

1. Introduction
Much has been made about J. L. W. Thudichum’s colorful, and one could say clairvoyant, naming of sphingosine “in commemoration of the many enigmas which it presented to the inquirer” in his 1884 treatise The Chemistry of the Brain(1) because many of the riddles of sphingolipids (as the broader field was later named)(2) remained unanswered for the following century. This changed radically over the past several decades as researchers explored, and ultimately established, what seemed at the time to be radical concepts: that sphingolipids are not just structural elements of cells but also participate in intra- and extracellular signaling; that not only the complex glycan headgroups, but also the lipid backbones, are highly specified metabolically and have selective biochemical functions; and that even the longest known function of these lipids, as structural components of the “fluid mosaic” of cell membrane lipids, is not so simple, and often involves the dynamic clustering of sphingolipids in nontraditional microdomains referred to as rafts. We still know only a fraction of their secrets, but this enlightenment has defined models for thinking about these compounds that remove them from their enigmatic “black box.”
Now, a major challenge is to keep up with the rapid growth in knowledge about the sphingolipidome, that is, the ensemble of all sphingolipids.(3) A major goal of the review is to help the reader more easily grasp the metabolic interrelationships that account for the tens of thousands of molecular subspecies (and perhaps more) that appear in nature, with a focus on mammals. The magnitude of this subject precludes the inclusion of all of the enzymes and metabolites, and the author apologizes for the omission of many interesting topics. To put this information in context, there is a brief background discussion of their structures and functions, which have been dealt with also in a recent Chemical Reviews article(4) on the chemicophysical features of sphingolipids and raft formation, and by excellent reviews on sphingolipid signaling5,6 and the biological functions of complex glycosphingolipids.7−10
2. An Overview of Sphingolipid Structure and Function
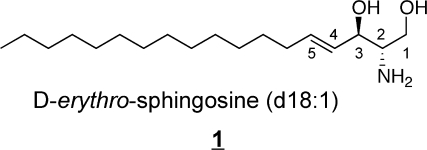
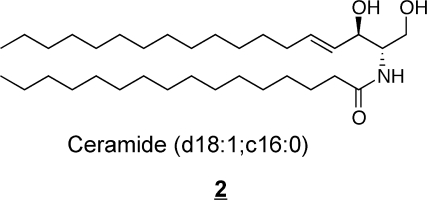
Sphingolipids share the common structural feature that all are comprised of backbones called “long-chain-” or “sphingoid” bases, which are represented by sphingosine, (2S,3R,4E)-2-aminooctadec-4-ene-1,3-diol (also referred to as (E)-sphing-4-enine) 1, the major sphingoid base found in mammals. Free sphingoid bases (i.e., underivatized) are typically present in very small amounts because most are amide-linked with a long- or very-long-chain fatty acid to form ceramides 2 that can be further derivatized by addition of a headgroup (at C1 in 2) to form more complex sphingolipids such as sphingomyelin (SM), glucosylceramide (GlcCer), galactosylceramides (GalCer) and more complex glycosphingolipids with a few to dozens of sugar residues.(3) There are also small amounts of “lyso-”sphingolipids (i.e., sphingoid bases plus a headgroup but lacking the N-acyl-substituent, such as sphingosine 1-phosphate, sphingosine 1-phosphocholine, and lyso-glycosphingolipids), N-methyl-derivatives, and covalent adducts with proteins.
2.1. Backbone and Headgroup Nomenclature
Because organisms usually have more than one type of sphingoid base (e.g., not just sphingosine 1, but also sphingoid bases with more or fewer hydroxyls, somewhat shorter or longer alkyl chains, and other structural variations),(11) a convenient short-hand nomenclature to distinguish them by these features is to give the number of hydroxyl groups (m for one, mono-, d for the two, di-, and t for three, tri-) followed by the chain length and number of double bonds (with their position, if necessary). Therefore, sphingosine 1 is usually abbreviated d18:1, with the double bond assumed to be at position 4, or specified by a prefix (4E-d18:1) or superscripted suffix (d18:1Δ4t). The addition of an amide-linked fatty acid to form a ceramide can be designated by a semicolon or slash followed by the carbon chain length and number of double bonds for the fatty acid. Using this nomenclature, the Cer 2 (N-palmitoylsphingosine) would be abbreviated d18:1;C16:0 (or d18:1/C16:0, or 4E-d18:1;C16:0, etc.). This can be added to the name of the sphingolipid headgroup subcategory (as shown in Figure 1 for ganglioside GM1a) to provide explicit information about the molecule that is being described.
Figure 1.

Basic structures of mammalian sphingolipids. The upper left panel summarizes the categories of complex sphingolipids, and the upper right panel displays the root structures of the glycosphingolipid families using the glycan symbols defined by the key in the lower panel (the letter and number within the symbols convey the nature of the glycosidic linkage between that carbohydrate and the species to its right, for example: “β4” represents a β1–4 linkage). The abbreviations are: Glc, glucose; GlcNAc, N-acetylglucosamine; Gal, galactose; GalNAc, N-acetylgalactosamine; Neu5Ac, N-acetylneuraminic acid; Fuc, fucose. The lower panel displays the structure of ganglioside GM1a using both ChemDraw and glycan symbols, the Roman numbering system for the positions of the glucans (i.e., beginning with the first carbohydrate attached to ceramide), and a comparison with two other gangliosides (GM1b and fucosyl-GM1a) using the glycan symbol system.
The major headgroup types and some aspects of their nomenclature are shown in Figure 1. They can be categorized as having substituents that are nonpolar (H- and O-acyl) versus polar (phospho- and glyco-) or polar with an ionic group (phosphate, carboxyl for N-acetylneuraminic acid and glucuronic acid, or sulfate). The phosphosphingolipids of mammals are ceramide 1-phosphate (Cer1P), sphingomyelins (SM) and ceramide phosphoethanolamines (CerPE) (plus the “lyso” forms of these, that is, with the sphingoid base but not amide linked fatty acid). The glycosphingolipids are divided into multiple subcategories: first by whether they have glucose (GlcCer) or galactose (GalCer) as the carbohydrate attached in β-linkage to Cer, then by the nature of the additional substituents (for example, sulfated glycosphingolipids are referred to as sulfatides). GlcCer is followed by addition of Gal to form Galβ1–4Glcβ1Cer (lactosylceramide, LacCer), which is at a branchpoint for formation of the so-called root structure families (globo-, isoglobo-, lacto-, neolacto-, and ganglio-) shown in Figure 1. Likewise, the order and position of addition of other substituents, in particular the addition of N-acetylneuraminic acid (Neu5Ac,which is also called sialic acid) defines branching families of glycosphingolipids (in this case, gangliosides), with ganglioside GM1a shown as an example in Figure 1 as both a chemdraw structural diagram and commonly used symbols (see key in Figure 1).(12) It is worth mentioning that there are structural differences in the repertoire of carbohydrates that are used among even fairly closely related mammals, for example, the sialic acid of human gangliosides is primarily comprised of N-acetylneuraminic acid (Neu5Ac) as shown for GM1a in Figure 1 whereas other mammals have both Neu5Ac and N-glycolylneuraminic acid (abbreviated Neu5Gc), which cannot be made by humans.(13)
Many of the glycosphingolipids are comprised of the same units attached in different combinations and arrangements, as exemplified in Figure 1 by the root structure families globo- versus isoglobo- and lacto- versus neolacto- (as well as by the two gangliosides GM1a and GM1b) and, thus, glycosphingolipid biosynthesis has been described as nature’s version of combinatorial chemistry.(14)
There are several nomenclature systems for glycosphingolipids, and many compounds are still referred to by their historically assigned names (such as gangliosides GM1a and GM1b shown in Figure 1). Using IUPAC-IUB guidelines for systematic naming of glycosphingolipids,15,16 these gangliosides would be described as Neu5Acα2–3(Galβ1–3GalNAcβ1–4)Galβ1–4Glcβ1Cer (d18:1/C18:0) for GM1a, and Neu5Acα2–3Galβ1–3GalNacβ1–4Galβ1–4Glcβ1Cer (with the same Cer backbone specification, if it applied) for GM1b. These compounds could also be named starting with the Ganglio (Gg) root structure that they both share (see insert in the upper right of Figure 1) with designation of the location of the Neu5NAc along the chain using Roman numerals (this numbering system is shown for GM1a) and Arabic superscripts to designate the hydroxyl- to which the Neu5NAc is linked. By this system, GM1a is described as II3-α-Neu5NAc-Gg4Cer (which would be read “II3-α-N-acetylneuraminosyl-gangliotetraosylCer”) and GM1b is IV3-α-Neu5NAc-Gg4Cer (IV3-α-N-acetylneuraminosyl-gangliotetraosylCer). When there are additional modifications, such as 9-O-acetylation of sialic acid13,17 or formation of an intramolecular lactone,(18) these are added to the name. Some other glycans that are still referred to by historic names are the Lewis blood group antigens (Figure 2).19,20
Figure 2.
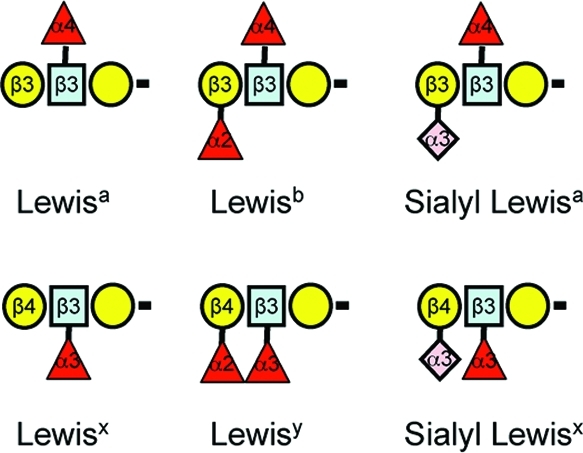
Representative structures of Lewis epitopes. The key for the glycan symbols is the same as for Figure 1.
The biological rationale behind this structural complexity is no mystery when one considers the sophistication of the functions of sphingolipids, as represented schematically in Figure 3. Complex sphingolipids (shown here as SM, in black, and gangliosides GM3 and GM1a, using the headgroup key from Figure 1) help form lipid bilayers with unique surface characteristics (charge, polarity and morphology) and fluidity, which contribute to the clustering of sphingolipids and cholesterol (and some proteins) in rafts.4,21 Also depicted are interactions between surface glycosphingolipids and proteins on the same cell, the extracellular matrix, neighboring cells, and other entities (such as bacterial toxins and viruses), which have been referred to as the “glycosynapse.”(22) Sphingolipids additionally contribute to membrane dynamics(23) and cell regulation through metabolic interconversions (shown for SM and Cer, which can occur via sphingomyelinases and SM synthase)(24) and membrane trafficking,21,25 and this can lead to production of additional bioactive metabolites (Cer1P, sphingosine and S1P)(26) that act in situ, inside, or outside of the cell (as shown for S1P and an S1P receptor).(6) Although these functions (both of the complex sphingolipids and the “signaling” lipid moieties) are shown at the plasma membrane, where they certainly can occur,(27) the lipid backbones from de novo sphingolipid biosynthesis also contribute to signaling(28) (sometimes with deleterious consequences),(29) and sphingolipids have functions in previously unexpected organelles such as the nucleus,(30) and in some cases using enzymes classically thought to be active only for “housekeeping” turnover of sphingolipids, such as acid sphingomyelinase.(31) It will be a challenge to figure out which specific sphingolipid molecules (and combinations of molecules) are present and interacting with which specific targets to achieve the sphingolipid-regulated steps in a biological process.
Figure 3.

Schematic representation of sphingolipid functions. This diagram depicts a hypothetical plasma membrane with representative categories of sphingolipids with the black headgroup representing sphingomyelin, the colored headgroups the glycosphingolipids as in Figure 1, and the lipid backbones with the sphingoid base in blue and the amide-linked fatty acid in gray; phosphoglycerolipids and cholesterol are depicted in gray. The diagram illustrates the clustering of a portion of the sphingolipids (and cholesterol) in membrane “rafts,” the binding of ganglioside GM3 (left) and GM1 (right) to proteins, and the metabolic interconversions of some of the sphingolipids (shown in the box, in the order ceramide 1-phosphate, ceramide, sphingosine, and sphingosine 1-phosphate, S1P), which alters both the biophysical properties of the membrane and generates signaling molecules, such as S1P, which is involved in both intracellular signaling and extracellular signaling (represented by the green arrow).
2.2. Variation in the Lipid Moieties
Some of the breakthroughs in understanding the functions of sphingolipids, especially with respect to cell signaling, have come from having the capacity to measure more than one bioactive subspecies so the correct signaling pathways can be sorted out,(32) especially when the metabolites have opposite effects, such as ceramide versus S1P.(33) In recent years, the analytical technology of choice has been mass spectrometry;34,35 however, even when the analysis of the lipid moieties of sphingolipids was quite laborious (for example, using chemical degradation to determine sphingoid base composition),36,37 the few biological samples that were examined in depth gave an astonishing result, that is, that a given class of sphingolipid is comprised of dozens of different backbones, not just the handful that are usually discussed.(38) Indeed, a recent analysis of human plasma SM using a mass spectrometry protocol that is able to distinguish the isobaric and isomeric subspecies (using a technique we refer to as “ion trap facilitated fragmentation”)(39) identified ∼100 different lipid subspecies,(40) and other types of mass spectrometry have uncovered an equivalent level of structural diversity with mammalian samples.(41)
2.2.1. Sphingoid Base Diversity
Sphingoid bases vary in type (such as sphingosines versus sphinganines) (Figure 1) and chain length. Two of the most common chain length variants of sphingosine (d18:1) 1 are d16:1, which has been found, for examples, in plasma sphingolipids40,42 and bovine milk,43,44 and d20:1 (eicosasphingosine), which is present in substantial amounts in brain gangliosides, especially with advanced age.(45) Other locations include human stomach and intestinal mucosa,(46) skin ceramides,(47) sulfatides,(48) and perhaps most puzzlingly, in host liver SM from rats bearing Morris hepatoma 7777.(49) Mammalian sphingoid bases also include odd chain length variants (e.g., linear d17- and d19-, but odd carbon numbers are sometimes due to branched alkyl chains) and shorter chain-length subspecies (which are in trace amounts in mammals, but more common in other organisms, such as Drosophila(50)).(11) This variation has important implications for analysis of sphingolipids by mass spectrometry, which follows specific molecular ions and fragmentation products (often as precursor-product pairs)(40) so the bookkeeping of how much of a particular category of sphingolipid is present (for example, all the SM’s) will depend on successful inclusion of all of the subspecies in the analysis protocol (within the detection limits selected by the investigator).
Little is known about the biological significance of this seemingly subtle backbone chain-length variation, however, the alkyl-chain length mismatch has substantial biophysical consequences.(51) And if selective anatomical localization of d20:1 sphingosine is an expression of the adage that “form follows function,” it is noteworthy that gangliosides from sensory nerve contain larger proportions of d18:1 than motor nerve gangliosides (which have higher d20:1).(52) The very powerful technique of tissue-imaging mass spectrometry has established that d20:1 gangliosides are selectively localized along the entorhinal-hippocampus projections, especially in the molecular layer of the dentate gyrus, whereas those with the 18-carbon sphingoid base backbone are widely distributed throughout the frontal brain.(53)
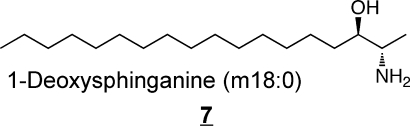
Other types of structural variations that have been found in humans are shown in 3 to 7 (these also appear in other alkyl chain lengths).(11)
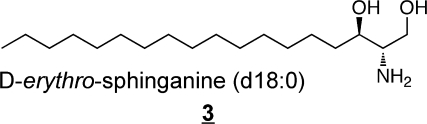
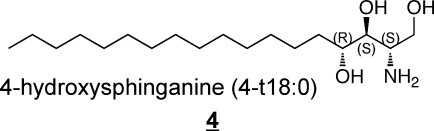
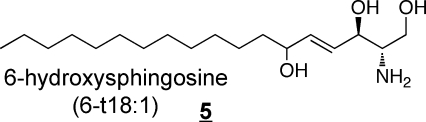
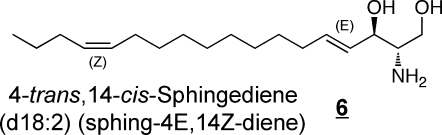
Sphinganine (also referred to as dihydrosphingosine) 3 is an intermediate of sphingoid base biosynthesis and is found in most complex sphingolipids in at least small amounts; 4-hydroxysphinganine 4, which is often referred to as phytosphingosine, is commonly found in sphingolipids from epithelial tissues(54) and skin (which also has another trihydroxy-sphingoid base with the extra hydroxylation at postion 6 rather than 4, 5).47,55−58 In addition to these, the diene 6 has been noted in plasma(40) and brain,59,60 and other mammalian sources.(61) Sphingadienes with double bonds at other positions62−64 (and trienes)(65) are found in plants, and have been reported in SM from human breast milk.(66)
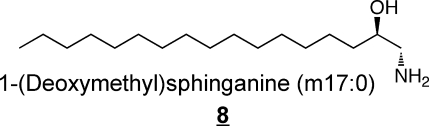
Sphingoid bases 7 and 8 have been found in mammals only recently,67,68 and are very intriguing because they lack the 1-hydroxyl-group that is found on all of the other sphingoid bases, which means they (or the N-acyl-“1-deoxydihydroceramide” derivatives) can not be metabolized to more complex sphingolipids by headgroup addition. They are mainly present as the N-acylated (1-deoxydihydroceramide) metabolites,(67) which will be extremely hydrophobic. It is not clear how they are catabolized since degradation of the typical sphingoid bases proceeds via the 1-phosphates.(69)
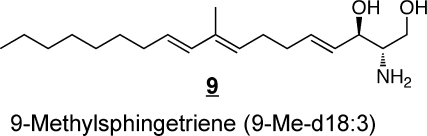
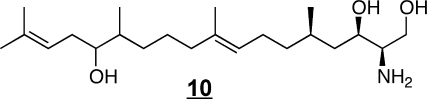
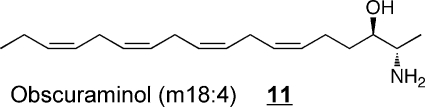
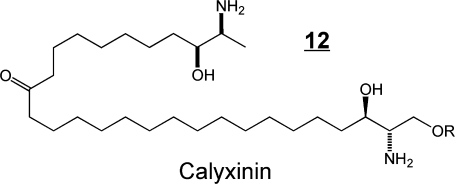
Some of the structural variety found with other organisms11,36,37 is illustrated by examples 9–12. The branched triene 9 has been identified in squid nerve sphingomyelin,(70) aplidiasphingosine 10 has been isolated from the marine tunicate Aplidium sp.(71,72) and noted to have antimicrobial and antitumorial activity,71,73 and obscuraminol 11 was isolated from a chloroform extract of Pseudodistoma obscurum(74) that was cytotoxic for various tumor cell lines (but the isolated compound was only mildly cytotoxic). Calyxinin 12 is a member of a fascinating series of compounds that resemble “two-headed” sphingoid bases, that is, two sphingoid bases connected tail-to-tail (note that the lower portion of calyxinin resembles sphinganine the upper portion a 1-deoxysphinganine at the other end with threo- stereochemistry).(75) These and other extraordinary sphingoid bases(11) warrant attention because they might be useful tools for studies of sphingolipid metabolism or functions (as will be discussed below for two stellar examples, fumonisin B113 and myriocin 14). Furthermore, some might appear in humans if consumed in the diet (or, perhaps, produced de novo but previously overlooked). Indeed, both apply to 1-deoxysphinganine 7, which was first named spisulosine upon its isolation from Spisula polynyma,(76) a clam that is consumed by humans as sushi, chowder and “clam strips” (appearing in recipes as the Arctic surf clam or Stimpson’s surf clam). It was later found to be made by mammals.67,68
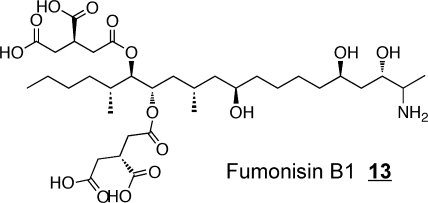
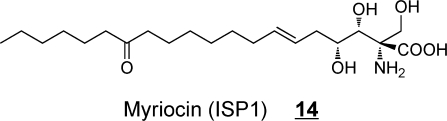
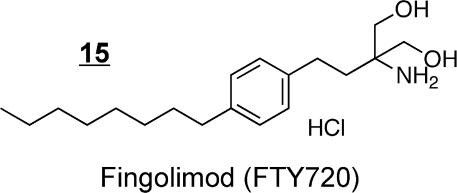
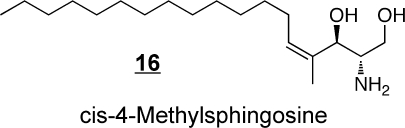
The fungal secondary metabolites fumonisin B113 and myriocin 14 are the two most widely studied extraordinary sphingoid bases. Soon after the structure of fumonisin B1 was elucidated,(77) its similarity to sphinganine led to Ron Riley and my laboratories to explore if it might affect sphingolipid metabolism and the discovery that fumonisins are potent inhibitors of ceramide synthase.(78) Furthermore, this inhibition is thought to be a major contributor to the diseases caused by this family of mycotoxins, including a recent association with birth defects.(79) Fumonisin B1 has been employed in hundreds of studies of sphingolipid metabolism, and is a useful tool if the investigator bears in mind that it also causes accumulation of sphingoid bases and often their 1-phosphates.(80) Likewise, myriocin (also called ISP-1) has been of tremendous value in sphingolipid research as a potent inhibitor of serine palmitoyltransferase,81−83 and studies of immunosuppression by myriocin81,83,84 led to the development of FTY720 (Fingolimod) 15, a compound that undergoes phosphorylation and disrupts lymphocyte trafficking by binding to S1P receptor(s).(85) FTY720 has shown promise in treatment of a number of diseases, including multiple sclerosis.85,86 Interestingly, cis-4-methylsphingosine 16 is another sphingoid base analog that is readily taken up by cells, undergoes phosphorylation, and affects S1P receptors;(87) it also inhibits de novo sphingolipid biosynthesis.(88)
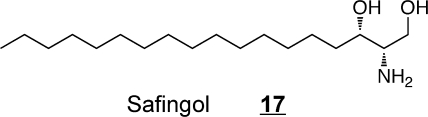
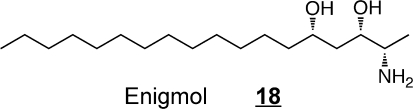
Thus, many of these compounds might serve as pharmacophors for development of novel therapeutic agents. The sphingoid base safingol (l-threo-sphinganine) 17 was one of the first sphingoid base analogs to be tested as a potential anticancer agent because it inhibits protein kinase C and has a longer half-life than naturally occurring sphingoid bases (and is now of interest also because it inhibits sphingosine kinase and induces autophagy).89,90 Safingol has been evaluated in a phase I clinical trial alone and in combination with cisplatin and, in addition to defining the dosages that can be administered safely, the studies found that Safingol caused a dose-dependent reduction in S1P, as predicted.(91) A synthetic 1-deoxy-sphingoid base analog, Enigmol(92)18, has shown efficacy against using colon and prostate cancer in mouse models. And, phase I clinical trials have also been conducted with 1-deoxysphinganine 7 (under the name ES-285),93,94 which surfaced in a screen of lipid extracts from aquatic organisms for potential anticancer compounds.(95)
The mechanisms of action of sphingoid bases have been difficult to pin down because they affect many targets, which include receptors, protein kinases and ion transporters,96−98 and because they are metabolized to and from other highly bioactive compounds (Cer, S1P, and others) (as depicted in Figure 3). Sphingolipids are also produced by yeast, and an understanding of signaling by free sphingoid bases is becoming clearer for that organism.(99)
2.2.2. N-Acyl-sphingoid Bases (Ceramides)
Acylation of the amino group of sphingoid bases with a fatty acid produces compounds broadly referred to as “ceramides” 2, although another current convention is to use this term specifically for N-acylsphingosines, and to apply other descriptors when a different sphingoid base is present, such as dihydroceramides for N-acylsphinganines and 4-hydroxyceramides or phytoceramides for N-acyl-4-hydroxysphinganines. The fatty acid chains are predominately 14 to 36 carbon atoms in length, and usually saturated, or with a single double bond or an α-hydroxyl group. Some of the most structurally complex ceramides are found in skin, which includes the presence of a very-long-chain fatty acid (C30 to 32) with an ω-hydroxyl group that is esterified to another fatty acid,100−102 and in testis, which contains neutral glycosphingolipids with very-long-chain (C26 to C32) polyunsaturated (4 to 6 double bonds) fatty acids.103,104 Ceramides with very short fatty acids, as short as two carbons (acetyl-, C2-Cer), have also been found in mammals(105) and suggested to arise from transfer of the acetyl group from platelet-activating factor.(106)
Ceramide nomenclature follows the conventions already discussed. If the fatty acid is not stated explicitly (e.g., N-palmitoylsphingosine), the fatty acyl-chain length is usually presented as a prefix, such as C16-Cer for N-palmitoylsphingosine, or by the abbreviated nomenclature described in section 2.1.1. (d18:1/C16:0, 2).
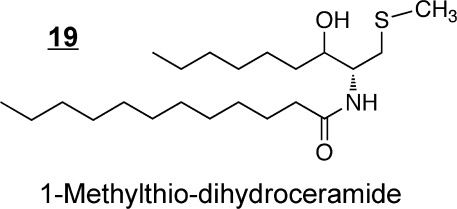
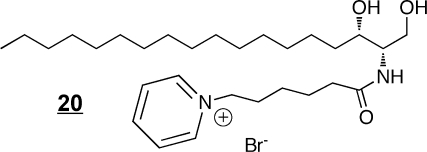
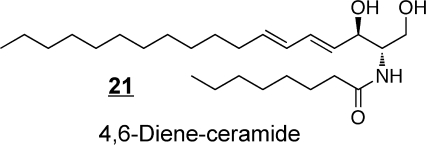
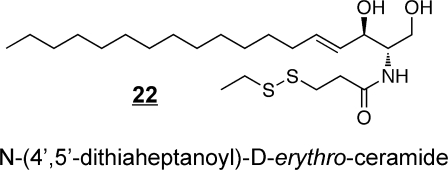
Synthetic ceramide analogs have been prepared for a wide range of purposes, including the production of species that are more readily taken up by cells (e.g., C2-ceramides),(107) for exploration of structure–function relationships in cell signaling,(108) as inhibitors of enzymes of ceramide metabolism(109) (including an interesting case where 1-methylthiodihydroceramide 19 inhibits Cer biosynthesis by inducing sphingosine kinase),(110) and development of novel compounds that have shown activity as potential anticancer agents, such as l-threo-C6-pyridinium-ceramide-bromide 20 (which targets the nucleus and mitochondria),(111) the 4,6-diene-Cer 21(112) (i.e., which contains an additional trans-double bond between carbons 6 and 7, like the 14-carbon sphingoid base from Drosophila that has been reported to prevent intestinal tumorigenesis(98)) and N-(4′,5′-dithiaheptanoyl)-D-erythro-Cer(113)22. Novel methods of delivery of ceramides (namely, C6-ceramide) have been developed by preparation of nanoliposomal particles to facilitate solubility(114) and are showing efficacy in cancer chemotherapy.115,116
The biophysical properties of ceramides include many interesting features,(51) most notably that the alkyl chains are largely saturated and thus have high phase transition temperatures and give rise to rigid ceramide-enriched domains in membranes of otherwise more “fluid” components.(23) These properties are not generalizable to all “ceramides,” however, and raft stability is affected by the ceramide N-acyl chain,(117) among other factors. Ceramides also change membrane curvature,(118) transbilayer (flip-flop) movement of lipids(119) and other molecules,(120) appearing to form channels in mitochondrial outer membranes when present in sufficient concentrations.(120)
Cell signaling by ceramides has been elegantly reviewed many times5,31,121−125 (just to list a few) and its roles in regulation of cell growth, senescence and death account for the aforementioned interest in ceramide analogs and modulators of ceramide metabolism as potential anticancer agents.124,126,127 The regulation of autophagy by both ceramide128,129 and de novo synthesized dihydroceramide(97) is intriguing because this is different than for most of the other cellular processes regulated by ceramide (e.g., apoptosis), which require the 4,5-trans double bond. This raises the possibility that cells might use these relatively safe molecular subspecies for autophagy under conditions where comparable elevation of ceramides might be dangerous. This underscores how specific molecular subspecies are likely to be important for normal cell function, and the corollary that cells will have mechanisms to produce and localize the appropriate subspecies for the necessary structural and regulatory functions. In the words of Hannun and Obeid in a recent review: “First and foremost, the ‘Many Ceramides’ approach negates the current prevailing paradigm that ceramide can be understood in terms of regulation and function as a single entity... at the very least mechanistic studies on ceramide function and regulation should focus on specific pathways of formation.”(125) The mechanisms for formation of specific ceramide subspecies will be discussed in section .
2.3. Variation of the Complex Sphingolipid Headgroups
For the purpose of this review, complex sphingolipids will be defined as having both of the alkyl chains of the lipid moiety (i.e., “ceramide”) and a substituent at the hydroxyl at position 1. The major headgroup categories for mammalian sphingolipids (ceramides, sphingomyelins, glucosylceramides, galactosylceramides, etc.) are summarized in Figure 1, and this shows only a fraction of the glycan headgroups (Robert Yu has recently compiled structures for 174 neutral glycosphingolipids, 190 gangliosides and 24 sulfated glycosphingolipids);(19) the total estimate is closer to 600 if one adds likely biosynthetic intermediates that have not yet been characterized (for a depiction of these, see www.sphingomap.org).(3) The number expands considerably if one adds headgroups (and backbones) that are found in other organisms, such as plants,(130) fungi131−133 and other organisms.(134) Even this summation is likely to underestimate the total as more sensitive analytical methods allow us to see minor subspecies.
Fortunately (for the analytical chemist), the number of species that are produced biologically will be much lower than the number that could be theoretically made from these glycans (if all combinations and positional isomers are considered) due to the relatively limited number of synthases for the complex sphingolipids and their substrate specificities. For an idea of how many species might theoretically exist, Roger Laine estimated that six different hexoses could be combined to form >1012 different hexasaccharides, ∼1015 heptasaccharides, >1018 octasaccharides, and nearly Avogadro’s number for nonasaccharides.(135) Mind-boggling numbers, indeed! But, as one examines some of the largest mammalian glycosphingolipids, such as the placental tetrasialosylpoly-N-acetyllactosaminyl ganglioside 23 shown here,(136) it is striking that it is comprised of a few repeating units (for which these types of compounds have been named “polylactosaminoglycans”).(137)
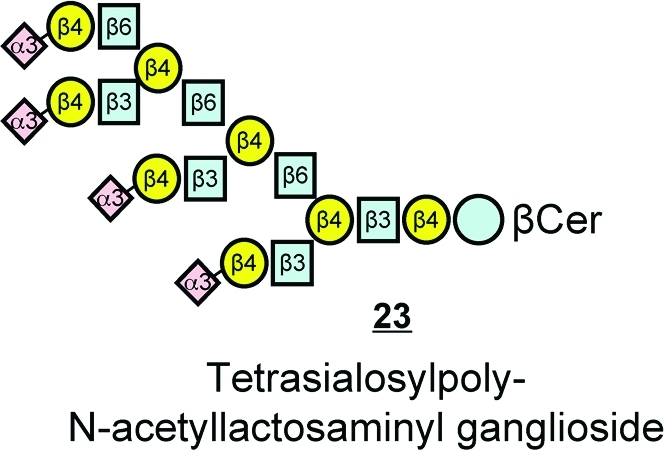
Therefore, one can imagine that there might be a relatively simple biosynthetic pathway for such compounds with a few enzymes that act repetitively on the growing chains. This illustrates how cells might make many complex glycosphingolipids using a relatively small number of glycosyltransferases, and conversely, how the existence of a finite number of glycosyltransferases determines that cells will produce only a fraction of the theoretical number of combinations and permutations of the glycans.
2.3.1. Phosphosphingolipids
The simplest complex phosphosphingolipid is ceramide 1-phosphate, which has not yet been studied much for molecular subspecies but methods for its analysis have been developed.39,138 The N-acyl-chain length of Cer1P is influenced by its site of synthesis, with the Cer1P that is made de novo being enriched in C16-subspecies because it acts on Cer that have been delivered by the ceramide transport protein (CERT),(139) which is selective for long-chain versus very-long-chain Cer. The biological functions of Cer1P are still being discovered, but include phagocytosis,(140) stimulation of DNA synthesis,(141) inhibition of apoptosis,(142) activation of mTOR and RhoA,(143) and activation of phospholipase A2(144) and production of eicosanoids(145) and lipid droplets.(146)
The most prevalent phosphosphingolipid in most mammalian tissues (and lipoproteins) is SM, and its chain length diversity has already been mentioned with respect to the ∼100 molecular subspecies in human plasma.(40) Besides its well-known membrane properties,147,148 it has been suggested that the N-acyl chain length affects endocytic trafficking of SM.(149) Bacteria produce a SM-binding protein (Lysenin) that is a pore-forming toxin(150) that has also been useful in studies of SM-mediated signal transduction.(151)
Mammals also produce small amounts of ceramide phosphoethanolamines,152−154 although these are found in more substantial amounts in other organisms, such as chickens (in liver)(152) and Drosophila melanogaster.(155) Fungi, plants, and other organisms have inositol phosphorylceramides and other types of glycophosphosphingolipids, often with novel lipid backbones,133,156,157 and they have been suggested to have functions in intracellular processes and cell-to-cell interactions, including between cells of different species in host–pathogen interactions.(158)
2.3.2. Glycosphingolipids
Mammalian glycosphingolipids begin with either glucose or galactose attached to the 1-hydroxyl of Cer via a β-glycosidic bond. In addition to being intermediates in the biosynthesis of more complex glycosphingolipids, these monohexosylceramides have also been suggested to have biochemical functions. GlcCer plays a critical role in skin (as a precursor that is hydrolyzed to skin ceramides to form the permeability barrier),(55) and is required for intracellular membrane transport,159,160 cell proliferation and survival,(161) multidrug resistance,162,163 and natural killer T cell functions.(164) In addition, the levels of GlcCer are altered by a wide spectrum of diseases, including cardiovascular disease, cancer, diabetes, and skin disorders.(161) Galactosylceramide (and its sulfated derivatives, termed sulfatides) are major components of myelin and have been reported to interact with each other by carbohydrate-carbohydrate interactions, perhaps on apposing surfaces of the multilayered myelin sheath.(165) Considerable attention has been given to α-GalCer, with an α-versus the β-glycosidic linkage, which was originally uncovered in studies using extracts from sponges and is now synthetically produced as KRN7000, because it is a potent activator of iNKT cells and promotes immunotolerance.(166) It is also of interest that the cytokine profile induced by GalCer has been found to be affected by the nature of the lipid backbone.167,168
GalCer are sulfated to produce the acidic glycosphingolipids referred to as “sulfatides” (GlcCer is also sulfated to 3′-sulfo-Glcβ1Cer, SM4s-Glc in some tissues).(4) 3-O-sulfogalactosylceramide (3′-sulfo-Galβ1Cer, also called cerebrosulfatide or GalCer-I3-sulfate) is a major component of the myelin sheath in the central and peripheral nervous system, kidney, gastrointestinal tract and endometrium.(169) Sulfatides are thought to be involved in neuronal cell differentiation, myelin formation and maintenance,(9) and it has been suggested that sulfatide interacts with GalCer in myelin through trans-carbohydrate–carbohydrate interactions.(165) Sulfatides additionally affect the behavior of macrophages,(170) participate in adhesion of leukocytes to selectins, and are thought to be involved in platelet aggregation via P-selectin (with inhibition of the P-selectin–sulfatide interaction leading to a reversal of platelet aggregation).(171) Other extracellular proteins that have been found to bind sulfatides include laminin and thrombospondin(172) and hepatocyte growth factor.(173) It should also be borne in mind, however, that some of the regulatory functions might be intracellular, because sulfatides bind to the N-terminal domain of sphingosine kinase 2.(174) Sulfatides are elevated in a wide range of cancers, including colorectal,(175) hepatocellular,(176) renal,(177) brain,(178) small-cell lung,(179) and ovarian(180) cancers, and are thought participate in metastasis.175,181
The major disaccharide (Galβ1–4Glcβ1-ceramide), lactosylceramide (LacCer), is a critical intermediate in the biosynthesis of all of the root structure families of more complex sphingolipids (Figure 1). LacCer has been proposed to function in cell signaling pathway(s) that affect cell proliferation, adhesion, migration, angiogenesis, phagocytosis and inflammation.182−185 In human neutrophils, the Src family kinase Lyn appears to be coupled with LacCer-enriched domains in the plasma membrane so that ligand (i.e., microorganism) binding to LacCer activates Lyn, triggering neutrophil functions, such as superoxide generation and cell migration.(186) Interestingly, the LacCer must have a very-long-chain fatty acid (C24:1 or C24:0) in the ceramide moiety, perhaps because that is necessary for proper membrane interdigitation and organization.186,187 It is also possible that LacCer participates in glycan–glycan interactions with other glycosphingolipids, such as GM3.(188)
One of the simplest glycosphingolipids (with three carbohydrates) is ganglioside GM3, Neu5Acα2–3Galβ1–4Glcβ1Cer. A function for GM3 in the regulation of cell proliferation was uncovered several decades ago by Hakomori and his colleages, who found that GM3 inhibits the stimulation of growth by epithelial growth factor (EGF) via inhibition of the activation of the EGF receptor tyrosine kinase.189,190 Subsequent studies revealed that the interaction at the surface appears to be via glycan–glycan binding involving multivalent GlcNAc termini on the EGF receptor,(191) and that the intracellular consequences are prevention of the autophosphorylation of the intracellular kinase domain and the allosteric structural transition to a signaling dimer.(192) This (and a similar finding that GM1 inhibits growth stimulation by platelet derived growth factor, PDGF)(193) defined the paradigm for ganglioside action illustrated in Figure 2, that is, that they not only help define the properties of the surface of the plasma membrane but also interact with surface proteins to modulate their function. Gangliosides are expressed on essentially all vertebrate cells, and typically with tissue-selective, and often developmentally related, profiles, that is, with varying types of headgroups and lipid backbones,(7) and in addition to modulating the way cells respond to a wide range of growth factors (EGF, PDGF, VEGF, and others), they interact with glycan-binding proteins on apposing cells via receptors called Siglecs that function in cell–cell recognition.7,194 Gangliosides have been found to regulate natural killer cell cytotoxicity via Siglec-7, myelin-axon interactions via Siglec-4 (also referred to as myelin-associated glycoprotein, MAG), and inflammation via E-selectin.(7) Some sialic acid-containing glycosphingolipids are very large, such as the tri- and tetra-sialosylpoly-N-acetyllactosaminyl gangliosides of human placenta that have >20 residues,(136) and might function to create a surface or barrier with particular biophysical properties.
The globo (Gb) and isoglobo (iGb) series trihexosylceramides are abbreviated Gb3 and iGb3, respectively. As shown in Figure 1, they differ only with respect to the terminal glycosidic linkage, which is α1–4 in Gb3 and α1–3 for iGb3. Gb3 has received much attention because it accumulates in Fabry’s disease due to defective α-galactosidase A(195) and because it is bound by (and receptor for) Shiga toxin,(196) verotoxins and the HIV adhesin gp120.(10) Interestingly, the lipid backbones Gb3 also have a substantial effect on the way these proteins behave in cells, and might be important to the eventual pathogenic outcome.(10) Gb3 is elevated in numerous cancers (colorectal adenoma, Burkitt’s lymphoma, breast cancer and testicular carcinoma),(197) and a correlation between Gb3 and metastasis has been seen for colorectal cancer.(198) The relationships are being explored as a way for cancer detection and targeting using Shiga toxin.199,200 The story for iGb3 is less clear because although it stimulates NKT cells and has been hypothesized to be a natural modulator of them, recent studies have found that the human iGb3 synthase gene contains several mutations that render its product nonfunctional (in constrast to rat, where iGb3 synthase is intact and iGb3 is found).(201) Therefore, iGb3 is unlikely to represent a primary natural ligand for NKT cells in humans and iGb3 itself would be expected to be recognized by the immune system as a foreign antigen, which might cause humans to reject transplanted tissues from animals that express this gene and iGb3, such as pigs.(201)
The first compound in the lacto-/neolacto- category, Lc3 (GlcNAcβ1–3Galβ1–4Glcβ1Cer), appears to be important for embryonic development and brain morphogenesis because knockout mice for Lc3 synthase gene display preimplantation lethality.(202) The animals that are successfully born have reduced survival and display pleiotropic phenotypic changes, including dwarfism, fur loss, and obesity.(203)
These examples illustrate how disruption of the production of one category of complex sphingolipids can impact survival and physiological functions. The reader is referred to the references already cited and others204−207 for more information about additional glycosphingolipid structures and functions. Online sources that are also useful include: (i) the Consortium for Functional Glycomics (http://www.functionalglycomics.org/); (ii) the Complex Carbohydrate Research Center at the University of Georgia (http://www.ccrc.uga.edu/∼moremen/glycomics/); (iii) GlycoForum (http://www.glycoforum.gr.jp/); (iv) the KEGG ontology for glycosyltransferases (http://www.genome.jp/kegg/glycan/GT.html); and (v) LIPID MAPS (www.lipidmaps.org).
2.4. Other Types of Compounds
The term lysosphingolipid usually refers to a complex sphingolipid without the N-acyl-fatty acid, such as sphingosylphosphocholine (sphingoid base 1-phosphocholines) from SM, sphingosine-1-β-glucoside or -galactoside (“psychosines”), and other lyso-glycosphingolipids. Not much is known about the origins and functions of these compounds, although they have been found in blood and tissues in varying amounts and tend to be highly bioactive.208,209 For example, sphingosylphosphocholine display behaviors that might implicate it as an important lipid mediator in tissues such as heart, blood vessels, skin, brain, and immune system.(210) It has also been strongly implicated as a player in atopic dermatitis(211) via a SM deacylase that also acts on GlcCer.(212) The accumulation of psychosines was one of the hypotheses for the unusual cellular and biochemical characteristics of globoid cell leukodystrophy (Krabbe disease), as has been discussed.(213)
Trace amounts of N- and O-methyl-sphingoid bases are sometimes found in mammalian sphingolipids and are thought mostly to be artifacts of the extraction and handling.214,215 Nonetheless, a sphingosine N-methyltransferase activity has been found in mouse brain,(216) and when mice have been treated with safingol, the metabolites included the N-methyl-, N,N-dimethyl- and N,N,N-trimethyl-derivatives (and methylated sphingosine and sphinganine were detected);(217) therefore, there is an in vivo capacity to methylate sphingoid bases.
Sphingolipids have also been found as covalent adducts in the cornified cell envelope of the skin,218−220 and yeast have been found to make glycosylphosphatidylinositol-anchored proteins with ceramide as the lipid moiety.(221)
3. Sphingolipid Metabolic Pathways
The major focus of this discussion of the sphingolipid metabolic pathways will be to explain how the different subspecies are produced and, in some cases, how defects in these metabolic steps result in disease, rather than how the pathways are regulated, which would be a more monumental task. This begins with how the sphingoid bases arise since, by definition, all sphingolipids are comprised of that backbone. Most organisms derive a significant portion of their sphingoid bases from de novo biosynthesis because the first enzyme of the pathway (serine palmitoyltranserase) is essential for survival of cells in culture, from yeast(222) to mammals,(223) unless exogenous sphingoid bases are provided, and elimination of this enzyme is embryonic lethal for animals large (i.e., mammals)(224) and small (e.g., fruit flies).(225) This requirement appears to be due to the efficient degradation of sphingoid bases taken up by the intestine (via phosphorylation at the 1-hydroxyl then cleavage to a fatty aldehyde and ethanolamine phosphate),226−229 which might exist to allow mammals to be selective in which species are in their repertoire, since a much wider variety of sphingoid base structural variants are found in other organisms (and, thus, in food).11,230 The fate of dietary sphingolipids warrants further investigation, nonetheless, when one considers that humans have been estimated to consume more than one hundred grams of sphingolipids per year.(230) Furthermore, dietary sphingolipids have been well established to be protective against cancers of the intestine231−236 and other sites(237) in studies of experimental animals, and recent studies of sphingoid base analogs reveal that some structural variants are well absorbed, as exemplified by findings with Enigmol 18,(92) a synthetic 1-deoxy- analog similar to compounds found in some foods.(11)
3.1. Biosynthesis of the Lipid Moieties de Novo
Approximately one decade after elucidation of the definitive structure of sphingosine by Herb Carter and colleagues in 1947,(238) its biosynthesis in vitro was achieved by Brady and co-workers.239,240 Another decade later, Braun and Snell(241) and Stoffel et al.(242) demonstrated that the initial biochemical reaction is the formation of 3-ketosphinganine by condensation of serine and palmitoyl-CoA followed by rapid reduction of the intermediate ketone to produce sphinganine, if NADPH is also present; thereby establishing the first steps of sphingoid base biosynthesis de novo (Figure 4). In the early 1990s, the genes for the enzyme that catalyzes the initial reaction, serine palmitoyltransferase (SPT), were identified in yeast (LCB1 and LCB2)243,244 and soon afterward for mammals (SPTLC1, SPTLC2, and SPTLC3),245−247 followed in relatively rapid succession by discovery of genes for most of the other enzymes of ceramide biosynthesis (as discussed below). Thus, the major steps for biosynthesis of the lipid moieties of sphingolipids are now fairly well mapped out biochemically and genetically, although additional features will undoubtedly surface over time, as for other pathways.
Figure 4.

De novo sphingolipid biosynthesis through lactosylceramide and sulfatide. Starting at top left, serine and palmitoyl-CoA are condensed by serine palmitoyltransferase (SPT) to form 3-ketosphinganine that is reduced to sphinganine, which is N-acylated by ceramide synthases (CerS) with the shown fatty acyl-CoA preferences, or phosphorylated by sphingosine kinase (SphK). The N-acylsphinganines (dihydroceramides, DHCer) can be incorporated into more complex dihydro-sphingomyelins, SM, from sphingomyelin synthases, SMS; -ceramide 1-phosphates, CerP, from ceramide kinase, CERK; -glucosylceramides, GlcCer, from GlcCer synthase; and -galactosylceramides, GalCer, from GalCer synthase). DHCer is also oxidized to Cer by dihydroceramide desaturase (DES1 and DES2; DES2 is also capable of hydroxylating the 4-position to form 4-hydroxydihydroceramides, t18:0) and incorporated into more complex sphingolipids as shown. The diagram also displays the formation of lactosylceramide (LacCer) from GlcCer and sulfatides (ST) from GalCer, and the turnover of DHCer to sphinganine (and Cer to sphingosine), which can be recycled or phosphorylated and cleaved to fatty aldehydes and ethanolamine phosphate. Not shown is ceramide phosphoethanolamine, which is present in mammalian cells in nearly trace amounts. The key is shown at the bottom, and is the same as for Figure 1 except that heavy black boxes represent SM, thin black for Cer1P, and (DH)Cer are represented by the green octagon.
3.1.1. Formation of the Sphingoid Base Backbones
SPT is a pyridoxal 5′-phosphate (PLP)-dependent enzyme that catalyzes the condensation of serine and palmitoyl-CoA (and other amino acid and fatty acyl-CoA cosubstrates, as will be discussed later). It is a member of the PLP-dependent α-oxoamine synthase (POAS) subfamily and, like most POAS members, shares a conserved motif (T[FL][GTS]K[SAG][FLV]G on SPT2) that contains an active site Lys that is responsible for formation of a Schiff’s base with PLP.(248) For most organisms,(249) SPT is comprised of at least two separate polypeptides (and perhaps higher aggregates)(250) that are located in the membrane of the endoplasmic reticulum. There is also evidence for SPT being present in other regions of the cell, such as focal adhesions(251) and the nucleus (and, interestingly, appearing to shift to the nucleus in proliferating cells).(252) In the endoplasmic reticulum, the active site appears to be oriented toward the cytoplasm,253,254 as for the other enzymes of ceramide biosynthesis.(253) It is likely that SPT interacts with other regulatory proteins. Yeast SPT requires an additional 10-kDa peptide for optimum activity,(255) and although a mammalian homologue of Tsc3 was not found,(256) several categories of proteins have been suggested to play a regulatory role for mammalian SPT, including two small SPT subunits, ssSPTa, and ssSPTb, that appear to influence the fatty acyl-CoA selectivity,(257) ER proteins that might enhance Ser utilization (termed Serinc1 to 5),(258) and ORM1.259,260 Using tandem-affinity purification and mass spectrometry) to discover protein–protein interactions, a substantial number of proteins have been identified as potential LCB2-associated proteins in Saccharomyces cerevisiae.(261) These proteins are involved in various biological processes such as vesicle transport, nuclear import and export, among others. A genome-wide yeast two-hybrid analysis in Drosophila(262) has suggested that SPT2 may interact with 13 proteins, which include a proton transporter, organic cation transporter, hsc-70, and ribosomal proteins, among others.
Elegant structural and spectroscopic studies have been conducted with SPT from the Gram-negative bacterium Sphingomonas paucimobilis,263−265 which is a soluble homodimer with ∼30% amino acid sequence identity with mammalian SPT1 and SPT2,(266) and Sphingobacterium multivorum.(267,268) These have supported the general mechanism shown in Figure 5. As for many PLP-dependent enzymes,(269) the amino acid substrate is covalently bound to PLP as a Schiff base 24 (which is often referred to as the “external aldimine” versus the “internal aldimine” that is produced by the enzyme-Lys-PLP Schiff base). Spectroscopic evidence has indicated that there is a structural rearrangement of this chromophoric species upon binding of the second substrate, a fatty acyl-CoA. The proposed steps for condensation of the substrates (Figure 5) are similar to what was deduced decades ago by isotope kinetics studies(270) and generally occurs with PAOS family enzymes: deprotonation at Cα of the external aldimine complex 25 to form a quinonoid intermediate 26 and a Claisen condensation with the acyl-CoA substrate and loss of free CoASH; this β-ketoacid intermediate 27 is doubly β,γ-unsaturated and undergoes decarboxylation to form another quinonoid intermediate 28 that rearranges to acquire a proton to form the product external aldimine 29 that is released from the enzyme as 3-ketosphinganine 30.
Figure 5.

Proposed reaction mechanism for serine palmitoyltransferase (modified from D. J. Campopiano and colleagues,263−265 see text). Starting with the enzyme with pyridoxal 5′-phosphate (PLP) bound as a Schiff’s base with an active site Lys (upper left), Ser is bound to make the external aldimine 24 then palmitoyl-CoA is bound and the reaction proceeds as shown until 3-ketosphinganine 30 is released.
It was once presumed that SPT is specific for l-serine, however, recent studies of the effects of fumonisin B1 on animals and cells in culture(67) and of the disease human hereditary sensory neuropathy type 1 (HSN1), which is caused by mutations in SPT,271−273 have found that wild type, and especially mutant,68,274−276 SPT is also able to utilize l-alanine and glycine to produce cytotoxic 1-deoxysphinganines and 1-(deoxymethyl)sphinganines, as shown in Figure 6. Like sphinganine, these “atypical” sphingoid bases are rapidly N-acylated,67,277 which might explain why their production even by wild type SPT had escaped previous notice. Studies of one of the disease-causing mutations (C133W in SPTLC1)(276) indicate that the wild-type and mutant enzymes are not altered in serine utilization and have similar apparent binding affinities for alanine, but the C133W mutation appears to enhance the condensation of alanine with the acyl-CoA substrate. It is very intriguing that SPT (even wild-type SPT)(67) is able to utilize this ensemble of metabolically interrelated substrates, Ser and Gly are interconverted via serine hydroxymethyltransferase, and Ser is catabolized to pyruvate (a precursor for Ala) via serine dehydratase,(278) at a crossroad of major metabolic pathways, which include glycolysis, amino acid metabolism, lipid metabolism and one-carbon metabolism (with implications for nucleotide biosynthesis) (Figure 6). Thus, many factors might affect their amounts and, indeed, elevated production of 1-deoxysphingolipids has recently been proposed to play a role in diabetes.(279) In another context, these compounds appear to have beneficial functions as an anti-cancer compound(280) that has been evaluated not only with cancer cells in culture(281) but also by phase I clinical trials.94,282 Surprisingly high dosages were tolerated in the trials, although the reported side effects included a case of peripheral motor and sensory neuropathy.(94)
Figure 6.

Comparison of the structures of the “typical” and “atypical” sphingoid bases and the interrelationships between intermediary metabolism and the precursor substrates for them. The interconversion of Ser and Gly are catalyzed by serine hydroxymethyltransferase, and Ser is converted to pyruvate by serine dehydratase. Ser, Ala, and Gly are related to other metabolic pathways as illustrated, and produce the shown sphingoid bases when utilized by serine palmitoyltransferase.
SPT also binds d-serine as a competitive inhibitor with an IC50 of ∼0.3 mM (which is similar to the Km for l-serine),256,283 but does not appear to be utilized as a substrate. d-serine is found in plasma284,285 and urine,(286) and has been shown to be nephrotoxic,(287) so inhibition of SPT by d-serine might occur under some in vivo circumstances.
As implied by its name, SPT is usually most active with palmitoyl-CoA (C16:0-) as the cosubstrate, but it can accommodate fatty acyl-CoAs that are longer and shorter by one carbon fairly well,248,256 but these are usually much less prevalent than palmitoyl-CoA in mammalian cells.(288) These factors probably account for the high proportions of 18-carbon-chain-length sphingoid bases in most mammalian sphingolipids. It appears that another SPT isoform (SPTLC3) has a preference for myristoyl-CoA (C14)(289) and the amounts of C16-sphingoid bases are more substantial when this SPT isoform is expressed.(290) Such shorter chain length sphingoid bases are common in insects such as Drosophila, which contain C14- and 16-sphingoid bases and differ in regions of SPT that might account for this difference.291,292 C20-sphingoid bases are found in human gangliosides,(293) and it is not clear why or how they are elevated although the production of C20-sphingoid bases might be determined by expression of ssSPTb.(257) Interestingly, in yeast, C20-sphingoid bases are elevated under certain stages of growth and stress, and are thought to have roles in cell signaling.(99) Enhanced de novo biosynthesis of sphingolipids when cells are treated with palmitate(294) might link this pathway and the lipotoxicity of this fatty acid for many cell types,(295) and perhaps through elevated sphingosine 1-phosphate.(296)
SPT is potently and selectively inhibited by several naturally occurring compounds,81,297−299 such as myriocin (ISP-1) 14 (which has obvious structural similarity to active site intermediates) (Figure 4), sphingofungins, lipoxamycin (neoenactin M1), and sulfamisterin, as well as by viridiofungins, which are also potent but additionally inhibit squalene synthase.(300) As would be predicted for an enzyme that utilizes PLP as a cofactor, SPT is inhibited by compounds such as β-chloro-l-alanine(301) and cycloserine,(265) and O-tert-butyl-l-serine methyl ester hydrochloride has also been reported to be inhibitory.(302) These inhibitors have been quite useful in studies of the roles of de novo synthesized sphingolipids in normal and abnormal cell functions, as has a mammalian cell line (CHO-LY-B cells)(303) that cannot make sphingoid bases due to loss of catalytic activity due to a G246R transformation in SPT1,(304) and knockout mice.(305)
After establishment of the chain length and subcategory of sphingoid base (i.e., traditional sphingoid base type versus 1-deoxy- or 1-(desoxymethyl)-sphingoid base), further modifications, such as introduction of the 4,5-trans double bond of sphingosine and the 4-hydroxylgroup of 4-hydroxysphinganine (phytosphingosine) generally take place after the 3-keto-sphingoid base has been reduced by an NADPH-dependent reductase(306) and N-acylated, as described in the following sections.
3.1.2. Ceramide Synthases
As shown in Figure 4, sphinganine is at the next key branchpoint in the pathway, where it is either acylated to different dihydroceramides by a family of Cer synthases (CerS)307,308 or phosphorylated to sphinganine 1-phosphate by sphingosine kinase(s).309,310
The first genes coding for Cer synthases (CerS), Lag1p and Lac1p, were found in Saccharomyces cerevisiae,(311,312) followed by identification of a lower molecular weight protein that is also required for activity.(313) Soon thereafter, mammalian homologues of Lag1p were characterized and the first cloned CerS (originally called lass1, and now referred to as CerS1) was found to be highly selective for stearoyl-CoA and to make C18-(DH)Cer.(314) This was followed by characterization of five additional CerS with distinct substrate selectivities (summarized in Figure 4) and other features, such as relative mRNA expression level and tissue distribution, that were consistent with the types of ceramides found in the respective source.307,308,314−319
CerS1 has been found to have an additional mode of regulation in that it is turned over rapidly under basal conditions, and even more rapidly under stress from agents such as UV light and chemotherapeutic drugs.(320) Turnover of CerS1 proceeds via ubiquitination and proteasomal processing, with translocation from the endoplasmic reticulum to the Golgi apparatus.(321) The subcellular localization of CerS1 might explain why administration of exogenous sphingosine to cells in culture disproportionately elevates C18-Cer.(309) Ogretmen and co-workers(322) have discovered that head and neck tumors have lower CerS1 and lower proportions of C18-Cer than neighboring normal tissue (consistent with the substrate specificity of CerS1 for C18-fatty acyl-CoA, as shown in Figure 4). In addition, decreased C18-Cer levels were significantly associated with the higher incidences of lymphovascular invasion, pathologic nodal metastasis, and the overall stage of the primary tumors.(323) These correlations were shown to have functional consequences by transfection of the CerS1 gene into head and neck tumor cells in culture, which restored the levels of C18-Cer and suppressed cell growth.(322) Therefore, CerS1 and C18-Cer appear to play important roles in the pathogenesis or progression of head and neck cancer. C18-Cer has been reported to result in repression of the hTERT promoter via deacetylation of Sp3 by histone deacetylase 1 (HDAC1) in A549 human lung adenocarcinoma cells.(324) Up-regulation of CerS1 has also been suggested to participate in the induction of apoptosis in chronic myeloid leukemia cells by dasatinib.(325) Studies of two mouse strains, flincher and toppler, with spontaneous recessive mutations that cause cerebellar ataxia and Purkinje cell degeneration have found that the mutations reside in the CerS1 gene, resulting in complete loss of CerS1 catalytic activity.(326) In addition to Purkinje cell death, there was also accumulation of lipofuscin, which is common with aging and in some neurodegenerative diseases, thus, might implicate CerS1/C18-Cer in these processes.(324)
CerS2 mRNA is found at the highest level of all CerS and has the broadest tissue distribution. It prefers the longer chain fatty acyl-CoAs, as shown in Figure 4, and there is a good correlation between CerS2 mRNA levels and the prevalence of those acyl chains in ceramide and sphingomyelin. Interestingly, CerS2 has an S1P receptor-like motif that raises the possibility that the activity of CerS2 might be regulated by S1P.(327) CerS2 is the only CerS for which there is currently a knockout mouse.328−330 The mice were essentially devoid of very-long-chain (C22 and C24)-Cer and downstream sphingolipids, which is also consistent with the substrate specificity of CerS2 toward these chain length fatty acyl-CoAs (Figure 4). Apparently as compensation for the lower very-long-chain sphingolipids, C16-Cer-sphingolipids were elevated, and differences were observed in the biophysical properties of lipid extracts isolated from liver microsomes, with membranes from CerS2 null mice displaying higher membrane fluidity and differences in morphology. As part of the “sphingolipidomic” analysis of these mice by our lab,329,330 we discovered that sphinganine was elevated, by up to 50-fold, which was reminiscent to inhibition of ceramide synthase by fumonisins.(80) This was striking because, as occurs when mice are exposed to fumonisins, the livers of the CerS2-knockout mice developed severe hepatopathy from about 30 days of age, and displayed increased rates of hepatocyte apoptosis and proliferation progressing to hepatomegaly and noninvasive hepatocellular carcinoma later in life.(330) These data suggest that CerS2 is important for the synthesis of dihydroceramide and prevention of the accumulation of sphinganine. It also appears to be particularly important for synthesis of myelin sphingolipids(331) because the mice displayed encephalopathy, which may be largely because of reduced galactosylceramide levels.(332) CerS2 mRNA expression has been noted to be significantly elevated in breast cancer tissue compared to paired normal tissue.(333)
CerS3 is particularly important in epidermal keratinocytes and male germ cells, which produce large amounts of sphingolipids with very-long-chain- (C26–C36) Cer.(334) Its expression in keratinocytes increases upon differentiation, and it can produce 2-hydroxy-Cer, which are common in the epidermis.(335) Studies of mouse embryonic stem cells and embryoid bodies have found that the latter have higher CerS3 mRNA and higher proportions of C18-, C24- and C26-, and less C16-dihydroceramides.(336) Treatment of a mantle cell lymphoma cell line (Rec-1) with the endocannabinoid analogue R(+)-methanandamide has been reported to increase C16-, C18-, C24-, and C24:1-Cer and found transcriptional induction of CerS3.(337) All of these are consistent with the fatty acyl-CoA selectivity for CerS3 shown in Figure 4.
CerS4 has been studied less than the other CerS, perhaps in part because the Cer subspecies that it makes (C20 ± 2 carbons)(315) are not prevalent in most sphingolipids. It is expressed at highest levels in skin, leukocytes, heart, and liver.(327) Studies with a pancreatic beta-cell model, INS-1 beta-cells, found that supplementation of the medium with high glucose and palmitate increased CerS4.(338)
CerS5 and CerS6 are often considered in concert since both make C16-Cer, with CerS6 also utilizing myristoyl-CoA to make C14-Cer, as shown in Figure 4. CerS5 was the first mammalian CerS that was purified and proven to be a genuine synthase for ceramide.(317) Co-immunoprecipitation studies suggest that CerS2, 5, and 6 might exist as heterocomplexes in HeLa cells.(339) A number of factors induce CerS5 and CerS6, such as development,(340) ionizing radiation,(339) the cyclooxygenase-2 (COX-2) inhibitor celecoxib,(341) and the death receptor ligand TRAIL (tumor necrosis factor-related apoptosis-inducing ligand).(342)
Despite having differences in fatty acyl-CoA-specificity, the CerS have similar apparent Km toward the sphingoid base substrate sphinganine (ranging from 2 to 5 μM).(343) This implies that as sphinganine is made de novo, its partitioning into different categories of (dihydro)Cer will be governed by the relative levels of the CerS in its vicinity. This has fairly consistently been supported by the studies described above, where particular CerS were varied in amount in relationship to the other isozymes, and by a study by Obeid and co-workers(344) where individual CerS were suppressed in MCF-7 cells using small-interfering RNA (siRNA).(344) As was seen in the CerS2 knockdown mouse,329,330 elimination of one CerS often resulted in counter-regulation of one or more of the other CerS and corresponding shifts in the chain lengths of the cellular ceramides such that overall levels of complex sphingolipids were generally maintained despite reduction of a particular CerS (however, free sphinganine was not elevated in the siRNA studies).(344) It is not clear if the components of this pathway are present in the ER as discrete polypeptides that release their products into the ER membrane to diffuse to the next enzyme, or if there are macromolecular complexes that position the active sites so the product of one enzyme is released near the active site for the next enzyme. There is precedent for this latter scenario in recent findings with ELOVL1, a fatty acyl-CoA elongase that is essential for production of very long-chain fatty acids that are used by CerS2.(345) This might also account for the elevation of sphinganine in the CerS2 knockout mouse.329,330
There are a large number of naturally occurring inhibitors of CerS,(11) with the best characterized (because of their public health relevance) being the fumonisins, a family of mycotoxins produced by Fusarium verticillioides(346) that cause a wide range of diseases of agriculture animals (equine leukoencephalomalacia and porcine pulmonary edema) and humans (cancer and birth defects).79,347 The structure of fumonisin B1 13 and the characteristics of the inhibition suggest that the aminopentol backbone competes for binding of the sphingoid base substrate, whereas the anionic tricarballylic acids may interfere with binding of the fatty acyl-CoA.(348) Inhibition of what appears to be all CerS (based on complete blockage of de novo sphingolipid biosynthesis) is accompanied by dramatic elevations in sphinganine and sphinganine 1-phosphate at early times, later elevation of sphingosine and S1P (from blockage of reutilization of the backbones of sphingolipids that are turning over), and depletion of complex sphingolipids --all of which are likely to contribute to fumonisin toxicity, carcinogenicity348,349 and teratogenicity.79,350 There is also an intriguing interplay between TNFα and fumonisins351−353 which might be related to the ability of cytokines to affect sphingolipid biosynthesis and turnover.354−356 Somewhat paradoxically, but of possible clinical importance, treatment with fumonisin B1 has been found to significantly reduce the systemic toxicity, weight loss, and mortality of zymosan-induced nonseptic shock in mice.(357)
Cer can also be made by reversal of acid ceramidase with a strict stereochemical requirement for d-erythro-sphingosine,358,359d-erythro-sphinganine, and d-erythro-phytosphingosine but can occur with a wide spectrum of fatty acids, including both saturated and unsaturated fatty acids(358) and chain lengths varying from C8 to C22.(359) Detergents, pH, and various lipids, such as cardiolipin, phosphatidylcholine, and lysophosphatidylcholine can affect the hydrolysis reverse activity of ceramidases.(359) This appears to contribute little to Cer synthesis in vivo (as discussed above), however, recent findings with neutral ceramidase-deficient mice indicate that it might play a role in ceramide formation in mitochondria.(360)
N-acetyl-sphingosine (C2-Cer) and -sphinganine (C2-DHCer) have been reported to be made by a platelet-activating factor (PAF)-dependent transacetylase(361) that is widely distributed among tissues and appears to be more active with sphingosine than sphinganine.(106) This transacetylase is a multifunctional enzyme with three catalytic activities (lysophospholipid transacetylase, sphingosine transacetylase, and acetylhydrolase) and its regulation differs for macrophages compared to monocytes.(362) C2-DHCer has also been found in cells and animals treated with fumonisin B1 (as well as the untreated controls),(348) but it is not clear if this is produced by the PAF transacetylase or a more generic acetyltransferase used in detoxification of xenobiotics.
3.1.3. Desaturation and Hydroxylation of Dihydroceramide to Form Ceramides and 4-Hydroxyceramides (Phytoceramides)
Ong and Brady first suggested that incorporation of the 4,5-trans-double bond of sphingosine occurrs at the DHCer level(363) as shown in Figure 4, but this was ignored for many years by textbooks (and even today by metabolic pathway wall charts) that showed direct conversion of sphinganine to sphingosine. Desaturation at the DHCer level in vivo was established conclusively by pulse chase labeling studies,(294) and confirmed by development of an in vitro assay for this highly labile enzyme.(364) DHCer desaturases were then cloned from plants,365,366 leading to the subsequent identification of the desaturase genes from many organisms, including humans.367−370 The two mammalian desaturases, DES1 and DES2, appear to have different functions, for DES1 to add the 4,5-trans double bond to make Cer,(367) and DES2 to hydroxylate DHCer at position 4 to produce the t18:0 backbone of phytoceramides.368−370
DES activity is influenced by the alkyl chain length of the sphingoid base and fatty acid, the stereochemistry of the sphingoid base (d-erythro versus l-threo-dihydroceramides), the nature of headgroup, and the ability to utilize alternative reductants.(364) Introduction of the 4,5-double bond can be analyzed using NBD-DHCer, which reveals interesting features about the stereoselectivity of the reaction and subsequent metabolism.(371) DES1 is a myristoylated protein and its activity appears to be affected by this post-translational modification.(372)
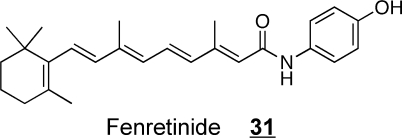
DES plays a very important role in cell regulation because the signaling targets of Cer typically are not affected by comparable levels of DHCer, which is a sensible mechanism to minimize accidental induction of apoptosis by this intermediate of de novo sphingolipid biosynthesis.(108) DHCer are bioactive, nonetheless, as inducers of autophagy, which surfaced in studies of the mechanisms of action of the anticancer drug fenretinide (4-hydroxyphenyl retinamide, 4HPR) 31.(97) Fenretinide had been thought previously to elevate Cer in studies of how this compound was toxic for numerous human cancer cell lines,373,374 both as an inducer of SPT and ceramide synthase.(375) However, when examined by mass spectrometry, the accumulating “Cer” was found to be DHCer, and fenretinide was deduced to inhibit DES,(97) which has been subsequently confirmed.(376) The sphingolipidomic studies that uncovered this novel mechanism of action of fenretinide also revealed that this agent elevated sphingoid bases and sphingoid base 1-phosphates,(97) which have the potential to mediate, or suppress, cancer cell killing, respectively; therefore, follow-up studies examined whether coadministration of a sphingosine kinase inhibitor would enhance the toxicity of fenretinide, and this was found to be the case.(377) Likewise, knockdown of ceramidase has the potential to decrease the production of free sphingoid bases and ameliorate the toxicity of fenretinide, and this too has been found.(378) A large number of inhibitors specifically targeted to DES have also been prepared and characterized.379−383
A number of physiological factors have also been found to modulate DES. For example, palmitate (but not oleate) increased mRNA encoding DES1 and Cer biosynthesis,(384) and oxidative stress decreased dihydroceramide desaturase activity in a time- and dose-dependent fashion (and elevated DHCer).(385) A recent comparison of breast cancer cell lines noted that they differed in the relative expression levels of DES1 versus DES2, and follow-up analysis of the sphingolipids of the cells found the correlating differences in Cer versus PhytoCer in the sphingolipids.(290)
The enzymes and genes have not yet been identified for the production of the mammalian sphingoid bases with a second double bond at carbon 14, or for the skin sphingoid base with a hydroxyl at position 6.
3.2. Complex Sphingolipid Biosynthesis
In mammals, Cer is at the branchpoint for biosynthesis of four major compounds (Figures 4 and 7): the two phosphosphingolipids, sphingomyelin (SM) and Cer 1-phosphate (Cer-P) and two glycosphingolipids, galactosylceramide (GalCer) and glucosylceramide (GlcCer), which are converted into hundreds of complex glycosphingolipids as discussed above and summarized in an excellent review by Furukawa and colleagues,(386) a comprehensive series of pathway maps prepared by Akemi Suzuki,(387) a web-based hypothetical pathway scheme (www.sphingomap.com), and this review. Pathway maps based on the known genes for these pathways have also been developed for use with gene expression data sets.290,388 In addition, two more headgroups have been found to be produced by mammals, ceramide phosphoethanolamine(153) and 1-O-acylceramide,(389) but in such small amounts that they have not been included in Figure 4 or 7.
Figure 7.

A scheme depicting the major headgroup additions to (dihydro)ceramides and subsequent metabolites that define the different categories (including root structure series) of more complex sphingolipids. Ceramides and dihydroceramides (one of which is depicted in the octagon at one o’clock in this diagram) are converted into sphingomyelin (SM), ceramide 1-phosphate (CerP), glucosylceramide (GlcCer), and galactosylceramide (GalCer), then to downstream metabolites as shown (see text). Ceramide phosphoethanolamine and 1-O-acyl-ceramides are not shown because they appear in mammalian cells in trace amounts. Each enclosed section represents a subcategory of glycosphingolipid, such as ST for sulfatides (red circles, as in Figure 4) (note that some of the sulfated glycosphingolipids fall into both the GalCer, that is, Gala, subcategory and others are derivatives of GlcCer). The arrow to the isoglobo family is less bold because that enzyme is not thought to be active in humans. The key for the symbols and coloring scheme is the same as in Figure 1 the earlier figures.
3.2.1. Sphingomyelin, Ceramide Phosphoethanolamine, and Ceramide Phosphate
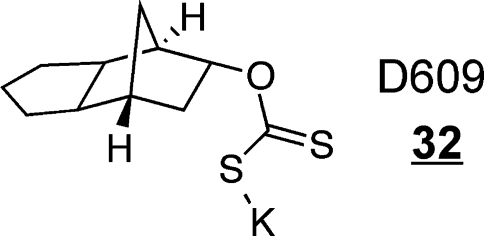
Cer is metabolized to SM in the Golgi390,391 and plasma membrane392,393 by SM synthases that catalyze the transfer of phosphorylcholine from phosphatidylcholine to the 1-hydroxyl of Cer with the liberation of diacylglycerol,394,395 with SMS1 localized to the Golgi, and SMS2 localized to the plasma membrane.(396) Because SM biosynthesis occurs at multiple sites and by more than one enzyme, as well as involves trafficking of the precursor Cer by more than one mechanism, it can be a difficult process to study.(397) This is probably also a manifestation of the multiple roles that these metabolites (SM, Cer and diacylglycerols) play in plasma membrane signaling.(27) A substantial number of studies have explored the biosynthesis and turnover of SM in cell signaling and disease, as reviewed recently by Hannun and colleagues.27,28,31 A useful tool in studies of SM synthesis has been the inhibitor D609 32.(398)
SMS2 knockout and SMS2 liver-specific transgenic mice have been prepared(399) and both had lower plasma SM than wild-type mice under usual dietary conditions, but differed when fed with high fat diets.(399) The SMS2 knockout mouse has also shown attenuated lung injury in response to lipopolysaccharide(400) and reduced atherogenesis,(401) among other interesting phenotypes.
Ceramide phosphoethanolamine biosynthesis involves the analogous transfer of the phosphoethanolamine group from phosphatidylethanolamine to Cer, which was first noted with microsomes and plasma membranes from rat brain and liver(393) (also with subsequent methylation using S-adenosylmethionine to produce SM).(402) The enzymes responsible for ceramide phosphoethanolamine biosynthesis have been reported to be a specific transferase, SMSr, that has only ceramide phosphoethanolamine synthase activity, and SMS2, which appears to be bifunctional enzyme that synthesizes both SM and ceramide phosphoethanolamine.(153) SMSr catalyzes the synthesis of ceramide phosphoethanolamine in the lumen of the endoplasmic reticulum, but in only trace amounts, and has been speculated to play a role in Cer homeostasis because blocking its catalytic activity causes a substantial rise in Cer.(154)
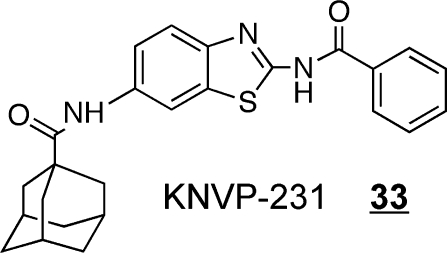
The other phosphosphingolipid made by mammals is ceramide 1-phosphate, which is produced by ceramide kinase (CERK) and possibly other yet-to-be-discovered enzymes because CERK knockout does not completely eliminate these compounds.(403) CERK is selective for Cer with a minimum fatty acyl chain length of 12 carbons, and the 4,5-trans double bond of the sphingoid base backbone is important for substrate recognition.(404) The production of ceramide 1-phosphate has been implicated in cell proliferation and survival,(405) and activation of the cytosolic phospholipase A2 (cPLA2) for inflammatory signaling.(406) KNVP-231 33 is a specific and reversible CerK inhibitor that is active in the low nanomolar range and useful in studies of this metabolic step.(407)
One of the factors that governs the biosynthesis of both SM and Cer1P is the delivery of Cer to the enzyme by a Cer transport protein (CERT) discovered by Hanada and co-workers.408,409 CERT mediates the ER-to-Golgi trafficking of ceramide,(410) and appears to act at membrane contact sites between the ER and the Golgi apparatus.(411)
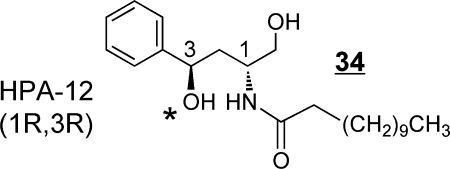
CERT most efficiently transfers Cer having C14- to C20- chain lengths (but not longer alkyl chains) as well as C16-dihydro- and phyto-Cer.(412) N-(3-Hydroxy-1-hydroxymethyl-3-phenylpropyl)dodecanamide (HPA-12 34) (see comment on stereochemistry)(413) inhibits ceramide trafficking by CERT.(414)
3.2.2. Other Non-Glycan Headgroups
The other known category of nonglycan headgroup modification by mammals is O-acylation,(389) which has been shown to be due to a group XV calcium-dependent, lysosomal phospholipase A that has the unique ability to transacylate short chain ceramides. It is highly expressed in alveolar macrophages, and mice lacking this enzyme develop a phenotype similar to human autoimmune disease.(415)
3.2.3. Glycosphingolipids
The core concepts for how cells biosynthesize hundreds of different headgroup categories of glycosphingolipids are summarized in Figures 7 and 8. Basically, the stage is set by there being a limited number of initial glycosyltransferases (mammals only add glucose and galactose directly to Cer even though several other types of carbohydrates are utilized later), followed by one major product from GlcCer (addition of Gal to form LacCer), then generation of further diversity by expansion to the root structure categories summarized in Figure 1. The Gala series (i.e., from GalCer) is simpler, although it contains somewhat more components than are illustrated in Figure 7 (such as the sulfated glucuronoglycolipids, which will be described later).
Figure 8.

Representative reactions of ganglioside biosynthesis. An illustration of the “combinatorial” nature of ganglioside biosynthesis by the indicated glycosyltransferases (note alternatives names for each enzyme). The key is the same as in Figure 1.
Glycosphingolipid biosynthesis (and especially ganglioside biosynthesis) has been referred to as a “combinatorial” process(14) because it produces many products from relatively few reactions (catalyzed by the glycosyltransferases) that are able to utilize a toolkit of precursors and intermediates to produce an ensemble of products. To a certain degree, the nature of the products are predictable based on the specificities of the enzymes, their locations, and the localization, amounts and types of the cosubstrates; however, since most of the components are membrane associated, all possibilities are not necessarily produced in detectable amounts.
The glycosyltransferases often transfer a specific carbohydrate from the appropriate sugar nucleotide (e.g., UDP-Glc, UDP-Gal, CMP-sialic acid) to a specific position on a particular type of acceptor (Cer or to the nonreducing end of the growing carbohydrate chain attached to Cer). For most of the enzymes presented in this review, sphingolipids are the preferred acceptors.(386) In large part, the structure feature of the acceptor that is recognized is the carbohydrate portion, however, there are instances where the backbone has been noted to be a factor, such as in the partitioning of α-hydroxy-Cer into downstream glycosphingolipids.(416)
3.2.3.1. Biosynthesis of GlcCer
GlcCer are synthesized by UDP-Glc:Cer glucosyltransferase (alternatively called GlcCer synthase and abbreviated GCS, UGCG and CGlcT-1),(417) and the only mechanism to produce GlcCer appears to be via this gene product based on studies with a mouse melanoma cell line (GM-95 cells) with mutated GCS.(418) Studies with this cell line have been very informative about the effects of eliminating GlcCer and downstream glycosphingolipids, which slowed their growth rate and altered cell morphology,(418) although the cells retained the ability to adhere to extracellular matrix (ECM) proteins such as fibronectin, collagen, and laminin.(419) Elimination of this enzyme in mice with a null mutation(420) was embryonic lethal, but embryogenesis proceeded well into gastrulation with differentiation into primitive germ layers and patterning of the embryo before death.
GlcCer biosynthesis can be blocked by inhibitors of this enzyme,(421) and when applied to mouse knockout model of Fabry disease (where a deficiency of the enzyme α-galactosidase causes Gb3 to accumulate), inhibitor treatment blocked accumulation of Gb3 in the kidney, liver, and heart without significant changes in body weight or organ weight, which was suggestive that such compounds might be promising as therapeutic agents for the treatment of glycosphingolipid storage disorders. The most recent generation of inhibitors of GlcCer synthase is (1R,2R)-nonanoic acid[2-(2′,3′-dihydro-benzo [1,4] dioxin-6′-yl)-2-hydroxy-1-pyrrolidin-1-ylmethyl-ethyl]-amide-L-tartaric acid salt (Genz-123346) 35.(422) This compound has also shown efficacy in mouse models for Fabry disease,(423) Gaucher disease (where there is accumulation of GlcCer due to defective lysosomal glucocerebrosidase/acid β-glucosidase),(424) and polycystic kidney disease, a family of genetic disorders characterized by renal cystic growth and progression to kidney failure.(425)
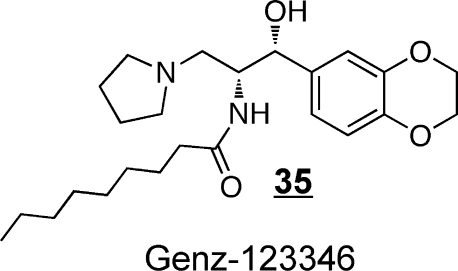
Use of GlcCer synthase inhibitors has revealed how decreases in cellular levels of neutral glycosphingolipids and gangliosides (and elevation of Cer) causes cell cycle arrest,(426) and how GlcCer synthesis appears to be a major determinant of survival of tumor cells.427,428 They also led to identification of a previously unknown pathway for ceramide metabolism, the formation of 1-O-acylceramide via a phospholipase A2.415,429
3.2.3.2. Biosynthesis of LacCer
GlcCer is next glycosylated to Galβ1–4Glcβ1Cer (LacCer) by two LacCer synthases (β4-galactosyltransferases), β4GalT-V and -VI,(430) with the former also being implicated in the synthesis of N-glycans of cell surface glycoproteins.(183) Before this can occur, however, the GlcCer must flip to the inside of the Golgi because GlcCer is made on the cytosolic aspect of the ER or early Golgi membranes,431,432 whereas LacCer and more complex glycosphingolipids are made in the lumen of the Golgi apparatus.(433) Studies with rat liver ER and Golgi membranes have found that transbilayer movement of spin-labeled GlcCer is rapid, saturable, and inhibitable by protease treatment, which suggests that the membranes contain a GlcCer flippase.(434) The mechanics of GlcCer delivery to the sites of higher glycolipid biosynthesis appears to be more complex than just flipping across the membrane because after GlcCer is made on the cytosolic leaflet of the Golgi, it is transported back into the ER (via Golgi-associated four-phosphate adaptor protein 2, FAPP2) before achieving access to the lumen of the Golgi.435,436 FAPP2 is a dimeric protein that has the capability to form tubules from membrane sheets (an activity that is dependent on the phosphoinositide-binding activity of the PH domain of FAPP2) and it has been suggested that FAPP2 functions directly in the formation of apical carriers in the trans-Golgi network.437,438
Some of the factors that have been reported to regulate LacCer synthase include growth factors, cytokines, lipids, lipoproteins, and hemodynamic factors, such as fluid shear stress.(439)
3.2.3.3. Biosynthesis of Ganglio-Series Glycosphingolipids
As shown in Figure 7, one of the fates of LacCer is conversion to the neutral and acidic members of the ganglio-root structure series glycosphingolipids (blue bordered box in Figure 7). The enzyme responsible for the first neutral metabolite GalNAcβ1–4Galβ1–4Glcβ1Cer (GA2, also called asialo-GM2) is GM2 synthase, which is also called β4GalNAcT, β1,4-N-acetylgalactosylaminyltransferase and GM2/GD2 synthase because it additionally converts gangliosides GM3 to GM2, GD3 to GD2, etc., as shown in Figures 7 and 8.(440) Therefore, this enzyme is critical for synthesis of all complex gangliosides enriched in the nervous system of vertebrates (GM1a, GD1a, GD1b, GT1b, GQ1b, etc.), as well as downstream neutral (asialo-) glycosphingolipids (GA1), which has been confirmed by studies with the knockout mouse.386,441 Interestingly, knockout of this gene did not affect brain morphology/histology, but there were effects on the maintenance and repair of nervous tissues, differentiation of spermatocytes, and regulation of interleukin-2 receptor complex.
The other major branch of metabolism of LacCer is its sialylation to ganglioside GM3 (Figures 7 and 8) by ST3Gal-V (SAT-I, CMP-N-acetyl-neuraminate:lactosylceramide α2,3-sialyltransferase, GM3 Synthase).(442) GM3 null mice are unable to synthesize GM3, as anticipated, and appear to be without major abnormalities, but have a greater sensitivity to insulin due to enhanced insulin receptor phosphorylation in skeletal muscle, are protected from high-fat diet-induced insulin resistance,(443) and have impaired hearing due to selective degeneration of the stereocilia of hair cells in the organ of Corti.(444) The relationship between GM3 and insulin signaling is provocative because it might provide better insight into type 2 diabetes, and it has been suggested that this involves interactions between insulin receptors and gangliosides in membrane microdomains, and might be a new paradigm for insulin receptor regulation.(444) Defects in GM3 synthase have also been found clinically,(445) wherein a nonsence mutation in the gene that would cause a premature termination caused loss of activity, GM3 and its derivatives, and developmental stagnation and blindness.
The other downstream metabolites in this pathway shown in Figures 7 and 8 are formed by analogous reactions, for example, GA2 is converted to GA1 by β3GalT-IV (also called GM1 synthase, β3GalT, and Gal-T2),(446) which can be in turn sialylated to ganglioside GM1b by GT1b/GD1a/GM1b synthase (also named ST3Gal-II, SAT-IV, CMP-N-acetylneuramininate: d-galactosyl-N-acetyl-d-galactosaminyl-(N-acetylneuraminyl)-d-galactoxyl-d-glucosylceramide α2,3-sialyltransferase).(447) Likewise, to form the disialo- (GD3) and trisialo- (GT3) gangliosides, the additional enzymes GD3 synthase (SAT-II, ST8Sia-I, CMP-N-acetylneuraminate: GM3 α2,8-sialyltransferase) and GT3 synthase (SAT-III) are invoked (with the products of each of these serving as substrates for the enzymes already described to synthesize GD2, GT2, etc., as shown in Figure 8). Thus, the profile of ganglio-series glycolipids that are made by a particular cell will depend on the particular glycosyltransferases that are expressed, their kinetic properties, and other issues such as localization, availability of the substrates, presence of enzymes that may be competing for the same intermediates, and the rates at which the precursors and products are trafficked through the Golgi.(14) As these relationships become better understood, one can begin to make computational predictions about what species will be made based on gene expression profiles and pathway maps,(448) and even predictions about glycan structures from genomic information about glycosyltransferases,449,450 although the outcomes still require experimental verification.
A reaction not shown in these diagrams is the addition of fucose to produce compounds such as fucosyl-GM1a shown in Figure 1. This is catalyzed by fucosyltransferases (α1,2-fucosyltransferase 1 and 2, FUT1 and FUT2;(451) α3/4-fucosyltransferase, FUT3, Lewis enzyme;(452) and others). Fucosyl-GM1, which can be made by both FUT1 and FUT2, is expressed in a variety of cancer tissues, and has been considered to be a tumor marker and target for immunotherapy.(453) Aberrant glycosphingolipid profiles are one of the hallmarks of cancer and over forty years ago, Hakomori and Murakami noted that “the structural remodeling of glycolipids and glycoproteins are undoubtedly a key to open a secret box of malignancy.”(454) Largely through the efforts of Sen-Itiroh Hakomori and his many collalborators and trainees,(455) a lot is now known about the links between glycosphingolipids and abnormal cell behavior in cancer, as well as tumor progression, metastasis, and invasivity.456,457 The underlying premise has been that some of these compounds might be useful biomarkers, and/or that restoration of a more normal composition might have clinical benefit. This latter idea has been supported, for example, by studies of gliomas, which have lower expression of several sialyltransferases (ST6Gal1, ST6, and ST6GalNAcV), so when U373MG glioma cells were stably transfected with ST6GalNAcV, this increased expression of GM2α and GM3 gangliosides, caused marked inhibition of in vitro invasivity, modified cellular adhesion to fibronectin and laminin, and inhibited tumor growth in vivo.(458) Therefore, the concept that normalization of sphingolipid profiles in cancer could be of therapeutic benefit is very appealing. It is worth mentioning that the links between cancer and glycosphingolipids have not been limited to the headgroups(459) and this also might be useful in identifying more unique biomarkers.
One other category of ganglioside derivative that is not shown in these pathway diagrams is the O-acetylation of the Neu5Ac, which is conducted by a 7- or 9-position sialic acid-specific O-acetyltransferase.(460) An enzyme has also been found that removes the acetyl-group from 9-O-acetyl-sialic acids.(461) There has been considerable interest in 9-O-acetylGD3 because it is found in tumors and appears to protect them from apoptosis.(462)
Regulation of ganglioside expression has been studied fairly extensively, particularly during brain development, where ganglioside biosynthesis switches between expressing simple and complex gangliosides or between different ganglioside series, and the factors that govern this “orchestration of glycosyltransferases”(463) have been reviewed.
3.2.3.4. Biosynthesis of Lacto-/Neolacto-Series Glycosphingolipids
Biosynthesis of the lacto-/neolacto-series glycosphingolipids begins with the formation of GlcNAcβ1–3Galβ1–4Glcβ1Cer (also referred to as Lc3 or amino-ceramide trihexoside, amino-CTH) by β-1,3-N-acetylglucosaminyltransferase (also named UDP-N-acetylglucosamine: β-galactose β1,3-N-acetylglucosaminyltransferase, amino-CTH synthase or β3GlcNAcT) (Figure 7). This gene has been cloned(464) and the gene named β3Gn-T5. The knockout mouse displays multiple phenotypic changes with some dying in less than 2 months, developing early stage growth retardation, and having shorter lifespan overall. Pathologies include splenomegaly and notably enlarged lymph nodes, fur loss, obesity, and reproductive defects.(203)
The distinction between the lacto- versus neolacto- series glycosphingolipids is determined by the next galactosyltransferases, which are β1,3GalT for Lc4 and β1,4GalT for nLc4 (Figure 7). Lc4 is a precursor for formation of Lewisa, Lewisb upon addition of fucoses, and sialyl Lewisa (by the action of ST3Gal-III), and nLc4 is a precursor for Lewisx, Lewisy, and sialyl-Lewisx (c.f., Figure 2 and Figure 7).
3.2.3.5. Biosynthesis of Globo-/Isoglobo-Series Glycosphingolipids
Biosynthesis of the Gb3 (Galα1–4Galβ1–4Glcβ1Cer) is catalyzed by Gb3 synthase (α1–4-galactosyltransferase, α1–4GalT), then to Gb4 (via β3GalNAcT), as shown in Figure 7. The next globoside in this series, Gb5 (synthesized by β3GalT-V), is also known as the stage-specific embryonic antigen-3 (SSEA-3),(465) a frequently used stem cell marker.(466) Although iGb3 synthase (α1–3GalT) is shown in Figure 7 by a faint line, the human iGb3 synthase gene contains several mutations that render its product nonfunctional,(201) and this has been supported by in vitro assays.(197)
As noted earlier, globosides have also received much attention as receptors for Shiga toxin,(196) verotoxins, and the HIV adhesin gp120,(10) and for their elevations in cancer(197) and Fabry’s disease.(195)
3.2.3.6. Biosynthesis of GalCer
GalCer are made by UDP-Gal:Cer galactosyltransferase, also called GalCer synthase or cerebroside synthase, and abbreviated CGT or CGalT (human gene, UGT8).(467) GalCer is synthesized in the lumen of the ER(468) using UDP-Gal that is transported to the lumen of the ER by UDP-Gal transporter 2 (UGT2), a splice variant of UGT1 (the transporter for UDP-Gal into the Golgi) that contains an ER locating dilysine motif.(469) Disruption of the mouse gene produced animals that did not synthesize GalCer or sulfatide but they formed myelin containing glucocerebroside. This did not substitute fully for GalCer, however, because the mice exhibited severe generalized tremoring and mild ataxia, and developed progressive hindlimb paralysis and extensive vacuolation of the ventral region of the spinal cord.(470) Transgenic mice overexpressing the this GalCer synthase had an increase in activity and monogalactosyl diglyceride and nonhydroxy fatty acid-containing GalCer, but the latter was accompanied by a concomitant decrease in α-hydroxylated GalCer; therefore, there must be some mechanism to maintain the total level of GalCer. Nonetheless, the transgenic mice developed progressive hindlimb paralysis and demyelination.(471)
3.2.3.7. Sulfated Glycosphingolipids
Sulfatides are formed by the transfer of a sulfate to one of the hydroxyls of a glycosphingolipid by using the activated sulfate donor 3′-phosphoadenosine-5′-phosphosulfate (Figure 7). Two of the more thoroughly studied sulfotransferases are 3′-phosphoadenosine 5′-phosphosulfate:galactosylceramide sulfotransferase (GalCer sulfotransferase), which produces 3′-sulfo-galactosylCer,(472) and a sulfotransferase from rat brain(473) that catalyzes the transfer of sulfate to glucuronylglycolipids (GGL),(474) such as GlcAβ1–3Galβ1–4GlcNAcβ1–3Galβl–4Glcβl-Cer to from sulfated glucuronylglycolipids (SGGL), 3′-sulfo-GlcAβ1–3Galβ1–4GlcNAcβ1–3Galβl–4Glcβl-Cer. Mass spectrometry has been helpful in mapping out the metabolic pathways for sulfatides of the ganglio-series.(475) A connection between vitamin K (or warfarin administration) and sulfatide biosynthesis has been suggested by several studies, but the mechanism has been elusive.(476)
The cDNA encoding GalCer sulfotransferase has been cloned,(477) and knockout mice have been generated to analyze the biological roles of sulfoglycolipids and pathophysiology of their deficiency, which included neurological disorders due to myelin dysfunction and amelioration of monocyte infiltration in the kidney after ureteral obstruction, which provides evidence that sulfatide is an endogenous ligand of l-selectin.478,479 Studies of sulfatide deficiency have also utilized mice that are genetically deficient in UDP-galactose: ceramide galactosyltransferase because the inability to synthesize galactosylceramide precludes the biosynthesis of sulfatide by sulfation of GalCer(480) and have compared the morphological features between the galactolipid-null and the sulfatide-null mice.(481) Reduced formation of sulfatides has been suggested to play a role in polycystic kidney disease.(482)
3.2.4. Integration of Backbone and Headgroup Biosynthetic Pathways
Figure 9 provides a symbolic representation of sphingolipid biosynthesis de novo from the perspective of both the lipid backbones and the headgroups.(3) Panel A explains the layout for the sphingolipids made from Ser, with the first node representing 3-ketosphinganine; the next, sphinganine; then fanning out to form a ring that represents the ensemble of N-acyl-sphinganines (with the fatty acid chain labeled in blue); from which the different categories of headgroups are added, as indicated in the blow-out in the upper portion of panel A. Thus, each fan blade represents all of the headgroup derivatives for one N-acyl-sphingoid base backbone. Panel B illustrates how this scheme expands as one includes all three types of sphingoid bases made by SPT using Ser (for sphinganine, d18:0), Ala (for 1-deoxysphinganine, m18:0) and Gly (for 1-desoxymethylsphinganine, m17:0). The initial from each of these represent the N-acyl-derivatives followed by headgroup addition, as shown in panel A (note that the blades of the fans for m18:0 and m17:0 do not extend beyond the first ring because neither of these undergo headgroup addition), and the lines that connect the inner rings to the next fans represent the desaturation of the sphingoid base backbones each of these N-acyl-sphingoid bases (e.g., converting DHCer, N-acyl-d18:0, to Cer with an N-acyl-d18:1 backbone, etc.). Also shown are the additional backbones from 4-hydroxylation of DHCer by DES2 to form the t18:0 backbone, and desaturation of Cer to produce the sphingadiene backbone (d18:2) (and others could be added for additional modifications to the sphingoid base chain). The 1-deoxy- and 1-desoxymethyl-sphingoid bases might also undergo these further backbone modifications, but we have not yet seen these products by mass spectrometric analysis of mammalian samples (unpublished observation), so they have not been added to the scheme. For a representation of all of the sphingolipids that can be made de novo, one would display several of these maps, with one for each fatty acyl-CoA that is utilized in the first step (for examples, myristoyl-CoA for the d16:0 sphingoid base chain length; stearoyl-CoA for d20:0, etc.). This type of diagram is mainly useful as a mental exercise to appreciate the pathways that would produce every individual molecular subspecies; however, it might be possible someday to populate it with colored pixels representing the relative amount of each subspecies (or differences in amounts between two sources, as in a gene expression heat map), to facilitate visualization of patterns or interrelationships that would otherwise be difficult to appreciate.
Figure 9.

Backbone and headgroup relational depiction of mammalian sphingolipid biosynthesis. This alternative depiction of de novo sphingolipid biosynthesis displays how the pathway can be envisioned to start with a fatty acyl-CoA (palmitoyl-CoA in lower portion of panel A) that is condensed with Ser to form 3-ketosphinganine then sphinganine (at the center of the fan), which is N-acylated to produce different chain-length dihydroceramides (represented by the ring, with examples of chain lengths labeled in blue). Each dihydroceramide subspecies can be converted into families of dihydro-complex sphingolipids, which are symbolized by the blades. The upper portion of panel A shows some of the complex sphingolipids within each wedge (which are only a fraction of the actual number of compounds that can be made, as illustrated by Figures 7 and 8, and the discussion in the text). Panel B displays further complexities related to the lipid backbones. The upper portion of panel B illustrates how the dihydroceramides from each sphingoid base backbone (in this case, d18:0 from palmitoyl-CoA) can be hydroxylated to phytoceramides (t18:0) and/or desaturated to ceramides (d18:1) (c.f., Figure 4); the latter is also presumed to undergo further desaturation to form N-acyl-sphingadienes (d18:2). The blades radiating from each N-acyl-chain subspecies represents the complex sphingolipids, as explained for panel A. The lower portion of panel B shows that Ala or Gly are alternatively used by serine palmitoyltransferase to form m18:0 and m17:0 which are N-acylated and, to some degree, desaturated to N-acyl-m18:1’s and N-acyl-m17:1 (to date, backbone hydroxylation has not been noted). Note that these do not radiate into larger blades because headgroups cannot be added. Not shown are the utilization of other fatty acyl-CoAs, which would constitute parallel schemes like these, nor pathways where sphingolipids are turned over to generate intermediates that are recycled or turned over (although one can envision this occurring within the blades to return to the hub, with the apex of the hub representing the free sphingoid base). The symbols and abbreviations are the same as have been used in the other figures in this Review.
The elements of this scheme occur in discrete (and often multiple) locations in the cell, where particular combinations of the enzymes, cosubstrates, trafficking proteins, etc., make a major contribute to the outcome. Further discussion of this facet of the biosynthetic pathway is beyond the scope of this review, except for the few instances that have been presented with a particular metabolic step, such as the compartmentation of GalCer biosynthesis in the lumen of the ER.
3.3. Sphingolipid Turnover, Trafficking, and Recycling
Metabolic homeostasis for the sphingolipidome is achieved by balancing biosynthesis, degradation, recycling, and processes that add exogenous sphingolipids to, and remove them from, the cell. This review is concerned mainly with sphingolipid biosynthesis but will also briefly address these other processes because they are interrelated and should be kept in mind. Metabolic turnover is defined as any process that hydrolyzes a complex sphingolipid to component parts. This usually occurs via lysosomal enzymes(483) and, if the intermediates are not recycled, is followed by the irreversible degradation of the sphingoid base to products that are no longer categorized as sphingolipids (e.g., a fatty aldehyde and ethanolamine phosphate) in the cytosol.(484) Hydrolytic enzymes for sphingolipids are also found in other locations in the cell to produce bioactive products for cell signaling5,6 and rearrangement of membrane architecture.23,485 The complete balance sheet for sphingolipids additionally includes uptake of sphingolipids from exogenous sources (such as albumin and lipoproteins)486,487 and losses by efflux,(488) lipoprotein secretion,354,489 and shedding of membrane vesicles containing sphingolipids.(490)
3.3.1. Metabolic Turnover
The major steps in the hydrolysis of complex sphingolipids are summarized in Figure 10, which has the same basic pathway layout as for the other figures for self-consistency. Most of these steps were discovered in the course of understanding genetic diseases that had been noted to involve accumulation of a particular category of sphingolipid.(483) As the lysosomal hydrolases were characterized and in some cases found not to be mutated despite the appearance of disease symptoms in some of the patients, accessory proteins (such as GM2 activator protein shown in Figure 10) were found also to be important. In addition to these, one disease that appeared to be due to defective sphingolipid turnover, Niemann–Pick type C disease (due to accumulation of sphingomyelin, although that was erratic), was found not to be due to a genetic defect in an enzyme of sphingomyelin metabolism but rather a lipid transporter that affects multiple categories of lipids.(491) If the reader is interested in more information about the diseases resulting from defects in sphingolipid metabolism, at least three outstanding reviews of that topic have been published recently.295,483,492
Figure 10.

Sphingolipid turnover and catabolism. Representative enzymes and intermediates are shown for the turnover of each complex sphingolipid family to the lipid moiety (Cer), and the insert displays the degradation of the sphingoid base by phosphorylation and lytic cleavage to a fatty aldehyde and ethanolamine phosphate. The symbols and abbreviations are the same as have been used in the other figures in this Review.
Over time, some sphingolipids were found to be hydrolyzed not only by lysosomal enzymes (with acidic pH optima) but also by enzymes with neutral or alkaline pH optima. These include sialidases in the plasma membrane (which are able to modulate cell regulation by gangliosides)(493) and nuclear membranes (the latter apparently to produce GM1 by hydrolysis of GD1a for nuclear function),(494) plasma membrane β-galactosidase and β-glucosidase (which is active without activator proteins and displays a trans activity in living cells),(495) alkaline,(496) and neutral497,498 sphingomyelinases as well as multiple ceramidases (at least five human ceramidases encoded by distinct genes: acid, neutral, and three alkaline ceramidase)(499) that have functions from sphingolipid digestion to cell signaling. Therefore, turnover of sphingolipids occurs in a large number of locations: in the extracellular environment (e.g., as in sphingolipid digestion in the lumen of the intestine), on the extracellular and intracellular surfaces of the plasma membrane, and associated with multiple intracellular organelles.
3.3.2. Sphingolipid Trafficking and Membrane Dynamics
The general scheme for sphingolipid biosynthesis and trafficking that has been in place for some time500,501 is that Cer is produced de novo in the ER(253) then transported to the cis-Golgi via vesicular trafficking or the trans-Golgi by CERT,(410) where more complex (glyco)sphingolipids are made (with the differential localization of CERK, SM synthases and specific glycosyltransferases influencing the partitioning of the intermediates into the endproducts)(463) and (mostly) delivered to the plasma membrane to enrich it with sphingolipids and cholesterol.(501) This traditional pathway is schematically represented in Figure 11 by black arrows. Other routes of sphingolipid relocation in cells include transbilayer movement by ATP-binding cassette (ABC) family of membrane-bound transporters,(502) which may also be a pathway for efflux of S1P,(503) intermembrane transfer via the Glycolipid Transfer Protein (GLTP) superfamily(504) and other transporters such as MDR2 (P-glycoprotein) and the cystic fibrosis transmembrane regulator (CFTR).(505)
Figure 11.

Schematic representation of the locations in and outside of the cell where sphingolipids are metabolized and trafficked. The black dashed lines show the traditional biosynthetic pathway beginning with biosynthesis of the lipid backbone in the ER and subsequent trafficking through the Golgi for further metabolism, leading ultimately to movement to the plasma membrane and other parts of the cell via vesicles and transport proteins (e.g., GLTP) or across the membrane via pumps (ABC, etc.). The green arrows reflect inward movement of sphingolipids destined to lysosomes or to the ER via retrograde motion. The red lines represent additional trafficking of sphingolipids; for examples, for autophagosome formation, formation of multivesicular endosomes and multivesicular bodies, and fusion with the plasma membrane as shown.
Inward trafficking of sphingolipids is illustrated by the green arrows in Figure 11. This was initially defined as “housekeeping” turnover of sphingolipids via internalization of membrane vesicles that are sorted into components for lysosomal hydrolysis, with the released sphingosine being degraded or recycled as summarized, respectively, in sections and . Cellular membranes additionally enter the lysosomal compartment by autophagy during phagocytosis,(506) which is thought to use autophagy components to facilitate acquisition of lysosomal enzymes by the phagosome.507−509 Since sphingolipids are components of autophagosomes (and are required for induction of autophagy),(510) they are likely to be hydrolyzed when the autophagosome becomes acidic and acquires lysosomal hydrolases. Retrograde trafficking from the plasma membrane to the Golgi and ER provides another pathway for inward movement of sphingolipids, and has been studied mostly from the perspective of how it provides a mechanism for bacterial toxins that bind glycosphingolipids (e.g., GM1 for cholera toxin;(511) Gb3 for Shiga toxin(512) and verotoxin513,514) to gain access to the ER.
In addition to these processes, there are many interesting and not-yet-fully explained observations that reveal that the metabolism and trafficking of sphingolipids is even more complicated. For examples, it has been noted: that at least one subunit of serine palmitoyltransferase appears in the nucleus and focal adhesions and affects cell morphology;(251) that ceramide synthesis (N-acylation) occurs not only in the ER but to some extent also in mitochondria,339,360 a site where Cer production or targeting can induce cell death;515,516 that GlcCer destined for glycolipid synthesis appears to be made in the Golgi but is transported back into the ER (via FAPP2) before achieving access to the lumen of the Golgi,(435) apparently because the FAPP proteins are involved in forming a tubular network that effects transport;(437) that cells make GalCer in the lumen of the ER,468,517 raising the possibility that slowed trafficking of Cer from the ER to Golgi (and, thus, more time for Cer to flip from the cytosolic to luminal leaflet) might contribute to the elevated biosynthesis of Gala series glycosphingolipids in stressed cells(518) and that enzymes of sphingolipid metabolism are being found in many other regions of the cell, including the nucleus(207) and the outer leaflet of the plasma membrane, where there are “ecto” glycohydrolases(519) and glycosyltransferases,(520) including a recently described ecto-sialyltransferase (ecto-Sial-T2) that is able to sialylate GM3 exposed on the membrane of neighboring cells using CMP-N-acetylneuraminic acid in the extracellular milieu.(521)
Other exported (secreted) enzymes include sphingosine kinase,522,523 neutral,524,525 and acidic(525) ceramidase, and acid sphingomyelinase.(31) Acid sphingomyelinase is a particularly interesting case because it appears to have multiple functions,526,527 including to be secreted by cells upon membrane wounding to facilitate endocytosis of the affected region of the membrane, perhaps by inducing membrane curvature.(528) This introduces the third category of trafficking processes, represented by the red arrows in Figure 11, which include vesicle fusion and trafficking,(529) formation of autophagosomes (discussed above) and multivesicular bodies (which have been proposed to provide a mechanism to release autophagosomes via “unconventional” vesicular secretion),(530) release of membrane particles (microvesicles, shed vesicles, exosomes and ectosomes), as well as the endocytotic processes discussed above. Many of these have already been found to involve sphingolipids, such as that Cer modulates the rate of ER to Golgi trafficking,(531) induces autophagy(532) (as also does dihydroceramides),(97) generates dynamic membrane asymmetry for promotion of membrane curvature,(533) and triggers budding of exosome vesicles into multivesicular endosomes.(534) Some of these processes might reflect a specific sorting process, as has been proposed to occur in a subapical compartment or common endosome,(535) or might be a manifestation of the biophysical properties of Cer, or perhaps an indicator of the broader process such as raft formation,536,537 with Cer serving as just one of many participants. In any event, these rapidly evolving subjects are very likely to change the way that we think about sphingolipid homeostasis.
3.3.3. Sphingolipid Recycling (Salvage Pathways)
This review has referred only briefly to the reutilization of sphingolipids after they have been turned over to the free sphingoid base or other intermediates, which has been discussed in at least two excellent reviews.538,539 This is clearly an aspect that requires attention not only with respect to the contribution of salvage pathways to overall homeostasis but also for its implications for cell signaling studies where exogenous sphingolipids have been added to cells in culture. Studies of the recycling of endogenous sphingolipids is extremely difficult because the precursors that would be used for the labeling (palmitic acid, serine, etc.) are themselves interconnected with numerous other metabolic pathways.
Studies of the fate of exogenously added sphingolipids have found that they are reutilized, but in complicated ways. Complex sphingolipids labeled in the sphingosine backbone are taken up by cells in culture and hydrolyzed and reutilized, but a substantial amount is rereleased into the culture medium, which implies that they receive special handling.(487) This loss to the medium is very intriguing and is consistent with the finding that microvesicles released from cells in culture are enriched in sphingolipids,540,541 and more recently that ceramide triggers budding of exosome vesicles into multivesicular endosomes.(534)
Exogenously added short chain (C6-) ceramides are extensively hydrolyzed and reacylated with long-chain fatty acids,(542) and studies with A549 human lung adenocarcinoma cells found that the generation of endogenous long-chain ceramide in response to exogenous C6-Cer was regulated by reactive oxygen species.(543) A comparison of C2- and C6-ceramides found that the former was not hydrolyzed and recycled like the latter, and proposed that this accounts for differences in the ability of these chain length subspecies to induce apoptosis (i.e., C6-Cer is more potent in inducing apoptosis).(544) Other types of exogenous ceramides, such as fluorescent derivatives with the fatty acyl position occupied by a 6-[N-(7-nitro-2,1,3-benzoxadiazol-4-yl)amino]hexanoyl- group (i.e., C6-NBD-), are also readily taken up and converted to more complex sphingolipids(545) (with interesting differences between different stereoisomers and NBD-dihydroceramides and NBD-ceramides).(371) Exogenously added sphingoid bases are rapidly taken up, phosphorylated,(546) acylated(277) and converted to more complex metabolites(92) depending on the structure of the sphingoid base. The story is also complicated (and perhaps even more so) for sphingosine 1-phosphate, which has been shown to be hydrolyzed by the extracellular lipid phosphatase LPP-1, which facilitated uptake of the sphingosine, followed by its intracellular rephosphorylation by sphingosine kinase (SphK1).(547) The Spiegel lab has shown that a similar phosphorylation-dephosphorylation cycle is involved in reutilization of sphingosine in mammalian cells and appears to take place in the endoplasmic reticulum via sphingosine-1-phosphate phosphohydrolase 1 (SPP-1) and sphingosine kinase 2 (SphK2).(548) SPP1 is an endoplasmic reticulum-resident enzyme that specifically dephosphorylates S1P, and its depletion has also been shown to induce ER-stress and autophagy, processes that alter ER/Golgi trafficking.(549)
Taken in concert, one wonders if it is technically feasible to add exogenous sphingolipids to cells and accurately deduce how endogenous sphingolipids behave because they not only follow different membrane trafficking pathways but also possess biological activities that perturb cell behavior. Similar concerns apply to studies using overexpression or knockout of genes for enzymes of the pathway. This is unsettlingly analogous to the Heisenberg uncertainty principle for quantum mechanics, therefore, one might consider referring to it as the “sphingolipid uncertainty principle”.
4. Analysis of Sphingolipid Metabolism by “Omic” Technologies
The sphingolipidome is theoretically defined as all of the molecular subspecies of sphingolipids in an organism or other system of interest, and as the discussion above has illustrated, this encompasses a large number of individual molecular species when both headgroup and backbone variation are taken into account. Many types of methods are available for studying sphingolipids;(550) however, sphingolipidomic analyses are usually conducted by mass spectrometry because both the sphingolipid category and molecular subspecies can be determined. But, as is the case for the other omics, current methods actually encompass only a fraction of the members of this family of compounds, therefore, a sphingolipidomic analysis describes compounds within a particular frame of reference (for example, all of the early metabolites of de novo sphingolipid biosynthesis, such as the ones shown in Figure 4). This can, nonetheless, provide very useful information about sphingolipid metabolism, especially when stable isotope-labeled precursors are used to track the newly made metabolites.(551)
4.1. Use of Mass Spectrometry for Sphingolipidomics
Mass spectrometry is not a “one size fits all” technology because different categories of compounds form ions more or less readily (and some are difficult to ionize without some degree of decomposition in the ion source, such as loss of sialic acid or dehydration), there are numerous instances where isomeric and isobaric compounds complicate the analysis and require the additional steps (chromatographic separation of GlcCer and GalCer, for example), and other components in the sample can interfere with the analysis by ionization suppression, clogging of columns and electrospray needles, and many other technical glitches. These considerations have been recently discussed from many perspectives.34,552−555 In general the major challenges in MS analysis of sphingolipids are (a) to obtain standards both for methods development and to serve as internal standards (at present, these are only available for a small fraction of the known sphingolipid subcategories); (b) to identify extraction conditions where the analytes of interest and selected internal standards are recovered in high yield whether highly polar (gangliosides and S1P), nonpolar (ceramides), vary in chain length and other biophysical features; (c) to develop complementary methods (usually chromatographic) to separate compounds that are not distinguished by MS alone, which includes the usually thought of isomers and isobars as well as occasions where relatively minor isotopomers (e.g., with several natural abundance 13C) of a major species overlaps with the m/z of a compound that is present in the biological sample in much lower abundance; (d) to optimize the ionization, fragmentation and detection parameters to gain maximal information from each compound, and to simply comparison of unknowns with the matched internal standards; (e) to have relatively rapid and facile ways to collect and analyze large data sets; and (f) to be able to display the data in ways that enable large amounts of information to be understood as easily and fully as possible.
Ideally, internal standards should have the same structure as the analyte and vary only in m/z—usually as a stable isotopically labeled version; however, this is not practical for lipidomic analysis and suitably selected representatives of subcategories of compounds are used. An internal standard cocktail has been developed by the LIPID MAPS Consortium and is commercially available from Avanti Polar Lipids (Alabaster, AL). It contains uncommon chain-length sphingoid bases (C17) for sphingosine, sphinganine and their 1-phosphates (S1P and Sa1P) and d18:1;C12:0-fatty acid Cer, Cer1P, SM, and mono- and dihexosylCer, and this can be supplemented with additional internal standards, as desired.
In our experience, it is difficult to extract both the nonpolar (e.g., Cer, SM, and hexosylCer) and the more polar sphingolipids (sphingoid base phosphates, Cer1P, etc.) using one solvent protocol; therefore, we divide the sample into two fractions: one that is later split into separate organic and aqueous phases (for the less polar sphingolipids) and one that is never divided into two phases (for the relatively water-soluble sphingolipids).(39) This protocol was subsequently altered to substitute methylene chloride for chloroform in all steps with free sphinogid bases to avoid the possibility of modification of the free amine via formation of carbene in basic conditions. The published extraction protocol gave high recoveries of all subspecies (i.e., SM, hexosylCer and Cer with C12- to C26-fatty acids); however, we have noted that when samples have a large amount of lipid (as is often encountered with plasma, liver and brain), extra effort may be required to redissolve all of the sphingolipids in the final extract in the LC solvent for LC-ESI-MS/MS, resulting in disproportionate losses of the very-long-chain subspecies.
Liquid chromatography39,552,556 is useful not only for the separation of isomeric and isobaric species (such as GlcCer from GalCer), but also tends to reduce ionization suppression. Reversed phase LC39,557−559 is used for separations based on the length and saturation of acyl chains (for example, to separate So and Sa), and normal phase LC39,220,557,560−562 to separate compounds primarily by their headgroup constituents (for example, distinguish Cer, GlcCer, LacCer, globotriaosylceramide, globotetraosylceramide, SM as well as cholesterol, etc.). LC-ESI MS/MS is the most popular analytical tool for sphingolipid analysis (as represented by the applications cited above, and more),563−567 because sensitivities are on the order of fmol (or less), which allows analysis of small biological samples (such as ∼105–106 cells in culture) while providing a wide dynamic range (typically several orders of magnitude), which allows analysis of both trace metabolites, such as the sphingoid base 1-phosphates and major structural species (SM). Results from a LC-ESI MS/MS method that has been developed following these principles (and the internal standard cocktail available from Avanti Polar Lipids, Alabaster, AL)(39) can be seen in a recent study of the sphingolipids in RAW264.7 cells activated by KDO2–Lipid A.(510) It warrants comment that thin-layer chromatography has also been combined with MALDI-MS/MS for analysis of some of the difficult to distinguish glycosphingolipids.568−570
Although quantitative analyses have most often used electrospray to ionize sphingolipids,39,550 other ionization methods have included atmospheric pressure chemical ionization, APCI,(562) desorption electrospray ionization (DESI),(571) as well as MALDI,(572) which is not always thought of as a quantitative method, but can be with the appropriate controls. MALDI has particularly aided the analysis of more complex glycosphingolipids,573−575 but might also be applied to smaller molecules (which typically have been obscured by background chemical noise from the MALDI matrix ions) new advances in matrix choices and high-pressure sources.572,576−580
Ion separation and mass analysis is most frequently conducted using triple quadrupole or tandem quadrupole-linear ion trap mass analyzers for MS/MS and MSn,39,133,552,581 respectively, or for higher mass accuracy, time-of-flight (TOF),(582) orbitrap(583) or Fourier transform (FT) instruments;584−586 ion mobility MS has also been recently applied to sphingolipids.(587) Fragmentation in MS/MS mode is achieved by a number of ways, depending on the type of compounds involved, and include collision induced dissociation (CID) with nitrogen for most applications and, in a novel approach to determine double bond position, ozone;(588) a recent use of the ion trap to favor backbone fragmentation of SM;(39) and electron transfer dissociation (ETD) for analysis of glycans.(589) Analysis of higher glycosphingolipids can also be conducted by removal of the lipid moiety using endoglycoceramidase,(590) followed by analysis of the glycans by mass spectrometric methods used to characterize O- and N-linked glycans from glycoproteins.(591)
Although sphingolipids are complex, many are relatively easily ionized and can be fragmented to ions that allow the sphingoid base and amide-linked fatty acid to be determined; therefore, stable-isotope labeled precursors (such as U–13C-palmitate) can be used to follow biosynthesis of the sphingoid base backbone as well as N-acylation.(551) In this example, 13C appears in three isotopomers and isotopologues: [M + 16 for the sphingoid base or N-acyl fatty acid, and [M + 32] for both), in addition to the unlabeled species (corrected for the natural abundance 13C species). In interpreting the data, one needs also to determine the isotopic enrichment of palmitoyl-CoA (i.e., the fraction with 13C- versus endogenous 12C-palmitoyl-CoA) and ideally also that for longer chain fatty acyl-CoA’s that are made by desaturation and/or elongation reactions before incorporation into N-acyl-sphingolipids, which is technically feasible by a recently developed method for LC-ESI-MS/MS analysis of fatty acyl-CoAs.(288) This study used 0.1 mM [U–13C]palmitate (added as the 1:1 complex with bovine-serum albumin) to try to remain within the concentration usually found in circulation,(592) in an attempt to cause minimal perturbation of the total cellular palmitoyl-CoA because this compound has been shown to affect gene expression,593,594 ion transport,(595) and sphingolipid biosynthesis.294,296 Nonetheless, even this low amount, which achieved about 50% labeling of the total cellular palmitoyl-CoA, elevated the amount in the cells by about 3-fold. An alternative approach might be to use [13C]acetate to label the endogenous palmitoyl-CoA pool, but quantitative analysis of the labeling is more complicated,596−598 or to use labeled serine, which has the disadvantage that it only shifts the m/z of the labeled sphingolipids by a few amu (not to mention that serine also participates in multiple metabolic pathways). For some applications, it is also useful to add an exogenous sphingolipid that can be tracked because it has an unusual structure, such as an odd chain length or fluorescence tag.(539)
4.2. Tissue-Imaging Mass Spectrometry of Sphingolipids
Studies of sphingolipid metabolism in vivo are complicated by the loss of information about histological localization of the compounds of interest after the tissues have been homogenized for extraction and analysis. This can be addressed, in some cases, by a more direct method of analysis that is broadly called “imaging mass spectrometry,”(599) and the specific application described below has been termed “MALDI imaging mass spectrometry.”(600) In this procedure (in general, and as applied to sphingolipids),(601) a tissue (and sometimes an entire animal)(602) is usually frozen and sliced (the thickness varies, but is usually on the order of ∼10 μm), and adjacent sections are often placed on a chilled MALDI plate and a glass slide, so the MALDI image can be compared to the histologic appearance of the tissue using traditional staining. MALDI matrix compound is imbedded in the sample as uniformly and nondisruptively as possible, then a laser beam is moved incrementally across the sample to generate ions and collect MS and sometimes MS/MS spectra for regions ∼50 μm in diameter (larger, and sometimes smaller, regions can be chosen for each spectrum; however, the technique is usually limited to a histological, that is, one or a few cells, rather than a subcellular scale; subcellular analysis requires a different method for generating ions that is under development).(603) MALDI-imaging MS produces thousands of spectra for samples even only 1 mm2 which are analyzed using imaging software to locate specific m/z of interest (representing compounds chosen by the user, or other criteria such as abundance, coclustering, and other features), and these are plotted in x,y-space to yield a virtual molecular image of the distribution of the ions, with color coding that reflects different compounds, or displays the relative abundance of a specific ion (in a heat map style). These images can be cross-referenced with adjacent slices to orient where the ions of interest are located with respect to more traditional histological markers.
An approach to enhance the sensitivity of imaging MS has been to use gold nanoparticles in place of the usual MALDI matrix compounds (in a technique called nanoparticle-assisted laser desorption/ionization MS, or nano-PALDI-imaging mass spectrometry).604,605 For much higher resolution (<1 μm), secondary ion mass spectrometry (SIMS) has been used,(606) but the high energy of the ion beams causes extensive fragmentation of lipids. This has been circumvented by using a focused buckminsterfullerene (C60) cluster ion beam that is less destructive to the lipids.(603)
This technique is highly informative when applied to lipids.607,608 Some of the findings for sphingolipids from application of tissue-imaging mass spectrometry have been: to localize the areas of accumulation of di- and trihexosylceramides in cutaneous biopsies from patients with Fabry’s disease (a study that used both MALDI and SIMS);(609) to profile the normal distribution of lipids within human skin;(610) to characterize the lipid composition of atheroma;(611) to describe the sphingolipids of the human lens with aging;(612) to discover and localize elevated sulfatides in ovarian cancer;(180) to examine the lipids of lung,(613) and lungs infected with C. neoformans and find that specific SM species are associated with neutrophil infiltration at the site of the infection;(614) and to study in some depth the distribution of sphingolipids in brain using MALDI and SIMS imaging MS,571,575,606,607,615−618 with the interesting findings including the distinct localization of gangliosides with the d20:1-sphingoid base backbone,(53) and accumulation of GM2 and GA2 in a mouse model for Tay Sachs/Sandhoff disease.(619)
As these examples show, imaging MS has already proven to be useful in identifying specific molecular subspecies and histological locations of sphingolipids under a wide range of normal and abnormal physiologic conditions; therefore, it is reasonable to think that it will become increasingly valuable as a tool for metabolic studies as the technology becomes better refined and there are more research centers with the instruments (and knowledgeable operators). Although they have thus far had only limited use, stable isotope-labeled sphingolipids can be discerned by this form of mass spectrometry, too, so imaging studies can add a dynamic component. There are still limitations with respect to its ability to resolve isomeric and isobaric compounds and to provide absolute quantitation, however, these can be addressed to some extent by using both standard and imaging mass spectrometry as part of the investigation.
5. Integration of “Omic” Data Sets for a Systems Biology of Sphingolipid Metabolism and Function
A typical analysis of the major sphingolipids of cells in culture, plasma and other sources using lipidomic methods generally produces hundreds to thousands of data points, and the number will expand by several orders of magnitude when methods are available to look at all of the subspecies, which puts the sphingolipidome on the scale of other omic data sets. Therefore, sphingolipid researchers face the challenge of all “omics” disciplines, to figure out how to handle and visualize such large amounts of data, mine large data sets for relationships that have not been previously seen by more focused approaches, and integrate what has been (and will continue to be) learned by traditional reductionist approaches with the data produced by metabolomic, transcriptomic, and other omic analyses. There are several ways to envision accomplishing these goals, such as to develop relatively facile ways to visualize the information and, ultimately, to develop mathematical models for all of the components of the system.
5.1. Visualization Tools
Graphic display is often the most effective way to communicate data, if done cautiously.620,621 This is particularly true for large data sets and complex pathways because, as has been well stated by Alan Aderem, Director of the Institute for Systems Biology: “Human minds are incapable of inferring the emergent properties of a system from thousands of data points, but we have evolved to intelligently interpret an enormous amount of visual information” (http://www.systemsbiology.org/technology/data_visualization_and_analysis).
A typical analysis of the major sphingolipids of cells in culture using lipidomic methods will generally produce hundreds to thousands of data points, which puts it on the scale of transcriptomic data sets, where use of heat maps and other types of visualization tools have become commonplace. In heat map format, lipidomic data are often displayed in the order of N-acyl-chain length or summed carbon number, mass (or m/z, if the data are from mass spectrometry), and sometimes divided into lipid subcategories(622) and/or hierarchial clustering.(623)
Several additional visualization schemes have been developed for mammalian sphingolipid metabolism(3) to display all of the molecular subspecies in a pathway format, similar to Figure 4 (as illustrated in Figure 1 from a recent analysis of de novo sphingolipid biosynthesis by activated RAW264.7 cells),(624) or using a platform of pathway tools prepared by LIPID MAPS (http://www.lipidmaps.org/pathways/index.html) that can also show time course data. Other display formats have been prepared for the glycosphingolipids by the Consortium for Functional Glycomics (www.functionalglycomics.org). One report describes a way to extend the visualization of complex sphingolipid pathways via an interactive visualization tool.(625)
An approach that we have found to be useful(290) allows visualization of both transcriptomic and metabolomic data sets using an open access pathway browser, Pathvisio v1.1,(626) and KEGG-style pathway maps (Kyoto Encyclopaedia of Genes and Genomes) that have been updated and expanded for sphingolipid metabolism.290,554 In illustrating the use of this tool, microarray data for two breast cancer cell lines (MDA-MB-231 versus MCF7 cells) were compared and based on differences in the apparent differences in mRNA abundances, possible differences in sphingolipid subspecies were made and evaluated by analysis of the sphingolipid compositions of the cells by mass spectrometry.(290) Two of the predicted differences that were thus confirmed were in the nature of the sphingoid bases in the cells, both with respect to chain length (i.e., higher proportions of C16-sphingosine in the cells with the relatively higher expression of SPT3) and 4-hydroxylation (i.e., higher proportions of 4-hydroxysphinganine, phytosphingosine, in the cells with the relatively higher expression of DES2). When data from a wide range of cancer cell lines, tumors and normal tissues were considered, there was a surprisingly high probability of match between the gene expression data and sphingolipid composition (73%),(290) considering that there are multiple mechanisms for regulation of metabolism beyond transcript amount.
A similar approach has been used to compare gene expression data from mouse embryoid bodies versus embryonic stem cells, determined by quantitative real-time PCR (qRT-PCR) with the sphingolipid composition determined by mass spectrometry.(336) And, to take this approach a further step, it was also used to interpret a gene expression data set for ovarian cancer cells obtained by laser capture microdissection (versus normal human ovarian epithelial cells), which led to the prediction that sulfatides are elevated in human ovarian cancer, which was first confirmed by LC-ESI-MS/MS and then the sulfatides were specifically localized to ovarian epithelial carcinoma cells versus the neighboring stromal cells by tissue-imaging MS.(180) This simple integration of two types of “omic” technologies (“transcriptomics” to direct “sphingolipidomics”) could facilitate the discovery of new facets of how sphingolipid metabolism is regulated, relationships between sphingolipid metabolism and disease, and possibly the identification of new biomarkers.
5.2. Mathematical Modeling
Mathematical modeling of metabolic pathways and functions is a rapidly developing science,(627) but is difficult to apply to the sphingolipidome because the pathways are not only complex but still have many yet-to-be-discovered elements. The most comprehensive attempts toward this objective, to date, have been made with yeast Saccharomyces cerevisiae because its sphingolipid biosynthetic pathway has fewer genes and metabolites.628−630 The models appear to be compatible with the available information about gene expression levels, the kinetic properties of the enzymes, metabolite amounts, etc.628−630 and how perturbations, such as drugs,(631) heat stress, carbon source utilization, sporulation, cell wall integrity, and others affect this system.(632) A combined integrative analysis of genomic, transcriptomic and lipidomic data revealed a signaling role for phytosphingosine-1-phosphate in regulating genes required for mitochondrial respiration.(633) Mathematical modeling has also been applied to Cryptococcus neoformans to explore sphingolipid metabolism in the organism under acidic conditions with the goal of better understanding fungal pathogenesis.(634) The applicability of this approach to other organisms has also been discussed,635,636 and attention has been given to the more complex glycomes (“systems glycobiology”).(637)
Two other mathematical approaches have been applied to data sets from studies of mammalian sphingolipid biosynthesis. One used model-reference adaptive control (MRAC) to investigate the dynamics of de novo sphingolipid synthesis by Hek cells stably transfected with serine palmitoyltransferase, and the MRAC simulations produced results that were comparable to simulations from a standard model using mass action kinetics, and suggested that there might be adaptive feedback from increased metabolite levels.(638) The other approach integrated lipidomics and transcriptomics data collected by the LIPID MAPS Consortium (www.lipidmaps.org) for RAW264.7 cells using a two-step matrix-based approach wherein the rate constants obtained from the first step were further refined using generalized constrained nonlinear optimization.(639) The primary focus of the analysis was the C16-ceramide backbone species, and the resulting model fit the experimental data, with the robustness of the model being validated through parametric sensitivity analysis.
Such efforts will not only provide a better understanding about how these molecules are made and function, but also, to help interpret (and ultimately predict) the outcomes of changes in precursors, effects of inhibitors, genetic mutations, etc. In addition, as has been noted by Voit et al.,(636) it also allows the investigator to test one’s “intuitive grasp of the system through simulation studies that represent What-If scenarios.” These are useful not only to test the model, but also to direct researchers toward potentially interesting directions for future investigation.
6. Perspective on the Current State of Sphingolipid Research
Research discoveries over the last several decades have, to a substantial extent, transformed sphingolipids from enigmas into intricate puzzles within the ultimate puzzle of life. And unlike an enigmatic riddle, which is usually perplexing until a simple (and retrospectively obvious) answer is found, puzzles are hard to understand until most of the pieces are in place. Finding all the sphingolipid pieces and their places is still a daunting task, but this quest is part and parcel of the omics/systems biology era. One looks forward with great expectations, and curiosity, for what will be understood next.
Acknowledgments
This chapter draws heavily on ideas that the author has absorbed from laboratory colleagues (students and postdoctoral fellows, long-time associate Elaine Wang, and collaborators M. Cameron Sullards and May Wang), and gleaned from works by, and fruitful conversations with, Tony Futerman, Yusuf Hannun, Konrad Sandhoff, Walt Shaw, Jim Shayman, Sarah Spiegel, Akemi Suzuki and too many other experts on the topics to cite them adequately. Therefore, heartfelt thanks are due to them all, as well as to the reviewers of this manuscript for Chemical Reviews. Any errors are, of course, the author’s fault. Support from the NIH is gratefully acknowledged (NIH GM069338 “Lipid Maps,” GM76217 and CA137812), as is the Smithgall Institute endowment for the chair in Molecular Cell Biology at Georgia Tech that has facilitated some of the studies described herein.
Glossary
List of Abbreviations
- Cer
ceramide
- CERK
ceramide kinase
- CerPE
ceramide phosphoethanolamine
- CerS
ceramide synthase
- CERT
ceramide transport protein
- DHCer
dihydroceramide
- DES
dihydroceramide desaturase
- ESI-MS/MS
electrospray tandem mass spectrometry
- ER
endoplasmic reticulum
- Fuc
fucose
- Gal
galactose
- GalCer synthase; CGalT
galactosylceramide synthase
- Gg, with subscript for the number of carbohydrates
ganglio-
- G, with subscript for the subclass
ganglioside
- Gb, with subscript for the number of carbohydrates
globo- or globo-series
- Glc
glucose
- GlcCer synthase; CGlcT
glucosylceramide synthase
- GlcA
glucuronic acid
- GM2-AP
GM2-activator protein
- Hex A or B
hexosaminidase A or B
- iGb, with subscript for the number of carbohydrates
isoglobo- or isoglobo-series
- Lc, with subscript for the number of carbohydrates
lacto-
- Lac
lactose
- GalNAc
N-acetylgalactosamine
- GlcNAc
N-acetylglucosamine
- Neu5Ac
N-acetylneuraminic acid
- nLc
neolacto-
- Pal-CoA
palmitoyl-CoA
- Sa1P
sphinganine 1-phosphate
- SphK
sphingosine kinase
- S1P
sphingosine 1-phosphate
- Ser
serine
- SPT
serine palmitoyltransferase
- SAP
sphingolipid activator protein
- SM
sphingomyelin
- SMase
sphingomyelinase
- UDP-sugar
uridine dinucleotide phosphate sugar
Biography

Al Merrill, born in 1951, trained at Virginia Tech, Cornell (Ph.D.), and Duke (postdoc with Robert M. Bell); then, he was on the Emory University faculty until 2001, when he moved to Georgia Tech as the Smithgall Institute Chair in Molecular Cell Biology in the School of Biology and Petit Institute for Bioengineering and Bioscience. His early research contributed to the understanding of how the sphingolipid backbones are made and serve as cell signals, and (with Elaine Wang and Ron Riley), discovered that fumonisins inhibit ceramide synthase, and thereby identified the first diseases attributable to disruption of sphingolipid biosynthesis. His lab is currently developing methods to analyze the “sphingolipidome” as part of the LIPID MAPS Consortium. The findings thus far (with M. Cameron Sullards, Tony Futerman, and other collaborators) include characterization of the first cloned mammalian ceramide synthase, discovery that fenretinide inhibits dihydroceramide desaturase, and the structural elucidation of 1-deoxysphingoid bases and their N-acyl-derivatives as products of mammalian serine palmitoyltransferase and ceramide synthases.
Funding Statement
National Institutes of Health, United States
References
- Thudichum J. L. W.A Treatise on the Chemical Constitution of Brain; Bailliere, Tindall, and Cox: London, 1884. [Google Scholar]
- Carter H. E.; Haines W. J.; Ledyard W. E.; Norris W. P. J. Biol. Chem. 1947, 169, 77. [PubMed] [Google Scholar]
- Merrill A. H. Jr.; Wang M. D.; Park M.; Sullards M. C. Trends Biochem. Sci. 2007, 32, 457. [DOI] [PubMed] [Google Scholar]
- Sonnino S.; Prinetti A.; Mauri L.; Chigorno V.; Tettamanti G. Chem. Rev. 2006, 106, 2111. [DOI] [PubMed] [Google Scholar]
- Bartke N.; Hannun Y. A. J. Lipid Res. 2009, 50 (Suppl), S91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strub G. M.; Maceyka M.; Hait N. C.; Milstien S.; Spiegel S. Adv. Exp. Med. Biol. 2010, 688, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez P. H.; Schnaar R. L. Curr. Opin. Struct. Biol. 2009, 19, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R. K.; Nakatani Y.; Yanagisawa M. J. Lipid Res. 2009, 50 (Suppl), S440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckhardt M. Mol. Neurobiol. 2008, 37, 93. [DOI] [PubMed] [Google Scholar]
- Lingwood C. A.; Binnington B.; Manis A.; Branch D. R. FEBS Lett. 2010, 584, 1879. [DOI] [PubMed] [Google Scholar]
- Pruett S. T.; Bushnev A.; Hagedorn K.; Adiga M.; Haynes C. A.; Sullards M. C.; Liotta D. C.; Merrill A. H. Jr. J. Lipid Res. 2008, 49, 1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A.; Cummings R. D.; Esko J. D.; Freeze H. H.; Stanley P.; Marth J. D.; Bertozzi C. R.; Hart G. W.; Etzler M. E. Proteomics 2009, 9, 5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A. Trends Mol. Med. 2008, 14, 351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolter T.; Proia R. L.; Sandhoff K. J. Biol. Chem. 2002, 277, 25859. [DOI] [PubMed] [Google Scholar]
- Chester M. A. Eur. J. Biochem. 1998, 257, 293. [DOI] [PubMed] [Google Scholar]
- Fahy E.; Subramaniam S.; Brown H. A.; Glass C. K.; Merrill A. H. Jr.; Murphy R. C.; Raetz C. R.; Russell D. W.; Seyama Y.; Shaw W.; Shimizu T.; Spener F.; van Meer G.; VanNieuwenhze M. S.; White S. H.; Witztum J. L.; Dennis E. A. J. Lipid Res. 2005, 46, 839. [DOI] [PubMed] [Google Scholar]
- Schauer R. Curr. Opin. Struct. Biol. 2009, 19, 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantu L.; Corti M.; Casellato R.; Acquotti D.; Sonnino S. Chem. Phys. Lipids 1991, 60, 111. [DOI] [PubMed] [Google Scholar]
- Yu R. K.; Yanagisawa M.; Ariga T. In Comprehensive Glycoscience. From Chemistry to Systems Biology; Kamerling J. P., Ed.; Elsevier: Oxford, U.K., 2007; Vol. 1. [Google Scholar]
- Nystrom K.; Grahn A.; Lindh M.; Brytting M.; Mandel U.; Larson G.; Olofsson S. Glycobiology 2007, 17, 355. [DOI] [PubMed] [Google Scholar]
- Simons K.; Gerl M. J. Nat. Rev. Mol. Cell Biol. 2010, 11, 688. [DOI] [PubMed] [Google Scholar]
- Hakomori Si, S. I. Proc. Natl. Acad. Sci. U. S. A. 2002, 99, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goni F. M.; Alonso A. Biochim. Biophys. Acta 2009, 1788, 169. [DOI] [PubMed] [Google Scholar]
- Luberto C.; Yoo D. S.; Suidan H. S.; Bartoli G. M.; Hannun Y. A. J. Biol. Chem. 2000, 275, 14760. [DOI] [PubMed] [Google Scholar]
- Singh R. D.; Marks D. L.; Pagano R. E.. Curr. Protoc. Cell Biol. 2007, Chapter 24, Unit 24 1. [DOI] [PubMed] [Google Scholar]
- Fyrst H.; Saba J. D. Nat. Chem. Biol. 2010, 6, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milhas D.; Clarke C. J.; Hannun Y. A. FEBS Lett. 2010, 584, 1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gault C. R.; Obeid L. M.; Hannun Y. A. Adv. Exp. Med. Biol. 2010, 688, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merrill A. H. Jr. J. Biol. Chem. 2002, 277, 25843. [DOI] [PubMed] [Google Scholar]
- Ledeen R. W.; Wu G. J. Lipid Res. 2008, 49, 1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidan Y. H.; Hannun Y. A. Curr. Mol. Med. 2010, 10, 454. [DOI] [PubMed] [Google Scholar]
- Nikolova-Karakashian M.; Morgan E. T.; Alexander C.; Liotta D. C.; Merrill A. H. Jr. J. Biol. Chem. 1997, 272, 18718. [DOI] [PubMed] [Google Scholar]
- Maceyka M.; Milstien S.; Spiegel S. Prostaglandins Other Lipid Mediators 2005, 77, 15. [DOI] [PubMed] [Google Scholar]
- Sullards M. C.; Merrill A. H. Jr. Sci STKE 2001, 2001, l1. [DOI] [PubMed] [Google Scholar]
- Sullards M. C.; Liu Y.; Chen Y.; Merrill A. H. Jr. Biochim. Biophys. Acta 2011, 10.1016/j.bbalip.2011.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson K. A. Chem. Phys Lipids 1970, 5, 6. [DOI] [PubMed] [Google Scholar]
- Karlsson K. A. Lipids 1970, 5, 878. [DOI] [PubMed] [Google Scholar]
- Samuelsson B.; Samuelsson L. J. Lipid Res. 1969, 10, 47. [PubMed] [Google Scholar]
- Shaner R. L.; Allegood J. C.; Park H.; Wang E.; Kelly S.; Haynes C. A.; Sullards M. C.; Merrill A. H. Jr. J. Lipid Res. 2009, 50, 1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quehenberger O.; Armando A. M.; Brown A. H.; Milne S. B.; Myers D. S.; Merrill A. H.; Bandyopadhyay S.; Jones K. N.; Kelly S.; Shaner R. L.; Sullards C. M.; Wang E.; Murphy R. C.; Barkley R. M.; Leiker T. J.; Raetz C. R.; Guan Z.; Laird G. M.; Six D. A.; Russell D. W.; McDonald J. G.; Subramaniam S.; Fahy E.; Dennis E. A. J. Lipid Res. 2010, 51, 3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K.; Yamada M.; Tamiya-Koizumi K.; Kannagi R.; Aoyama T.; Hara A.; Kyogashima M. Glycoconj. J. 2011, 28, 67. [DOI] [PubMed] [Google Scholar]
- Scherer M.; Bottcher A.; Schmitz G.; Liebisch G. Biochim. Biophys. Acta 2011, 1811, 68. [DOI] [PubMed] [Google Scholar]
- Fujino Y.; Fujishima T. J. Dairy Res. 1972, 39, 11. [DOI] [PubMed] [Google Scholar]
- Byrdwell W. C.; Perry R. H. J. Chromatogr. A 2007, 1146, 164. [DOI] [PubMed] [Google Scholar]
- Sonnino S.; Chigorno V. Biochim. Biophys. Acta 2000, 1469, 63. [DOI] [PubMed] [Google Scholar]
- Keranen A. Chem. Phys. Lipids 1976, 17, 14. [DOI] [PubMed] [Google Scholar]
- Kyogashima M.; Tadano-Aritomi K.; Aoyama T.; Yusa A.; Goto Y.; Tamiya-Koizumi K.; Ito H.; Murate T.; Kannagi R.; Hara A. J. Biochem. 2008, 144, 95. [DOI] [PubMed] [Google Scholar]
- Kyogashima M.; Tamiya-Koizumi K.; Ehara T.; Li G.; Hu R.; Hara A.; Aoyama T.; Kannagi R. Glycobiology 2006, 16, 719. [DOI] [PubMed] [Google Scholar]
- Merrill A. H. Jr.; Wang E.; Wertz P. W. Lipids 1986, 21, 529. [DOI] [PubMed] [Google Scholar]
- Fyrst H.; Herr D. R.; Harris G. L.; Saba J. D. J. Lipid Res. 2004, 45, 54. [DOI] [PubMed] [Google Scholar]
- Goni F. M.; Alonso A. Biochim. Biophys. Acta 2006, 1758, 1902–21. [DOI] [PubMed] [Google Scholar]
- Ogawa-Goto K.; Funamoto N.; Abe T.; Nagashima K. J. Neurochem. 1990, 55, 1486. [DOI] [PubMed] [Google Scholar]
- Sugiura Y.; Shimma S.; Konishi Y.; Yamada M. K.; Setou M. PLoS One 2008, 3, e3232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umesaki Y.; Takamizawa K.; Ohara M. Biochim. Biophys. Acta 1989, 1001, 157. [DOI] [PubMed] [Google Scholar]
- Holleran W. M.; Takagi Y.; Uchida Y. FEBS Lett. 2006, 580, 5456. [DOI] [PubMed] [Google Scholar]
- Stewart M. E.; Downing D. T. J. Invest. Dermatol. 1995, 105, 613. [DOI] [PubMed] [Google Scholar]
- Stewart M. E.; Downing D. T. J. Lipid Res. 1999, 40, 1434. [PubMed] [Google Scholar]
- Chun J.; Byun H. S.; Bittman R. J. Org. Chem. 2003, 68, 348. [DOI] [PubMed] [Google Scholar]
- Renkonen O.; Hirvisalo E. L. J. Lipid Res. 1969, 10, 687. [PubMed] [Google Scholar]
- Panganamala R. V.; Geer J. C.; Cornwell D. G. J. Lipid Res. 1969, 10, 445. [PubMed] [Google Scholar]
- Touboul D.; Roy S.; Germain D. P.; Baillet A.; Brion F.; Prognon P.; Chaminade P.; Laprevote O. Anal. Bioanal. Chem. 2005, 382, 1209. [DOI] [PubMed] [Google Scholar]
- Lynch D. V.; Caffrey M.; Hogan J. L.; Steponkus P. L. Biophys. J. 1992, 61, 1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullards M. C.; Lynch D. V.; Merrill A. H. Jr.; Adams J. J. Mass Spectrom. 2000, 35, 347. [DOI] [PubMed] [Google Scholar]
- Bartke N.; Fischbeck A.; Humpf H. U. Mol. Nutr. Food Res. 2006, 50, 1201. [DOI] [PubMed] [Google Scholar]
- Sugawara T.; Duan J.; Aida K.; Tsuduki T.; Hirata T.. Lipids, 45, 451. [DOI] [PubMed]
- Blaas N.; Schuurmann C.; Bartke N.; Stahl B.; Humpf H. U. J. Agric. Food Chem. 2011, 59, 6018. [DOI] [PubMed] [Google Scholar]
- Zitomer N. C.; Mitchell T.; Voss K. A.; Bondy G. S.; Pruett S. T.; Garnier-Amblard E. C.; Liebeskind L. S.; Park H.; Wang E.; Sullards M. C.; Merrill A. H. Jr.; Riley R. T. J. Biol. Chem. 2009, 284, 4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penno A.; Reilly M. M.; Houlden H.; Laura M.; Rentsch K.; Niederkofler V.; Stoeckli E. T.; Nicholson G.; Eichler F.; Brown R. H. Jr.; von Eckardstein A.; Hornemann T. J. Biol. Chem. 2010, 285, 11178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fyrst H.; Saba J. D. Biochim. Biophys. Acta 2008, 1781, 448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi Y.; Tanaka T.; Akashi S.; Morimoto S.; Kishimoto Y.; Nagai Y. J. Lipid Res. 2000, 41, 1118. [PubMed] [Google Scholar]
- Carter G. T.; Rinehart K. L. J. Am. Chem. Soc. 1978, 100, 7441. [Google Scholar]
- Umemura T.; Mori K. Agric. Biol. Chem. 1987, 51, 217. [Google Scholar]
- Molinski T. F. Curr. Med. Chem.: Anti-Infect. Agents 2000, 3, 197. [Google Scholar]
- Garrido L.; Zubia E.; Ortega M. J.; Naranjo S.; Salva J. Tetrahedron 2001, 57, 4579. [Google Scholar]
- Zhou B.-N.; Mattern M. P.; Johnson R. K.; Kingston D. G. I. Tetrahedron 2001, 57, 9549. [Google Scholar]
- Cuadros R.; de Garcini E. M.; Wandosell F.; Faircloth G.; Fernandez-Sousa J. M.; Avila J. Cancer Lett. 2000, 152, 23. [DOI] [PubMed] [Google Scholar]
- Gelderblom W. C.; Jaskiewicz K.; Marasas W. F.; Thiel P. G.; Horak R. M.; Vleggaar R.; Kriek N. P. Appl. Environ. Microbiol. 1988, 54, 1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E.; Norred W. P.; Bacon C. W.; Riley R. T.; Merrill A. H. Jr. J. Biol. Chem. 1991, 266, 14486. [PubMed] [Google Scholar]
- Marasas W. F.; Riley R. T.; Hendricks K. A.; Stevens V. L.; Sadler T. W.; Gelineau-van Waes J.; Missmer S. A.; Cabrera J.; Torres O.; Gelderblom W. C.; Allegood J.; Martinez C.; Maddox J.; Miller J. D.; Starr L.; Sullards M. C.; Roman A. V.; Voss K. A.; Wang E.; Merrill A. H. Jr. J. Nutr. 2004, 134, 711. [DOI] [PubMed] [Google Scholar]
- Merrill A. H. Jr.; Liotta D. C.; Riley R. T. Trends Cell Biol. 1996, 6, 218. [DOI] [PubMed] [Google Scholar]
- Miyake Y.; Kozutsumi Y.; Nakamura S.; Fujita T.; Kawasaki T. Biochem. Biophys. Res. Commun. 1995, 211, 396. [DOI] [PubMed] [Google Scholar]
- Hanada K.; Nishijima M.; Fujita T.; Kobayashi S. Biochem. Pharmacol. 2000, 59, 1211. [DOI] [PubMed] [Google Scholar]
- Ikushiro H.; Hayashi H.; Kagamiyama H. Biochemistry 2004, 43, 1082. [DOI] [PubMed] [Google Scholar]
- Fujita T.; Hirose R.; Yoneta M.; Sasaki S.; Inoue K.; Kiuchi M.; Hirase S.; Chiba K.; Sakamoto H.; Arita M. J. Med. Chem. 1996, 39, 4451. [DOI] [PubMed] [Google Scholar]
- Thangada S; Khanna K. M.; Blaho V. A.; Oo M. L.; Im D. S.; Guo C; Lefrancois L; Hla T. J Exp. Med. 2010, 207, 1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne S.; Pyne N. J. Trends Mol. Med. 2011, 17, 463. [DOI] [PubMed] [Google Scholar]
- Ter Braak M.; Claas R. F.; Hegen B.; Labocha S.; Ferreiros N.; Pfeilschifter J.; Huwiler A.; van Echten-Deckert G.; Meyer Zu Heringdorf D. Biochem. Pharmacol. 2011, 81, 617. [DOI] [PubMed] [Google Scholar]
- van Echten-Deckert G.; Zschoche A.; Bar T.; Schmidt R. R.; Raths A.; Heinemann T.; Sandhoff K. J. Biol. Chem. 1997, 272, 15825. [DOI] [PubMed] [Google Scholar]
- Schwartz G. K.; Ward D.; Saltz L.; Casper E. S.; Spiess T.; Mullen E.; Woodworth J.; Venuti R.; Zervos P.; Storniolo A. M.; Kelsen D. P. Clin. Cancer Res. 1997, 3, 537. [PubMed] [Google Scholar]
- Coward J.; Ambrosini G.; Musi E.; Truman J. P.; Haimovitz-Friedman A.; Allegood J. C.; Wang E.; Merrill A. H. Jr.; Schwartz G. K. Autophagy 2009, 5, 184. [DOI] [PubMed] [Google Scholar]
- Dickson M. A.; Carvajal R. D.; Merrill A. H. Jr.; Gonen M.; Cane L. M.; Schwartz G. K. Clin. Cancer Res. 2011, 17, 2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symolon H.; Bushnev A.; Peng Q.; Ramaraju H.; Mays S. G.; Allegood J. C.; Pruett S. T.; Sullards M. C.; Dillehay D. L.; Liotta D. C.; Merrill A. H. Jr. Mol. Cancer Ther. 2011, 10, 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baird R. D.; Kitzen J.; Clarke P. A.; Planting A.; Reade S.; Reid A.; Welsh L.; Lopez Lazaro L.; de las Heras B.; Judson I. R.; Kaye S. B.; Eskens F.; Workman P.; deBono J. S.; Verweij J. Mol. Cancer Ther. 2009, 8, 1430. [DOI] [PubMed] [Google Scholar]
- Schoffski P.; Dumez H.; Ruijter R.; Miguel-Lillo B.; Soto-Matos A.; Alfaro V.; Giaccone G. Cancer Chemother. Pharmacol. 2011, 10.1007/s00280-011-1612-1. [DOI] [PubMed] [Google Scholar]
- Faircloth G.; Cuevas C. Prog. Mol. Subcell. Biol. 2006, 43, 363. [DOI] [PubMed] [Google Scholar]
- Menaldino D. S.; Bushnev A.; Sun A.; Liotta D. C.; Symolon H.; Desai K.; Dillehay D. L.; Peng Q.; Wang E.; Allegood J.; Trotman-Pruett S.; Sullards M. C.; Merrill A. H. Jr. Pharmacol. Res. 2003, 47, 373. [DOI] [PubMed] [Google Scholar]
- Zheng W.; Kollmeyer J.; Symolon H.; Momin A.; Munter E.; Wang E.; Kelly S.; Allegood J. C.; Liu Y.; Peng Q.; Ramaraju H.; Sullards M. C.; Cabot M.; Merrill A. H. Jr. Biochim. Biophys. Acta 2006, 1758, 1864. [DOI] [PubMed] [Google Scholar]
- Fyrst H.; Oskouian B.; Bandhuvula P.; Gong Y.; Byun H. S.; Bittman R.; Lee A. R.; Saba J. D. Cancer Res. 2009, 69, 9457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K.; Zhang X.; Sumanasekera C.; Lester R. L.; Dickson R. C. Biochem. Soc. Trans. 2005, 33, 1170. [DOI] [PubMed] [Google Scholar]
- Wertz P. W. Acta Derm. Venereol. Suppl. (Stockh.) 2000, 208, 7. [DOI] [PubMed] [Google Scholar]
- Wartewig S.; Neubert R. H. Skin Pharmacol. Physiol. 2007, 20, 220. [DOI] [PubMed] [Google Scholar]
- Feingold K. R. J. Lipid Res. 2007, 48, 2531. [DOI] [PubMed] [Google Scholar]
- Walden C. M.; Sandhoff R.; Chuang C. C.; Yildiz Y.; Butters T. D.; Dwek R. A.; Platt F. M.; van der Spoel A. C. J. Biol. Chem. 2007, 282, 32655. [DOI] [PubMed] [Google Scholar]
- Rabionet M.; van der Spoel A. C.; Chuang C. C.; von Tumpling-Radosta B.; Litjens M.; Bouwmeester D.; Hellbusch C. C.; Korner C.; Wiegandt H.; Gorgas K.; Platt F. M.; Grone H. J.; Sandhoff R. J. Biol. Chem. 2008, 283, 13357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Overloop H.; Denizot Y.; Baes M.; Van Veldhoven P. P. Biol. Chem. 2007, 388, 315. [DOI] [PubMed] [Google Scholar]
- Lee T. C.; Ou M. C.; Shinozaki K.; Malone B.; Snyder F. J. Biol. Chem. 1996, 271, 209. [DOI] [PubMed] [Google Scholar]
- Bielawska A.; Linardic C. M.; Hannun Y. A. FEBS Lett. 1992, 307, 211. [DOI] [PubMed] [Google Scholar]
- Bielawska A.; Crane H. M.; Liotta D.; Obeid L. M.; Hannun Y. A. J. Biol. Chem. 1993, 268, 26226. [PubMed] [Google Scholar]
- Bielawska A.; Greenberg M. S.; Perry D.; Jayadev S.; Shayman J. A.; McKay C.; Hannun Y. A. J. Biol. Chem. 1996, 271, 12646. [DOI] [PubMed] [Google Scholar]
- van Echten-Deckert G.; Giannis A.; Schwarz A.; Futerman A. H.; Sandhoff K. J. Biol. Chem. 1998, 273, 1184. [DOI] [PubMed] [Google Scholar]
- Senkal C. E.; Ponnusamy S.; Rossi M. J.; Sundararaj K.; Szulc Z.; Bielawski J.; Bielawska A.; Meyer M.; Cobanoglu B.; Koybasi S.; Sinha D.; Day T. A.; Obeid L. M.; Hannun Y. A.; Ogretmen B. J. Pharmacol. Exp. Ther. 2006, 317, 1188. [DOI] [PubMed] [Google Scholar]
- Struckhoff A. P.; Bittman R.; Burow M. E.; Clejan S.; Elliott S.; Hammond T.; Tang Y.; Beckman B. S. J. Pharmacol. Exp. Ther. 2004, 309, 523. [DOI] [PubMed] [Google Scholar]
- Bittman R.; Li Z.; Samadder P.; Arthur G. Cancer Lett. 2007, 251, 53. [DOI] [PubMed] [Google Scholar]
- Stover T. C.; Kim Y. S.; Lowe T. L.; Kester M. Biomaterials 2008, 29, 359. [DOI] [PubMed] [Google Scholar]
- Liu X.; Ryland L.; Yang J.; Liao A.; Aliaga C.; Watts R.; Tan S. F.; Kaiser J.; Shanmugavelandy S. S.; Rogers A.; Loughran K.; Petersen B.; Yuen J.; Meng F.; Baab K. T.; Jarbadan N. R.; Broeg K.; Zhang R.; Liao J.; Sayers T. J.; Kester M.; Loughran T. P. Jr. Blood 2010, 116, 4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagaram H. R.; Divittore N. A.; Barth B. M.; Kaiser J. M.; Avella D.; Kimchi E. T.; Jiang Y.; Isom H. C.; Kester M.; Staveley-O’Carroll K. F. Gut 2011, 60, 695. [DOI] [PubMed] [Google Scholar]
- Megha; Sawatzki P.; Kolter T.; Bittman R.; London E. Biochim. Biophys. Acta 2007, 1768, 2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goni F. M.; Contreras F. X.; Montes L. R.; Sot J.; Alonso A. Biochem. Soc. Symp. 2005, 177. [DOI] [PubMed] [Google Scholar]
- Contreras F. X.; Basanez G.; Alonso A.; Herrmann A.; Goni F. M. Biophys. J. 2005, 88, 348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siskind L. J.; Kolesnick R. N.; Colombini M. Mitochondrion 2006, 6, 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannun Y. A.; Obeid L. M. J. Biol. Chem. 2002, 277, 25847. [DOI] [PubMed] [Google Scholar]
- Hannun Y. A.; Obeid L. M. Nat. Rev. Mol. Cell Biol. 2008, 9, 139. [DOI] [PubMed] [Google Scholar]
- Gangoiti P.; Camacho L.; Arana L.; Ouro A.; Granado M. H.; Brizuela L.; Casas J.; Fabrias G.; Abad J. L.; Delgado A.; Gomez-Munoz A. Prog. Lipid Res. 2010, 49, 316. [DOI] [PubMed] [Google Scholar]
- Ogretmen B.; Hannun Y. A. Nat. Rev. Cancer 2004, 4, 604. [DOI] [PubMed] [Google Scholar]
- Hannun Y. A.; Obeid L. M. J. Biol. Chem. 2011, 286, 27855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskouian B.; Saba J. D. Adv. Exp. Med. Biol. 2010, 688, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox T. E.; Finnegan C. M.; Blumenthal R.; Kester M. Cell. Mol. Life Sci. 2006, 63, 1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daido S.; Kanzawa T.; Yamamoto A.; Takeuchi H.; Kondo Y.; Kondo S. Cancer Res. 2004, 64, 4286. [DOI] [PubMed] [Google Scholar]
- Scarlatti F.; Bauvy C.; Ventruti A.; Sala G.; Cluzeaud F.; Vandewalle A.; Ghidoni R.; Codogno P. J. Biol. Chem. 2004, 279, 18384. [DOI] [PubMed] [Google Scholar]
- Zauner S.; Ternes P.; Warnecke D. Adv. Exp. Med. Biol. 2010, 688, 249. [DOI] [PubMed] [Google Scholar]
- Jennemann R.; Geyer R.; Sandhoff R.; Gschwind R. M.; Levery S. B.; Grone H. J.; Wiegandt H. Eur. J. Biochem. 2001, 268, 1190. [DOI] [PubMed] [Google Scholar]
- Levery S. B.; Toledo M. S.; Straus A. H.; Takahashi H. K. Rapid Commun. Mass Spectrom. 2001, 15, 2240. [DOI] [PubMed] [Google Scholar]
- Levery S. B. Methods Enzymol. 2005, 405, 300. [DOI] [PubMed] [Google Scholar]
- Itonori S.; Sugita M. In Comprehansive Glycoscience. From Chemistry to Systems Biology; Kamerling J. P., Ed.; Elsevier: Oxford, U.K., 2007; Vol. 3. [Google Scholar]
- Laine R. A. Glycobiology 1994, 4, 759. [DOI] [PubMed] [Google Scholar]
- Levery S. B.; Nudelman E. D.; Salyan M. E.; Hakomori S. Biochemistry 1989, 28, 7772. [DOI] [PubMed] [Google Scholar]
- Fukuda M.; Fukuda M. N.; Papayannopoulou T.; Hakomori S. Proc. Natl. Acad. Sci. U.S.A. 1980, 77, 3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijesinghe D. S.; Allegood J. C.; Gentile L. B.; Fox T. E.; Kester M.; Chalfant C. E. J. Lipid Res. 2010, 51, 641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamour N. F.; Stahelin R. V.; Wijesinghe D. S.; Maceyka M.; Wang E.; Allegood J. C.; Merrill A. H. Jr.; Cho W.; Chalfant C. E. J. Lipid Res. 2007, 48, 1293. [DOI] [PubMed] [Google Scholar]
- Hinkovska-Galcheva V.; Boxer L. A.; Kindzelskii A.; Hiraoka M.; Abe A.; Goparju S.; Spiegel S.; Petty H. R.; Shayman J. A. J. Biol. Chem. 2005, 280, 26612. [DOI] [PubMed] [Google Scholar]
- Gomez-Munoz A. Biochim. Biophys. Acta 2006, 1758, 2049. [DOI] [PubMed] [Google Scholar]
- Gomez-Munoz A.; Kong J. Y.; Salh B.; Steinbrecher U. P. J. Lipid Res. 2004, 45, 99. [DOI] [PubMed] [Google Scholar]
- Gangoiti P.; Arana L.; Ouro A.; Granado M. H.; Trueba M.; Gomez-Munoz A. Cell Signal 2011, 23, 27. [DOI] [PubMed] [Google Scholar]
- Lamour N. F.; Subramanian P.; Wijesinghe D. S.; Stahelin R. V.; Bonventre J. V.; Chalfant C. E. J. Biol. Chem. 2009, 284, 26897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettus B. J.; Chalfant C. E.; Hannun Y. A. Curr. Mol. Med. 2004, 4, 405. [DOI] [PubMed] [Google Scholar]
- Gubern A.; Barcelo-Torns M.; Barneda D.; Lopez J. M.; Masgrau R.; Picatoste F.; Chalfant C. E.; Balsinde J.; Balboa M. A.; Claro E. J. Biol. Chem. 2009, 284, 32359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barenholz Y.; Thompson T. E. Chem. Phys Lipids 1999, 102, 29. [DOI] [PubMed] [Google Scholar]
- Barenholz Y. Subcell Biochem 2004, 37, 167. [DOI] [PubMed] [Google Scholar]
- Koivusalo M.; Jansen M.; Somerharju P.; Ikonen E. Mol. Biol. Cell 2007, 18, 5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogomori H.; Kobayashi T. Biochim. Biophys. Acta 2008, 1780, 612. [DOI] [PubMed] [Google Scholar]
- Kiyokawa E.; Baba T.; Otsuka N.; Makino A.; Ohno S.; Kobayashi T. J. Biol. Chem. 2005, 280, 24072. [DOI] [PubMed] [Google Scholar]
- Muehlenberg B. A.; Sribney M.; Duffe M. K. Can. J. Biochem. 1972, 50, 166. [DOI] [PubMed] [Google Scholar]
- Ternes P.; Brouwers J. F.; van den Dikkenberg J.; Holthuis J. C. J. Lipid Res. 2009, 50, 2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacaru A. M.; Tafesse F. G.; Ternes P.; Kondylis V.; Hermansson M.; Brouwers J. F.; Somerharju P.; Rabouille C.; Holthuis J. C. J. Cell Biol. 2009, 185, 1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masood M. A.; Yuan C.; Acharya J. K.; Veenstra T. D.; Blonder J. Anal. Biochem. 2010, 400, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester R. L.; Dickson R. C. Adv Lipid Res 1993, 26, 253. [PubMed] [Google Scholar]
- Dickson R. C.; Lester R. L. Biochim. Biophys. Acta 1999, 1426, 347. [DOI] [PubMed] [Google Scholar]
- Warnecke D.; Heinz E. Cell. Mol. Life Sci. 2003, 60, 919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillence D. J.; Puri V.; Marks D. L.; Butters T. D.; Dwek R. A.; Pagano R. E.; Platt F. M. J. Lipid Res. 2002, 43, 1837. [DOI] [PubMed] [Google Scholar]
- van Meer G.; Wolthoorn J.; Degroote S. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2003, 358, 869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messner M. C.; Cabot M. C. Adv. Exp. Med. Biol. 2010, 688, 156. [DOI] [PubMed] [Google Scholar]
- Lavie Y.; Cao H.; Bursten S. L.; Giuliano A. E.; Cabot M. C. J. Biol. Chem. 1996, 271, 19530. [DOI] [PubMed] [Google Scholar]
- Liu Y. Y.; Patwardhan G. A.; Xie P.; Gu X.; Giuliano A. E.; Cabot M. C. Int. J. Oncol. 2011, 39, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalazar G.; Ben Ya’acov A.; Livovsky D. M.; El Haj M.; Pappo O.; Preston S.; Zolotarov L.; Ilan Y. Am. J. Pathol. 2009, 174, 1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boggs J. M.; Gao W.; Zhao J.; Park H. J.; Liu Y.; Basu A. FEBS Lett. 2010, 584, 1771. [DOI] [PubMed] [Google Scholar]
- Venkataswamy M. M.; Porcelli S. A. Semin. Immunol. 2010, 22, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff R. D.; Gao Y.; Mattner J.; Zhou D.; Yin N.; Cantu C. 3rd; Teyton L.; Bendelac A.; Savage P. B. J. Am. Chem. Soc. 2004, 126, 13602. [DOI] [PubMed] [Google Scholar]
- Yu K. O.; Im J. S.; Molano A.; Dutronc Y.; Illarionov P. A.; Forestier C.; Fujiwara N.; Arias I.; Miyake S.; Yamamura T.; Chang Y. T.; Besra G. S.; Porcelli S. A. Proc. Natl. Acad. Sci. U.S.A. 2005, 102, 3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka I. Prog. Lipid Res. 1997, 36, 245. [DOI] [PubMed] [Google Scholar]
- Popovic Z. V.; Sandhoff R.; Sijmonsma T. P.; Kaden S.; Jennemann R.; Kiss E.; Tone E.; Autschbach F.; Platt N.; Malle E.; Grone H. J. J. Immunol. 2007, 179, 6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merten M.; Thiagarajan P. Z. Kardiol. 2004, 93, 855. [DOI] [PubMed] [Google Scholar]
- Shikata K.; Suzuki Y.; Wada J.; Hirata K.; Matsuda M.; Kawashima H.; Suzuki T.; Iizuka M.; Makino H.; Miyasaka M. J. Pathol. 1999, 188, 93. [DOI] [PubMed] [Google Scholar]
- Kobayashi T.; Honke K.; Miyazaki T.; Matsumoto K.; Nakamura T.; Ishizuka I.; Makita A. J. Biol. Chem. 1994, 269, 9817. [PubMed] [Google Scholar]
- Don A. S.; Rosen H. Biochem. Biophys. Res. Commun. 2009, 380, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morichika H.; Hamanaka Y.; Tai T.; Ishizuka I. Cancer 1996, 78, 43. [DOI] [PubMed] [Google Scholar]
- Hiraiwa N.; Fukuda Y.; Imura H.; Tadano-Aritomi K.; Nagai K.; Ishizuka I.; Kannagi R. Cancer Res. 1990, 50, 2917. [PubMed] [Google Scholar]
- Sakakibara N.; Gasa S.; Kamio K.; Makita A.; Nonomura K.; Togashi M.; Koyanagi T.; Hatae Y.; Takeda K. Cancer Lett. 1991, 57, 187. [DOI] [PubMed] [Google Scholar]
- Li J.; Pearl D. K.; Pfeiffer S. E.; Yates A. J. J. Neurosci. Res. 1994, 39, 148. [DOI] [PubMed] [Google Scholar]
- Gnewuch C.; Jaques G.; Havemann K.; Wiegandt H. Int. J. Cancer Suppl 1994, 8, 125. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Chen Y.; Momin A.; Shaner R.; Wang E.; Bowen N. J.; Matyunina L. V.; Walker L. D.; McDonald J. F.; Sullards M. C.; Merrill A. H. Jr. Mol. Cancer 2010, 9, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris J. F.; Beaton D. W. Clin. Exp. Metastasis 1990, 8, 361. [DOI] [PubMed] [Google Scholar]
- Iwabuchi K.; Nagaoka I. Blood 2002, 100, 1454. [PubMed] [Google Scholar]
- Chatterjee S.; Pandey A. Biochim. Biophys. Acta 2008, 1780, 370. [DOI] [PubMed] [Google Scholar]
- Won J. S.; Singh A. K.; Singh I. J. Neurochem. 2007, 103 (Suppl 1), 180. [DOI] [PubMed] [Google Scholar]
- Nakayama H.; Yoshizaki F.; Prinetti A.; Sonnino S.; Mauri L.; Takamori K.; Ogawa H.; Iwabuchi K. J. Leukocyte Biol. 2008, 83, 728. [DOI] [PubMed] [Google Scholar]
- Sonnino S.; Prinetti A.; Nakayama H.; Yangida M.; Ogawa H.; Iwabuchi K. Glycoconj. J. 2009, 26, 615. [DOI] [PubMed] [Google Scholar]
- Iwabuchi K.; Nakayama H.; Iwahara C.; Takamori K. FEBS Lett. 2010, 584, 1642. [DOI] [PubMed] [Google Scholar]
- Seah N.; Santacroce P. V.; Basu A. Org. Lett. 2009, 11, 559. [DOI] [PubMed] [Google Scholar]
- Bremer E. G.; Schlessinger J.; Hakomori S. J. Biol. Chem. 1986, 261, 2434. [PubMed] [Google Scholar]
- Zhou Q.; Hakomori S.; Kitamura K.; Igarashi Y. J. Biol. Chem. 1994, 269, 1959. [PubMed] [Google Scholar]
- Yoon S. J.; Nakayama K.; Hikita T.; Handa K.; Hakomori S. I. Proc. Natl. Acad. Sci. U.S.A. 2006, 103, 18987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coskun U.; Grzybek M.; Drechsel D.; Simons K. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 9044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremer E. G.; Hakomori S.; Bowen-Pope D. F.; Raines E.; Ross R. J. Biol. Chem. 1984, 259, 6818. [PubMed] [Google Scholar]
- von Gunten S.; Bochner B. S. Ann. N.Y. Acad. Sci. 2008, 1143, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekri S.; Lidove O.; Jaussaud R.; Knebelmann B.; Barbey F. Cardiovasc. Hematol. Agents Med. Chem. 2006, 4, 289. [DOI] [PubMed] [Google Scholar]
- Utskarpen A.; Massol R.; van Deurs B.; Lauvrak S. U.; Kirchhausen T.; Sandvig K. PLoS One 2010, 5, e10944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adlercreutz D.; Weadge J. T.; Petersen B. O.; Duus J. O.; Dovichi N. J.; Palcic M. M. Carbohydr. Res. 2010, 345, 1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falguieres T.; Maak M.; von Weyhern C.; Sarr M.; Sastre X.; Poupon M. F.; Robine S.; Johannes L.; Janssen K. P. Mol. Cancer Ther. 2008, 7, 2498. [DOI] [PubMed] [Google Scholar]
- Johannes L.; Romer W. Nat. Rev. Microbiol. 2010, 8, 105. [DOI] [PubMed] [Google Scholar]
- Engedal N.; Skotland T.; Torgersen M. L.; Sandvig K. Microb. Biotechnol. 2011, 4, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christiansen D.; Milland J.; Mouhtouris E.; Vaughan H.; Pellicci D. G.; McConville M. J.; Godfrey D. I.; Sandrin M. S. PLoS Biol. 2008, 6, e172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biellmann F.; Hulsmeier A. J.; Zhou D.; Cinelli P.; Hennet T. BMC Dev. Biol. 2008, 8, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuan C. T.; Chang J.; Mansson J. E.; Li J.; Pegram C.; Fredman P.; McLendon R. E.; Bigner D. D. BMC Dev. Biol. 2010, 10, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerlund B.; Slotte J. P. Biochim. Biophys. Acta 2009, 1788, 194. [DOI] [PubMed] [Google Scholar]
- Wennekes T.; van den Berg R. J.; Boot R. G.; van der Marel G. A.; Overkleeft H. S.; Aerts J. M. Angew. Chem., Int. Ed. Engl. 2009, 48, 8848. [DOI] [PubMed] [Google Scholar]
- Degroote S.; Wolthoorn J.; van Meer G. Semin. Cell Dev. Biol. 2004, 15, 375. [DOI] [PubMed] [Google Scholar]
- Ledeen R. W.; Wu G. Biochim. Biophys. Acta 2006, 1761, 588. [DOI] [PubMed] [Google Scholar]
- Dyatlovitskaya E. V. Biochemistry (Moscow) 2007, 72, 479. [DOI] [PubMed] [Google Scholar]
- Meyer zu Heringdorf D.; Jakobs K. H. Biochim. Biophys. Acta 2007, 1768, 923. [DOI] [PubMed] [Google Scholar]
- Nixon G. F.; Mathieson F. A.; Hunter I. Prog. Lipid Res. 2008, 47, 62. [DOI] [PubMed] [Google Scholar]
- Okamoto R.; Arikawa J.; Ishibashi M.; Kawashima M.; Takagi Y.; Imokawa G. J. Lipid Res. 2003, 44, 93. [DOI] [PubMed] [Google Scholar]
- Imokawa G. J. Dermatol. Sci. 2009, 55, 1. [DOI] [PubMed] [Google Scholar]
- Suzuki K. J. Neurochem. Res. 1998, 23, 251. [DOI] [PubMed] [Google Scholar]
- Carter H. E.; Nalbandov O.; Tavormina P. A. J. Biol. Chem. 1951, 192, 197. [PubMed] [Google Scholar]
- Kisic A.; Tsuda M.; Kulmacz R. J.; Wilson W. K.; Schroepfer G. J. Jr. J. Lipid Res. 1995, 36, 787. [PubMed] [Google Scholar]
- Igarashi Y.; Hakomori S. Biochem. Biophys. Res. Commun. 1989, 164, 1411. [DOI] [PubMed] [Google Scholar]
- Morales P. R.; Dillehay D. L.; Moody S. J.; Pallas D. C.; Pruett S.; Allgood J. C.; Symolon H.; Merrill A. H. Jr. Drug Chem. Toxicol. 2007, 30, 197. [DOI] [PubMed] [Google Scholar]
- Doering T.; Proia R. L.; Sandhoff K. FEBS Lett. 1999, 447, 167. [DOI] [PubMed] [Google Scholar]
- Stewart M. E.; Downing D. T. J. Lipid Res. 2001, 42, 1105. [PubMed] [Google Scholar]
- Farwanah H.; Pierstorff B.; Schmelzer C. E.; Raith K.; Neubert R. H.; Kolter T.; Sandhoff K. J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2007, 852, 562. [DOI] [PubMed] [Google Scholar]
- Bosson R.; Guillas I.; Vionnet C.; Roubaty C.; Conzelmann A. Eukaryot. Cell 2009, 8, 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto W. J.; Srinivasan B.; Shepherd S.; Schmidt A.; Dickson R. C.; Lester R. L. J. Bacteriol. 1992, 174, 2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada K.; Nishijima M.; Akamatsu Y. J. Biol. Chem. 1990, 265, 22137. [PubMed] [Google Scholar]
- Hojjati M. R.; Li Z.; Jiang X. C. Biochim. Biophys. Acta 2005, 1737, 44. [DOI] [PubMed] [Google Scholar]
- Adachi-Yamada T.; Gotoh T.; Sugimura I.; Tateno M.; Nishida Y.; Onuki T.; Date H. Mol. Cell. Biol. 1999, 19, 7276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson A. Biochim. Biophys. Acta 1968, 164, 575. [DOI] [PubMed] [Google Scholar]
- Nilsson A. Biochim. Biophys. Acta 1969, 187, 113. [DOI] [PubMed] [Google Scholar]
- Schmelz E. M.; Crall K. J.; Larocque R.; Dillehay D. L.; Merrill A. H. Jr. J. Nutr. 1994, 124, 702. [DOI] [PubMed] [Google Scholar]
- Nilsson A.; Duan R. D. J. Lipid Res. 2006, 47, 154. [DOI] [PubMed] [Google Scholar]
- Vesper H.; Schmelz E. M.; Nikolova-Karakashian M. N.; Dillehay D. L.; Lynch D. V.; Merrill A. H. Jr. J. Nutr. 1999, 129, 1239. [DOI] [PubMed] [Google Scholar]
- Dillehay D. L.; Webb S. K.; Schmelz E. M.; Merrill A. H. Jr. J. Nutr. 1994, 124, 615. [DOI] [PubMed] [Google Scholar]
- Schmelz E. M.; Roberts P. C.; Kustin E. M.; Lemonnier L. A.; Sullards M. C.; Dillehay D. L.; Merrill A. H. Jr. Cancer Res. 2001, 61, 6723. [PubMed] [Google Scholar]
- Symolon H.; Schmelz E. M.; Dillehay D. L.; Merrill A. H. Jr. J. Nutr. 2004, 134, 1157. [DOI] [PubMed] [Google Scholar]
- Inamine M.; Suzui M.; Morioka T.; Kinjo T.; Kaneshiro T.; Sugishita T.; Okada T.; Yoshimi N. Cancer Sci. 2005, 96, 876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon K. W.; Roberts P. C.; Vespremi M. J.; Manchen S.; Schmelz E. M. Mol. Nutr. Food Res. 2009, 53, 332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzei J. C.; Zhou H.; Brayfield B. P.; Hontecillas R.; Bassaganya-Riera J.; Schmelz E. M. J Nutr Biochem 2011, 10.1016/j.jnutbio.2010.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujiwara K.; Kitatani K.; Fukushima K.; Yazama H.; Umehara H.; Kikuchi M.; Igarashi Y.; Kitano H.; Okazaki T. Int. J. Clin. Oncol. 2011, 16, 133. [DOI] [PubMed] [Google Scholar]
- Carter H. E.; Glick F. J.; Norris W. P.; Phillips G. E. J. Biol. Chem. 1947, 170, 285. [Google Scholar]
- Brady R. O.; Koval G. J. J. Biol. Chem. 1958, 233, 26. [PubMed] [Google Scholar]
- Brady R. O.; Formica J. V.; Koval G. J. J. Biol. Chem. 1958, 233, 1072. [PubMed] [Google Scholar]
- Braun P. E.; Snell E. E. Proc. Natl. Acad. Sci. U.S.A. 1967, 58, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffel W.; LeKim D.; Sticht G. Hoppe Seylers Z. Physiol. Chem. 1968, 349, 664. [DOI] [PubMed] [Google Scholar]
- Buede R.; Rinker-Schaffer C.; Pinto W. J.; Lester R. L.; Dickson R. C. J. Bacteriol. 1991, 173, 4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagiec M. M.; Baltisberger J. A.; Wells G. B.; Lester R. L.; Dickson R. C. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 7899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss B.; Stoffel W. Eur. J. Biochem. 1997, 249, 239. [DOI] [PubMed] [Google Scholar]
- Nagiec M. M.; Lester R. L.; Dickson R. C. Gene 1996, 177, 237. [DOI] [PubMed] [Google Scholar]
- Hanada K.; Hara T.; Nishijima M.; Kuge O.; Dickson R. C.; Nagiec M. M. J. Biol. Chem. 1997, 272, 32108. [DOI] [PubMed] [Google Scholar]
- Williams R. D.; Wang E.; Merrill A. H. Jr. Arch. Biochem. Biophys. 1984, 228, 282. [DOI] [PubMed] [Google Scholar]
- Hanada K. Biochim. Biophys. Acta 2003, 1632, 16. [DOI] [PubMed] [Google Scholar]
- Hornemann T.; Wei Y.; von Eckardstein A. Biochem. J. 2007, 405, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J.; Yerokun T.; Leipelt M.; Haynes C. A.; Radhakrishna H.; Momin A.; Kelly S.; Park H.; Wang E.; Carton J. M.; Uhlinger D. J.; Merrill A. H. Jr. Biochim. Biophys. Acta 2009, 1791, 746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carton J. M.; Uhlinger D. J.; Batheja A. D.; Derian C.; Ho G.; Argenteri D.; D’Andrea M. R. J. Histochem. Cytochem. 2003, 51, 715. [DOI] [PubMed] [Google Scholar]
- Mandon E. C.; Ehses I.; Rother J.; van Echten G.; Sandhoff K. J. Biol. Chem. 1992, 267, 11144–8. [PubMed] [Google Scholar]
- Yasuda S.; Nishijima M.; Hanada K. J. Biol. Chem. 2003, 278, 4176. [DOI] [PubMed] [Google Scholar]
- Gable K.; Slife H.; Bacikova D.; Monaghan E.; Dunn T. M. J. Biol. Chem. 2000, 275, 7597. [DOI] [PubMed] [Google Scholar]
- Hanada K.; Hara T.; Nishijima M. J. Biol. Chem. 2000, 275, 8409. [DOI] [PubMed] [Google Scholar]
- Han G.; Gupta S. D.; Gable K.; Niranjanakumari S.; Moitra P.; Eichler F.; Brown R. H. Jr.; Harmon J. M.; Dunn T. M. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inuzuka M.; Hayakawa M.; Ingi T. J. Biol. Chem. 2005, 280, 35776. [DOI] [PubMed] [Google Scholar]
- Breslow D. K.; Collins S. R.; Bodenmiller B.; Aebersold R.; Simons K.; Shevchenko A.; Ejsing C. S.; Weissman J. S. Nature 2010, 463, 1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han S.; Lone M. A.; Schneiter R.; Chang A. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavin A. C.; Bosche M.; Krause R.; Grandi P.; Marzioch M.; Bauer A.; Schultz J.; Rick J. M.; Michon A. M.; Cruciat C. M.; Remor M.; Hofert C.; Schelder M.; Brajenovic M.; Ruffner H.; Merino A.; Klein K.; Hudak M.; Dickson D.; Rudi T.; Gnau V.; Bauch A.; Bastuck S.; Huhse B.; Leutwein C.; Heurtier M. A.; Copley R. R.; Edelmann A.; Querfurth E.; Rybin V.; Drewes G.; Raida M.; Bouwmeester T.; Bork P.; Seraphin B.; Kuster B.; Neubauer G.; Superti-Furga G. Nature 2002, 415, 141. [DOI] [PubMed] [Google Scholar]
- Giot L.; Bader J. S.; Brouwer C.; Chaudhuri A.; Kuang B.; Li Y.; Hao Y. L.; Ooi C. E.; Godwin B.; Vitols E.; Vijayadamodar G.; Pochart P.; Machineni H.; Welsh M.; Kong Y.; Zerhusen B.; Malcolm R.; Varrone Z.; Collis A.; Minto M.; Burgess S.; McDaniel L.; Stimpson E.; Spriggs F.; Williams J.; Neurath K.; Ioime N.; Agee M.; Voss E.; Furtak K.; Renzulli R.; Aanensen N.; Carrolla S.; Bickelhaupt E.; Lazovatsky Y.; DaSilva A.; Zhong J.; Stanyon C. A.; Finley R. L. Jr.; White K. P.; Braverman M.; Jarvie T.; Gold S.; Leach M.; Knight J.; Shimkets R. A.; McKenna M. P.; Chant J.; Rothberg J. M. Science 2003, 302, 1727. [DOI] [PubMed] [Google Scholar]
- Yard B. A.; Carter L. G.; Johnson K. A.; Overton I. M.; Dorward M.; Liu H.; McMahon S. A.; Oke M.; Puech D.; Barton G. J.; Naismith J. H.; Campopiano D. J. J. Mol. Biol. 2007, 370, 870. [DOI] [PubMed] [Google Scholar]
- Raman M. C.; Johnson K. A.; Yard B. A.; Lowther J.; Carter L. G.; Naismith J. H.; Campopiano D. J. J. Biol. Chem. 2009, 284, 17328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowther J.; Yard B. A.; Johnson K. A.; Carter L. G.; Bhat V. T.; Raman M. C.; Clarke D. J.; Ramakers B.; McMahon S. A.; Naismith J. H.; Campopiano D. J. Mol. Biosyst. 2010, 6, 1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikushiro H.; Hayashi H.; Kagamiyama H. J. Biol. Chem. 2001, 276, 18249. [DOI] [PubMed] [Google Scholar]
- Ikushiro H.; Islam M. M.; Tojo H.; Hayashi H. J. Bacteriol. 2007, 189, 5749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikushiro H.; Islam M. M.; Okamoto A.; Hoseki J.; Murakawa T.; Fujii S.; Miyahara I.; Hayashi H. J. Biochem. 2009, 146, 549. [DOI] [PubMed] [Google Scholar]
- Ikushiro H.; Hayashi H. Biochim. Biophys. Acta 2011, 10.1016/j.bbapap.2011.02.005. [DOI] [PubMed] [Google Scholar]
- Krisnangkura K.; Sweeley C. C. J. Biol. Chem. 1976, 251, 1597. [PubMed] [Google Scholar]
- Dawkins J. L.; Hulme D. J.; Brahmbhatt S. B.; Auer-Grumbach M.; Nicholson G. A. Nat. Genet. 2001, 27, 309. [DOI] [PubMed] [Google Scholar]
- Bejaoui K.; Uchida Y.; Yasuda S.; Ho M.; Nishijima M.; Brown R. H. Jr.; Holleran W. M.; Hanada K. J. Clin. Invest. 2002, 110, 1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotthier A.; Auer-Grumbach M.; Janssens K.; Baets J.; Penno A.; Almeida-Souza L.; Van Hoof K.; Jacobs A.; De Vriendt E.; Schlotter-Weigel B.; Loscher W.; Vondracek P.; Seeman P.; De Jonghe P.; Van Dijck P.; Jordanova A.; Hornemann T.; Timmerman V. Am. J. Hum. Genet. 2010, 87, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler F. S.; Hornemann T.; McCampbell A.; Kuljis D.; Penno A.; Vardeh D.; Tamrazian E.; Garofalo K.; Lee H. J.; Kini L.; Selig M.; Frosch M.; Gable K.; von Eckardstein A.; Woolf C. J.; Guan G.; Harmon J. M.; Dunn T. M.; Brown R. H. Jr. J. Neurosci. 2009, 29, 14646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotthier A.; Penno A.; Rautenstrauss B.; Auer-Grumbach M.; Stettner G. M.; Asselbergh B.; Van Hoof K.; Sticht H.; Levy N.; Timmerman V.; Hornemann T.; Janssens K. Hum. Mutat 2011, 32, E2211. [DOI] [PubMed] [Google Scholar]
- Gable K.; Gupta S. D.; Han G.; Niranjanakumari S.; Harmon J. M.; Dunn T. M. J. Biol. Chem. 2010, 285, 22846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humpf H. U.; Schmelz E. M.; Meredith F. I.; Vesper H.; Vales T. R.; Wang E.; Menaldino D. S.; Liotta D. C.; Merrill A. H. Jr. J. Biol. Chem. 1998, 273, 19060. [DOI] [PubMed] [Google Scholar]
- Xue H. H.; Fujie M.; Sakaguchi T.; Oda T.; Ogawa H.; Kneer N. M.; Lardy H. A.; Ichiyama A. J. Biol. Chem. 1999, 274, 16020. [DOI] [PubMed] [Google Scholar]
- Bertea M.; Rutti M. F.; Othman A.; Marti-Jaun J.; Hersberger M.; von Eckardstein A.; Hornemann T. Lipids Health Dis. 2010, 9, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokvis E.; Nan-Offeringa L.; Rosing H.; Lopez-Lazaro L.; Acena J. L.; Miranda E.; Lyubimov A.; Levine B. S.; D’Aleo C.; Schellens J. H.; Beijnen J. H. J. Mass Spectrom. 2003, 38, 548. [DOI] [PubMed] [Google Scholar]
- Sanchez A. M.; Malagarie-Cazenave S.; Olea N.; Vara D.; Cuevas C.; Diaz-Laviada I. Eur. J. Pharmacol. 2008, 584, 237. [DOI] [PubMed] [Google Scholar]
- Vilar E.; Grunwald V.; Schoffski P.; Singer H.; Salazar R.; Iglesias J. L.; Casado E.; Cullell-Young M.; Baselga J.; Tabernero J. Invest. New Drugs 2010, 10.1007/s10637-010-9529-9. [DOI] [PubMed] [Google Scholar]
- Hanada K.; Hara T.; Nishijima M. FEBS Lett. 2000, 474, 63. [DOI] [PubMed] [Google Scholar]
- Nagata Y.; Masui R.; Akino T. Experientia 1992, 48, 986. [DOI] [PubMed] [Google Scholar]
- Grant S. L.; Shulman Y.; Tibbo P.; Hampson D. R.; Baker G. B. J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2006, 844, 278. [DOI] [PubMed] [Google Scholar]
- Huang Y.; Nishikawa T.; Satoh K.; Iwata T.; Fukushima T.; Santa T.; Homma H.; Imai K. Biol. Pharm. Bull. 1998, 21, 156. [DOI] [PubMed] [Google Scholar]
- Williams R. E.; Lock E. A. Toxicology 2004, 201, 231. [DOI] [PubMed] [Google Scholar]
- Haynes C. A.; Allegood J. C.; Sims K.; Wang E. W.; Sullards M. C.; Merrill A. H. Jr. J. Lipid Res. 2008, 49, 1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornemann T.; Penno A.; Rutti M. F.; Ernst D.; Kivrak-Pfiffner F.; Rohrer L.; von Eckardstein A. J. Biol. Chem. 2009, 284, 26322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momin A. A.; Park H.; Portz B. J.; Haynes C. A.; Shaner R. L.; Kelly S. L.; Jordan I. K.; Merrill A. H. Jr. J. Lipid Res. 2011, 52, 1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Acharya U.; Acharya J. K. Cell. Mol. Life Sci. 2005, 62, 128. [DOI] [PubMed] [Google Scholar]
- Fyrst H.; Zhang X.; Herr D. R.; Byun H. S.; Bittman R.; Phan V. H.; Harris G. L.; Saba J. D. J. Lipid Res. 2008, 49, 597. [DOI] [PubMed] [Google Scholar]
- Merrill A. H. Jr. & Sandhoff K In New Comprehensive Biochemistry: Biochemistry of Lipids,Lipoproteins,and Membranes; Vance D. E., Vance J. E., Eds.; Elsevier Science Publ.: Amsterdam, 2002; Vol. Chapter 14. [Google Scholar]
- Merrill A. H. Jr.; Wang E.; Mullins R. E. Biochemistry 1988, 27, 340. [DOI] [PubMed] [Google Scholar]
- Kolter T. Chem. Phys. Lipids 2011, 164, 590. [DOI] [PubMed] [Google Scholar]
- Hu W.; Bielawski J.; Samad F.; Merrill A. H. Jr.; Cowart L. A. J. Lipid Res. 2009, 50, 1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaji-Hasegawa A.; Takahashi A.; Tetsuka Y.; Senoh Y.; Kobayashi T. Biochemistry 2005, 44, 268. [DOI] [PubMed] [Google Scholar]
- Zweerink M. M.; Edison A. M.; Wells G. B.; Pinto W.; Lester R. L. J. Biol. Chem. 1992, 267, 25032. [PubMed] [Google Scholar]
- Mandala S. M.; Frommer B. R.; Thornton R. A.; Kurtz M. B.; Young N. M.; Cabello M. A.; Genilloud O.; Liesch J. M.; Smith J. L.; Horn W. S. J. Antibiot. (Tokyo) 1994, 47, 376. [DOI] [PubMed] [Google Scholar]
- Mandala S. M.; Thornton R. A.; Frommer B. R.; Dreikorn S.; Kurtz M. B. J. Antibiot. (Tokyo) 1997, 50, 339. [DOI] [PubMed] [Google Scholar]
- Medlock K. A.; Merrill A. H. Jr. Biochemistry 1988, 27, 7079. [DOI] [PubMed] [Google Scholar]
- Bahtiar A.; Matsumoto T.; Nakamura T.; Akiyama M.; Yogo K.; Ishida-Kitagawa N.; Ogawa T.; Takeya T. J. Biol. Chem. 2009, 284, 34157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada K.; Hara T.; Fukasawa M.; Yamaji A.; Umeda M.; Nishijima M. J. Biol. Chem. 1998, 273, 33787. [DOI] [PubMed] [Google Scholar]
- Momin A. A.; Park H.; Allegood J. C.; Leipelt M.; Kelly S. L.; Merrill A. H. Jr.; Hanada K. Lipids 2009, 44, 725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta E.; Ohira T.; Matsue K.; Ikeda Y.; Fujii K.; Ohwaki K.; Osuka S.; Hirabayashi Y.; Sasaki M. Exp. Anim. 2009, 58, 515. [DOI] [PubMed] [Google Scholar]
- Kihara A.; Igarashi Y. J. Biol. Chem. 2004, 279, 49243. [DOI] [PubMed] [Google Scholar]
- Levy M.; Futerman A. H. IUBMB Life 2010, 62, 347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiban J.; Tidhar R.; Futerman A. H. Adv. Exp. Med. Biol. 2010, 688, 60. [DOI] [PubMed] [Google Scholar]
- Maceyka M.; Sankala H.; Hait N. C.; Le Stunff H.; Liu H.; Toman R.; Collier C.; Zhang M.; Satin L. S.; Merrill A. H. Jr.; Milstien S.; Spiegel S. J. Biol. Chem. 2005, 280, 37118. [DOI] [PubMed] [Google Scholar]
- Siow D. L.; Anderson C. D.; Berdyshev E. V.; Skobeleva A.; Pitson S. M.; Wattenberg B. W. J. Lipid Res. 2010, 51, 2546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillas I.; Kirchman P. A.; Chuard R.; Pfefferli M.; Jiang J. C.; Jazwinski S. M.; Conzelmann A. EMBO J. 2001, 20, 2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schorling S.; Vallee B.; Barz W. P.; Riezman H.; Oesterhelt D. Mol. Biol. Cell 2001, 12, 3417–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallee B.; Riezman H. EMBO J. 2005, 24, 730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman K.; Riebeling C.; Bodennec J.; Riezman H.; Allegood J. C.; Sullards M. C.; Merrill A. H. Jr.; Futerman A. H. J. Biol. Chem. 2002, 277, 35642. [DOI] [PubMed] [Google Scholar]
- Riebeling C.; Allegood J. C.; Wang E.; Merrill A. H. Jr.; Futerman A. H. J. Biol. Chem. 2003, 278, 43452. [DOI] [PubMed] [Google Scholar]
- Mizutani Y.; Kihara A.; Igarashi Y. Biochem. J. 2005, 390, 263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri S.; Futerman A. H. J. Biol. Chem. 2005, 280, 33735. [DOI] [PubMed] [Google Scholar]
- Pewzner-Jung Y.; Ben-Dor S.; Futerman A. H. J. Biol. Chem. 2006, 281, 25001. [DOI] [PubMed] [Google Scholar]
- Guillas I.; Jiang J. C.; Vionnet C.; Roubaty C.; Uldry D.; Chuard R.; Wang J.; Jazwinski S. M.; Conzelmann A. J. Biol. Chem. 2003, 278, 37083. [DOI] [PubMed] [Google Scholar]
- Sridevi P.; Alexander H.; Laviad E. L.; Pewzner-Jung Y.; Hannink M.; Futerman A. H.; Alexander S. Biochim. Biophys. Acta 2009, 1793, 1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sridevi P.; Alexander H.; Laviad E. L.; Min J.; Mesika A.; Hannink M.; Futerman A. H.; Alexander S. Exp. Cell Res. 2010, 316, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koybasi S.; Senkal C. E.; Sundararaj K.; Spassieva S.; Bielawski J.; Osta W.; Day T. A.; Jiang J. C.; Jazwinski S. M.; Hannun Y. A.; Obeid L. M.; Ogretmen B. J. Biol. Chem. 2004, 279, 44311. [DOI] [PubMed] [Google Scholar]
- Karahatay S.; Thomas K.; Koybasi S.; Senkal C. E.; Elojeimy S.; Liu X.; Bielawski J.; Day T. A.; Gillespie M. B.; Sinha D.; Norris J. S.; Hannun Y. A.; Ogretmen B. Cancer Lett. 2007, 256, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wooten-Blanks L. G.; Song P.; Senkal C. E.; Ogretmen B. FASEB J. 2007, 21, 3386. [DOI] [PubMed] [Google Scholar]
- Gencer E. B.; Ural A. U.; Avcu F.; Baran Y. Ann. Hematol. 2011, 10.1007/s00277-011-1212-5. [DOI] [PubMed] [Google Scholar]
- Zhao L.; Spassieva S. D.; Jucius T. J.; Shultz L. D.; Shick H. E.; Macklin W. B.; Hannun Y. A.; Obeid L. M.; Ackerman S. L. PLoS Genet. 2011, 7, e1002063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laviad E. L.; Albee L.; Pankova-Kholmyansky I.; Epstein S.; Park H.; Merrill A. H. Jr.; Futerman A. H. J. Biol. Chem. 2008, 283, 5677. [DOI] [PubMed] [Google Scholar]
- Imgrund S.; Hartmann D.; Farwanah H.; Eckhardt M.; Sandhoff R.; Degen J.; Gieselmann V.; Sandhoff K.; Willecke K. J. Biol. Chem. 2009, 284, 33549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pewzner-Jung Y.; Park H.; Laviad E. L.; Silva L. C.; Lahiri S.; Stiban J.; Erez-Roman R.; Brugger B.; Sachsenheimer T.; Wieland F.; Prieto M.; Merrill A. H. Jr.; Futerman A. H. J. Biol. Chem. 2010, 285, 10902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pewzner-Jung Y.; Brenner O.; Braun S.; Laviad E. L.; Ben-Dor S.; Feldmesser E.; Horn-Saban S.; Amann-Zalcenstein D.; Raanan C.; Berkutzki T.; Erez-Roman R.; Ben-David O.; Levy M.; Holzman D.; Park H.; Nyska A.; Merrill A. H. Jr.; Futerman A. H. J. Biol. Chem. 2010, 285, 10911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker I.; Wang-Eckhardt L.; Yaghootfam A.; Gieselmann V.; Eckhardt M. Histochem. Cell Biol. 2008, 129, 233. [DOI] [PubMed] [Google Scholar]
- Ben-David O.; Pewzner-Jung Y.; Brenner O.; Laviad E. L.; Kogot-Levin A.; Weissberg I.; Biton I. E.; Pienik R.; Wang E.; Kelly S.; Alroy J.; Raas-Rothschild A.; Friedman A.; Brugger B.; Merrill A. H. Jr.; Futerman A. H. J. Biol. Chem. 2011, 10.1074/jbc.M111.261206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erez-Roman R.; Pienik R.; Futerman A. H. Biochem. Biophys. Res. Commun. 2010, 391, 219. [DOI] [PubMed] [Google Scholar]
- Sandhoff R. FEBS Lett. 2010, 584, 1907. [DOI] [PubMed] [Google Scholar]
- Mizutani Y.; Kihara A.; Chiba H.; Tojo H.; Igarashi Y. J. Lipid Res. 2008, 49, 2356. [DOI] [PubMed] [Google Scholar]
- Park H.; Haynes C. A.; Nairn A. V.; Kulik M.; Dalton S.; Moremen K.; Merrill A. H. Jr. J. Lipid Res. 2010, 51, 480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson K.; Sander B.; Bielawski J.; Hannun Y. A.; Flygare J. Mol. Cancer Res. 2009, 7, 1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veret J.; Coant N.; Berdyshev E.; Skobeleva A.; Therville N.; Bailbe D.; Gorshkova I.; Natarajan V.; Portha B.; Le Stunff H. Biochem. J. 2011, 438, 177. [DOI] [PubMed] [Google Scholar]
- Mesicek J.; Lee H.; Feldman T.; Jiang X.; Skobeleva A.; Berdyshev E. V.; Haimovitz-Friedman A.; Fuks Z.; Kolesnick R. Cell Signal 2010, 22, 1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novgorodov S. A.; Chudakova D. A.; Wheeler B. W.; Bielawski J.; Kindy M. S.; Obeid L. M.; Gudz T. I. J. Biol. Chem. 2011, 286, 4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffmann S.; Ziebell S.; Sandner J.; Birod K.; Deckmann K.; Hartmann D.; Rode S.; Schmidt H.; Angioni C.; Geisslinger G.; Grosch S. Biochem. Pharmacol. 2010, 80, 1632. [DOI] [PubMed] [Google Scholar]
- White-Gilbertson S.; Mullen T.; Senkal C.; Lu P.; Ogretmen B.; Obeid L.; Voelkel-Johnson C. Oncogene 2009, 28, 1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahiri S.; Lee H.; Mesicek J.; Fuks Z.; Haimovitz-Friedman A.; Kolesnick R. N.; Futerman A. H. FEBS Lett. 2007, 581, 5289. [DOI] [PubMed] [Google Scholar]
- Mullen T. D.; Spassieva S.; Jenkins R. W.; Kitatani K.; Bielawski J.; Hannun Y. A.; Obeid L. M. J. Lipid Res. 2010, 52, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno Y.; Suto S.; Yamanaka M.; Mizutani Y.; Mitsutake S.; Igarashi Y.; Sassa T.; Kihara A. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 18439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marasas W. F. Environ. Health Perspect. 2001, 109 (Suppl 2), 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelineau-van Waes J.; Voss K. A.; Stevens V. L.; Speer M. C.; Riley R. T. Adv. Food Nutr. Res. 2009, 56, 145. [DOI] [PubMed] [Google Scholar]
- Merrill A. H. Jr.; Sullards M. C.; Wang E.; Voss K. A.; Riley R. T. Environ. Health Perspect. 2001, 109 (Suppl 2), 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley R. T.; Enongene E.; Voss K. A.; Norred W. P.; Meredith F. I.; Sharma R. P.; Spitsbergen J.; Williams D. E.; Carlson D. B.; Merrill A. H. Jr. Environ. Health Perspect. 2001, 109 (Suppl 2), 301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelineau-van Waes J.; Starr L.; Maddox J.; Aleman F.; Voss K. A.; Wilberding J.; Riley R. T. Birth Defects Res. A Clin. Mol. Teratol. 2005, 73, 487. [DOI] [PubMed] [Google Scholar]
- Sharma R. P.; Bhandari N.; He Q.; Riley R. T.; Voss K. A. Toxicology 2001, 159, 69. [DOI] [PubMed] [Google Scholar]
- Voss K. A.; Riley R.; Dunn C.; Christopher Corton J. Toxicology 2006, 222, 165. [DOI] [PubMed] [Google Scholar]
- He Q.; Kim J.; Sharma R. P. Toxicol. Sci. 2004, 80, 335. [DOI] [PubMed] [Google Scholar]
- Memon R. A.; Holleran W. M.; Moser A. H.; Seki T.; Uchida Y.; Fuller J.; Shigenaga J. K.; Grunfeld C.; Feingold K. R. Arterioscler., Thromb., Vasc. Biol. 1998, 18, 1257. [DOI] [PubMed] [Google Scholar]
- Meyer S. G.; de Groot H. Biochim. Biophys. Acta 2003, 1643, 1. [DOI] [PubMed] [Google Scholar]
- Osawa Y.; Uchinami H.; Bielawski J.; Schwabe R. F.; Hannun Y. A.; Brenner D. A. J. Biol. Chem. 2005, 280, 27879. [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S.; Genovese T.; Mazzon E.; Esposito E.; Crisafulli C.; Di Paola R.; Bramanti P.; Salvemini D. Shock 2009, 31, 170. [DOI] [PubMed] [Google Scholar]
- Kita K.; Okino N.; Ito M. Biochim. Biophys. Acta 2000, 1485, 111. [DOI] [PubMed] [Google Scholar]
- Okino N.; He X.; Gatt S.; Sandhoff K.; Ito M.; Schuchman E. H. J. Biol. Chem. 2003, 278, 29948. [DOI] [PubMed] [Google Scholar]
- Novgorodov S. A.; Wu B. X.; Gudz T. I.; Bielawski J.; Ovchinnikova T. V.; Hannun Y. A.; Obeid L. M. J. Biol. Chem. 2011, 286, 25352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T. Adv. Exp. Med. Biol. 1996, 416, 113. [DOI] [PubMed] [Google Scholar]
- Servillo L.; Balestrieri C.; Giovane A.; Pari P.; Palma D.; Giannattasio G.; Triggiani M.; Balestrieri M. L. FASEB J. 2006, 20, 1015. [DOI] [PubMed] [Google Scholar]
- Ong D. E.; Brady R. N. J. Biol. Chem. 1973, 248, 3884. [PubMed] [Google Scholar]
- Michel C.; van Echten-Deckert G.; Rother J.; Sandhoff K.; Wang E.; Merrill A. H. Jr. J. Biol. Chem. 1997, 272, 22432. [DOI] [PubMed] [Google Scholar]
- Sperling P.; Zahringer U.; Heinz E. J. Biol. Chem. 1998, 273, 28590. [DOI] [PubMed] [Google Scholar]
- Sperling P.; Blume A.; Zahringer U.; Heinz E. Biochem. Soc. Trans. 2000, 28, 638. [PubMed] [Google Scholar]
- Ternes P.; Franke S.; Zahringer U.; Sperling P.; Heinz E. J. Biol. Chem. 2002, 277, 25512. [DOI] [PubMed] [Google Scholar]
- Omae F.; Miyazaki M.; Enomoto A.; Suzuki M.; Suzuki Y.; Suzuki A. Biochem. J. 2004, 379, 687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Omae F.; Miyazaki M.; Enomoto A.; Suzuki A. FEBS Lett. 2004, 576, 63. [DOI] [PubMed] [Google Scholar]
- Mizutani Y.; Kihara A.; Igarashi Y. FEBS Lett. 2004, 563, 93. [DOI] [PubMed] [Google Scholar]
- Kok J. W.; Nikolova-Karakashian M.; Klappe K.; Alexander C.; Merrill A. H. Jr. J. Biol. Chem. 1997, 272, 21128. [DOI] [PubMed] [Google Scholar]
- Beauchamp E.; Goenaga D.; Le Bloc’h J.; Catheline D.; Legrand P.; Rioux V. Biochimie 2007, 89, 1553. [DOI] [PubMed] [Google Scholar]
- Wang H.; Charles A. G.; Frankel A. J.; Cabot M. C. Urology 2003, 61, 1047. [DOI] [PubMed] [Google Scholar]
- Reynolds C. P.; Maurer B. J.; Kolesnick R. N. Cancer Lett. 2004, 206, 169. [DOI] [PubMed] [Google Scholar]
- Wang H.; Maurer B. J.; Reynolds C. P.; Cabot M. C. Cancer Res. 2001, 61, 5102. [PubMed] [Google Scholar]
- Rahmaniyan M.; Curley R. W. Jr.; Obeid L. M.; Hannun Y. A.; Kraveka J. M. J. Biol. Chem. 2011, 286, 24754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Maurer B. J.; Liu Y. Y.; Wang E.; Allegood J. C.; Kelly S.; Symolon H.; Liu Y.; Merrill A. H. Jr.; Gouaze-Andersson V.; Yu J. Y.; Giuliano A. E.; Cabot M. C. Mol. Cancer Ther. 2008, 7, 2967. [DOI] [PubMed] [Google Scholar]
- Mao Z.; Sun W.; Xu R.; Novgorodov S.; Szulc Z. M.; Bielawski J.; Obeid L. M.; Mao C. J. Biol. Chem. 2010, 285, 29078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Triola G.; Fabrias G.; Llebaria A. Angew. Chem., Int. Ed. 2001, 40, 1960. [PubMed] [Google Scholar]
- Triola G.; Fabrias G.; Casas J.; Llebaria A. J. Org. Chem. 2003, 68, 9924. [DOI] [PubMed] [Google Scholar]
- Triola G.; Fabrias G.; Dragusin M.; Niederhausen L.; Broere R.; Llebaria A.; van Echten-Deckert G. Mol. Pharmacol. 2004, 66, 1671. [DOI] [PubMed] [Google Scholar]
- Bedia C.; Triola G.; Casas J.; Llebaria A.; Fabrias G. Org. Biomol. Chem. 2005, 3, 3707. [DOI] [PubMed] [Google Scholar]
- Munoz-Olaya J. M.; Matabosch X.; Bedia C.; Egido-Gabas M.; Casas J.; Llebaria A.; Delgado A.; Fabrias G. Chem. Med. Chem. 2008, 3, 946. [DOI] [PubMed] [Google Scholar]
- Hu W.; Ross J.; Geng T.; Brice S. E.; Cowart L. A. J. Biol. Chem. 2011, 286, 16596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Idkowiak-Baldys J.; Apraiz A.; Li L.; Rahmaniyan M.; Clarke C. J.; Kraveka J. M.; Asumendi A.; Hannun Y. A. Biochem. J. 2010, 427, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa K.; Tsuchida A.; Furukawa K. In Comprehansive Glycoscience. From Chemistry to Systems Biology; Kamerling J. P., Ed.; Elsevier: Oxford, U.K., 2007; Vol. 3. [Google Scholar]
- Suzuki A.Map 3. Biosynthetic Pathways of Glycosphingolipids; Springer-Verlag: Tokyo, 2002. [Google Scholar]
- Nairn A. V.; York W. S.; Harris K.; Hall E. M.; Pierce J. M.; Moremen K. W. J. Biol. Chem. 2008, 283, 17298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe A.; Hiraoka M.; Shayman J. A. J. Lipid Res. 2007, 48, 2255. [DOI] [PubMed] [Google Scholar]
- Futerman A. H.; Stieger B.; Hubbard A. L.; Pagano R. E. J. Biol. Chem. 1990, 265, 8650. [PubMed] [Google Scholar]
- Jeckel D.; Karrenbauer A.; Birk R.; Schmidt R. R.; Wieland F. FEBS Lett. 1990, 261, 155. [DOI] [PubMed] [Google Scholar]
- van den Hill A.; van Heusden G. P.; Wirtz K. W. Biochim. Biophys. Acta 1985, 833, 354. [DOI] [PubMed] [Google Scholar]
- Malgat M.; Maurice A.; Baraud J. J. Lipid Res. 1986, 27, 251. [PubMed] [Google Scholar]
- Ullman M. D.; Radin N. S. J. Biol. Chem. 1974, 249, 1506. [PubMed] [Google Scholar]
- Voelker D. R.; Kennedy E. P. Biochemistry 1982, 21, 2753. [DOI] [PubMed] [Google Scholar]
- Huitema K.; van den Dikkenberg J.; Brouwers J. F.; Holthuis J. C. EMBO J. 2004, 23, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holthuis J. C.; Luberto C. Adv. Exp. Med. Biol.. 2010, 688, 72. [DOI] [PubMed] [Google Scholar]
- Luberto C.; Hannun Y. A. J. Biol. Chem. 1998, 273, 14550. [DOI] [PubMed] [Google Scholar]
- Liu J.; Zhang H.; Li Z.; Hailemariam T. K.; Chakraborty M.; Jiang K.; Qiu D.; Bui H. H.; Peake D. A.; Kuo M. S.; Wadgaonkar R.; Cao G.; Jiang X. C. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gowda S.; Yeang C.; Wadgaonkar S.; Anjum F.; Grinkina N.; Cutaia M.; Jiang X. C.; Wadgaonkar R. Am. J. Physiol. Lung Cell Mol. Physiol. 2011, 300, L430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J.; Huan C.; Chakraborty M.; Zhang H.; Lu D.; Kuo M. S.; Cao G.; Jiang X. C. Circ. Res. 2009, 105, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolova-Karakashian M. Methods Enzymol. 2000, 311, 31. [DOI] [PubMed] [Google Scholar]
- Bornancin F. Cell Signal 2011, 23, 999. [DOI] [PubMed] [Google Scholar]
- Wijesinghe D. S.; Massiello A.; Subramanian P.; Szulc Z.; Bielawska A.; Chalfant C. E. J. Lipid Res. 2005, 46, 2706. [DOI] [PubMed] [Google Scholar]
- Gomez-Munoz A.; Gangoiti P.; Granado M. H.; Arana L.; Ouro A. Adv. Exp. Med. Biol.. 2010, 688, 118. [DOI] [PubMed] [Google Scholar]
- Chalfant C. E.; Spiegel S. J. Cell Sci. 2005, 118, 4605. [DOI] [PubMed] [Google Scholar]
- Graf C.; Klumpp M.; Habig M.; Rovina P.; Billich A.; Baumruker T.; Oberhauser B.; Bornancin F. Mo Pharmacol. 2008, 74, 925. [DOI] [PubMed] [Google Scholar]
- Funakoshi T.; Yasuda S.; Fukasawa M.; Nishijima M.; Hanada K. J. Biol. Chem. 2000, 275, 29938. [DOI] [PubMed] [Google Scholar]
- Hanada K.; Kumagai K.; Yasuda S.; Miura Y.; Kawano M.; Fukasawa M.; Nishijima M. Nature 2003, 426, 803. [DOI] [PubMed] [Google Scholar]
- Hanada K.; Kumagai K.; Tomishige N.; Kawano M. Biochim. Biophys. Acta 2007, 1771, 644. [DOI] [PubMed] [Google Scholar]
- Hanada K. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2010, 86, 426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumagai K.; Yasuda S.; Okemoto K.; Nishijima M.; Kobayashi S.; Hanada K. J. Biol. Chem. 2005, 280, 6488. [DOI] [PubMed] [Google Scholar]
- Duris A.; Wiesenganger T.; Moravcikova D.; Baran P.; Kozisek J.; Daich A.; Berkes D. Org. Lett. 2011, 13, 1642. [DOI] [PubMed] [Google Scholar]
- Yasuda S.; Kitagawa H.; Ueno M.; Ishitani H.; Fukasawa M.; Nishijima M.; Kobayashi S.; Hanada K. J. Biol. Chem. 2001, 276, 43994. [DOI] [PubMed] [Google Scholar]
- Shayman J. A.; Kelly R.; Kollmeyer J.; He Y.; Abe A. Prog. Lipid Res. 2011, 50, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Genderen I.; van Meer G. J. Cell Biol.. 1995, 131, 645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirabayashi Y.; Ichikawa S. In Handbook of Glycosyltransferases and Related Genes; Taniguchi N., Honke K., Fukuda M., Eds.; Springer-Verlag Publishers: Tokyo, 2002. [Google Scholar]
- Ichikawa S.; Nakajo N.; Sakiyama H.; Hirabayashi Y. Proc. Natl. Acad. Sci. U.S.A. 1994, 91, 2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hidari K.; Ichikawa S.; Fujita T.; Sakiyama H.; Hirabayashi Y. J. Biol. Chem. 1996, 271, 14636. [DOI] [PubMed] [Google Scholar]
- Yamashita T.; Wada R.; Sasaki T.; Deng C.; Bierfreund U.; Sandhoff K.; Proia R. L. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 9142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe A.; Wild S. R.; Lee W. L.; Shayman J. A. Curr. Drug Metab. 2001, 2, 331. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Przybylska M.; Wu I. H.; Zhang J.; Siegel C.; Komarnitsky S.; Yew N. S.; Cheng S. H. Diabetes 2007, 56, 1210. [DOI] [PubMed] [Google Scholar]
- Marshall J.; Ashe K. M.; Bangari D.; McEachern K.; Chuang W. L.; Pacheco J.; Copeland D. P.; Desnick R. J.; Shayman J. A.; Scheule R. K.; Cheng S. H. PLoS One 2010, 5, e15033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shayman J. A.; Mahdiyoun S.; Deshmukh G.; Barcelon F.; Inokuchi J.; Radin N. S. J. Biol. Chem. 1990, 265, 12135. [PubMed] [Google Scholar]
- Natoli T. A.; Smith L. A.; Rogers K. A.; Wang B.; Komarnitsky S.; Budman Y.; Belenky A.; Bukanov N. O.; Dackowski W. R.; Husson H.; Russo R. J.; Shayman J. A.; Ledbetter S. R.; Leonard J. P.; Ibraghimov-Beskrovnaya O. Nat. Med. 2010, 16, 788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rani C. S.; Abe A.; Chang Y.; Rosenzweig N.; Saltiel A. R.; Radin N. S.; Shayman J. A. J. Biol. Chem. 1995, 270, 2859. [DOI] [PubMed] [Google Scholar]
- Liu Y. Y.; Han T. Y.; Giuliano A. E.; Cabot M. C. FASEB J. 2001, 15, 719. [DOI] [PubMed] [Google Scholar]
- Gouaze-Andersson V.; Cabot M. C. Biochim. Biophys. Acta 2006, 1758, 2096. [DOI] [PubMed] [Google Scholar]
- Shayman J. A.; Abe A.; Hiraoka M. Glycoconj. J. 2004, 20, 25. [DOI] [PubMed] [Google Scholar]
- Furukawa K.; Clausen H. In Handbook of Glycosyltransferases and Related Genes; Taniguchi N., Honke K., Fukuda M., Eds.; Springer-Verlag Publishers: Tokyo, 2002. [Google Scholar]
- Futerman A. H.; Pagano R. E. Biochem. J. 1991, 280 (Pt 2), 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeckel D.; Karrenbauer A.; Burger K. N.; van Meer G.; Wieland F. J. Cell Biol.. 1992, 117, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannert H.; Bunning C.; Jeckel D.; Wieland F. T. FEBS Lett. 1994, 342, 91. [DOI] [PubMed] [Google Scholar]
- Buton X.; Herve P.; Kubelt J.; Tannert A.; Burger K. N.; Fellmann P.; Muller P.; Herrmann A.; Seigneuret M.; Devaux P. F. Biochemistry 2002, 41, 13106. [DOI] [PubMed] [Google Scholar]
- Halter D.; Neumann S.; van Dijk S. M.; Wolthoorn J.; de Maziere A. M.; Vieira O. V.; Mattjus P.; Klumperman J.; van Meer G.; Sprong H. J. Cell Biol. 2007, 179, 101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaji T.; Kumagai K.; Tomishige N.; Hanada K. IUBMB Life 2008, 60, 511. [DOI] [PubMed] [Google Scholar]
- Cao X.; Coskun U.; Rossle M.; Buschhorn S. B.; Grzybek M.; Dafforn T. R.; Lenoir M.; Overduin M.; Simons K. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 21121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenoir M.; Coskun U.; Grzybek M.; Cao X.; Buschhorn S. B.; James J.; Simons K.; Overduin M. EMBO Rep 2010, 11, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee S.; Kolmakova A.; Rajesh M. Curr. Drug Targets 2008, 9, 272. [DOI] [PubMed] [Google Scholar]
- Furukawa K. In Handbook of Glycosyltransferases and Related Genes; Taniguchi N., Honke K., Fukuda M., Eds.; Springer-Verlag Publishers: Tokyo, 2002. [Google Scholar]
- Takamiya K.; Yamamoto A.; Furukawa K.; Yamashiro S.; Shin M.; Okada M.; Fukumoto S.; Haraguchi M.; Takeda N.; Fujimura K.; Sakae M.; Kishikawa M.; Shiku H.; Aizawa S. Proc. Natl. Acad. Sci. U.S.A. 1996, 93, 10662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito M.; Ishii A. In Handbook of Glycosyltransferases and Related Genes; Taniguchi N., Honke K., Fukuda M., Eds.; Springer-Verlag Publishers: Tokyo, 2002. [Google Scholar]
- Yamashita T.; Hashiramoto A.; Haluzik M.; Mizukami H.; Beck S.; Norton A.; Kono M.; Tsuji S.; Daniotti J. L.; Werth N.; Sandhoff R.; Sandhoff K.; Proia R. L. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inokuchi J. Proc. Jpn. Acad., Ser. B 2011, 87, 179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpson M. A.; Cross H.; Proukakis C.; Priestman D. A.; Neville D. C.; Reinkensmeier G.; Wang H.; Wiznitzer M.; Gurtz K.; Verganelaki A.; Pryde A.; Patton M. A.; Dwek R. A.; Butters T. D.; Platt F. M.; Crosby A. H. Nat. Genet. 2004, 36, 1225. [DOI] [PubMed] [Google Scholar]
- Furukawa K. In Handbook of Glycosyltransferases and Related Genes; Taniguchi N., Honke K., Fukuda M., Eds.; Springer-Verlag Publishers: Tokyo, 2002. [Google Scholar]
- Hamamoto T.; Tsuji S. In Handbook of Glycosyltransferases and Related Genes; Taniguchi N., Honke K., Fukuda M., Eds.; Springer-Verlag Publishers: Tokyo, 2002. [Google Scholar]
- Hashimoto K.; Goto S.; Kawano S.; Aoki-Kinoshita K. F.; Ueda N.; Hamajima M.; Kawasaki T.; Kanehisa M. Glycobiology 2006, 16, 63R. [DOI] [PubMed] [Google Scholar]
- Hashimoto K.; Tokimatsu T.; Kawano S.; Yoshizawa A. C.; Okuda S.; Goto S.; Kanehisa M. Carbohydr. Res. 2009, 344, 881. [DOI] [PubMed] [Google Scholar]
- Kawano S.; Hashimoto K.; Miyama T.; Goto S.; Kanehisa M. Bioinformatics 2005, 21, 3976. [DOI] [PubMed] [Google Scholar]
- Oriol R.; Mollicone R. In Handbook of Glycosyltransferases and Related Genes; Taniguchi N., Honke K., Fukuda M., Eds.; Springer-Verlag Publishers: Tokyo, 2002. [Google Scholar]
- Narimatsu H. In Handbook of Glycosyltransferases and Related Genes; Taniguchi N., Honke K., Fukuda M., Eds.; Springer-Verlag Publishers, 2002. [Google Scholar]
- Tokuda N.; Zhang Q.; Yoshida S.; Kusunoki S.; Urano T.; Furukawa K. Glycobiology 2006, 16, 916. [DOI] [PubMed] [Google Scholar]
- Hakomori S. I.; Murakami W. T. Proc. Natl. Acad. Sci. U.S.A. 1968, 59, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igarashi Y.; Kannagi R. J. Biochem. 2010, 147, 3. [DOI] [PubMed] [Google Scholar]
- Hakomori S. Adv. Cancer Res. 1973, 18, 265. [DOI] [PubMed] [Google Scholar]
- Hakomori S. Proc. Natl. Acad. Sci. U.S.A. 2002, 99, 10231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroes R. A.; He H.; Emmett M. R.; Nilsson C. L.; Leach F. E. 3rd; Amster I. J.; Marshall A. G.; Moskal J. R. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 12646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J.; Miyazaki K.; Shaner R. L.; Merrill A. H. Jr.; Kannagi R. FEBS Lett. 2010, 584, 1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W. X.; Chammas R.; Varki A. Glycobiology 1998, 8, 199. [DOI] [PubMed] [Google Scholar]
- Cariappa A.; Takematsu H.; Liu H.; Diaz S.; Haider K.; Boboila C.; Kalloo G.; Connole M.; Shi H. N.; Varki N.; Varki A.; Pillai S. J. Exp. Med. 2009, 206, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kniep B.; Kniep E.; Ozkucur N.; Barz S.; Bachmann M.; Malisan F.; Testi R.; Rieber E. P. Int. J. Cancer 2006, 119, 67. [DOI] [PubMed] [Google Scholar]
- Yu R. K.; Bieberich E.; Xia T.; Zeng G. J. Lipid Res. 2004, 45, 783. [DOI] [PubMed] [Google Scholar]
- Togayachi A.; Akashima T.; Ookubo R.; Kudo T.; Nishihara S.; Iwasaki H.; Natsume A.; Mio H.; Inokuchi J.; Irimura T.; Sasaki K.; Narimatsu H. J. Biol. Chem. 2001, 276, 22032. [DOI] [PubMed] [Google Scholar]
- Zhou D.; Henion T. R.; Jungalwala F. B.; Berger E. G.; Hennet T. J. Biol. Chem. 2000, 275, 22631. [DOI] [PubMed] [Google Scholar]
- Yu R. K.; Yanagisawa M. CNS Neurol Disord. Drug Targets 2006, 5, 415. [DOI] [PubMed] [Google Scholar]
- Stoffel W.GalCer Synthase (Ceramide Galactosyltransferase, CGT); Springer-Verlag Publishers: Tokyo, 2002. [Google Scholar]
- Sprong H.; Kruithof B.; Leijendekker R.; Slot J. W.; van Meer G.; van der Sluijs P. J. Biol. Chem. 1998, 273, 25880. [DOI] [PubMed] [Google Scholar]
- Kabuss R.; Ashikov A.; Oelmann S.; Gerardy-Schahn R.; Bakker H. Glycobiology 2005, 15, 905. [DOI] [PubMed] [Google Scholar]
- Coetzee T.; Fujita N.; Dupree J.; Shi R.; Blight A.; Suzuki K.; Popko B. Cell 1996, 86, 209. [DOI] [PubMed] [Google Scholar]
- Fewou S. N.; Bussow H.; Schaeren-Wiemers N.; Vanier M. T.; Macklin W. B.; Gieselmann V.; Eckhardt M. J. Neurochem. 2005, 94, 469. [DOI] [PubMed] [Google Scholar]
- Jungalwala F. B.; Koul O.; Stoolmiller A.; Sapirstein V. S. J. Neurochem. 1985, 45, 191. [DOI] [PubMed] [Google Scholar]
- Chou D. K.; Jungalwala F. B. J. Biol. Chem. 1993, 268, 330. [PubMed] [Google Scholar]
- Chou D. K.; Ilyas A. A.; Evans J. E.; Costello C.; Quarles R. H.; Jungalwala F. B. J. Biol. Chem. 1986, 261, 11717. [PubMed] [Google Scholar]
- Sandhoff R.; Hepbildikler S. T.; Jennemann R.; Geyer R.; Gieselmann V.; Proia R. L.; Wiegandt H.; Grone H. J. J. Biol. Chem. 2002, 277, 20386. [DOI] [PubMed] [Google Scholar]
- Sundaram K. S.; Lev M. J. Lipid Res. 1988, 29, 1475. [PubMed] [Google Scholar]
- Honke K.; Tsuda M.; Hirahara Y.; Ishii A.; Makita A.; Wada Y. J. Biol. Chem. 1997, 272, 4864. [DOI] [PubMed] [Google Scholar]
- Honke K.; Zhang Y.; Cheng X.; Kotani N.; Taniguchi N. Glycoconj. J. 2004, 21, 59. [DOI] [PubMed] [Google Scholar]
- Ogawa D.; Shikata K.; Honke K.; Sato S.; Matsuda M.; Nagase R.; Tone A.; Okada S.; Usui H.; Wada J.; Miyasaka M.; Kawashima H.; Suzuki Y.; Suzuki T.; Taniguchi N.; Hirahara Y.; Tadano-Aritomi K.; Ishizuka I.; Tedder T. F.; Makino H. J. Biol. Chem. 2004, 279, 2085. [DOI] [PubMed] [Google Scholar]
- Suzuki K.; Vanier M. T.; Coetzee T.; Popko B. Neurochem. Res. 1999, 24, 471. [DOI] [PubMed] [Google Scholar]
- Marcus J.; Honigbaum S.; Shroff S.; Honke K.; Rosenbluth J.; Dupree J. L. Glia 2006, 53, 372. [DOI] [PubMed] [Google Scholar]
- Deshmukh G. D.; Radin N. S.; Gattone V. H. 2nd; Shayman J. A. J. Lipid Res. 1994, 35, 1611. [PubMed] [Google Scholar]
- Kolter T.; Sandhoff K. FEBS Lett. 2010, 584, 1700. [DOI] [PubMed] [Google Scholar]
- Serra M.; Saba J. D. Adv. Enzyme Regul 2010, 50, 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnino S.; Aureli M.; Loberto N.; Chigorno V.; Prinetti A. FEBS Lett. 2010, 584, 1914. [DOI] [PubMed] [Google Scholar]
- Stoeckelhuber M.; Dobner P.; Baumgartner P.; Ehlert J.; Brandt E.; Mentele R.; Adam D.; Engelmann B. J. Biol. Chem. 2000, 275, 37365. [DOI] [PubMed] [Google Scholar]
- Chigorno V.; Giannotta C.; Ottico E.; Sciannamblo M.; Mikulak J.; Prinetti A.; Sonnino S. J. Biol. Chem. 2005, 280, 2668. [DOI] [PubMed] [Google Scholar]
- Kobayashi A.; Takanezawa Y.; Hirata T.; Shimizu Y.; Misasa K.; Kioka N.; Arai H.; Ueda K.; Matsuo M. J. Lipid Res. 2006, 47, 1791. [DOI] [PubMed] [Google Scholar]
- Merrill A. H. Jr.; Lingrell S.; Wang E.; Nikolova-Karakashian M.; Vales T. R.; Vance D. E. J. Biol. Chem. 1995, 270, 13834. [DOI] [PubMed] [Google Scholar]
- Jales A.; Falahati R.; Mari E.; Stemmy E. J.; Shen W.; Southammakosane C.; Herzog D.; Ladisch S.; Leitenberg D. Immunology 2011, 132, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance J. E.; Peake K. B. Curr. Opin. Lipidol. 2011, 22, 204. [DOI] [PubMed] [Google Scholar]
- Xu Y. H.; Barnes S.; Sun Y.; Grabowski G. A. J. Lipid Res. 2010, 51, 1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnino S.; Chigorno V.; Aureli M.; Masilamani A. P.; Valsecchi M.; Loberto N.; Prioni S.; Mauri L.; Prinetti A. Adv. Exp. Med. Biol. 2011, 705, 297. [DOI] [PubMed] [Google Scholar]
- Ledeen R. W.; Wu G. Neurochem. Res. 2010, 35, 1867. [DOI] [PubMed] [Google Scholar]
- Aureli M.; Masilamani A. P.; Illuzzi G.; Loberto N.; Scandroglio F.; Prinetti A.; Chigorno V.; Sonnino S. FEBS Lett. 2009, 583, 2469. [DOI] [PubMed] [Google Scholar]
- Duan R. D. Biochim. Biophys. Acta 2006, 1761, 281. [DOI] [PubMed] [Google Scholar]
- Wu B. X.; Clarke C. J.; Hannun Y. A. Neuromolecular Med. 2010, 12, 320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke C. J.; Wu B. X.; Hannun Y. A. Adv. Enzyme Regul 2011, 51, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C.; Obeid L. M. Biochim. Biophys. Acta 2008, 1781, 424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futerman A. H.; Riezman H. Trends Cell Biol. 2005, 15, 312. [DOI] [PubMed] [Google Scholar]
- Hoetzl S.; Sprong H.; van Meer G. J. Neurochem. 2007, 103 (Suppl 1), 3. [DOI] [PubMed] [Google Scholar]
- Aye I. L.; Singh A. T.; Keelan J. A. Chem. Biol. Interact 2009, 180, 327. [DOI] [PubMed] [Google Scholar]
- Kim R. H.; Takabe K.; Milstien S.; Spiegel S. Biochim. Biophys. Acta 2009, 1791, 692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown R. E.; Mattjus P. Biochim. Biophys. Acta 2007, 1771, 746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rosa M.; Park H. J.; Mylvaganum M.; Binnington B.; Lund N.; Branch D. R.; Lingwood C. A. Biochim. Biophys. Acta 2008, 1780, 347. [DOI] [PubMed] [Google Scholar]
- Schulze H.; Kolter T.; Sandhoff K. Biochim. Biophys. Acta 2009, 1793, 674. [DOI] [PubMed] [Google Scholar]
- Delgado M. A.; Deretic V. Cell Death Differ. 2009, 16, 976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Jagannath C.; Liu X. D.; Sharafkhaneh A.; Kolodziejska K. E.; Eissa N. T. Immunity 2007, 27, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado M. A.; Elmaoued R. A.; Davis A. S.; Kyei G.; Deretic V. EMBO J. 2008, 27, 1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims K.; Haynes C. A.; Kelly S.; Allegood J. C.; Wang E.; Momin A.; Leipelt M.; Reichart D.; Glass C. K.; Sullards M. C.; Merrill A. H. J. Biol. Chem. 2010, 285, 38568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lencer W. I.; Hirst T. R.; Holmes R. K. Biochim. Biophys. Acta 1999, 1450, 177. [DOI] [PubMed] [Google Scholar]
- Raa H.; Grimmer S.; Schwudke D.; Bergan J.; Walchli S.; Skotland T.; Shevchenko A.; Sandvig K. Traffic 2009, 10, 868. [DOI] [PubMed] [Google Scholar]
- Arab S.; Lingwood C. A. J. Cell Physiol. 1998, 177, 646. [DOI] [PubMed] [Google Scholar]
- Tam P.; Mahfoud R.; Nutikka A.; Khine A. A.; Binnington B.; Paroutis P.; Lingwood C. J. Cell Physiol. 2008, 216, 750. [DOI] [PubMed] [Google Scholar]
- Hou Q.; Jin J.; Zhou H.; Novgorodov S. A.; Bielawska A.; Szulc Z. M.; Hannun Y. A.; Obeid L. M.; Hsu Y. T. J. Lipid Res. 2011, 52, 278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.; Rotolo J. A.; Mesicek J.; Penate-Medina T.; Rimner A.; Liao W. C.; Yin X.; Ragupathi G.; Ehleiter D.; Gulbins E.; Zhai D.; Reed J. C.; Haimovitz-Friedman A.; Fuks Z.; Kolesnick R. PLoS One 2011, 6, e19783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprong H.; Degroote S.; Nilsson T.; Kawakita M.; Ishida N.; van der Sluijs P.; van Meer G. Mol. Biol. Cell 2003, 14, 3482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niimura Y.; Moue T.; Takahashi N.; Nagai K. Glycobiology 2010, 20, 710. [DOI] [PubMed] [Google Scholar]
- Aureli M.; Loberto N.; Lanteri P.; Chigorno V.; Prinetti A.; Sonnino S. J. Neurochem. 2011, 116, 891. [DOI] [PubMed] [Google Scholar]
- Crespo P. M.; Demichelis V. T.; Daniotti J. L. J. Biol. Chem. 2010, 285, 29179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilcaes A. A.; Torres Demichelis V.; Daniotti J. L. J. Biol. Chem. 2011, 10.1074/jbc.M111.257196. [Google Scholar]
- Venkataraman K.; Thangada S.; Michaud J.; Oo M. L.; Ai Y.; Lee Y. M.; Wu M.; Parikh N. S.; Khan F.; Proia R. L.; Hla T. Biochem. J. 2006, 397, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigogliuso S.; Donati C.; Cassara D.; Taverna S.; Salamone M.; Bruni P.; Vittorelli M. L. J. Oncol. 2010, 509329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romiti E.; Meacci E.; Donati C.; Formigli L.; Zecchi-Orlandini S.; Farnararo M.; Ito M.; Bruni P. Arch. Biochem. Biophys. 2003, 417, 27. [DOI] [PubMed] [Google Scholar]
- Romiti E.; Meacci E.; Tani M.; Nuti F.; Farnararo M.; Ito M.; Bruni P. Biochem. Biophys. Res. Commun. 2000, 275, 746. [DOI] [PubMed] [Google Scholar]
- Schuchman E. H. FEBS Lett. 2010, 584, 1895. [DOI] [PubMed] [Google Scholar]
- Zeidan Y. H.; Hannun Y. A. Curr. Mol. Med. 2010, 10, 454. [DOI] [PubMed] [Google Scholar]
- Tam C.; Idone V.; Devlin C.; Fernandes M. C.; Flannery A.; He X.; Schuchman E.; Tabas I.; Andrews N. W. J. Cell Biol. 2010, 189, 1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohrbough J.; Rushton E.; Palanker L.; Woodruff E.; Matthies H. J.; Acharya U.; Acharya J. K.; Broadie K. J. Neurosci. 2004, 24, 7789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjithaya R.; Subramani S. Trends Cell Biol. 2011, 21, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenwald A. G.; Pagano R. E. J. Biol. Chem. 1993, 268, 4577. [PubMed] [Google Scholar]
- Lavieu G.; Scarlatti F.; Sala G.; Carpentier S.; Levade T.; Ghidoni R.; Botti J.; Codogno P. Methods Mol. Biol. 2008, 445, 159. [DOI] [PubMed] [Google Scholar]
- van Meer G. Cold Spring Harb Perspect Biol 2011, 10.1101/cshperspect.a004671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trajkovic K.; Hsu C.; Chiantia S.; Rajendran L.; Wenzel D.; Wieland F.; Schwille P.; Brugger B.; Simons M. Science 2008, 319, 1244. [DOI] [PubMed] [Google Scholar]
- Ait Slimane T.; Hoekstra D. FEBS Lett. 2002, 529, 54. [DOI] [PubMed] [Google Scholar]
- Helms J. B.; Zurzolo C. Traffic 2004, 5, 247. [DOI] [PubMed] [Google Scholar]
- Lingwood D.; Simons K. Science 2010, 327, 46. [DOI] [PubMed] [Google Scholar]
- Tettamanti G.; Bassi R.; Viani P.; Riboni L. Biochimie 2003, 85, 423. [DOI] [PubMed] [Google Scholar]
- Kitatani K.; Idkowiak-Baldys J.; Hannun Y. A. Cell Signal 2008, 20, 1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trams E. G.; Lauter C. J.; Salem N. Jr.; Heine U. Biochim. Biophys. Acta 1981, 645, 63. [DOI] [PubMed] [Google Scholar]
- Subra C.; Laulagnier K.; Perret B.; Record M. Biochimie 2007, 89, 205. [DOI] [PubMed] [Google Scholar]
- Ogretmen B.; Pettus B. J.; Rossi M. J.; Wood R.; Usta J.; Szulc Z.; Bielawska A.; Obeid L. M.; Hannun Y. A. J. Biol. Chem. 2002, 277, 12960. [DOI] [PubMed] [Google Scholar]
- Sultan I.; Senkal C. E.; Ponnusamy S.; Bielawski J.; Szulc Z.; Bielawska A.; Hannun Y. A.; Ogretmen B. Biochem. J. 2006, 393, 513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S.; Mitsutake S.; Tsuji K.; Igarashi Y. J. Biochem. 2006, 139, 255. [DOI] [PubMed] [Google Scholar]
- Rosenwald A. G.; Pagano R. E. Adv. Lipid Res. 1993, 26, 101. [PubMed] [Google Scholar]
- Zhang H.; Desai N. N.; Olivera A.; Seki T.; Brooker G.; Spiegel S. J. Cell Biol. 1991, 114, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Kalari S. K.; Usatyuk P. V.; Gorshkova I.; He D.; Watkins T.; Brindley D. N.; Sun C.; Bittman R.; Garcia J. G.; Berdyshev E. V.; Natarajan V. J. Biol. Chem. 2007, 282, 14165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Stunff H.; Giussani P.; Maceyka M.; Lepine S.; Milstien S.; Spiegel S. J. Biol. Chem. 2007, 282, 34372. [DOI] [PubMed] [Google Scholar]
- Lepine S.; Allegood J. C.; Park M.; Dent P.; Milstien S.; Spiegel S. Cell Death Differ. 2011, 18, 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes C. A.; Allegood J. C.; Park H.; Sullards M. C. J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2009, 877, 2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haynes C. A.; Allegood J. C.; Wang E. W.; Kelly S. L.; Sullards M. C.; Merrill A. H. Jr. J. Lipid Res. 2011, 52, 1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullards M. C.; Allegood J. C.; Kelly S.; Wang E.; Haynes C. A.; Park H.; Chen Y.; Merrill A. H. Jr. Methods Enzymol. 2007, 432, 83–115. [DOI] [PubMed] [Google Scholar]
- Stahlman M.; Ejsing C. S.; Tarasov K.; Perman J.; Boren J.; Ekroos K. J Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2009, 877, 2664. [DOI] [PubMed] [Google Scholar]
- Merrill A. H. Jr.; Stokes T. H.; Momin A.; Park H.; Portz B. J.; Kelly S.; Wang E.; Sullards M. C.; Wang M. D. J. Lipid Res. 2009, 50 (Suppl), S97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han X. Biochim. Biophys. Acta 2010, 1801, 774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer U.; Herscovitz H.; Welty F. K.; Costello C. E. J. Lipid Res. 2006, 47, 804. [DOI] [PubMed] [Google Scholar]
- Merrill A. H. Jr.; Sullards M. C.; Allegood J. C.; Kelly S.; Wang E. Methods 2005, 36, 207. [DOI] [PubMed] [Google Scholar]
- Kaga N.; Kazuno S.; Taka H.; Iwabuchi K.; Murayama K. Anal. Biochem. 2005, 337, 316. [DOI] [PubMed] [Google Scholar]
- Lee M. H.; Lee G. H.; Yoo J. S. Rapid Commun. Mass Spectrom. 2003, 17, 64. [DOI] [PubMed] [Google Scholar]
- Pacetti D.; Boselli E.; Hulan H. W.; Frega N. G. J. Chromatogr. 2005, 1097, 66. [DOI] [PubMed] [Google Scholar]
- Pettus B. J.; Kroesen B. J.; Szulc Z. M.; Bielawska A.; Bielawski J.; Hannun Y. A.; Busman M. Rapid Commun. Mass Spectrom. 2004, 18, 577. [DOI] [PubMed] [Google Scholar]
- Farwanah H.; Wirtz J.; Kolter T.; Raith K.; Neubert R. H. H.; Sandhoff K. J. Chromatogr., B Anal. Technol. Biomed. Life Sci. 2009, 877, 2976. [DOI] [PubMed] [Google Scholar]
- Pettus B. J.; Bielawska A.; Kroesen B. J.; Moeller P. D.; Szulc Z. M.; Hannun Y. A.; Busman M. Rapid Commun. Mass Spectrom. 2003, 17, 1203. [DOI] [PubMed] [Google Scholar]
- Yoo H. H.; Son J.; Kim D.-H. J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2006, 843, 327. [DOI] [PubMed] [Google Scholar]
- Bielawski J.; Szulc Z. M.; Hannun Y. A.; Bielawska A. Methods 2006, 39, 82. [DOI] [PubMed] [Google Scholar]
- Merrill AH, J.; Sullards M. C.; Allegood J. C.; Kelly S; Wang E Methods 2005, 36, 207. [DOI] [PubMed] [Google Scholar]
- Ikeda K.; Shimizu T.; Taguchi R. J. Lipid Res. 2008, 49, 2678. [DOI] [PubMed] [Google Scholar]
- Ivleva V. B.; Sapp L. M.; O’Connor P. B.; Costello C. E. J. Am. Soc. Mass Spectrom. 2005, 16, 1552. [DOI] [PubMed] [Google Scholar]
- Muthing J.; Distler U. Mass. Spectrom. Rev. 2009, 29, 425. [DOI] [PubMed] [Google Scholar]
- Miyazaki M.; Yonesige A.; Matsuda J.; Kuroda Y.; Kojima N.; Suzuki A. J. AOAC Int. 2008, 91, 1218. [PubMed] [Google Scholar]
- Manicke N. E.; Wiseman J. M.; Ifa D. R.; Cooks R. G. J. Am. Soc. Mass Spectrom. 2008, 19, 531. [DOI] [PubMed] [Google Scholar]
- Goto-Inoue N.; Hayasaka T.; Sugiura Y.; Taki T.; Li Y. T.; Matsumoto M.; Setou M. J. Chromatogr., B: Anal. Technol. Biomed. Life Sci. 2008, 870, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki Y.; Suzuki M.; Ito E.; Goto-Inoue N.; Miseki K.; Iida J.; Yamazaki Y.; Yamada M.; Suzuki A. J. Biochem. (Tokyo) 2006, 139, 771. [DOI] [PubMed] [Google Scholar]
- Landoni M.; Duschak V. G.; Erra-Balsells R.; Couto A. S. J. Am. Soc. Mass Spectrom. 2008, 19, 923. [DOI] [PubMed] [Google Scholar]
- Chan K.; Lanthier P.; Liu X.; Sandhu J. K.; Stanimirovic D.; Li J. Anal. Chim. Acta 2009, 639, 57. [DOI] [PubMed] [Google Scholar]
- Stuebiger G.; Belgacem O. Anal. Chem. 2007, 79, 3206. [DOI] [PubMed] [Google Scholar]
- Zarei M.; Bindila L.; Souady J.; Dreisewerd K.; Berkenkamp S.; Muething J.; Peter-Katalinic J. J. Mass Spectrom. 2008, 43, 716. [DOI] [PubMed] [Google Scholar]
- Nakamura K.; Suzuki Y.; Goto-Inoue N.; Yoshida-Noro C.; Suzuki A. Anal. Chem. 2006, 78, 5736. [DOI] [PubMed] [Google Scholar]
- Cha S.; Yeung E. S. Anal. Chem. 2007, 79, 2373. [DOI] [PubMed] [Google Scholar]
- Astigarraga E.; Barreda-Gomez G.; Lombardero L.; Fresnedo O.; Castano F.; Giralt M. T.; Ochoa B.; Rodriguez-Puertas R.; Fernandez J. A. Anal. Chem. 2008, 80, 9105. [DOI] [PubMed] [Google Scholar]
- Serb A.; Schiopu C.; Flangea C.; Sisu E.; Zamfir A. D. J. Mass Spectrom. 2009, 44, 1434. [DOI] [PubMed] [Google Scholar]
- Kirsch S.; Zarei M.; Cindric M.; Muthing J.; Bindila L.; Peter-Katalinic J. Anal. Chem. 2008, 80, 4711. [DOI] [PubMed] [Google Scholar]
- Ejsing C. S.; Moehring T.; Bahr U.; Duchoslav E.; Karas M.; Simons K.; Shevchenko A. J. Mass Spectrom. 2006, 41, 372. [DOI] [PubMed] [Google Scholar]
- McFarland M. A.; Marshall A. G.; Hendrickson C. L.; Nilsson C. L.; Fredman P.; Mansson J. E. J. Am. Soc. Mass Spectrom. 2005, 16, 752. [DOI] [PubMed] [Google Scholar]
- Vukelic Z.; Kalanj-Bognar S.; Froesch M.; Bindila L.; Radic B.; Allen M.; Peter-Katalinic J.; Zamfir A. D. Glycobiology 2007, 17, 504. [DOI] [PubMed] [Google Scholar]
- Pol J.; Vidova V.; Kruppa G.; Kobliha V.; Novak P.; Lemr K.; Kotiaho T.; Kostiainen R.; Havlicek V.; Volny M. Anal. Chem. 2009, 81, 8479. [DOI] [PubMed] [Google Scholar]
- Jackson S. N.; Colsch B.; Egan T.; Lewis E. K.; Schultz J. A.; Woods A. S. Analyst 2011, 136, 463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas M. C.; Mitchell T. W.; Harman D. G.; Deeley J. M.; Murphy R. C.; Blanksby S. J. Anal. Chem. 2007, 79, 5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han L.; Costello C. E. J. Am. Soc. Mass Spectrom. 2011, 22, 997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson H.; Johansson L.; Miller-Podraza H.; Karlsson K. A. Glycobiology 1999, 9, 765. [DOI] [PubMed] [Google Scholar]
- Parry S.; Ledger V.; Tissot B.; Haslam S. M.; Scott J.; Morris H. R.; Dell A. Glycobiology 2007, 17, 646. [DOI] [PubMed] [Google Scholar]
- Patterson B. W.; Zhao G.; Elias N.; Hachey D. L.; Klein S. J. Lipid Res. 1999, 40, 2118. [PubMed] [Google Scholar]
- Black P. N.; Faergeman N. J.; DiRusso C. C. J. Nutr. 2000, 130, 305S. [DOI] [PubMed] [Google Scholar]
- Hertz R.; Magenheim J.; Berman I.; Bar-Tana J. Nature 1998, 392, 512. [DOI] [PubMed] [Google Scholar]
- Hamming K. S.; Riedel M. J.; Soliman D.; Matemisz L. C.; Webster N. J.; Searle G. J.; MacDonald P. E.; Light P. E. Mol. Endocrinol. 2008, 22, 2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellerstein M. K.; Neese R. A. Am. J. Physiol. 1999, 276, E1146. [DOI] [PubMed] [Google Scholar]
- Hellerstein M. K.; Schwarz J. M.; Neese R. A. Annu. Rev. Nutr. 1996, 16, 523. [DOI] [PubMed] [Google Scholar]
- Hellerstein M. K.; Neese R. A. Am. J. Physiol. 1992, 263, E988. [DOI] [PubMed] [Google Scholar]
- Todd P. J.; Schaaff T. G.; Chaurand P.; Caprioli R. M. J. Mass Spectrom. 2001, 36, 355. [DOI] [PubMed] [Google Scholar]
- Cornett D. S.; Reyzer M. L.; Chaurand P.; Caprioli R. M. Nat. Methods 2007, 4, 828. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Liu Y.; Sullards M. C.; Merrill A. H. Jr. Neuromolecular Med. 2010, 12, 306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaurand P.; Cornett D. S.; Angel P. M.; Caprioli R. M. Mol. Cell. Proteomics 2011, 10, O110.004259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winograd N.; Garrison B. J. Annu. Rev. Phys. Chem. 2010, 61, 305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ageta H.; Asai S.; Sugiura Y.; Goto-Inoue N.; Zaima N.; Setou M. Med. Mol. Morphol. 2009, 42, 16. [DOI] [PubMed] [Google Scholar]
- Goto-Inoue N.; Hayasaka T.; Zaima N.; Kashiwagi Y.; Yamamoto M.; Nakamoto M.; Setou M. J. Am. Soc. Mass Spectrom. 2010, 21, 1940. [DOI] [PubMed] [Google Scholar]
- Sjovall P.; Lausmaa J.; Johansson B. Anal. Chem. 2004, 76, 4271. [DOI] [PubMed] [Google Scholar]
- Murphy R. C.; Hankin J. A.; Barkley R. M. J. Lipid Res. 2009, 50 (Suppl), S317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto-Inoue N.; Hayasaka T.; Zaima N.; Setou M. Biochim. Biophys. Acta 2011, 10.1016/j.bbalip.2011.03.004. [DOI] [PubMed] [Google Scholar]
- Roy S.; Touboul D.; Brunelle A.; Germain D. P.; Prognon P.; Laprevote O.; Chaminade P. Ann. Pharm. Fr. 2006, 64, 328. [DOI] [PubMed] [Google Scholar]
- Hart P. J.; Francese S.; Claude E.; Woodroofe M. N.; Clench M. R. Anal. Bioanal. Chem. 2011, 401, 115. [DOI] [PubMed] [Google Scholar]
- Manicke N. E.; Nefliu M.; Wu C.; Woods J. W.; Reiser V.; Hendrickson R. C.; Cooks R. G. Anal. Chem. 2009, 81, 8702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeley J. M.; Hankin J. A.; Friedrich M. G.; Murphy R. C.; Truscott R. J.; Mitchell T. W.; Blanksby S. J. J. Lipid Res. 2010, 51, 2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry K. A.; Li B.; Reynolds S. D.; Barkley R. M.; Gijon M. A.; Hankin J. A.; Henson P. M.; Murphy R. C. J. Lipid Res. 2011, 52, 1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi A.; Subathra M.; Grey A.; Schey K.; Del Poeta M.; Luberto C. PLoS One 2011, 5, e15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borner K.; Nygren H.; Hagenhoff B.; Malmberg P.; Tallarek E.; Mansson J. E. Biochim. Biophys. Acta 2006, 1761, 335. [DOI] [PubMed] [Google Scholar]
- Woods A. S.; Jackson S. N. AAPS J 2006, 8, E391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson S. N.; Ugarov M.; Egan T.; Post J. D.; Langlais D.; Albert Schultz J.; Woods A. S. J. Mass Spectrom. 2007, 42, 1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. Y.; Jackson S. N.; Post J.; Woods A. S. Int. J. Mass Spectrom. 2008, 278, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y.; Allegood J.; Liu Y.; Wang E.; Cachon-Gonzalez B.; Cox T. M.; Merrill A. H. Jr.; Sullards M. C. Anal. Chem. 2008, 80, 2780. [DOI] [PubMed] [Google Scholar]
- Tufte E. R.The Visual Display of Quantitative Information; 2nd ed.; Graphics Press: Cheshire, CT, 2001. [Google Scholar]
- Tufte E. R.The Cognitive Style of Powerpoint: Pitching out Corrupts Within; 2nd ed.; Graphics Press LLC: Cheshire, CT, 2006. [Google Scholar]
- Andreyev A. Y.; Fahy E.; Guan Z.; Kelly S.; Li X.; McDonald J. G.; Milne S.; Myers D.; Park H.; Ryan A.; Thompson B. M.; Wang E.; Zhao Y.; Brown H. A.; Merrill A. H.; Raetz C. R.; Russell D. W.; Subramaniam S.; Dennis E. A. J. Lipid Res. 2010, 51, 2785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis E. A.; Deems R. A.; Harkewicz R.; Quehenberger O.; Brown H. A.; Milne S. B.; Myers D. S.; Glass C. K.; Hardiman G.; Reichart D.; Merrill A. H. Jr.; Sullards M. C.; Wang E.; Murphy R. C.; Raetz C. R.; Garrett T. A.; Guan Z.; Ryan A. C.; Russell D. W.; McDonald J. G.; Thompson B. M.; Shaw W. A.; Sud M.; Zhao Y.; Gupta S.; Maurya M. R.; Fahy E.; Subramaniam S. J. Biol. Chem. 2010, 285, 39976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims K.; Haynes C. A.; Kelly S.; Allegood J. C.; Wang E.; Momin A.; Leipelt M.; Reichart D.; Glass C. K.; Sullards M. C.; Merrill A. H. Jr. J. Biol. Chem. 2010, 285, 38568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoor S.; Quo C. F.; Merrill A. H. Jr.; Wang M. D. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2008, 2423. [DOI] [PubMed] [Google Scholar]
- van Iersel M. P.; Kelder T.; Pico A. R.; Hanspers K.; Coort S.; Conklin B. R.; Evelo C. BMC Bioinformatics 2008, 9, 399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldridge B. B.; Burke J. M.; Lauffenburger D. A.; Sorger P. K. Nat. Cell Biol. 2006, 8, 1195. [DOI] [PubMed] [Google Scholar]
- Alvarez-Vasquez F.; Sims K. J.; Hannun Y. A.; Voit E. O. J. Theor. Biol. 2004, 226, 265. [DOI] [PubMed] [Google Scholar]
- Sims K. J.; Spassieva S. D.; Voit E. O.; Obeid L. M. Biochem. Cell Biol. 2004, 82, 45. [DOI] [PubMed] [Google Scholar]
- Alvarez-Vasquez F.; Sims K. J.; Cowart L. A.; Okamoto Y.; Voit E. O.; Hannun Y. A. Nature 2005, 433, 425. [DOI] [PubMed] [Google Scholar]
- Ozbayraktar F. B.; Ulgen K. O. J. Biomed. Inform 2010, 43, 537. [DOI] [PubMed] [Google Scholar]
- Alvarez-Vasquez F.; Sims K. J.; Voit E. O.; Hannun Y. A. Theor. Biol. Med. Model 2007, 4, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowart L. A.; Shotwell M.; Worley M. L.; Richards A. J.; Montefusco D. J.; Hannun Y. A.; Lu X. Mol. Syst. Biol. 2010, 6, 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia J.; Shea J.; Alvarez-Vasquez F.; Qureshi A.; Luberto C.; Voit E. O.; Del Poeta M. Mol. Syst. Biol. 2008, 4, 183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims K. J.; Alvarez-Vasquez F.; Voit E. O.; Hannun Y. A. Methods Enzymol. 2007, 432, 319. [DOI] [PubMed] [Google Scholar]
- Voit E. O.; Alvarez-Vasquez F.; Hannun Y. A. Adv. Exp. Med. Biol. 2010, 688, 264. [DOI] [PubMed] [Google Scholar]
- Neelamegham S.; Liu G. Glycobiology 2011, 10.1093/glycob/cwr036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quo C. F.; Moffitt R. A.; Merrill A. H.; Wang M. D. J. Comput. Biol. 2011, 18, 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S.; Maurya M. R.; Merrill A. H. Jr.; Glass C. K.; Subramaniam S. BMC Syst. Biol. 2011, 5, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]