

Abstract
The axon initial segment is a unique neuronal subregion involved in the initiation of action potentials and in the control of axonal identity. Recent work has helped our understanding of how this specialised structure develops, not least in identifying possible mechanisms leading to the localisation of the AIS’s master organiser protein, ankyrin-G. The most exciting current work, however, focuses on later aspects of AIS function and plasticity. Recent studies have shown that the AIS is subdivided into distinct structural and functional domains, have demonstrated how the AIS acts as a cytoplasmic barrier for axonal transport, and have discovered that the AIS can be surprisingly plastic in its responses to alterations in neuronal activity.
Introduction
The axon initial segment (AIS) is a highly specialised region of the neuron that clusters cytoplasmic and transmembrane proteins to a section of the axon located near the cell body. To date, the AIS has been attributed two important roles: it acts as the site for action potential initiation and it provides a molecular barrier that helps establish axonal identity. The former role makes the AIS a master integrator of subthreshold synaptic events, converting them into all-or-nothing action potentials. The latter describes its critical structural role in neuronal polarity, where complex protein scaffolds regulate transport along the axon to form a tight boundary with the somatodendritic compartment. Both roles are key to the proper function and development of a neuron and highlight the AIS as a crucial site controlling neuronal polarity and excitability.
For such an important part of a neuron, our knowledge of AIS development, maintenance and subsequent plasticity is relatively poor. However, recent work reviewed here is fast improving this situation. In this review, we focus first on our current knowledge of how the AIS is built during development and the molecules involved in this process. Particularly exciting is the observation that the AIS is not a uniform structure, but is formed of sub-domains of clustered proteins that differ in their molecular composition and corresponding functional roles. We go on to present recent findings on the role of the AIS in setting up and maintaining neuronal polarity, with special emphasis on the role it plays as a selectivity filter for protein trafficking. And we finish with exciting new data that point towards considerable post-developmental AIS modifications – this large, dense neuronal sub-compartment turns out to be surprisingly plastic.
Development of the AIS
After post-mitotic commitment to a neuronal cell fate, the initial development of the AIS is a rather early maturational event, occurring alongside axon/dendrite specification and prior to the formation of most synaptic connections. In dissociated cultured neurons, the AIS is first detected around 3-4 days in vitro (div)[1,2]. The process of maturation is then relatively rapid, with a large majority of cells showing ‘mature’ AISs by the start of the second in vitro week [1,2]. Nevertheless, developmental modelling and re-modelling of the AIS, as discussed below, can extend a good deal further.
Ankyrin-G
The consensus in the field of AIS development is that the structure is utterly dependent upon the scaffolding protein ankyrin-G (AnkG). In neurons AnkG is targeted with a high degree of specificity to the AIS and nodes of Ranvier, is one of the earliest proteins localised to a segment of proximal axon [1,3], and, through its multiple ankyrin repeats, targets proteins containing AnkG-binding domains to the AIS. These include voltage-dependent sodium and potassium channels, as well as other transmembrane and scaffolding proteins [2-5]. Indeed, transgenic mice lacking AnkG also lack an AIS [6], and cultured neurons where AnkG has been knocked-down never develop an AIS [7]. But, although the precise localization of AnkG to a specific portion of the proximal axon is a crucial step in AIS formation, very little is known about this process. What localises AnkG to the proximal axon in the first place? And how is it restricted to a single discrete zone? One thing that is clear is that these processes are cell-autonomous – in comparison to nodes of Ranvier, where multiple Schwann cell-dependent mechanisms operate to restrict AnkG and other specialised proteins to node regions [8,9], the AIS can develop perfectly in the absence of any glial influences [10,11].
Recent evidence implicates the NFκB inhibitor IκBα in early AIS specification [•12]. IκBα interacts with the transcription factor NFκB and inhibits its translocation to the nucleus. Phosphorylation of specific serine residues on IκBα disrupts the interaction and allows NFκB to travel to the nucleus and control the transcription profile of specific genes. IκBα, together with the kinases responsible for its phosphorylation, is localised to the proximal axon, along with AnkG, in very young cultured neurons, and blocking its phosphorylation prevents the formation of an AnkG-containing AIS. However, although this suggests a role for IκBα in AIS formation, it still does not provide a mechanism for how AnkG position is established. To tackle this issue, more fundamental research into very early stages of axon specification is needed: for example, is AIS formation a necessary consequence of axonal differentiation? Could AnkG and/or IκBα boundaries be set by a combination of cytoplasmic molecular gradients along the axon, which define an optimal region for AIS formation? If so, what is the identity of the substrate(s) used? One attractive possibility is that the cytoskeleton may provide the necessary information. Microtubules in the proximal region of the axon are preferentially tyrosinated (similar to dendrites) and less stable than those in the remainder of the axon, which are preferentially acetylated [•13]. In fact, it is known that microtubule stabilization is needed for axon formation and drugs that produce this stabilization result in the formation of multiple axons, even in mature neurons [•13]. However, it still unclear whether the microtubule network drives AIS formation or vice-versa. The interactions between these two complexes have yet to be fully understood.
β-IV-spectrin
AnkG is accompanied at the AIS by another specific scaffolding protein, β-IV-spectrin, which binds to AnkG and, like other spectrins, to the actin cytoskeleton. Mutation studies have shown that the ankyrinG-binding domain of β-IV-spectrin is crucial for its AIS localisation [2], similar to many other AIS-targeted proteins. In vivo studies in β-IV-spectrin KO mice showed neurons lacked a clear AIS in adult animals, which led to speculation that β-IV-spectrin may also be crucial for AIS development [6,14]. However, RNAi knockdown of β-IV-spectrin expression in cultured neurons does not prevent the formation of (AnkG-based) AISs [7]. The current thinking is that β-IV-spectrin, though not crucial for AIS development, may be vital for maintaining and stabilising the AIS, through its interactions with the cytoskeleton by its actin-binding domains[7,15]. Importantly, quivering mice (qv), which carry a loss-of-function mutation in β-IV-spectrin, show severe auditory and motor neuropathies, including the mistargetting of voltage gated Na+ and K+ channels and a loss of synchronised neuronal responses, further underscoring the importance of this protein in AIS function [16].
Nav channels
The AIS owes its functional role as the site of action potential initiation to the high concentration of voltage-gated sodium (Nav) channels that cluster there [17]. Sodium channels are recruited to the AIS following the appearance of AnkG [3], and by means of an intracellular AnkG-binding domain present in the cytoplasmic II-III loop [4,18] . In fact, along with this specific AnkG binding site, the II-III loop of all alpha channel subunits also contains a membrane internalization sequence that removes sodium channels from compartments other than the AIS [19]. In this way, Nav channels use two motifs, working in concert, to localise themselves to AnkG-containing zones [4,19]. However, the AnkG binding site found in Nav channels is not specific and also allows binding to ankyrin-B, a scaffolding protein found throughout the length of the axon. How, then, do sodium channels localise specifically to the AIS? Recent evidence found that specific residues in the II-III loop are phosphorylated by protein kinase CK2 (CK2), an enzyme localised specifically to the AIS. Blocking CK2 activity disrupted Nav channel AIS localisation, suggesting phosphorylation plays an important role for AnkG-specific Nav channel interactions [20]. However, the mechanisms producing CK2’s AIS localisation remain unknown. To complicate matters further, there is also evidence showing that knocking down Nav channels disrupts AISs in some cell types [21] but not others [7], suggesting that Nav channels may be an important structural component for AIS development/maintenance, but only in certain tissues. This view is further complicated by developmental changes that occur in Nav channel subunit content at the AIS. In retinal ganglion cells, for example, Nav 1.2 subunits are predominantly found in early development, but are gradually supplemented by increased expression of Nav 1.6 subunits as the AIS matures [22,23]. Whether this simply reflects changes in overall cell expression from one subunit to another, or whether there are developmental changes in AnkG or other protein-protein interactions at the developing AIS to allow more Nav 1.6 binding, is currently unclear. Finally, alpha Nav channel subunits co-localise and are modulated by accessory beta subunits and modulatory proteins such as FGF14. Compromising these associated proteins is known to affect neuronal firing characteristics for example [24,25], but any developmental role for beta subunits or atypical FGFs in AIS formation is presently unclear.
Other voltage-gated channels
Although action potentials are initiated at the AIS through the action of Nav channels, they are shaped and modulated by local voltage-gated K+ (Kv) and Ca2+ channels [26]. A recent study has implicated T- and R-type Ca2+ channels in the generation of single and complex spikes using a pharmacological approach [•27], but little is known about the mechanisms of localisation at the AIS, nor their developmental timecourse. Rather more clear is the role played by specific AIS Kv channels, which can directly modulate action potential width and repetitive spiking [26,28,29]. KCNQ2/3 channels share the same AnkG binding site as Nav1 channels [5], although this sequence likely evolved some time after the appearance of the Nav-AnkG binding site in early chordates [30]. The two channel types may therefore compete for limited AnkG space within the AIS, although whether KCNQ channels also possess the membrane removal sequences and/or CK2 phosphorylation sites that are necessary for targeting sodium channels to the AIS is presently unclear. Interestingly, the localisation of KCNQ2/3 heteromers requires the presence of an AnkG motif on the KCNQ3 subunit, but not on the KCNQ2 subunit, indicating subunit specificity in AIS targeting [31]. Finally, Kv1 channels appear to use different mechanisms for AIS localisation that involve other scaffolding proteins, such as PSD-93, since an RNAi knockdown of this protein disrupted Kv1 channel AIS clustering [32]. How PSD-93 is localised to the AIS, though, is presently not known.
Cell adhesion molecules, extracellular matrix, and synapses
In contrast to development at nodes of Ranvier, where glial-dependent cell adhesion molecules direct the initial stages in maturation [8,9], extracellular signalling molecules such as NrCAM and NF186 appear relatively late at the AIS [1,3,7], and are dependent upon AnkG for their localisation. Both NrCAM and NF186 bind to AnkG through a FIGQY motif in their cytoplasmic domain [18,33], and while RNAi knockdown of either protein does not affect the development of an AnkG-based AIS in cultured hippocampal neurons, AnkG knockdown prevents either CAM from localising to the proximal axon [7]. NF186 does appear vital, however, for recruiting extracellular components to the AIS. At relatively late stages in development the AIS is surrounded by a dense extracellular matrix (ECM) of unique composition, including the chondroitin sulphate proteoglycan brevican which shows preferential AIS localisation [34]. NF186 binds to brevican through its extracellular domain, and is necessary for its AIS targeting [7]. NF186 is also vital for localising specific synaptic inputs to the AIS. In many projection neuron cell types in vivo, the AIS is specifically targeted by GABAergic inputs from specialised interneurons, with current controversy existing over whether this priviledged input at the site of action potential initiation is actually inhibitory or excitatory [35-37]. Regardless of their function, GABAergic inputs to cerebellar Purkinje cell proximal axons precisely follow the distribution of NF186, which in turn is dependent upon the localisation of AnkG [38].
The initial formation of the AIS therefore follows quite a clear sequence (Figure 1). First, just after or during axon specification, and in a phosphorylated-IκBα-dependent manner, AnkG localises to a domain in the proximal axon. Through various ankG binding sites, β-IV-spectrin, Nav channels, Kv channels, NrCAM and NF186, among other proteins [39] are all then recruited to form the basic AIS intracellular/membrane complex. Finally, NF186 directs the formation of an AIS-specific ECM, and targeted GABAergic synaptic inputs.
Figure 1.

Simplified stages in AIS development. (a) During and shortly after axon specification, phosphorylated IkBa in the proximal axon is needed for localisation of AnkG. (b) AnkG, now localised to a single band in the proximal axon, binds to and localises β-IV-spectrin, Nav and Kv channels, and transmembrane proteins, amongst other molecules. (c) NF186 is then required for the development of a specialised brevican-containing AIS ECM, and for the formation of AIS-specific GABAergic synapses.
Sub-domains within the AIS
Recent studies have shown that the AIS is not a uniform structure. Its molecular composition, and in consequence its functional role, can change significantly between its proximal and distal compartments. What is more, this subdivision within the AIS varies according to cell-type: different neurons possess AISs that are subdivided in different ways, allowing for a high degree of specificity and variety in the way cells initiate, propagate and shape an action potential [•40,41]. One of the clearest examples of this AIS subdivision is in different isoforms of Nav alpha subunits. The Nav1.1 channel, when expressed in neurons, is usually found in a small, tightly-localised band at the very proximal edge of the AIS [23,•40,42]. This contrasts markedly with the distribution of Nav1.6 channels, which do not co-localise with Nav1.1 subunits, and which instead increase in density towards the distal end of the AIS [23,•40]. An important recent study described a similar subdivision between proximal Nav1.2 and distal Nav1.6 subunits in the AIS of cortical pyramidal neurons [••43]. Through a combination of patch-clamp recordings and mathematical simulations, these authors went on to show that the Nav1.2- and Nav1.6-containing portions of the AIS subserve distinct functional roles. The low voltage threshold of Nav1.6 channels makes the distal AIS the site of action potential initiation, a role backed up by other recent imaging, recording and modelling studies [17,••43,44,45]. In contrast, the concentration of higher-threshold Nav1.2 channels in the proximal AIS ensures backpropagation of action potentials into the somatodendritic compartment [••43]. AIS subdivision can therefore allow the structure to play multiple distinct roles in action potential firing.
As well as different Nav classes targeting to different AIS regions, recent immunohistochemical evidence reveals that in certain cell types Kv1.1 and Kv1.2 channels also preferentially target the distal AIS. There, they always co-localise with each other, and with Nav1.6 [•40], suggesting that these channels could play a specialised role in the control of AP initiation.
Developmentally, how do neurons build a subdivided AIS? All Nav subunits appear to use the same conserved II-III loop sequence to bind AnkG, and AnkG, along with all other AIS scaffolding proteins described thus far, is present along the entirety of the AIS. Different preferential binding partners could underlie AIS subdivision, then, but no candidate domains nor any possible partners have yet been identified. Different channel subtypes could also compete with each other for AIS space, but this would not on its own produce two distinct, discrete AIS zones, and besides, Nav1.1 channels are still restricted to proximal AIS in RGCs even when Nav1.6 expression levels are very low [23]. The molecular and developmental mechanisms of AIS sub-localisation should form a very interesting field of study for the future.
A role for the AIS in neuronal polarity
The precise complex of proteins that makes up the AIS seems designed to bring together high densities and particular arrangements of voltage-gated ion channels, along with specific synaptic input, to enable action potential initiation, propagation, and modulation. But the protein matrix that evolved to enable precise control over cell excitability [cf. 30] may have a very different secondary function in delimiting the boundary between major neuronal compartments. Previous findings have shown that the AIS presents a significant barrier for the movement of molecules along the plasma membrane – proteins that diffuse freely within somatic or axonal membrane become more restricted as they pass through the AIS [e.g. 46]. A recent study has shown that this barrier role for the AIS extends to molecular movement within the axonal cytoplasm. Song et al. [••47] introduced different sized fluorescent dextrans into cultured hippocampal neurons and found that diffusion from the soma to the axon was restricted for large (70 kDa) molecules. Importantly, they showed the emergence of this axonal ‘filter’ occured at ~5div, a time when the AIS forms. A combination of RNAi and pharmacology showed the filter was dependent upon AnkG and F-actin; this, along with its location and the developmental time course over which it appears, strongly implicates the AIS in a filtering role. Furthermore, the authors went on to show that the ability of vesicles carrying membrane proteins to pass efficiently through the AIS depends on the transport efficacy of specific KIF motor proteins. Whereas KIF5 normally transports VAMP2-containing vesicles efficiently through the AIS, KIF17 driven vesicles that contain dendritic proteins such as NR2B do not cross the AIS. Interestingly, when the cargo binding domains for KIF5 and KIF17 were swapped, KIF5 was able to carry NR2B across the AIS more efficiently [••47].
The presence of a selective cytoplasmic filter in the proximal axon raised the possibility that the AIS may be actively involved in differentiating somatodendritic from axonal compartments. Indeed, data from two recent studies – one in vitro and one in vivo – suggest that this is precisely the case. Hedstrom et al. [48] targeted the knockdown of AnkG in cultured hippocampal neurons at a stage when the AIS had already formed. They found not only that this loss dismantled the AIS entirely, but also that the longest neuronal process – normally the axon – now showed multiple dendritic characteristics, including MAP2 expression and excitatory synapses that contacted dendritic-like spine protrusions. Very similar results were seen in cerebellar-specific AnkG KO mice, where the basal processes of Purkinje cells, normally smooth axons, developed dendrite-like spine protrusions that received bona fide synaptic contacts [49]. Together, these results suggest that the AIS plays an important role in confering axonal identity. Furthermore, recent work on the axonal regeneration of severed axons has provided some interesting findings on neurite identity: severing an axon within 35 μm of the soma resulted in a nearby dendrite changing its identity to become an axon, whereas a more distal cut caused the re-growth of the same axon [•13]. Although not explored, an intriguing possibility is that this 35 μm cut-off point represents the AIS, which, when removed, induces the re-specification of axonal identity to a dendrite.
These intriguing results raise a number of questions. Is the AIS, as a membrane and cytoplasmic diffusion barrier, the structure that precisely delineates where dendrites stop and the axon begins? If so, given that the molecular complex comprising the AIS often starts tens of microns away from the soma, is the stretch of neurite (including the axon hillock) between the cell body and the AIS actually axonal at all? Are different cellular compartments created by the AIS-like dendritic ‘hotspots’ that exist in certain neurons that lack an axon [50]? And, on the other hand, how does clear axon-dendrite specification occur in the neurons of species that lack Nav-dense AISs, such as C.elegans or Drosophila [cf. 51] ? In all likelihood the AIS is probably a major contributing factor towards neuronal polarity, rather than the one mechanism which defines the dendritic-axonal boundary. After all, the basal processes in AnkG knockout Purkinje cells still look far more like Purkinje cell axons than like the characteristic planar dendritic tree of this cell type [49], and it is possible to dissociate axon specification from AIS formation, as with blockade of IκBα phosphorylation, for example [•12]. The precise interplay between axonal identity and AIS formation will undoubtedly be a focus of much work in the near future.
AIS plasticity
Once the AIS has formed with its specialised sub-domains, how static a structure is it? The few experiments that have examined protein dynamics at the AIS suggest that under normal conditions its components are actually rather stable. After its initial development, the specific knockdown of several AIS component proteins, including NF-186, β-IV-spectrin and Nav channels, showed that they had a long half-life of at least 2 weeks. Similar findings were also observed using repetitive live labelling of extracellular NF-186 [48]. Both approaches indicate that AIS proteins undergo very slow baseline turnover. However, experiments looking at the mobility of Kv2.1 channels within the AIS found it to be quite dynamic. Using fluorescence recovery after photobleaching (FRAP), mixing of Kv2.1 channels at the AIS was shown to occur with a time-constant of around 11 seconds [52]. This mixing appears to be at odds with the existence of restricted sub-domains within the AIS, and raises interesting questions about the mobility of transmembrane proteins and possible crosstalk between different AIS sub-compartments.
AIS composition can certainly change over longer timescales. During postnatal development, as discussed above, the AISs of retinal ganglion cells switch their subunit composition from being dominated by Nav1.2 to Nav1.6 [22,23], and both AIS number and AIS length decrease significantly with development in monkey prefrontal cortex [53]. Chronic diseases are associated with AIS abnormalities, too: a decrease in AnkG-labelled AISs is seen in prefrontal cortex of people with schizophrenia [54], while the number of AIS-targeting GABAergic synapses is reduced in the cortex following long-term epilepsy [55,56].
Recent evidence has also revealed much more rapid and dramatic changes at the AIS as a response to neuronal injury [••57]. Following ischaemia in vivo or oxygen glucose deprivation (OGD) in culture, neurons rapidly degrade the AIS protein scaffold, producing a huge decrease in AIS component proteins after only ~2 hours. This response is independent of cell death or axon degeneration, is dependent upon the calcium-activated degredation enzyme calpain, and, as might be expected from the data discussed above, results in former axonal processes taking on dendritic characteristics [••57]. Importantly, since AIS degredation appears to be irreversible, it could be prevented both in vivo and in vitro injury models by pharmacological calpain inhibition [••57]. Although this could represent a promising new avenue for drug treatment of brain injury, if AIS degredation is a drastic attempt by dying neurons to escape the consequences of pathological over-excitation, such treatment could do more harm than good.
Dynamic changes at the AIS are certainly not restricted to conditions of neuronal injury, however, nor to mechanisms of scaffold disassembly. Recent work has shown that long-term changes in electrical activity can result in significant changes in AIS location. The precise position of the AIS within the axon is known to vary significantly both across [58] and within [59] different cell types, and modelling studies have shown that this variation can produce significant differences in neuronal excitability [59,60] and sensory response properties [59]. The mechanisms determining AIS location within the proximal axon, however, were entirely unknown. Recent findings have shown that chronic two day depolarisation of mature cultured hippocampal neurons produces a significant distal shift in the position of the AIS, moving the molecular complex up to 17 μm further away from the soma [••61]. This relocation is reversible upon return to control conditions, depends on calcium entry through T- and/or L-type channels, and, under conditions of chronic stimulation using the light-gated channel channelrhodopsin-2, requires specific bursting patterns of electrical activity. Activity-dependent AIS relocation also seems linked to changes in neuronal excitability, as predicted [59]: cells with more distal AISs have higher current thresholds for action potential initiation [••61].
Conclusions
Recent studies are beginning to give us a new view of the AIS. Rather than being a uniform, static structure involved solely in action potential initiation, the AIS is actually a diverse, dynamic neuronal compartment that also plays a key role in axon-dendrite partitioning. Future studies should focus on the molecular mechanisms that first establish AnkG localisation in the proximal axon – this event is utterly vital to AIS formation, and may turn out to be intimately linked to processes of axon-dendrite specification. Information on the development, maintenance, and potential plasticity of AIS subdivision would also be extremely welcome. How exactly does the structure become functionally divided? And can these subdivisions change according to recent neuronal activity? Finally, plasticity at the AIS is a fascinating new area to explore, especially the similarities and differences between injury-induced AIS degradation [••57] and activity-dependent AIS relocation [••61] (Figure 2), and the possible relationships of both phenomena to modification of local ion channel densities and/or distributions [41]. It may turn out that multiple features of the AIS can be modified in parallel to produce exquisite fine-tuning of neuronal excitability.
Figure 2.
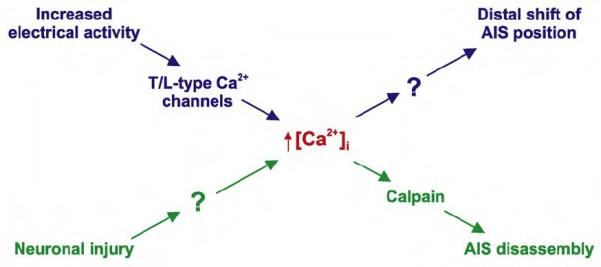
AIS plasticity requires elevated intracellular calcium. Are any other pathway components common?
References
- 1.Boiko T, Vakulenko M, Ewers H, Yap CC, Norden C, Winckler B. Ankyrin-dependent and - independent mechanisms orchestrate axonal compartmentalization of L1 family members neurofascin and L1/neuron-glia cell adhesion molecule. J Neurosci. 2007;27:590–603. doi: 10.1523/JNEUROSCI.4302-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yang Y, Ogawa Y, Hedstrom KL, Rasband MN. betaIV spectrin is recruited to axon initial segments and nodes of Ranvier by ankyrinG. J Cell Biol. 2007;176:509–519. doi: 10.1083/jcb.200610128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jenkins SM, Bennett V. Ankyrin-G coordinates assembly of the spectrin-based membrane skeleton, voltage-gated sodium channels, and L1 CAMs at Purkinje neuron initial segments. J Cell Biol. 2001;155:739–746. doi: 10.1083/jcb.200109026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garrido JJ, Giraud P, Carlier E, Fernandes F, Moussif A, Fache MP, Debanne D, Dargent B. A targeting motif involved in sodium channel clustering at the axonal initial segment. Science. 2003;300:2091–2094. doi: 10.1126/science.1085167. [DOI] [PubMed] [Google Scholar]
- 5.Pan Z, Kao T, Horvath Z, Lemos J, Sul JY, Cranstoun SD, Bennett V, Scherer SS, Cooper EC. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J Neurosci. 2006;26:2599–2613. doi: 10.1523/JNEUROSCI.4314-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou D, Lambert S, Malen PL, Carpenter S, Boland LM, Bennett V. AnkyrinG is required for clustering of voltage-gated Na channels at axon initial segments and for normal action potential firing. J Cell Biol. 1998;143:1295–1304. doi: 10.1083/jcb.143.5.1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hedstrom KL, Xu X, Ogawa Y, Frischknecht R, Seidenbecher CI, Shrager P, Rasband MN. Neurofascin assembles a specialized extracellular matrix at the axon initial segment. J Cell Biol. 2007;178:875–886. doi: 10.1083/jcb.200705119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Voas MG, Glenn TD, Raphael AR, Talbot WS. Schwann cells inhibit ectopic clustering of axonal sodium channels. J Neurosci. 2009;29:14408–14414. doi: 10.1523/JNEUROSCI.0841-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Feinberg K, Eshed-Eisenbach Y, Frechter S, Amor V, Salomon D, Sabanay H, Dupree JL, Grumet M, Brophy PJ, Shrager P, et al. A glial signal consisting of gliomedin and NrCAM clusters axonal Na+ channels during the formation of nodes of Ranvier. Neuron. 2010;65:490–502. doi: 10.1016/j.neuron.2010.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X, Bennett V. Restriction of 480/270-kD ankyrin G to axon proximal segments requires multiple ankyrin G-specific domains. J Cell Biol. 1998;142:1571–1581. doi: 10.1083/jcb.142.6.1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dzhashiashvili Y, Zhang Y, Galinska J, Lam I, Grumet M, Salzer JL. Nodes of Ranvier and axon initial segments are ankyrin G-dependent domains that assemble by distinct mechanisms. J Cell Biol. 2007;177:857–870. doi: 10.1083/jcb.200612012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •12.Sanchez-Ponce D, Tapia M, Munoz A, Garrido JJ. New role of IKK alpha/beta phosphorylated I kappa B alpha in axon outgrowth and axon initial segment development. Mol Cell Neurosci. 2008;37:832–844. doi: 10.1016/j.mcn.2008.01.010. [DOI] [PubMed] [Google Scholar]; Some of the first evidence for a factor involved in localising AnkG to the AIS. In cultured hippocampal neurons phosphorylated IκBα is found in the proximal presumptive axon very early in development. Pharmacologically blocking its phosphorylation shortly after axonal specification prevents the formation of an AnkG-containing AIS.
- •13.Gomis-Ruth S, Wierenga CJ, Bradke F. Plasticity of polarization: changing dendrites into axons in neurons integrated in neuronal circuits. Curr Biol. 2008;18:992–1000. doi: 10.1016/j.cub.2008.06.026. [DOI] [PubMed] [Google Scholar]; An intruiging study of axon specification. Cutting the longest neurite of cultured hippocampal neurons was followed by axonal regeneration at the same site if the cut was >35 μm from the soma, but caused axonal growth from a different process if the cut was <35 μm away. Is the AIS the key boundary point here?
- 14.Lacas-Gervais S, Guo J, Strenzke N, Scarfone E, Kolpe M, Jahkel M, De Camilli P, Moser T, Rasband MN, Solimena M. BetaIVSigma1 spectrin stabilizes the nodes of Ranvier and axon initial segments. J Cell Biol. 2004;166:983–990. doi: 10.1083/jcb.200408007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ogawa Y, Rasband MN. The functional organization and assembly of the axon initial segment. Curr Opin Neurobiol. 2008;18:307–313. doi: 10.1016/j.conb.2008.08.008. [DOI] [PubMed] [Google Scholar]
- 16.Parkinson NJ, Olsson CL, Hallows JL, McKee-Johnson J, Keogh BP, Noben-Trauth K, Kujawa SG, Tempel BL. Mutant beta-spectrin 4 causes auditory and motor neuropathies in quivering mice. Nat Genet. 2001;29:61–65. doi: 10.1038/ng710. [DOI] [PubMed] [Google Scholar]
- 17.Kole MH, Ilschner SU, Kampa BM, Williams SR, Ruben PC, Stuart GJ. Action potential generation requires a high sodium channel density in the axon initial segment. Nat Neurosci. 2008;11:178–186. doi: 10.1038/nn2040. [DOI] [PubMed] [Google Scholar]
- 18.Lemaillet G, Walker B, Lambert S. Identification of a conserved ankyrin-binding motif in the family of sodium channel alpha subunits. J Biol Chem. 2003;278:27333–27339. doi: 10.1074/jbc.M303327200. [DOI] [PubMed] [Google Scholar]
- 19.Fache MP, Moussif A, Fernandes F, Giraud P, Garrido JJ, Dargent B. Endocytotic elimination and domain-selective tethering constitute a potential mechanism of protein segregation at the axonal initial segment. J Cell Biol. 2004;166:571–578. doi: 10.1083/jcb.200312155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brechet A, Fache MP, Brachet A, Ferracci G, Baude A, Irondelle M, Pereira S, Leterrier C, Dargent B. Protein kinase CK2 contributes to the organization of sodium channels in axonal membranes by regulating their interactions with ankyrin G. J Cell Biol. 2008;183:1101–1114. doi: 10.1083/jcb.200805169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xu X, Shrager P. Dependence of axon initial segment formation on Na+ channel expression. J Neurosci Res. 2005;79:428–441. doi: 10.1002/jnr.20378. [DOI] [PubMed] [Google Scholar]
- 22.Boiko T, Van Wart A, Caldwell JH, Levinson SR, Trimmer JS, Matthews G. Functional specialization of the axon initial segment by isoform-specific sodium channel targeting. J Neurosci. 2003;23:2306–2313. doi: 10.1523/JNEUROSCI.23-06-02306.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Van Wart A, Trimmer JS, Matthews G. Polarized distribution of ion channels within microdomains of the axon initial segment. J Comp Neurol. 2007;500:339–352. doi: 10.1002/cne.21173. [DOI] [PubMed] [Google Scholar]
- 24.Laezza F, Gerber BR, Lou JY, Kozel MA, Hartman H, Craig AM, Ornitz DM, Nerbonne JM. The FGF14(F145S) mutation disrupts the interaction of FGF14 with voltage-gated Na+ channels and impairs neuronal excitability. J Neurosci. 2007;27:12033–12044. doi: 10.1523/JNEUROSCI.2282-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brackenbury WJ, Calhoun JD, Chen C, Miyazaki H, Nukina N, Oyama F, Ranscht B, Isom LL. Functional reciprocity between Na+ channel Nav1.6 and beta1 subunits in the coordinated regulation of excitability and neurite outgrowth. Proc Natl Acad Sci U S A. 107:2283–2288. doi: 10.1073/pnas.0909434107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clark BD, Goldberg EM, Rudy B. Electrogenic tuning of the axon initial segment. Neuroscientist. 2009;15:651–668. doi: 10.1177/1073858409341973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •27.Bender KJ, Trussell LO. Axon initial segment Ca2+ channels influence action potential generation and timing. Neuron. 2009;61:259–271. doi: 10.1016/j.neuron.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; An elegant initial characterisation of the T- and R-type voltage-gated calcium channels located at the AIS, and their role in influencing action potential initiation and firing.
- 28.Kole MH, Letzkus JJ, Stuart GJ. Axon initial segment Kv1 channels control axonal action potential waveform and synaptic efficacy. Neuron. 2007;55:633–647. doi: 10.1016/j.neuron.2007.07.031. [DOI] [PubMed] [Google Scholar]
- 29.Shah MM, Migliore M, Valencia I, Cooper EC, Brown DA. Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 2008;105:7869–7874. doi: 10.1073/pnas.0802805105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hill AS, Nishino A, Nakajo K, Zhang G, Fineman JR, Selzer ME, Okamura Y, Cooper EC. Ion channel clustering at the axon initial segment and node of Ranvier evolved sequentially in early chordates. PLoS Genet. 2008;4:e1000317. doi: 10.1371/journal.pgen.1000317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rasmussen HB, Frokjaer-Jensen C, Jensen CS, Jensen HS, Jorgensen NK, Misonou H, Trimmer JS, Olesen SP, Schmitt N. Requirement of subunit co-assembly and ankyrin-G for M-channel localization at the axon initial segment. J Cell Sci. 2007;120:953–963. doi: 10.1242/jcs.03396. [DOI] [PubMed] [Google Scholar]
- 32.Ogawa Y, Horresh I, Trimmer JS, Bredt DS, Peles E, Rasband MN. Postsynaptic density-93 clusters Kv1 channels at axon initial segments independently of Caspr2. J Neurosci. 2008;28:5731–5739. doi: 10.1523/JNEUROSCI.4431-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Davis JQ, Bennett V. Ankyrin binding activity shared by the neurofascin/L1/NrCAM family of nervous system cell adhesion molecules. J Biol Chem. 1994;269:27163–27166. [PubMed] [Google Scholar]
- 34.John N, Krugel H, Frischknecht R, Smalla KH, Schultz C, Kreutz MR, Gundelfinger ED, Seidenbecher CI. Brevican-containing perineuronal nets of extracellular matrix in dissociated hippocampal primary cultures. Mol Cell Neurosci. 2006;31:774–784. doi: 10.1016/j.mcn.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 35.Szabadics J, Varga C, Molnar G, Olah S, Barzo P, Tamas G. Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science. 2006;311:233–235. doi: 10.1126/science.1121325. [DOI] [PubMed] [Google Scholar]
- 36.Khirug S, Yamada J, Afzalov R, Voipio J, Khiroug L, Kaila K. GABAergic depolarization of the axon initial segment in cortical principal neurons is caused by the Na-K-2Cl cotransporter NKCC1. J Neurosci. 2008;28:4635–4639. doi: 10.1523/JNEUROSCI.0908-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Glickfeld LL, Roberts JD, Somogyi P, Scanziani M. Interneurons hyperpolarize pyramidal cells along their entire somatodendritic axis. Nat Neurosci. 2009;12:21–23. doi: 10.1038/nn.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ango F, di Cristo G, Higashiyama H, Bennett V, Wu P, Huang ZJ. Ankyrin-based subcellular gradient of neurofascin, an immunoglobulin family protein, directs GABAergic innervation at purkinje axon initial segment. Cell. 2004;119:257–272. doi: 10.1016/j.cell.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 39.Martin PM, Carnaud M, del Cano G Garcia, Irondelle M, Irinopoulou T, Girault JA, Dargent B, Goutebroze L. Schwannomin-interacting protein-1 isoform IQCJ-SCHIP-1 is a late component of nodes of Ranvier and axon initial segments. J Neurosci. 2008;28:6111–6117. doi: 10.1523/JNEUROSCI.1044-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- •40.Lorincz A, Nusser Z. Cell-type-dependent molecular composition of the axon initial segment. J Neurosci. 2008;28:14329–14340. doi: 10.1523/JNEUROSCI.4833-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]; Careful and technically proficient immunocytochemical labelling allowed these authors to visualise AIS subdomains characterised by different combinations of Nav and Kv channels, and to show that this sub-compartmentalisation varies significantly between different types of neuron.
- 41.Nusser Z. Variability in the subcellular distribution of ion channels increases neuronal diversity. Trends Neurosci. 2009;32:267–274. doi: 10.1016/j.tins.2009.01.003. [DOI] [PubMed] [Google Scholar]
- 42.Duflocq A, Le Bras B, Bullier E, Couraud F, Davenne M. Nav1.1 is predominantly expressed in nodes of Ranvier and axon initial segments. Mol Cell Neurosci. 2008;39:180–192. doi: 10.1016/j.mcn.2008.06.008. [DOI] [PubMed] [Google Scholar]
- ••43.Hu W, Tian C, Li T, Yang M, Hou H, Shu Y. Distinct contributions of Na(v)1.6 and Na(v)1.2 in action potential initiation and backpropagation. Nat Neurosci. 2009;12:996–1002. doi: 10.1038/nn.2359. [DOI] [PubMed] [Google Scholar]; These authors used an impressive combination of immunocytochemistry, electrophysiology and mathematical modelling to reveal distinct functional roles for proximal and distal AIS sub-compartments. The Nav1.6-rich distal domain is the site of action potential initiation, while the Nav1.2-rich proximal domain allows action potentials to back-propagate to the soma and beyond. But how does this functional subdivision develop?
- 44.Palmer LM, Stuart GJ. Site of action potential initiation in layer 5 pyramidal neurons. J Neurosci. 2006;26:1854–1863. doi: 10.1523/JNEUROSCI.4812-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Meeks JP, Mennerick S. Action potential initiation and propagation in CA3 pyramidal axons. J Neurophysiol. 2007;97:3460–3472. doi: 10.1152/jn.01288.2006. [DOI] [PubMed] [Google Scholar]
- 46.Winckler B, Forscher P, Mellman I. A diffusion barrier maintains distribution of membrane proteins in polarized neurons. Nature. 1999;397:698–701. doi: 10.1038/17806. [DOI] [PubMed] [Google Scholar]
- ••47.Song AH, Wang D, Chen G, Li Y, Luo J, Duan S, Poo MM. A selective filter for cytoplasmic transport at the axon initial segment. Cell. 2009;136:1148–1160. doi: 10.1016/j.cell.2009.01.016. [DOI] [PubMed] [Google Scholar]; A comprehensive set of experiments showing for the first time that the AIS directs axonal cytoplasmic transport, and thereby regulates axonal identity.
- 48.Hedstrom KL, Ogawa Y, Rasband MN. AnkyrinG is required for maintenance of the axon initial segment and neuronal polarity. J Cell Biol. 2008;183:635–640. doi: 10.1083/jcb.200806112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sobotzik JM, Sie JM, Politi C, Del Turco D, Bennett V, Deller T, Schultz C. AnkyrinG is required to maintain axo-dendritic polarity in vivo. Proc Natl Acad Sci U S A. 2009;106:17564–17569. doi: 10.1073/pnas.0909267106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kosaka T, Komada M, Kosaka K. Sodium channel cluster, betaIV-spectrin and ankyrinG positive “hot spots” on dendritic segments of parvalbumin-containing neurons and some other neurons in the mouse and rat main olfactory bulbs. Neurosci Res. 2008;62:176–186. doi: 10.1016/j.neures.2008.08.002. [DOI] [PubMed] [Google Scholar]
- 51.Katsuki T, Ailani D, Hiramoto M, Hiromi Y. Intra-axonal patterning: intrinsic compartmentalization of the axonal membrane in Drosophila neurons. Neuron. 2009;64:188–199. doi: 10.1016/j.neuron.2009.08.019. [DOI] [PubMed] [Google Scholar]
- 52.Sarmiere PD, Weigle CM, Tamkun MM. The Kv2.1 K+ channel targets to the axon initial segment of hippocampal and cortical neurons in culture and in situ. BMC Neurosci. 2008;9:112. doi: 10.1186/1471-2202-9-112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cruz DA, Lovallo EM, Stockton S, Rasband M, Lewis DA. Postnatal development of synaptic structure proteins in pyramidal neuron axon initial segments in monkey prefrontal cortex. J Comp Neurol. 2009;514:353–367. doi: 10.1002/cne.22006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cruz DA, Weaver CL, Lovallo EM, Melchitzky DS, Lewis DA. Selective alterations in postsynaptic markers of chandelier cell inputs to cortical pyramidal neurons in subjects with schizophrenia. Neuropsychopharmacology. 2009;34:2112–2124. doi: 10.1038/npp.2009.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Marco P, Sola RG, Ramon y Cajal S, DeFelipe J. Loss of inhibitory synapses on the soma and axon initial segment of pyramidal cells in human epileptic peritumoural neocortex: implications for epilepsy. Brain Res Bull. 1997;44:47–66. doi: 10.1016/s0361-9230(97)00090-7. [DOI] [PubMed] [Google Scholar]
- 56.Ribak CE. Axon terminals of GABAergic chandelier cells are lost at epileptic foci. Brain Res. 1985;326:251–260. doi: 10.1016/0006-8993(85)90034-4. [DOI] [PubMed] [Google Scholar]
- ••57.Schafer DP, Jha S, Liu F, Akella T, McCullough LD, Rasband MN. Disruption of the axon initial segment cytoskeleton is a new mechanism for neuronal injury. J Neurosci. 2009;29:13242–13254. doi: 10.1523/JNEUROSCI.3376-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]; A large array of in vivo and in vitro approaches converge on a mechanism by which neuronal injury, through calpain activation, causes rapid irreversible AIS disassembly. A last-gasp drastic effort by neurons to save themselves?
- 58.Fried SI, Lasker AC, Desai NJ, Eddington DK, Rizzo JF., 3rd Axonal sodium-channel bands shape the response to electric stimulation in retinal ganglion cells. J Neurophysiol. 2009;101:1972–1987. doi: 10.1152/jn.91081.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kuba H, Ishii TM, Ohmori H. Axonal site of spike initiation enhances auditory coincidence detection. Nature. 2006;444:1069–1072. doi: 10.1038/nature05347. [DOI] [PubMed] [Google Scholar]
- 60.Kress GJ, Dowling MJ, Eisenman LN, Mennerick S. Axonal sodium channel distribution shapes the depolarized action potential threshold of dentate granule neurons. Hippocampus. 2010;20:558–571. doi: 10.1002/hipo.20667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ••61.Grubb MS, Burrone J. Activity-dependent relocation of the axon initial segment fine-tunes neuronal excitability. Nature. 2010;465:1070–1074. doi: 10.1038/nature09160. [DOI] [PMC free article] [PubMed] [Google Scholar]; Evidence for a surprising degree of plasticity in the precise axonal position of the AIS. The authors show that particular patterns of electrical activity in neurons can produce shifts in AIS location, and that these shifts are associated with subtle alterations in neuronal excitability.