin as a standard.
2.7. Evaluations of the effect of TNFα siRNA in vitro
A glial cell line was used to verify the efficiency of TNFα small interfering RNA (siRNA) in vitro. TNFα siRNA (forward 5′-GCCCGUAGCCCACGUCGUAdTdT-3′, reverse-5′-UACGACGUGGGCUACGGGCdTdT-3′), and mismatch RNA (forward-5′-GCCCGUAGAACACGUCGUAdTdT-3′, reverse-5′-UACGACGUGUUCUACGGGCdTdT-3′) were synthesized by Invitrogen (Invitrogen, Camarillo, CA, USA). We have shown that HAPI cells (a glial cell line) treated with LPS, release TNFα (Cheepsunthorn et al., 2001; Zhou et al., 2008). HAPI cells were seeded into 6-well plates at 2 ×105 cells/well 24 h before transfection. Transient transfection procedures were performed according to Lipofectamine RNAiMAX reagent instructions (Invitrogen, Camarillo, CA, USA). Briefly, HAPI cells were incubated with 100 pmol siRNA and 6 μl Lipofectamine RNAiMAX complexes, in Opti-MEM I reduced serum medium (Invitrogen, Camarillo, CA, USA). Twenty four hours after siRNA application, the cells were stimulated by 1μg/ml of LPS (Sigma, St. Louis, MO, USA). Six hours after LPS, the cells were harvested and the expression of TNFα was measured using western blots.
2.8. Immunohistochemistry
Immunohistochemical expression of GFAP, TNFα and NeuN in the spinal cord and the L4/5 DRG in rats with ddC treatment was investigated as described previously (Hao et al., 2011). For immunofluorescence detection, cryosections were probed overnight with rabbit anti-GFAP polyclonal antibody (1: 2000, DakoCytomation, Glostrup, Denmark), goat anti-rat TNFα antibody (1: 100; R&D systems, Minneapolis, MN), and mouse anti-NeuN monoclonal antibody (A60) (1: 5000, Millipore, Billerica, MA). Then followed by incubation with complementary secondary antibodies labeled with blue-fluorescent Alexa Fluor 350, green-fluorescent Alexa Fluor 488, or red-fluorescent Alexa Fluor 594 (1: 2000, Molecular Probes, Eugene, OR), 2 h at room temperature and photographed using a fluorescence microscope. Sections were selected and scanned using a fluorescence microscope.
2.9. Drugs and Data Analysis
Agent ddC and pentoxifylline purchased from Sigma (Sigma, St. Louis, MO) were dissolved in saline. Recombinant rat soluble TNF receptor I were purchased from PeproTech and dissolved in 0.1% RSA. The doses of drug were selected on the basis of our preliminary studies. To facilitate siRNA into cells, we used polyethyleneimine (PEI), a cationic polymer, as a delivery vehicle to prevent degradation and enhance cell membrane penetration of siRNA (Tan et al., 2005). Small interfering RNA was purchased from Invitrogen, and dissolved in RNase-free water at the concentration of 1 μg/μl as a stock solution. Ten min before injection, 10 μl siRNA was mixed with 1.8 μl PEI. Intrathecal drugs were injected through the implanted polyethylene tubing (PE-10) with 10 μl, followed by 10 μl of saline; the injection lasted 30s by means of a Hamilton syringe. The behavioral testing was carried out by a blinded operator. The statistical significances of the differences were determined by a t test. The difference between the time-course curves of the behavioral testing was determined using a General Linear Model and repeated measure with SPSS software. p-values of less than 0.05 were considered to be statistically significant.
3. Results
3.1. Mechanical allodynia induced by systemic ddC
The baseline of the mechanical threshold in rats was around 9.5 gram in the left hindpaw. A single dose of ddC produced a significant reduction in the paw-withdrawal threshold from day 3 and remained lower through day 49 after the administration of ddC (Figure 1), which was similar to the original report in rats treated with a single intravenous administration of ddC (Joseph et al., 2004). In the vehicle group treated with saline, rats showed no significant changes in mechanical threshold throughout the 7-week testing period. The difference in the threshold was significant in the two groups (Figure 1), which was consistent with the previous report (Joseph et al., 2004).
Figure 1.

The time course of mechanical threshold in the model of systemic ddC. Rats exposed to systemic ddC developed a persistent, mechanical allodynia of the ipsilateral hind paw compared to the vehicle group, F(1,12) =84.489, p < 0.001, n=6–8, general linear model, repeated measure, SPSS.
3.2. Systemic ddC upregulated the expression of spinal GFAP and TNFα in the mRNA and the protein levels
Previous studies show that ddC induced a significant decrease in the mechanical threshold and a marked increase in immunoreactivity of glia in the dorsal horn of the L5 spinal cord at 21 days (Wallace et al., 2007). In our study, two weeks after ddC, lumbar spinal dorsal horn was harvested; mRNA expression of GFAP and TNFα was tested using RT-PCR. Administration of ddC significantly induced the upregulation of mRNA of GFAP and TNFα (Figure 2). Similarly, we found that ddC significantly increased the expression of GFAP and TNFα protein in the spinal dorsal horn compared to the sham group using Western blots (Figure 3). To our knowledge, we are the first to observe that single ddC increases the expression of TNFα in the spinal dorsal horn.
Figure 2.

The expression of mRNA of spinal GFAP and TNFα induced by systemic ddC. Two weeks after ddC, left L4/5 spinal dorsal horns were harvested under anesthesia, and mRNA expression was examined using RT-PCR. Systemic ddC significantly upregulated mRNA of spinal GFAP and TNF3. * p < 0.05 vs sham, n=3–5, t test.
Figure 3.

The upregulation of spinal GFAP and TNFα induced by ddC. Two weeks after ddC, left L4/5 spinal dorsal horns were harvested under anesthesia, and protein expression of spinal GFAP and TNFα was tested using western blots. Systemic ddC significantly induced the upregulation of GFAP and TNFα in the spinal cord level. *p < 0.05 vs sham, t test.
3.3 Determination of cellular localization of TNFα in the spinal dorsal horn in ddC-induced neuropathic pain
Activated glia induced by nerve injury release proinflammatory cytokines (e.g., TNFα and IL-1β) in the spinal level (Raghavendra et al., 2002; Watkins and Maier, 2003). To determine the cellular localization of TNFα induced by ddC, double-label immunostaining of spinal GFAP and TNFα was carried out. There was an almost complete colocalization between GFAP (blue) and TNFα (red) imaging, which suggested that TNFα was located in astrocytes (Figure 4).
Figure 4.

Determination of cellular localization of TNFα in the spinal dorsal horn in rats treated with ddC for 2 weeks. Immunostaining of GFAP and TNF3 was carried out. There was an almost complete colocalization between GFAP and TNFα imaging, but TNFα did not colocalize with NeuN (data not shown), which suggested that TNFα was located in the astrocytes, but not neurons. Arrow shows the colocalization. Scale bar, 100μm.
3.4 The expression of TNFα in the DRG
Previous studies have shown that TNFα plays a role in the neuropathic pain induced by nerve injury (He et al., 2010; Homma et al., 2002). In our current study, immunohistochemistry showed a strong immunoreactivity of TNFα, which was observed in the DRG in rats treated with ddC, but not saline. We also found that systemic ddC for 2 weeks significantly increased the expression of TNFα in the DRG using Western blots, compared to the control group (treated with saline) (Figure 5). To our knowledge, we are the first to report that TNFα in the DRG was increased in the neuropathic pain induced by antiretroviral therapy. To determine the cellular localization of TNFα, double-label immunostaining was carried out in the DRG sections from animals injected with ddC. We found that almost all TNFα immunostaining colocalized with NeuN, but not GFAP, indicating that TNFα was expressed in neurons, but not GFAP-positive satellite cells in the DRG (Figure 6).
Figure 5.
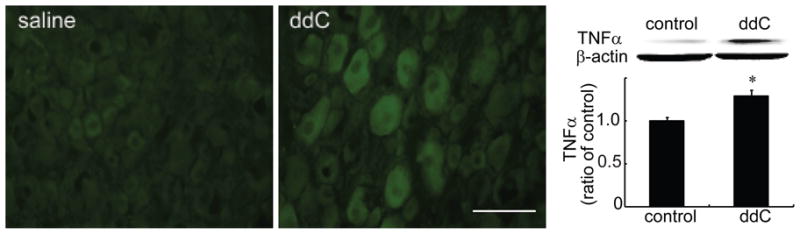
The systemic injection of ddC increased TNFα in the rat lumbar DRG sections. Animals received a single ddC injection and tissue was collected at 14 days after ddC injection. The systemic injection of ddC induced clear immunoreactivity of TNFα compared to the saline group. Bar graph showed that systemic injection of ddC significantly increased TNFα in the DRG using a Western blot, compared to the saline group, *p < 0.05 vs saline, t test. n=3–6.
Figure 6.
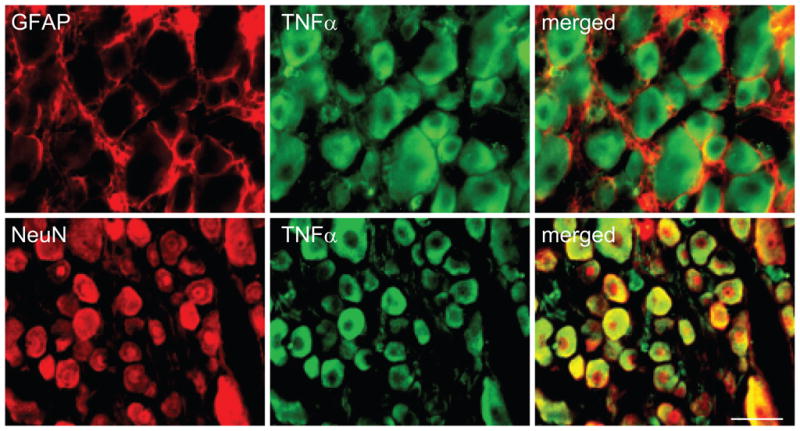
Colocalization of TNFα and GFAP (upper panel) or NeuN (lower panel) in the DRG. Animals received a single ddC injection, and the DRG was collected at 14 days after ddC injection. Double-label immunostaining showed that TNFα was colocalized with NeuN, but not GFAP immunostaining in the DRG. Scale bar, 50 μm.
3.5. The antinociceptive effect of glial inhibitor on the ddC-induced mechanical allodynia
The results above suggest that TNFα contributes ddC-induced neuropathic pain. Furthermore, we investigated the antinociceptive effect of the glial inhibitor on the ddC-induced mechanical allodynia. Intrathecal administration of pentoxifylline, a glial inhibitor (Mika et al., 2009; Shohami et al., 1996) significantly reversed the mechanical allodynia in the model (Figure 7), F(1,8)=29.002, p = 0.001, n=5, General Linear Model, repeated measure, SPSS, which was consistent with that reported in other neuropathic pain models (Mika et al., 2009; Saito et al., 2010).
Figure 7.

The effect of spinal glial inhibitor on the mechanical threshold in rats treated with ddC. At 2 weeks after ddC, rats received intrathecal injection of pentoxifilline or vehicle. Administration of glial inhibitor, but not vehicle reversed the mechanical threshold, F(1,8)=29.002, p = 0.001 vs saline (vehicle), n=5, General Linear Model, repeated measure, SPSS.
3.6. The antinociceptive effect of TNF soluble receptor on the ddC-induced mechanical allodynia
TNF soluble receptor may block TNFα from binding to a membrane TNF receptor on the cell surface, to neutralize the biological effect of TNFα. Overexpression of TNF soluble receptor by HSV vectors, reversed the increase in TNFα and the mechanical allodynia in the neuropathic pain models (Hao et al., 2007a; Peng et al., 2006). In this study, 12 days after ddC, recombinant TNF soluble receptor I or vehicle was administered intrathecally 3 times at 12-hour intervals. Mechanical threshold was tested after the last injection. The mechanical threshold was increased significantly in rats with TNF soluble receptor, but not vehicle, F(1,14)=25.923, p < 0.001, n=8, General Linear Model, repeated measure, SPSS (Figure 8).
Figure 8.

The antinociceptive effect of recombinant soluble TNF receptor on the mechanical threshold in rats treated with ddC. At 2 weeks after ddC, rats received intrathecal injection of recombinant soluble TNF receptor or vehicle. Administration of siRNA of TNFα, but not vehicle reversed the mechanical threshold, F(1,14)=25.923, p < 0.001 vs vehicle, n=8, General Linear Model, repeated measure, SPSS.
3.7. The suppression of TNFα by siRNA of TNFα in both the in vitro and in vivo studies
To identify the efficacy of TNFα siRNA that we ordered, we used cultured cells. We have found that LPS effectively stimulates HAPI cells (one of glial cell lines) to release TNFα (Zhou et al., 2008). HAPI cells were pretreated with TNFα siRNA or mismatch RNA for 24 hours, then stimulated with LPS for 6 hours. The cells were collected for testing the expression of TNFα using Western blots. Pretreatment with TNFα siRNA, but not mismatch RNA suppressed the expression of TNFα in the cultured cells (Figure 9A). In the control group without LPS, non TNFα was detected (data not shown). To test if intrathecal administration of siRNA of TNFα suppresses the expression of TNFα in the spinal cord and DRG in the ddC-induced neuropathic pain, we injected intrathecally siRNA of TNFα or mismatch RNA 2 times at 24-hour intervals at 12 days after ddC. The lumbar spinal cord and L4/5 DRG were harvested 24 hours after last injection of siRNA. We tested the expression of TNFα using Western blots. We found that either the expression of TNFα in the DRG (Figure 9B) or spinal cord (Figure 9C) was significantly lowered in rats receiving siRNA of TNFα compared to that receiving mismatch RNA. Thus, we think that siRNA of TNFα effectively reduced the synthesis of TNFα in the lumbar DRG and spinal cord level.
Figure 9.

(A) The effect of TNFα siRNA on the expression of TNFα induced by LPS in the in vitro study. Cultured HAPI cells were treated with TNFα siRNA or mismatch RNA 24 hours prior to the treatment with LPS. Cells were collected 6 hours after LPS and then homogenized with lysis buffer. TNFα was measured using a Western blot. The expression of TNFα in cells treated with TNFα siRNA was significantly lower than that in cells treated with mismatch RNA, ** p < 0.01 vs mmRNA, n=3, t test. The effect of TNFα siRNA on the expression of TNFα induced by ddC in the DRG (B) and spinal dorsal horn (C) was tested. At 12 days after ddC, rats received intrathecal injection of siRNA of TNFα or mismatch RNA once a day for 2 days. The DRG and the spinal cord of animals were harvested, and Western blots were carried out. The expression of TNFα in rats treated with TNFα siRNA was significantly lower than that in rats treated with mismatch RNA, *p < 0.05 vs mmRNA, n=4, t test. (D)The antinociceptive effect of siRNA of TNFα on the mechanical threshold in rats treated with ddC. At 12 days after ddC, rats received intrathecal injection of siRNA of TNFα or mismatch RNA. Administration of siRNA of TNFα, but not mismatch RNA, reversed the mechanical threshold, F(1,12)=11.733, p = 0.005 vs mismatch RNA, n=7, General Linear Model, repeated measure, SPSS.
3.8. The antinociceptive effect of siRNA of TNFα in the ddC-induced neuropathic pain
In the chronic constriction injury model of peripheral neuropathic pain, neutralizing antibodies directed against TNFα or the p55 TNF receptor (TNFR), reduce thermal hyperalgesia and mechanical allodynia (Homma et al., 2002; Sommer et al., 1998). We investigated whether TNFα knockdown with siRNA reduced mechanical allodynia induced by ddC. At 12 days after ddC, intrathecal siRNA of TNFα or mismatch RNA was given 2 times at 24-hour intervals. The mechanical threshold was tested after the last injection. The mechanical threshold was increased significantly in rats with siRNA of TNFα, but not mismatch RNA, F(1,12)=11.733, p = 0.005 vs mismatch RNA, n=7, General Linear Model, repeated measure, SPSS (Figure 9D).
4. Discussion
In the current study, we found 1) that intraperitoneal ddC induced mechanical allodynia lasting more than 7 weeks, 2) that ddC significantly upregulated both mRNA and protein expression of spinal GFAP and TNFα, 3) that knockdown of TNFα with intrathecal siRNA reversed mechanical allodynia and the expression of TNFα protein in the DRG and spinal cord, and 4) that intrathecal either glial inhibitor or TNF soluble receptor decreased mechanical allodynia.
Substantial evidence indicates that peripheral nerve damage or inflammation results in the activation of glia in the dorsal horn, which plays an important role in the pathogenesis of neuropathic pain (Hashizume et al., 2000; Homma et al., 2002; Raghavendra et al., 2002). In the setting of peripheral nerve damage, activated glia express proinflammatory cytokines (e.g., TNFα)(Homma et al., 2002). Administration of drugs either that block the effects of these cytokines (Schafers et al., 2003; Sweitzer et al., 2001a) or that block glial activation (Sweitzer et al., 2001b) can be used to prevent or reverse neuropathic pain.
Several lines of evidence indicate that TNFα plays a key role in chronic pain. In response to either peripheral nerve injury or after spinal cord injury, TNFα is increased in the spinal cord (Hao et al., 2007b; Peng et al., 2006; Raghavendra et al., 2002). In the chronic constriction injury model of peripheral neuropathic pain, neutralizing antibodies directed against TNFα or the p55 TNF receptor (TNFR) reduce thermal hyperalgesia and mechanical allodynia (Sommer et al., 1998). Intrathecal administration of the recombinant soluble TNFR (sTNFR) peptide (etanercept) prior to selective spinal nerve ligation decreases mechanical allodynia (Zou et al., 2007). Immunocytochemical staining for spinal TNFα is increased in rats with nerve injury (DeLeo et al., 1997; Hao et al., 2007b). Deleo et al reports that mechanical allodynia was significantly enhanced in the transgenic mice in which expression of murine TNF is targeted to astrocytes using a GFAP-TNF fusion gene (DeLeo et al., 2000). The critical role of TNFα in neuropathic pain is supported by evidence from animal studies demonstrating that intrathecal TNFRII fragment reduces mechanical allodynia in the model of neuropathic pain(Svensson et al., 2005), and that gene transfer of a soluble fragment of TNFRI to DRG from skin delivery using an HSV-based vector, reduces mechanical allodynia in the spinal nerve ligation and spinal hemisection models (Peng et al., 2006) (Hao et al., 2007b).
HIV-associated sensory neuropathy (HIV-SN) is one of the most common forms of peripheral neuropathy, affecting about 30% of people with acquired immune deficiency syndrome (AIDS) (Schifitto et al., 2002; Simpson et al., 2006). The symptoms of HIV-SN are dominated by neuropathic pain, which is often excruciating (Dorsey and Morton, 2006; Newshan, 1997). Patients with HIV/AIDS have benefited greatly from the introduction of HAART (Sacktor, 2002), but NRTIs also induce toxic neuropathies by inhibiting mitochondrial DNA, and by releasing mitochondrial reactive oxygen species (Lewis et al., 2003). Neuropathic pain, induced by NRTIs is clinically quite common, and is recognized as an important source of morbidity in HIV-infected individuals (Berger et al., 1993; Luciano et al., 2003), though the mechanisms underlying this phenomenon are yet to be determined. It is possible that ddC-induced release of inflammatory molecules in the peripheral and/or central nervous systems may play an important role in this model.
In the in vitro studies, Schwann cells through CXCR4 results in the release of RANTES, which induces TNFα production by DRG, and subsequent TNFR1-mediated neurotoxicity in an autocrine/paracrine fashion (Keswani et al., 2003). It was observed that rats treated with ddC produced a substantial upregulation of chemokine CXCR4 mRNA expression in the DRG (Bhangoo et al., 2007). In addition, there is an increase in levels of inflammatory SDF-1 mRNA in glial cells (Bhangoo et al., 2007). Although these data implicate CXCR4 in the genesis of ddC-related neuropathic pain, the precise sequence of cellular events through which the drug increases SDF-1/CXCR4 expression is not entirely clear. In addition, inflammatory chemokine monocyte chemoattractant protein-1 (MCP-1, CCL2) in the DRG also is involved in neuropathic pain induced by ddC (Wallace et al., 2007). MCP-1 is strongly induced by the TNFα signal (Gao et al., 2009). Our results showed that ddC induced the overexpression in the DRG, and that knockdown with TNFα siRNA reduced the expression of TNFα in the DRG. The relationship between TNFα and CXCR4 or MCP-1 in the DRG needs to be elucidated.
Mitochondrial dysfunction has been implicated in neurological disorders of the central and peripheral nervous (Szewczyk and Wojtczak, 2002). The mitochondrial electron transport chain (mETC) plays a role in some forms of pain; the contribution of mETC in neuropathic pain is inflammatory factor ATP dependent (Joseph and Levine, 2006). Inhibitors of mETC complexes are antinociceptive in ddC-induced neuropathic pain. In addition, inhibitors of mETC complexes inhibit the hyperalgesia induced by TNFα (Joseph and Levine, 2006). TNFα, which acts on the dorsal root ganglion neurons and the spinal cord, is a well-established inflammatory mediator in neuropathic pain. The mETC contributed to TNFα hyperalgesia(Joseph and Levine, 2006). Thus, it is possible that ddC induces release of TNFα to enhance the activation of mETC in the DRG and the spinal cord. Reactive oxygen species (ROS) is a signaling molecule involved in the increased formation of proinflammatory cytokines (e.g., TNFα, IL-1β, and IL-6) (Muscoli et al., 2007). The initial site of action of NRTIs is thought to be the mitochondria significantly increasing ROS generation (Opii et al., 2007). It is certainly possible that these cellular elements are linked in the regulation of TNFα in the present case. In the in vitro study ddC significantly induce release of TNFα in the cultured mononuclear cells (Mattioli et al., 2004). We are the first to show that ddC induced upregulation of TNFα in the both DRG and spinal cord. Thus, from the description above, it is possible that TNFα induced by ddC plays an important role in the neuropathic pain.
In summary, the detailed mechanisms underlying ddC-related chronic pain are poorly understood. Peripheral nerve degeneration results in a state of sensitization in which normal activation of peripheral sensory neurons is perceived as painful (Costigan et al., 2009). There is abundant evidence to suggest that one of important elements is neuroimmune activation (DeLeo et al., 2004; Marchand et al., 2005; Watkins and Maier, 2003). In the present study, we found that TNFα might play a role in neuropathic pain induced by ddC. The current evidence showed that TNFα did is involved in neuropathic pain induced by ddC. The results of the current investigation provided an important insight into the pathogenesis of ddC-induced neuropathic pain.
Highlights.
The ddC increased TNFα, and inhibition of TNFα reversed painful behavior in rats, suggesting TNFα plays a role in the HIV/AIDS-related pain.
Acknowledgments
This work was supported by grants from the NIH DA026734 (S.H.), DA025527 (S.H.) and NS066792 (S.H.), Department of Veterans Affairs and the NINDS NS038850 and NIDDK DK044935 (D.J.F and M.M), Jilin University (W.Z.), and China Scholarship Council (X.Z.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arenas-Pinto A, Bhaskaran K, Dunn D, Weller IV. The risk of developing peripheral neuropathy induced by nucleoside reverse transcriptase inhibitors decreases over time: evidence from the Delta trial. Antivir Ther. 2008;13:289–295. [PubMed] [Google Scholar]
- Beck EJ, Mandalia S, Williams I, Power A, Newson R, Molesworth A, Barlow D, Easterbrook P, Fisher M, Innes J, Kinghorn G, Mandel B, Pozniak A, Tang A, Tomlinson D. Decreased morbidity and use of hospital services in English HIV-infected individuals with increased uptake of anti-retroviral therapy 1996–1997. National Prospective Monitoring System Steering Group. Aids. 1999;13:2157–2164. doi: 10.1097/00002030-199910220-00020. [DOI] [PubMed] [Google Scholar]
- Berger AR, Arezzo JC, Schaumburg HH, Skowron G, Merigan T, Bozzette S, Richman D, Soo W. 2′,3′-dideoxycytidine (ddC) toxic neuropathy: a study of 52 patients. Neurology. 1993;43:358–362. doi: 10.1212/wnl.43.2.358. [DOI] [PubMed] [Google Scholar]
- Bhangoo SK, Ren D, Miller RJ, Chan DM, Ripsch MS, Weiss C, McGinnis C, White FA. CXCR4 chemokine receptor signaling mediates pain hypersensitivity in association with antiretroviral toxic neuropathy. Brain Behav Immun. 2007;21:581–591. doi: 10.1016/j.bbi.2006.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Cheepsunthorn P, Radov L, Menzies S, Reid J, Connor JR. Characterization of a novel brain-derived microglial cell line isolated from neonatal rat brain. Glia. 2001;35:53–62. doi: 10.1002/glia.1070. [DOI] [PubMed] [Google Scholar]
- Choi HC, Song P, Xie Z, Wu Y, Xu J, Zhang M, Dong Y, Wang S, Lau K, Zou MH. Reactive nitrogen species is required for the activation of the AMP-activated protein kinase by statin in vivo. J Biol Chem. 2008;283:20186–20197. doi: 10.1074/jbc.M803020200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. 2009;32:1–32. doi: 10.1146/annurev.neuro.051508.135531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Liao XX, Liu W, Guo RX, Wu ZZ, Zhao CM, Chen PX, Feng JQ. A novel role of minocycline: attenuating morphine antinociceptive tolerance by inhibition of p38 MAPK in the activated spinal microglia. Brain Behav Immun. 2008;22:114–123. doi: 10.1016/j.bbi.2007.07.014. [DOI] [PubMed] [Google Scholar]
- Dalakas MC. Peripheral neuropathy and antiretroviral drugs. J Peripher Nerv Syst. 2001;6:14–20. doi: 10.1046/j.1529-8027.2001.006001014.x. [DOI] [PubMed] [Google Scholar]
- Dalakas MC, Semino-Mora C, Leon-Monzon M. Mitochondrial alterations with mitochondrial DNA depletion in the nerves of AIDS patients with peripheral neuropathy induced by 2′3′-dideoxycytidine (ddC) Lab Invest. 2001;81:1537–1544. doi: 10.1038/labinvest.3780367. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Colburn RW, Rickman AJ. Cytokine and growth factor immunohistochemical spinal profiles in two animal models of mononeuropathy. Brain Res. 1997;759:50–57. doi: 10.1016/s0006-8993(97)00209-6. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Rutkowski MD, Stalder AK, Campbell IL. Transgenic expression of TNF by astrocytes increases mechanical allodynia in a mouse neuropathy model. Neuroreport. 2000;11:599–602. doi: 10.1097/00001756-200002280-00033. [DOI] [PubMed] [Google Scholar]
- DeLeo JA, Tanga FY, Tawfik VL. Neuroimmune activation and neuroinflammation in chronic pain and opioid tolerance/hyperalgesia. Neuroscientist. 2004;10:40–52. doi: 10.1177/1073858403259950. [DOI] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Dorsey SG, Morton PG. HIV peripheral neuropathy: pathophysiology and clinical implications. AACN Clin Issues. 2006;17:30–36. doi: 10.1097/00044067-200601000-00004. [DOI] [PubMed] [Google Scholar]
- Dubinsky RM, Yarchoan R, Dalakas M, Broder S. Reversible axonal neuropathy from the treatment of AIDS and related disorders with 2′,3′-dideoxycytidine (ddC) Muscle Nerve. 1989;12:856–860. doi: 10.1002/mus.880121012. [DOI] [PubMed] [Google Scholar]
- Gao YJ, Zhang L, Samad OA, Suter MR, Yasuhiko K, Xu ZZ, Park JY, Lind AL, Ma Q, Ji RR. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci. 2009;29:4096–4108. doi: 10.1523/JNEUROSCI.3623-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas DW, Geraghty DE, Andersen J, Mar J, Motsinger AA, D’Aquila RT, Unutmaz D, Benson CA, Ritchie MD, Landay A. Immunogenetics of CD4 lymphocyte count recovery during antiretroviral therapy: An AIDS Clinical Trials Group study. J Infect Dis. 2006;194:1098–1107. doi: 10.1086/507313. [DOI] [PubMed] [Google Scholar]
- Hao S, Liu S, Zheng X, Zheng W, Ouyang H, Mata M, Fink DJ. The role of TNFalpha in the periaqueductal gray during naloxone-precipitated morphine withdrawal in rats. Neuropsychopharmacology. 2011;36:664–676. doi: 10.1038/npp.2010.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S, Mata M, Fink DJ. Viral vector-based gene transfer for treatment of chronic pain. Int Anesthesiol Clin. 2007a;45:59–71. doi: 10.1097/AIA.0b013e318034199c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao S, Mata M, Glorioso JC, Fink DJ. Gene transfer to interfere with TNFalpha signaling in neuropathic pain. Gene Ther. 2007b;14:1010–1016. doi: 10.1038/sj.gt.3302950. [DOI] [PubMed] [Google Scholar]
- Hao S, Wolfe D, Glorioso JC, Mata M, Fink DJ. Effects of transgene-mediated endomorphin-2 in inflammatory pain. Eur J Pain. 2009;13:380–386. doi: 10.1016/j.ejpain.2008.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashizume H, DeLeo JA, Colburn RW, Weinstein JN. Spinal glial activation and cytokine expression after lumbar root injury in the rat. Spine (Phila Pa 1976) 2000;25:1206–1217. doi: 10.1097/00007632-200005150-00003. [DOI] [PubMed] [Google Scholar]
- He XH, Zang Y, Chen X, Pang RP, Xu JT, Zhou X, Wei XH, Li YY, Xin WJ, Qin ZH, Liu XG. TNF-alpha contributes to up-regulation of Nav1.3 and Nav1.8 in DRG neurons following motor fiber injury. Pain. 2010;151:266–279. doi: 10.1016/j.pain.2010.06.005. [DOI] [PubMed] [Google Scholar]
- Homma Y, Brull SJ, Zhang JM. A comparison of chronic pain behavior following local application of tumor necrosis factor alpha to the normal and mechanically compressed lumbar ganglia in the rat. Pain. 2002;95:239–246. doi: 10.1016/S0304-3959(01)00404-3. [DOI] [PubMed] [Google Scholar]
- Joseph EK, Chen X, Khasar SG, Levine JD. Novel mechanism of enhanced nociception in a model of AIDS therapy-induced painful peripheral neuropathy in the rat. Pain. 2004;107:147–158. doi: 10.1016/j.pain.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Joseph EK, Levine JD. Mitochondrial electron transport in models of neuropathic and inflammatory pain. Pain. 2006;121:105–114. doi: 10.1016/j.pain.2005.12.010. [DOI] [PubMed] [Google Scholar]
- Kakuda TN. Pharmacology of nucleoside and nucleotide reverse transcriptase inhibitor-induced mitochondrial toxicity. Clin Ther. 2000;22:685–708. doi: 10.1016/S0149-2918(00)90004-3. [DOI] [PubMed] [Google Scholar]
- Keswani SC, Polley M, Pardo CA, Griffin JW, McArthur JC, Hoke A. Schwann cell chemokine receptors mediate HIV-1 gp120 toxicity to sensory neurons. Ann Neurol. 2003;54:287–296. doi: 10.1002/ana.10645. [DOI] [PubMed] [Google Scholar]
- Lewis W, Day BJ, Copeland WC. Mitochondrial toxicity of NRTI antiviral drugs: an integrated cellular perspective. Nat Rev Drug Discov. 2003;2:812–822. doi: 10.1038/nrd1201. [DOI] [PubMed] [Google Scholar]
- Lopez OL, Becker JT, Dew MA, Caldararo R. Risk modifiers for peripheral sensory neuropathy in HIV infection/AIDS. Eur J Neurol. 2004;11:97–102. doi: 10.1046/j.1351-5101.2003.00713.x. [DOI] [PubMed] [Google Scholar]
- Luciano CA, Pardo CA, McArthur JC. Recent developments in the HIV neuropathies. Curr Opin Neurol. 2003;16:403–409. doi: 10.1097/01.wco.0000073943.19076.98. [DOI] [PubMed] [Google Scholar]
- Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Nat Rev Neurosci. 2005;6:521–532. doi: 10.1038/nrn1700. [DOI] [PubMed] [Google Scholar]
- Mattioli B, Giordani L, Quaranta MG, Viora M. Effect of indinavir used alone or in double or triple combination with AZT and ddC on human immune functions. Life Sci. 2004;74:2291–2300. doi: 10.1016/j.lfs.2003.09.052. [DOI] [PubMed] [Google Scholar]
- McCarthy WF, Gable J, Lawrence J, Thompson M. A retrospective study to determine if hydroxyurea augmentation of antiretroviral drug regimens that contain ddl and/or d4T increases the risk of developing peripheral neuropathy in HIV-1 infected individuals. Pharmacoepidemiol Drug Saf. 2000;9:49–53. doi: 10.1002/(SICI)1099-1557(200001/02)9:1<49::AID-PDS465>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Mika J, Osikowicz M, Rojewska E, Korostynski M, Wawrzczak-Bargiela A, Przewlocki R, Przewlocka B. Differential activation of spinal microglial and astroglial cells in a mouse model of peripheral neuropathic pain. Eur J Pharmacol. 2009;623:65–72. doi: 10.1016/j.ejphar.2009.09.030. [DOI] [PubMed] [Google Scholar]
- Muscoli C, Cuzzocrea S, Ndengele MM, Mollace V, Porreca F, Fabrizi F, Esposito E, Masini E, Matuschak GM, Salvemini D. Therapeutic manipulation of peroxynitrite attenuates the development of opiate-induced antinociceptive tolerance in mice. J Clin Invest. 2007;117:3530–3539. doi: 10.1172/JCI32420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newshan G. Pain in human immunodeficiency virus disease. Semin Oncol Nurs. 1997;13:36–41. doi: 10.1016/s0749-2081(97)80048-4. [DOI] [PubMed] [Google Scholar]
- Opii WO, Sultana R, Abdul HM, Ansari MA, Nath A, Butterfield DA. Oxidative stress and toxicity induced by the nucleoside reverse transcriptase inhibitor (NRTI)--2′3′-dideoxycytidine (ddC): relevance to HIV-dementia. Exp Neurol. 2007;204:29–38. doi: 10.1016/j.expneurol.2006.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo CA, McArthur JC, Griffin JW. HIV neuropathy: insights in the pathology of HIV peripheral nerve disease. J Peripher Nerv Syst. 2001;6:21–27. doi: 10.1046/j.1529-8027.2001.006001021.x. [DOI] [PubMed] [Google Scholar]
- Peng XM, Zhou ZG, Glorioso JC, Fink DJ, Mata M. Tumor necrosis factor-alpha contributes to below-level neuropathic pain after spinal cord injury. Ann Neurol. 2006;59:843–851. doi: 10.1002/ana.20855. [DOI] [PubMed] [Google Scholar]
- Raghavendra V, Rutkowski MD, DeLeo JA. The role of spinal neuroimmune activation in morphine tolerance/hyperalgesia in neuropathic and sham-operated rats. J Neurosci. 2002;22:9980–9989. doi: 10.1523/JNEUROSCI.22-22-09980.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reliquet V, Mussini JM, Chennebault JM, Lafeuillade A, Raffi F. Peripheral neuropathy during stavudine-didanosine antiretroviral therapy. HIV Med. 2001;2:92–96. doi: 10.1046/j.1468-1293.2001.00066.x. [DOI] [PubMed] [Google Scholar]
- Sacktor N. The epidemiology of human immunodeficiency virus-associated neurological disease in the era of highly active antiretroviral therapy. J Neurovirol 8 Suppl. 2002;2:115–121. doi: 10.1080/13550280290101094. [DOI] [PubMed] [Google Scholar]
- Saito O, Svensson CI, Buczynski MW, Wegner K, Hua XY, Codeluppi S, Schaloske RH, Deems RA, Dennis EA, Yaksh TL. Spinal glial TLR4-mediated nociception and production of prostaglandin E(2) and TNF. Br J Pharmacol. 2010;160:1754–1764. doi: 10.1111/j.1476-5381.2010.00811.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafers M, Svensson CI, Sommer C, Sorkin LS. Tumor necrosis factor-alpha induces mechanical allodynia after spinal nerve ligation by activation of p38 MAPK in primary sensory neurons. J Neurosci. 2003;23:2517–2521. doi: 10.1523/JNEUROSCI.23-07-02517.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schifitto G, McDermott MP, McArthur JC, Marder K, Sacktor N, Epstein L, Kieburtz K. Incidence of and risk factors for HIV-associated distal sensory polyneuropathy. Neurology. 2002;58:1764–1768. doi: 10.1212/wnl.58.12.1764. [DOI] [PubMed] [Google Scholar]
- Shohami E, Bass R, Wallach D, Yamin A, Gallily R. Inhibition of tumor necrosis factor alpha (TNFalpha) actvity in rat brain is associated with cerebroprotection after closed head injury. J Cereb Blood Flow Metab. 1996;16:378–384. doi: 10.1097/00004647-199605000-00004. [DOI] [PubMed] [Google Scholar]
- Simpson DM. Selected peripheral neuropathies associated with human immunodeficiency virus infection and antiretroviral therapy. J Neurovirol 8 Suppl. 2002;2:33–41. doi: 10.1080/13550280290167939. [DOI] [PubMed] [Google Scholar]
- Simpson DM, Kitch D, Evans SR, McArthur JC, Asmuth DM, Cohen B, Goodkin K, Gerschenson M, So Y, Marra CM, Diaz-Arrastia R, Shriver S, Millar L, Clifford DB. HIV neuropathy natural history cohort study: assessment measures and risk factors. Neurology. 2006;66:1679–1687. doi: 10.1212/01.wnl.0000218303.48113.5d. [DOI] [PubMed] [Google Scholar]
- Sommer C, Schmidt C, George A. Hyperalgesia in experimental neuropathy is dependent on the TNF receptor 1. Exp Neurol. 1998;151:138–142. doi: 10.1006/exnr.1998.6797. [DOI] [PubMed] [Google Scholar]
- Sun J, Liu S, Mata M, Fink DJ, Hao S. Transgene-mediated expression of tumor necrosis factor soluble receptor attenuates morphine tolerance in rats. Gene Ther. 2011 doi: 10.1038/gt.2011.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson CI, Schafers M, Jones TL, Powell H, Sorkin LS. Spinal blockade of TNF blocks spinal nerve ligation-induced increases in spinal P-p38. Neurosci Lett. 2005;379:209–213. doi: 10.1016/j.neulet.2004.12.064. [DOI] [PubMed] [Google Scholar]
- Sweitzer S, Martin D, DeLeo JA. Intrathecal interleukin-1 receptor antagonist in combination with soluble tumor necrosis factor receptor exhibits an anti-allodynic action in a rat model of neuropathic pain. Neuroscience. 2001a;103:529–539. doi: 10.1016/s0306-4522(00)00574-1. [DOI] [PubMed] [Google Scholar]
- Sweitzer SM, Schubert P, DeLeo JA. Propentofylline, a glial modulating agent, exhibits antiallodynic properties in a rat model of neuropathic pain. J Pharmacol Exp Ther. 2001b;297:1210–1217. [PubMed] [Google Scholar]
- Szewczyk A, Wojtczak L. Mitochondria as a pharmacological target. Pharmacol Rev. 2002;54:101–127. doi: 10.1124/pr.54.1.101. [DOI] [PubMed] [Google Scholar]
- Tan PH, Yang LC, Shih HC, Lan KC, Cheng JT. Gene knockdown with intrathecal siRNA of NMDA receptor NR2B subunit reduces formalin-induced nociception in the rat. Gene Ther. 2005;12:59–66. doi: 10.1038/sj.gt.3302376. [DOI] [PubMed] [Google Scholar]
- Wallace VC, Blackbeard J, Segerdahl AR, Hasnie F, Pheby T, McMahon SB, Rice AS. Characterization of rodent models of HIV-gp120 and anti-retroviral-associated neuropathic pain. Brain. 2007;130:2688–2702. doi: 10.1093/brain/awm195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Maier SF. Glia: a novel drug discovery target for clinical pain. Nat Rev Drug Discov. 2003;2:973–985. doi: 10.1038/nrd1251. [DOI] [PubMed] [Google Scholar]
- Zheng W, Ouyang H, Zheng X, Liu S, Mata M, Fink DJ, Hao S. Glial TNFalpha regulates neuropathic pain induced by HIV gp120 application in rats. Mol Pain. 2011;7:40. doi: 10.1186/1744-8069-7-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Peng X, Hao S, Fink DJ, Mata M. HSV-mediated transfer of interleukin-10 reduces inflammatory pain through modulation of membrane tumor necrosis factor alpha in spinal cord microglia. Gene Ther. 2008;15:183–190. doi: 10.1038/sj.gt.3303054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Zou W, Kim BO, Zhou BY, Liu Y, Messing A, He JJ. Protection against human immunodeficiency virus type 1 Tat neurotoxicity by Ginkgo biloba extract EGb 761 involving glial fibrillary acidic protein. Am J Pathol. 2007;171:1923–1935. doi: 10.2353/ajpath.2007.070333. [DOI] [PMC free article] [PubMed] [Google Scholar]