

Abstract
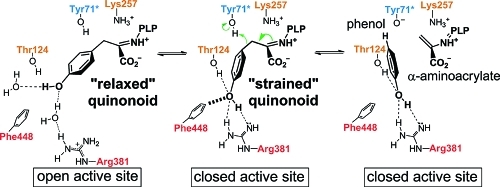
The key step in the enzymatic reaction catalyzed by tyrosine phenol-lyase (TPL) is reversible cleavage of the Cβ–Cγ bond of l-tyrosine. Here, we present X-ray structures for two enzymatic states that form just before and after the cleavage of the carbon–carbon bond. As for most other pyridoxal 5′-phosphate-dependent enzymes, the first state, a quinonoid intermediate, is central for the catalysis. We captured this relatively unstable intermediate in the crystalline state by introducing substitutions Y71F or F448H in Citrobacter freundii TPL and briefly soaking crystals of the mutant enzymes with a substrate 3-fluoro-l-tyrosine followed by flash-cooling. The X-ray structures, determined at ∼2.0 Å resolution, reveal two quinonoid geometries: “relaxed” in the open and “tense” in the closed state of the active site. The “tense” state is characterized by changes in enzyme contacts made with the substrate’s phenolic moiety, which result in significantly strained conformation at Cβ and Cγ positions. We also captured, at 2.25 Å resolution, the X-ray structure for the state just after the substrate’s Cβ–Cγ bond cleavage by preparing the ternary complex between TPL, alanine quinonoid and pyridine N-oxide, which mimics the α-aminoacrylate intermediate with bound phenol. In this state, the enzyme–ligand contacts remain almost exactly the same as in the “tense” quinonoid, indicating that the strain induced by the closure of the active site facilitates elimination of phenol. Taken together, structural observations demonstrate that the enzyme serves not only to stabilize the transition state but also to destabilize the ground state.
Introduction
Tyrosine phenol-lyase (TPL; E.C. 4.1.99.2) is a pyridoxal 5′-phosphate (PLP)-dependent enzyme, which catalyzes the reversible cleavage of the Cβ–Cγ bond of l-Tyr to produce phenol, ammonium, and pyruvate (Scheme 1). In addition to its physiological reaction, in vitro TPL catalyzes a plethora of other reactions including β-elimination (and its reversal) of a number of other α-amino acids and several structurally similar compounds,(1) as well as β-substitutions by phenol derivatives and their heterocyclic analogues.(2) Some of these TPL-catalyzed reactions have been utilized for biotechnological production of l-Tyr and several related compounds like l-DOPA, a top-selling drug prescribed for treatment of Parkinson’s disease.(3) In addition, TPL was modified by mutagenesis to have better performance in specific nonphysiological catalytic tasks, which proved to be useful for “green” organic synthesis of l-Tyr derivatives, including several pharmaceuticals and their precursors.(4)
Scheme 1. β-Elimination Reaction of l-Tyr Catalyzed by TPL.

The phenolic moiety is not a good leaving group, so its elimination from l-Tyr is a mechanistically intriguing process.(5) The postulated mechanism, catalyzed by TPL, consists of several intermediate steps (Scheme 1): (a) formation of the external aldimine by the reaction of the internal aldimine and l-Tyr; (b) Cα-proton abstraction performed by the PLP-binding Lys257 (amino acid numbering is for Citrobacter freundii TPL) resulting in the quinonoid intermediate; and (c) protonation of the substrate’s Cγ atom by Tyr71 of the adjacent subunit and the concerted elimination of phenol along with the formation of α-aminoacrylate.(6) We note that Scheme 1 shows protonation of Cγ and Cβ–Cγ bond cleavage as occurring in a stepwise mechanism; however, multiple isotope effects on the reaction of tryptophan indole-lyase, a structurally and mechanistically very similar enzyme, suggest a mechanism with concerted protonation and elimination.(7)
TPL is a homotetramer with four active sites, two per catalytic dimer, located at the monomer–monomer interface.8,9 Rearrangement of the large and small domains in a TPL subunit results in two different active-site conformations: open and closed.10,11 A quinonoid species is the key intermediate in almost all reactions catalyzed by PLP-dependent enzymes,12,13 but due to its low stability, there are only a few reports on structures of this important intermediate.11,14 Recently, we reported X-ray structures for TPL quinonoid intermediates prepared by soaking holoenzyme crystals with inhibitors l-Ala and l-Met.(11) While in the crystals of holoenzyme both subunits of the asymmetric part were in the “open” state, in the crystals of quinonoid intermediates one of the two subunits adopted the closed conformation. Two lines of evidence indicated that differences between the two states were not induced by lattice forces affecting the individual subunits in the crystal. First, spectroscopic measurements indicated equivalence of complexes formed in crystal to complexes formed in solution.(15) Second, the two states, open and closed, were earlier observed in the structure of apoenzyme, which did not involve soaking, suggesting that the two states represent two energetically favorable states. Furthermore, a changing pattern of molecular contacts observed in crystals for different states demonstrated plasticity in molecular interactions, suggesting that domain movements were not restricted by crystal contacts. Taken together, these observations indicated that structures of TPL complexes in crystallo were directly related to complexes that form in solution.
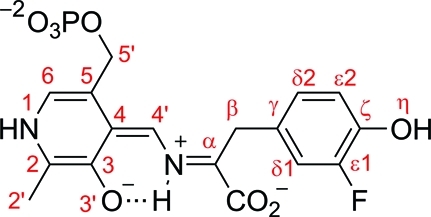
Despite previous structural data, the molecular mechanism of the reaction and in particular the detailed structural changes accompanying and facilitating the cleavage of the Cβ–Cγ bond of the physiological substrate l-Tyr and the subsequent elimination of phenol remained elusive. Herein, we show that the closure of TPL active site does not only bring the catalytically important Thr124 and Arg381 into positions suitable for catalysis, but also causes a significant strain in the quinonoid intermediate formed with the substrate. The resulting “tense” quinonoid intermediate is less stable and thus more liable to the cleavage of the Cβ–Cγ bond in the final stage of the β-elimination. These notions are based on the crystal structures of the quinonoid intermediates formed with the substrate 3-fluoro-l-tyrosine (3-F-l-Tyr; Chart 1). 3-F-l-Tyr was used for preparation of quinonoid intermediates because its kinetic properties are very similar to l-Tyr,16−18 but it is at least 5 times more soluble in the solution used for preparation of complexes (see the Experimental Section). The quinonoid intermediates were “captured” in the crystalline state of C. freundii TPL by introducing replacements Y71F or F448H. These mutant enzymes lack the β-elimination activity with l-Tyr or 3-F-l-Tyr as a substrate; instead, they accumulate quinonoid intermediates when incubated in the solution of either of these two amino acids.15−17 We used this property to prepare crystals of quinonoid complexes. Soaking crystals of the Y71F or F448H mutant holoenzymes with 3-F-l-Tyr for about 15 s resulted in a quick color change, from yellow to orange, suggesting formation of the quinonoid intermediate. However, unlike for quinonoid complexes formed with l-Ala and l-Met,(11) diffraction quality of these crystals deteriorated very rapidly, necessitating short soaking times followed by flash-cooling in liquid nitrogen. The structures, determined at 2.04 Å (Y71F) and 2.00 Å (F448H) resolution, reveal two different geometries of the quinonoid intermediate: “relaxed” in the open and “tense” in the closed conformation of the active site. In addition, we determined the structure of the wild-type TPL in complex with alanine quinonoid and pyridine N-oxide. This structure mimics the state just after the Cβ–Cγ bond cleavage, with the α-aminoacrylate intermediate and a phenol molecule bound in the closed active site.
Chart 1. 3-Fluoro-l-tyrosine.

Results and Discussion
Quinonoid Intermediate in the Open Active Site of Y71F TPL
The crystalline quinonoid intermediate of Y71F TPL formed with 3-F-l-Tyr has two crystallographically independent active sites in the same catalytic dimer. One active site of this 2.04 Å resolution structure is found in the open conformation. It is occupied by the 3-F-Tyr quinonoid (λmax = 502 nm in crystal)(15) formed after the Cα-proton abstraction from the external aldimine of 3-F-l-Tyr (Figure 1a; Chart 2). The corresponding quinonoid geometry is characterized by the sp2-hybridized Cα and Cγ atoms, each in a trigonal planar environment, and the phenolic moiety rotated with respect to the Cα–Cβ bond (torsion angle Cα–Cβ–Cγ–Cδ1) by 73° (Figure S1 in the Supporting Information). All H-bonds and salt bridges with the enzyme observed for the quinonoid intermediates formed with l-Ala or l-Met(11) are also found in the open active site of the 3-F-Tyr quinonoid (Figure 1a). Additionally, the hydroxyl group of the 3-F-Tyr quinonoid complex forms H-bonds with two water molecules, while the fluorine atom is H-bonded to only one of them. The substrate’s hydroxyl is at the distance of 4.1 Å from the hydroxyl of Thr124 and at 4.8 Å from the closest nitrogen atom in the guanidine of Arg381. Because in this open conformation there is no strain on the quinonoid molecule induced by the enzyme, we refer to this quinonoid geometry as the “relaxed” state.
Figure 1.

Quinonoid intermediates of the Cβ–Cγ bond cleavage trapped in crystal structures (stereo views). The σA-weighted |Fo| – |Fc| omit electron density maps contoured at 3.0σ are in blue. Residues from the large domain are colored in orange, those from the small domain are in pink, and residues from the adjacent subunit are depicted in blue and labeled with an asterisk. Hydrogen bonds are denoted by dashed lines. (a) Open active site (B) of Y71F TPL with the “relaxed” 3-F-Tyr quinonoid. (b) One of the four closed active sites of F448H TPL with the “tense” 3-F-Tyr quinonoid. (c) The disordered active site (A) of Y71F TPL occupied mostly by the “tense” (gray; 0.67 occupancy) and partially by the “relaxed” 3-F-Tyr quinonoid (yellow; 0.33 occupancy). Only the closed active-site conformation could be modeled; a green hashed cylinder indicates a close contact between Phe448 and the “tense” quinonoid.
Chart 2. Quinonoid Intermediate with 3-Fluoro-l-tyrosine with the Corresponding Atom-Numbering Scheme.
Quinonoid Intermediate of F448H TPL
The 2.0 Å resolution structure of F448H TPL complexed with 3-F-l-Tyr contains four crystallographically independent active sites, all found in the closed conformation (Figures 1b and S4–S7). It was shown by rapid-scanning stopped-flow spectroscopy that F448H TPL accumulates the quinonoid intermediate (λmax = 515 nm) when incubated with 3-F-l-Tyr.(17) We note, however, that the quinonoid intermediate can be modeled in the F448H TPL active sites only in a “tense” (strained) geometry with the Cα–Cβ–Cγ angle of 103–106° (in contrast to 109° in the “relaxed” state) and the Cγ atom in a pyramidal geometry with the corresponding pyramidalization angle of 18–23° (Figure 2a; Table 1). Because the estimated coordinate error for this structure was 0.15 Å,(20) such distorted geometry was validated using several approaches. First, we noted that the structure of the “tense” quinonoid could be refined satisfactorily only when the planarity restraints at the Cγ atom were released (Figure 2b). In contrast, refinement with the standard geometrical restraints keeping the Cγ atom in a planar environment reduced fit into electron density maps (Figures S3 and S11 in the Supporting Information) and resulted in the reduction of the tetrahedral Cα–Cβ–Cγ angle to 90–96° (depending on a particular subunit) from 109° observed in the “relaxed” quinonoid state. Such significant reduction is highly improbable, and indeed the Ph–CH2–C angles of less than 105° have not been observed in small molecule structures.(21) Although absorption spectra obtained in solution(17) did not indicate formation of the alternative, external aldimine intermediate, we have attempted modeling and refining this intermediate (Figures S9 and S11). Refinement with the standard geometrical restraints resulted in external aldimine models with multiple geometrical distortions: the Cα–Cβ–Cγ angles of 102–106°, a slight pyramidalization of Cγ (2–4°), flattening of a tetrahedral Cα (a chiral volume of 2.1–2.2 Å3 in comparison with 2.5 Å3 in l-Tyr), and the N–C4′–C4–C3 torsion angles of 23–27°. These observations indicated that the closed active sites did not contain the external aldimine intermediate, in agreement with spectroscopic data.(17) The final models were further validated using several computational approaches as detailed in the Experimental Section.
Figure 2.

(a) Geometry of a 3-F-Tyr quinonoid molecule in the “tense” conformation observed in the closed active sites of F448H and Y71F TPL. (b) A stereo view of a 3-F-l-Tyr quinonoid in a closed active site of F448H refined using the standard (yellow) and relaxed (gray) geometrical restraints. The undistorted quinonoid geometry (thin black lines) is shown for comparison. The σA-weighted |Fo| – |Fc| omit electron density map is shown at 3.0σ.
Table 1. Key Angles in the Quinonoid Structures (deg).
| structure: | Y71F TPL–3-F-l-Tyr |
F448H TPL–3-F-l-Tyr |
|||||
|---|---|---|---|---|---|---|---|
| conformation: | “relaxed” |
“tense” | “tense” |
||||
| active site: | A | B | A | A | B | C | D |
| Cα–Cβ–Cγ | 105 | 109 | 108 | 106 | 105 | 104 | 103 |
| Cα–Cβ–Cγ–Cδ1 (torsion angle)a | 77 | 73 | 77 | 81 | 83 | 80 | 81 |
| pyramidalization angle at Cγb | 0.6 | 2 | 27 | 18 | 23 | 20 | 20 |
Cα–Cβ–Cγ–Cδ1 refers to the torsion angle defining rotation around the Cβ–Cγ bond.
Pyramidalization angle at Cγ, defined as an angle between the Cβ–Cγ line and the Cδ1–Cγ–Cδ2 plane, was calculated using PLATON.(19)
Distortion of the benzene moiety (including bond length alternations and out-of-plane deformations) is a well-known phenomenon observed in sterically constrained aromatic molecules (due to the bulky substituents or the crystal-packing interactions).(22) DFT calculations in the gas phase showed that the “tense” molecule of 3-F-Tyr quinonoid is by 12–21 kcal mol–1 less favorable than the “relaxed” form. However, closure of the active site results in a significant increase in the number of enzyme–quinonoid contacts (as described in the next paragraph), which might compensate for the observed distortion and thus stabilize the quinonoid molecule in the “tense” geometry. It is important to note that the “tense” quinonoid molecule appears to be the species “prepared” for the attack of a proton at the Cγ atom and consecutive Cβ–Cγ bond cleavage. The phenol elimination should proceed through the transition structure with (at least) a partially sp3-hybridized Cγ atom, similar to the ketoquinonoid structure shown in Scheme 1. In agreement with this, the proton-donating hydroxyl group of Tyr71 is in a suitable position (Figure 1b), being at a distance of 3.8–3.9 Å in four crystallographically independent subunits from the Cγ atom of the quinonoid.
Quinonoid Intermediate in the Closed Active Site of Y71F TPL
The second subunit of the catalytic dimer in the Y71F TPL complex is statistically disordered (details provided in the Supporting Information), but it is found predominantly in the closed conformation (Figure 1c). Like in the case of F448H TPL, the closed active-site state of Y71F TPL is occupied by the quinonoid molecule in the “tense” conformation, with the Cα–Cβ–Cγ angle of 108° and the Cγ pyramidalization angle of 27° (Figure 3; Table 1). In the case of Y71F TPL, spectra were previously measured both in solution(16) and in crystallo,(15) revealing formation of the quinonoid intermediate upon addition of 3-F-l-Tyr and ruling out the existence of the external aldimine. Significantly, spectra obtained in crystallo were essentially identical to those recorded from solution. In agreement with spectroscopic observations, refinement of the external aldimine intermediate results in a highly distorted structure (Figures S8 and S10 in the Supporting Information), like in the case of F448H TPL. These observations further support the model with the “tense” quinonoid in the closed active site.
Figure 3.

Ligands modeled in the disordered active site of Y71F TPL. A stereo view with the “relaxed” (yellow) and “tense” (gray) quinonoid molecules superimposed with the corresponding σA-weighted |Fo| – |Fc| omit electron density maps contoured at 3.0σ.
All H-bonding and salt-bridge contacts with the enzyme found in the “relaxed” conformation of the Y71F TPL complex are also present in the “tense” conformation of the 3-F-Tyr quinonoid (Figure 1). However, the “tense” conformation is additionally stabilized by H-bonds formed between the substrate’s hydroxyl group and the hydroxyl group of Thr124 as well as the guanidine group of Arg381. The phenyl ring and the hydroxyl group of the substrate in the closed active site of the Y71F TPL complex are in close contacts with the side chain of Phe448 (the shortest distance is 3.1 Å). These structural observations are corroborated by the earlier biochemical studies, which showed that both Thr124 and Phe448 are required for substrate specificity of TPL,(17) while the deprotonated side chain of Arg381 has an unusual role as a catalytic base (Scheme 1).(9) In both open and closed conformations, Arg381 is part of a triad of H-bonded residues involving the guanidine of Arg381, the hydroxyl of Ser385, and the carboxylate of Glu380 (Figures 5 and S13). Via this H-bond network, the negatively charged carboxylate of Glu380 could additionally activate Arg381 to act as the catalytic base. In the F448H TPL complex, the substrate’s phenolic group is not only H-bonded to the side chains of Arg381 and Thr124, but also to the imidazole group of His448 (mutated Phe448). The additional H-bond further stabilizes the closed conformation and thus prevents the completion of the enzymatic reaction. This explains why F448H TPL exhibits very low β-elimination activity with l-Tyr, but retains significant activity toward the substrates with good leaving groups, which cannot form H-bonds with His448 in the mutated enzyme.(17) In the wild-type TPL, the phenyl group of Phe448 induces further strain on the substrate as evidenced by the larger Cγ pyramidalization angle in the closed active site of the Y71F TPL complex.
Figure 5.

Protein–substrate interactions during the three intermediate steps of the Cβ–Cγ bond cleavage observed in crystal structures. Hydrogen bonds are shown as dashed lines. A short van der Waals contact between the side chain of Phe448 and the substrate phenolic group is denoted by a hashed line. Label colors correspond to residue colors in Figure 1.
TPL Complex with Alanine and Pyridine N-Oxide
Previous spectroscopic23,24 and crystallographic(11) studies showed that wild-type TPL forms the Ala quinonoid intermediate when incubated with l-Ala. Pyridine N-oxide (PNO) binds selectively to the Ala quinonoid intermediate.(24) We used this knowledge to prepare the ternary complex by soaking the wild-type holo-TPL crystals with a mixture of l-Ala and PNO. The structure of this complex, determined at 2.25 Å resolution, mimics the α-aminoacrylate intermediate with the phenol molecule bound in the active site (Scheme 1). While the open active site of this structure is occupied only by the internal aldimine, the closed active site contains a mixture of different ligands in a statistical disorder (details provided in the Supporting Information; Figure 4). A PNO molecule is bound in the pocket of the closed active site otherwise occupied by the substrate’s phenolic moiety in Y71F and F448H TPL complexes. The C4 atom of the PNO molecule (the equivalent of Cγ in l-Tyr) is located only 3.0 Å from the Cβ of the Ala quinonoid, so this structure models a phenol molecule and the α-aminoacrylate intermediate just after the cleavage of the substrate’s Cβ–Cγ bond. Mean planes of the PNO molecule and the “alanine” moiety (atoms Cα, Cβ, imino N, and carboxylic C) in the quinonoid make an angle of only 26° (Figure 4). We note that the equivalent angle in the “relaxed” quinonoid molecule is 110°, in contrast to 78° found for the “tense” quinonoid in the Y71F TPL complex and 81–84° observed for the F448H TPL complex. Like in the “tense” quinonoid, the O atom of the PNO makes H-bonds with the side chains of Thr124 and Arg381, with a close contact (∼2.9 Å) being made between the C4 atom of PNO and the proton-donating hydroxyl of Tyr71. The close resemblance of enzyme–ligand contacts in the “tense” quinonoid to contacts observed in this complex, representing the state just after the Cβ–Cγ bond cleavage, indicates that the strained conformation induced by the closure of the active site facilitates elimination of phenol.
Figure 4.

Stereo view of the closed active site of the wild-type TPL with bound PNO. The σA-weighted |Fo| – |Fc| omit electron density maps are contoured at 3.0σ. Residues are colored as in Figure 1. Hydrogen bonds are denoted by dashed lines. The alanine quinonoid, the phosphate anion, and the Wat2 solvent molecule were modeled with 0.5 occupancy.
After completion of the β-elimination reaction, the active site has to open to release the phenol molecule. As the closed conformation of the active site and a bound phenol molecule protect the α-aminoacrylate intermediate from the solvent, we speculate that the α-aminoacrylate intermediate can undergo the transaldimination reaction only after a phenol molecule is released and the active site becomes solvent accessible. In that way, the other product of this reaction, an iminopyruvate molecule, can be released and become available for the nonenzymatic hydrolysis (Scheme 1).
The bonds being broken as the result of the catalytic act are oriented trans (anti) to each other on opposite sides of the PLP plane. Thus, the mechanism that is operative in the case of TPL should be classified as anti-E1cb. Until now, this anti elimination was reported only in the case of O-acetylserine sulfhydrylase(25) where the concerted E2 mechanism was observed. The data indicate, therefore, that the traditional notion that most PLP-dependent enzymes catalyze their elimination reactions via an E1cb mechanism with syn-geometry(12) is somewhat exaggerated. It is noteworthy that in many cases the transfer of the hydrogen isotope label from the α-position of the substrate to the leaving group is considered to be the strongest argument for this mechanism. However, the internal return may be associated with the concomitant isotopic exchange reaction, especially under the conditions where a single turnover in the main elimination reaction corresponds to several turnovers in the exchange reaction.
Conclusion
Taken together, the presented structures demonstrate the importance of the closed conformation for the enzymatic activity of TPL. It is evident that the closure of the active site induces a significant strain in the quinonoid intermediate, facilitating distortion of the substrate’s Cα–Cβ–Cγ angle and pyramidalization of the Cγ atom. Structural observations indicate that the “tense” structure is stabilized by the H-bonds between the substrate’s phenol hydroxyl and Arg381 and Thr124, as well as van der Waals contacts with Phe448 (Figure 5). To release the strain, the “tense” quinonoid is more susceptible to the Cγ-protonation and the Cβ–Cγ bond cleavage, which ultimately result in the elimination of phenol. Thus, the closure of the active site serves as the main driving force for the β-elimination reaction of l-Tyr.
The remarkable rate accelerations seen in enzyme catalysis have been often attributed primarily to transition-state stabilization, as was first proposed by Pauling.(26) However, it has been suggested that ground-state destabilization may contribute as much or even more to the decrease in activation energy as transition-state stabilization.(27) In fact, on the basis of the structure of hen egg white lysozyme, the first enzyme crystal structure to be obtained, it was proposed that the reactive D-ring of the substrate is forced to adopt a higher energy half-chair conformation on binding, introducing strain that would be relieved on formation of the oxocarbenium ion intermediate.(27) Evidence for electronic strain of π-bonds in bound substrates was obtained by vibrational spectroscopy by Carey for thiol proteases,(28) by Belasco and Knowles with triose phosphate isomerase,(29) and more recently by Anderson for enoyl-CoA hydratase.(30) Evidence for ground-state strain was very recently obtained by Zhang and Schramm from examination of equilibrium isotope effects on binding of substrates to orotate phosphoribosyltransferase.(31) However, there appear to be no crystal structures that have shown a clear evidence for substrate strain. The results of the present crystallographic study demonstrate that TPL induces significant strain in the quinonoid complex of the substrate, distorting it in the ground state toward the transition state geometry required for cleavage of the formally unactivated Cβ–Cγ bond.
Experimental Section
Preparation of Quinonoid Complexes
Wild-type, Y71F, and F448H C. freundii TPL were produced in Escherichia coli SVS 370 cells transformed with the plasmid pTZTPL containing either the wild-type or the mutant TPL gene. All proteins were purified as described before.17,18 Crystals of the wild-type and mutant holo-TPL were grown using previously established conditions.10,11,15 The alanine quinonoid complex with pyridine N-oxide (PNO) was prepared by soaking wild-type TPL crystals in the stabilization solution containing 40% (w/v) poly(ethylene glycol) 5000 monomethyl ether (PEG 5000 MME), 50 mM triethanolamine pH 8.0, 0.25 M KCl, 0.2 mM PLP, 0.5 mM dithiothreitol, with addition of 100 mM l-Ala and a saturating concentration of PNO. Because the crystals’ quality deteriorated rapidly, the soaking time had to be restricted to ∼20 s. Complexes of the mutant TPL proteins with the substrate 3-fluoro-l-tyrosine (3-F-l-Tyr) were prepared by soaking crystals in the same stabilizing solution but with addition of 10 mM 3-F-l-Tyr. To prevent crystal deterioration, soaking time had to be restricted to ∼30 s.
Structure Determination and Analysis
All crystals were flash-cooled in liquid nitrogen directly from the soaking conditions. Diffraction data were collected at 100 K and processed using DENZO and SCALEPACK,(32) Table 2. Most crystallographic calculations were carried out using the CCP4 program package.(33) The structure of the F448H TPL–3-F-l-Tyr was determined by molecular replacement (PHASER)(34) using the complete tetramer of the C. freundii TPL apoenzyme (PDB ID: 2ez2)(10) as a search model. All other structures were refined using the TPL apoenzyme dimer as a starting model (PDB ID: 2ez2).(10) Models were rebuilt by COOT,(35) and water molecules were added using ARP/wARP.(36) The refinement was performed by REFMAC(37) with the small (residues 19–44, 346–404, 434–456) and large (residues 1–13, 45–345, 405–422) domains(10) of each subunit treated as separate TLS groups.(38) Models of ligand molecules were generated by SKETCHER, and the corresponding library files for the refinement were created by LIBCHECK.(33) The final refined models were further validated using MolProbity.(39) Geometries of the 3-F-Tyr quinonoid molecules were cross-checked using the MAESTRO program suite.(40) The initial model of the 3-F-Tyr quinonoid molecule was optimized by molecular mechanics using the all-atom OPLS force field in MACROMODEL.(41) The optimized ligand model was docked into the active sites and refined in real space against the corresponding omit σA-weighted |Fo| – |Fc| electron density maps using program PRIMEX.(42) The all-atom OPLS force field, the default electron density weights (120 for energy and 50 for scoring), and the low planar group restraints were used throughout the refinement in PRIMEX. DFT calculations on the quinonoid models refined in PRIMEX were performed with GAMESS(43) at the B3LYP level, using the 6-31G(d) basis set in the gas phase. Figures were made by PyMOL.(44)
Table 2. Crystallographic Data Processing and Refinement Statistics.
| structure | Y71F TPL–3-F-l-Tyr | F448H TPL–3-F-l-Tyr | TPL–l-Ala–PNO |
|---|---|---|---|
| Data Collection and Processing | |||
| space group | P21212 | P212121 | P21212 |
| unit cell parameters | |||
| a (Å) | 133.6 | 136.4 | 134.0 |
| b (Å) | 144.4 | 143.8 | 143.8 |
| c (Å) | 59.7 | 118.5 | 60.1 |
| radiation source | ESRF BM14 | ESRF BM30A | Rigaku Ru200 with rotating anode (Cu-Kα) |
| wavelength (Å) | 0.980 | 0.980 | 1.5418 |
| resolution rangea (Å) | 17.0–2.04 (2.09–2.04) | 30.0–2.00 (2.07–2.00) | 20.0–2.25 (2.29–2.25) |
| unique reflections | 71 319 (4260) | 154 478 (13 859) | 55 230 (2668) |
| Rmerge (%) | 4.6 (34.6) | 9.1 (53.1) | 4.8 (17.2) |
| average I/σ(I) | 13.7 (3.2) | 7.5 (2.5) | 13.8 (4.5) |
| redundancy | 3.0 (1.9) | 4.7 (4.5) | 3.4 (2.8) |
| completeness (%) | 96.5 (83.6) | 99.0 (99.2) | 98.8 (94.3) |
| Wilson B-factor (Å2) | 28.2 | 22.4 | 34.6 |
| Refinement and Model Correlation | |||
| no. of atoms | |||
| protein | 7 306 | 14 634 | 7281 |
| ligand | 80 | 393 | 101 |
| solvent | 745 | 1863 | 791 |
| reflections used in refinement | 70 272 (4193) | 152 931 (13 724) | 54 235 (2623) |
| Rcryst (%) | 16.3 (20.1) | 14.1 (17.8) | 13.9 (17.0) |
| Rfree (%) | 20.9 (23.3) | 17.5 (22.9) | 18.1 (22.9) |
| reflections used for Rfree | 1047 (69) | 1547 (135) | 995 (45) |
| average B-factor (Å2) | |||
| polypeptide chain A/B/C/D | 38.9/34.7/–/– | 22.3/24.1/22.4/23.2 | 29.8/28.4/–/– |
| ligand | 27.2 | 18.1 | 18.9 |
| solvent | 26.6 | 29.0 | 26.3 |
| overall | 36.8 | 23.9 | 28.9 |
| deviations from ideal geometryb | |||
| bond lengths (Å) | 0.014 (0.022) | 0.013 (0.022) | 0.014 (0.022) |
| bond angles (deg) | 1.4 (2.0) | 1.3 (2.0) | 1.3 (2.0) |
| estimated coordinate errorc (Å) | 0.15 | 0.12 | 0.16 |
| Ramachandran plot d (%) | |||
| favored | 97.6 | 97.8 | 98.1 |
| outliers | 0.0 | 0.2e | 0.0 |
| PDB accession code | 2ycn | 2ycp | 2yct |
Values in parentheses are for the outer resolution shell.
Target values are given in parentheses.
The diffraction-component precision index (DPI) based on Rfree.(19)
Analyzed by MolProbity.(39)
The only Ramachandran plot outliers are Met121(A), Met121(B), and Met121(C) with the φ and ψ values next to the borderline of the allowed region(45) and very similar to those of Met121(D). All of these residues are clearly defined in the electron density maps.
Acknowledgments
We thank Dr. Martin Walsh and Dr. Richard Kahn for help during the data collection at ESRF beamlines BM14 and BM30A, Prof. Zlatko Mihalić and Ivan Kodrin for help with the MacroModel software, and Dr. Ivan Halasz for critically reading the manuscript and for useful suggestions. This work was funded by Grant 119-1193079-1084 of the Ministry of Science, Education and Sports of the Republic of Croatia (D.M. and D.M.-Č.), Grants 11-04-00220-a (T.V.D.) and 11-04-01535 (N.G.F.) of the Russian Foundation for Basic Research, and Wellcome Trust fellowship 081916 (A.A.A.).
Supporting Information Available
Detailed description of X-ray structures and stereo views of ligand models superimposed with the electron density maps. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Kumagai H.; Yamada H.; Matsui H.; Ohkishi H.; Ogata K. J. Biol. Chem. 1970, 245, 1767–1772. [PubMed] [Google Scholar]; Phillips R. S. Arch. Biochem. Biophys. 1987, 256, 302–310. [DOI] [PubMed] [Google Scholar]; Enei H.; Nakazawa H.; Matsui H.; Okumura S.; Yamada H. FEBS Lett. 1972, 21, 39–41. [DOI] [PubMed] [Google Scholar]; Yamada H.; Kumagai H. Adv. Appl. Microbiol. 1975, 19, 249–288. [DOI] [PubMed] [Google Scholar]; Yamada H.; Kumagai H.; Kashima N.; Torii H.; Enei H.; Okumura S. Biochem. Biophys. Res. Commun. 1972, 46, 370–374. [DOI] [PubMed] [Google Scholar]; Kim K.; Cole P. A. Bioorg. Med. Chem. Lett. 1999, 9, 1205–1208. [DOI] [PubMed] [Google Scholar]; Faleev N. G.; Zhukov Y. N.; Khurs E. N.; Gogoleva O. I.; Barbolina M. V.; Bazhulina N. P.; Belikov V. M.; Demidkina T. V.; Khomutov R. M. Eur. J. Biochem. 2000, 267, 6897–6902. [DOI] [PubMed] [Google Scholar]
- Kumagai H.; Matsui H.; Ohgishi H.; Ogata K.; Yamada H.; Ueno T.; Fukami H. Biochem. Biophys. Res. Commun. 1969, 34, 266–270. [DOI] [PubMed] [Google Scholar]; Ueno T.; Fukami H.; Ohkishi H.; Kumagai H.; Yamada H. Biochim. Biophys. Acta 1970, 206, 476–479. [DOI] [PubMed] [Google Scholar]; Phillips R. S.; Ravichandran K.; Von Tersch R. L. Enzyme Microb. Technol. 1989, 11, 80–83. [Google Scholar]; Watkins E. B.; Phillips R. S. Bioorg. Med. Chem. Lett. 2001, 11, 2099–2100. [DOI] [PubMed] [Google Scholar]; Lee S.-G.; Ro H.-S.; Hong S.-P.; Kim E.-H.; Sung M.-H. J. Microbiol. Biotechnol. 1996, 6, 98–102. [Google Scholar]
- Koyanagi T.; Katayama T.; Suzuki H.; Nakazawa H.; Yokozeki K.; Kumagai H. J. Biotechnol. 2005, 115, 303–306. [DOI] [PubMed] [Google Scholar]; Lütke-Eversloh T.; Santos C. N. S.; Stephanopoulos G. Appl. Microbiol. Biotechnol. 2007, 77, 751–762. [DOI] [PubMed] [Google Scholar]
- Mouratou B.; Kasper P.; Gehring H.; Christen P. J. Biol. Chem. 1999, 274, 1320–1325. [DOI] [PubMed] [Google Scholar]; Seisser B.; Zinkl R.; Gruber K.; Kaufmann F.; Hafner A.; Kroutil W. Adv. Synth. Catal. 2010, 352, 731–736. [Google Scholar]; Kim J. H.; Song J. J.; Kim B. G.; Sung M. H.; Lee S. C. J. Microbiol. Biotechnol. 2004, 14, 153–157. [Google Scholar]; Lee S.-G.; Hong S.-P.; Kim D. Y.; Song J. J.; Ro H.-S.; Sung M.-H. FEBS J. 2006, 273, 5564–5573. [DOI] [PubMed] [Google Scholar]; Rha E.; Kim S.; Choi S. L.; Hong S. P.; Sung M. H.; Song J. J.; Lee S. G. FEBS J. 2009, 276, 6187–6194. [DOI] [PubMed] [Google Scholar]
- Phillips R. S.; Demidkina T. V.; Faleev N. G. Biochim. Biophys. Acta 2003, 1647, 167–172. [DOI] [PubMed] [Google Scholar]; Demidkina T. V.; Antson A. A.; Faleev N. G.; Phillips R. S.; Zakomirdina L. N. Mol. Biol. 2009, 43, 269–283. [PubMed] [Google Scholar]
- Phillips R. S.; Chen H. Y.; Faleev N. G. Biochemistry 2006, 45, 9575–9583. [DOI] [PubMed] [Google Scholar]
- Phillips R. S.; Sundararaju B.; Faleev N. G. J. Am. Chem. Soc. 2000, 122, 1008–1014. [Google Scholar]
- Antson A. A.; Demidkina T. V.; Gollnick P.; Dauter Z.; Von Tersch R. L.; Long J.; Berezhnoy S. N.; Phillips R. S.; Harutyunyan E. H.; Wilson K. S. Biochemistry 1993, 32, 4195–4206. [DOI] [PubMed] [Google Scholar]
- Sundararaju B.; Antson A. A.; Phillips R. S.; Demidkina T. V.; Barbolina M. V.; Gollnick P.; Dodson G. G.; Wilson K. S. Biochemistry 1997, 36, 6502–6510. [DOI] [PubMed] [Google Scholar]
- Milić D.; Matković-Čalogović D.; Demidkina T. V.; Kulikova V. V.; Sinitzina N. I.; Antson A. A. Biochemistry 2006, 45, 7544–7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milić D.; Demidkina T. V.; Faleev N. G.; Matković-Čalogović D.; Antson A. A. J. Biol. Chem. 2008, 283, 29206–29214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunathan H. C. Proc. Natl. Acad. Sci. U.S.A. 1966, 55, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toney M. D. Arch. Biochem. Biophys. 2005, 433, 279–287. [DOI] [PubMed] [Google Scholar]
- Szebenyi D. M. E.; Liu X.; Kriksunov I. A.; Stover P. J.; Thiel D. J. Biochemistry 2000, 39, 13313–13323. [DOI] [PubMed] [Google Scholar]; Barends T. R. M.; Domratcheva T.; Kulik V.; Blumenstein L.; Niks D.; Dunn M. F.; Schlichting I. ChemBioChem 2008, 9, 1024–1028. [DOI] [PubMed] [Google Scholar]; Lai J.; Niks D.; Wang Y.; Domratcheva T.; Barends T. R. M.; Schwarz F.; Olsen R. A.; Elliott D. W.; Fatmi M. Q.; Chang C.-e. A.; Schlichting I.; Dunn M. F.; Mueller L. J. J. Am. Chem. Soc. 2011, 133, 4–7. [DOI] [PubMed] [Google Scholar]
- Phillips R. S.; Demidkina T. V.; Zakomirdina L. N.; Bruno S.; Ronda L.; Mozzarelli A. J. Biol. Chem. 2002, 277, 21592–21597. [DOI] [PubMed] [Google Scholar]
- Chen H. Y.; Demidkina T. V.; Phillips R. S. Biochemistry 1995, 34, 12276–12283. [DOI] [PubMed] [Google Scholar]
- Demidkina T. V.; Barbolina M. V.; Faleev N. G.; Sundararaju B.; Gollnick P. D.; Phillips R. S. Biochem. J. 2002, 363, 745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Gollnick P.; Phillips R. S. Eur. J. Biochem. 1995, 229, 540–549. [PubMed] [Google Scholar]
- Spek A. Acta Crystallogr., Sect. D 2009, 65, 148–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The diffraction-component precision index (DPI) based on Rfree: Cruickshank D. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1999, 55, 583–601. [DOI] [PubMed] [Google Scholar]
- The search of the Cambridge Structural Database (CSD; Version 5.31) revealed 3744 entries containing the C–CH2–Ph noncyclic fragment, not disordered, not solved from powder data, and with R-factor less than 5%. The mean value of the tetrahedral C–C–C angle was 113.8° with the sample s.u. of 2.3° (the minimal and maximal values were 96.5° and 131.9°, respectively). Only two entries that satisfy the above criteria have the C–CH2–Ph angle smaller than 105°. Closer inspection of these two structures (CSD codes: REGGAM01 and RUYDOE) revealed an error in the reported unit-cell parameters, which accounts for the distorted geometrical parameters and the observed C–CH2–Ph angle values of 101.0(7)° and 96(1)° for REGGAM01 and RUYDOE, respectively.
- Wynberg H.; Nieuwpoort W. C.; Jonkman H. T. Tetrahedron Lett. 1973, 14, 4623–4628. [Google Scholar]; Dijkstra F.; Van Lenthe J. H. Int. J. Quantum Chem. 1999, 74, 213–221. [Google Scholar]; Endo Y.; Songkram C.; Ohta K.; Kaszynski P.; Yamaguchi K. Tetrahedron Lett. 2005, 46, 699–702. [Google Scholar]; Datta A.; Pati S. K. Chem. Phys. Lett. 2006, 433, 67–70. [Google Scholar]; Feixas F.; Matito E.; Poater J.; Sola M. J. Phys. Chem. A 2007, 111, 4513–4521. [DOI] [PubMed] [Google Scholar]; Shishkin O. V.; Pichugin K. Y.; Gorb L.; Leszczynski J. J. Mol. Struct. 2002, 616, 159–166. [Google Scholar]
- Kumagai H.; Kashima N.; Yamada H. Biochem. Biophys. Res. Commun. 1970, 39, 796–801. [DOI] [PubMed] [Google Scholar]; Muro T.; Nakatani H.; Hiromi K.; Kumagai H.; Yamada H. J. Biochem. (Tokyo) 1978, 84, 633–640. [DOI] [PubMed] [Google Scholar]; Demidkina T. V.; Myagkikh I. V.; Azhayev A. V. Eur. J. Biochem. 1987, 170, 311–316. [DOI] [PubMed] [Google Scholar]
- Chen H.; Phillips R. S. Biochemistry 1993, 32, 11591–11599. [DOI] [PubMed] [Google Scholar]
- Tai C.-H.; Cook P. F. Acc. Chem. Res. 2001, 34, 49–59. [DOI] [PubMed] [Google Scholar]
- Pauling L. Am. Sci. 1948, 36, 50–58. [PubMed] [Google Scholar]; Radzicka A.; Wolfenden R.; Daniel L. P. Methods Enzymol. 1995, 249, 284–312. [DOI] [PubMed] [Google Scholar]; Schramm V. L. Annu. Rev. Biochem. 1998, 67, 693–720. [DOI] [PubMed] [Google Scholar]
- Blake C. C. F.; Johnson L. N.; Mair G. A.; North A. C. T.; Phillips D. C.; Sarma V. R. Proc. R. Soc. London, Ser. B 1967, 167, 378–388. [DOI] [PubMed] [Google Scholar]
- Carey P. R.; Carriere R. G.; Lynn K. R.; Schneider H. Biochemistry 1976, 15, 2387–2393. [DOI] [PubMed] [Google Scholar]; Carey P. R.; Carriere R. G.; Phelps D. J.; Schneider H. Biochemistry 1978, 17, 1081–1087. [DOI] [PubMed] [Google Scholar]
- Belasco J. G.; Knowles J. R. Biochemistry 1980, 19, 472–477. [DOI] [PubMed] [Google Scholar]; Belasco J. G.; Knowles J. R. Biochemistry 1983, 22, 122–129. [DOI] [PubMed] [Google Scholar]
- D’Ordine R. L.; Tonge P. J.; Carey P. R.; Anderson V. E. Biochemistry 1994, 33, 12635–12643. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Schramm V. L. Biochemistry 2011, 50, 4813–4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. Methods Enzymol. 1997, 276, 307–326. [DOI] [PubMed] [Google Scholar]
- Collaborative Computational Project, Number 4. Acta Crystallogr., Sect. D 1994, 50, 760–763.15299374 [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. J. Appl. Crystallogr. 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Acta Crystallogr., Sect. D 2004, 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Perrakis A.; Morris R.; Lamzin V. S. Nat. Struct. Biol. 1999, 6, 458–463. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Acta Crystallogr., Sect. D 1997, 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Winn M. D.; Isupov M. N.; Murshudov G. N. Acta Crystallogr., Sect. D 2001, 57, 122–133. [DOI] [PubMed] [Google Scholar]
- Davis I. W.; Leaver-Fay A.; Chen V. B.; Block J. N.; Kapral G. J.; Wang X.; Murray L. W.; Arendall W. B. III; Snoeyink J.; Richardson J. S.; Richardson D. C. Nucleic Acids Res. 2007, 35, W375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maestro, version 9.1; Schrödinger, LLC: New York, NY, 2010.
- MacroModel, version 9.5; Schrödinger, LLC: New York, NY, 2007.
- PrimeX, version 1.7; Schrödinger, LLC: New York, NY, 2010.
- Schmidt M. W.; Baldridge K. K.; Boatz J. A.; Elbert S. T.; Gordon M. S.; Jensen J. H.; Koseki S.; Matsunaga N.; Nguyen K. A.; Su S.; Windus T. L.; Dupuis M.; Montgomery J. A. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar]; Gordon M. S.; Schmidt M. W. In Theory and Applications of Computational Chemistry, the first forty years; Dykstra C. E., Frenking G., Kim K. S., Scuseria G. E., Eds.; Elsevier: Amsterdam, 2005; pp 1167–1189. [Google Scholar]
- DeLano W. L.DeLano Scientific: Palo Alto, CA, 2002; http://www.pymol.org.
- Lovell S. C.; Davis I. W.; Arendall W. B. III; de Bakker P. I. W.; Word J. M.; Prisant M. G.; Richardson J. S.; Richardson D. C. Proteins: Struct., Funct., Genet. 2003, 50, 437–450. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.