

Abstract
Airway hyperreactivity (AHR), lung inflammation, and atopy are clinical signs of allergic asthma. Gestational exposure to cigarette smoke (CS) markedly increases the risk for childhood allergic asthma. Muscarinic receptors regulate airway smooth muscle tone, and asthmatics exhibit increased AHR to muscarinic agonists. We have previously reported that in a murine model of bronchopulmonary aspergillosis maternal exposure to mainstream CS increases AHR after acute intratracheal administration of Aspergillus fumigatus extract (Af). However, the mechanism by which gestational CS induces allergic asthma is unclear. We now show for the first time that, compared to controls, mice exposed prenatally to secondhand CS exhibit increased lung inflammation (predominant infiltration by eosinophils and polymorphs), atopy, and airway resistance, and produce proinflammatory cytokines (IL-4, IL-5, IL-6, and IL-13, but not IL-2 or IFN-γ). These changes, which occur only after an allergen (Af) treatment, are correlated with marked upregulated lung expression of M1, M2, and M3 muscarinic receptors and phosphodiesterase-4D5 (PDE4D5) isozyme. Interestingly, the PDE4-selective inhibitor rolipram attenuates the increase in AHR, muscarinic receptors, and PDE4D5, but fails to downregulate lung inflammation, Th2 cytokines, or serum IgE levels. Thus, the fetus is extraordinarily sensitive to CS, inducing allergic asthma after postnatal exposure to allergens. While the increased AHR might reflect increased PDE4D5 and muscarinic receptor expression, the mechanisms underlying atopy and lung inflammation are unrelated to the PDE4 activity. Thus, PDE4 inhibitors might ease AHR, but are unlikely to attenuate lung inflammation and atopy associated with childhood allergic asthma.
Introduction
The adverse health effects of cigarette smoke (CS) are well recognized, and smoking is associated with increased risk for lung cancer and respiratory infections (1). Increasing evidence suggests that chronic exposure to environmental or secondhand tobacco smoke (SS) also causes significant health effects (2–4). Moreover, strong epidemiological evidence indicates parental smoking, particularly maternal smoking during pregnancy, increases the risk of allergic asthma in children (4–10). Yet in the USA alone, nearly 12% of prospective mothers continue to smoke during pregnancy (11). Interestingly, prenatal and postnatal exposure to CS may affect immune and inflammatory responses differently (12, 13). For example, some allergic diseases and ulcerative colitis are less common in adult smokers than nonsmokers (1, 14–18), whereas ex-smokers are more likely to develop asthma than current smokers (19, 20). Moreover, in animal models, chronic exposure of adult animals to mainstream CS or nicotine suppresses innate and adaptive immune responses (1, 21–24), and even SS moderates some parameters of allergic asthma in mice (25). In Brown Norway rats, chronic exposure to nicotine attenuates the ragweed/house dust mite-induced lung inflammation and atopy (13). Thus, in adult humans and animals, chronic CS/nicotine exposure may attenuate some parameters of allergic inflammation in the lung. On the other hand, in utero exposure to mainstream CS exacerbates allergic and inflammatory responses in the offspring (26), and it is likely that the mechanisms by which CS modulates the allergic responses in utero and during adult life do not totally overlap.
The mechanism(s) by which gestational exposure to CS affects the lung function in children is not clearly understood. In an established murine model of bronchopulmonary aspergillosis, where Af-extract induce allergic asthma (26, 27), we have shown that exposure of mothers to mainstream CS throughout the gestational period increases airway hyperreactivity (AHR) after an acute exposure to the allergen – Af and the increased AHR is related to elevated expression of phosphodiesterase-4 (PDE4) in the lungs of the progeny (26). However, unlike chronic Af sensitization (27), single exposure to the allergen did not induce significant lung inflammation and atopy (26). Therefore, in addition to the mechanism of allergen-induced increase in AHR, the effects of gestational CS exposure on lung inflammation and atopy are unknown. In this communication, we show that fetuses are highly sensitive to CS, and maternal exposure to evensecondhand cigarette smoke (SS) strongly exacerbates the allergen-induced AHR through upregulated expression of M1, M2, and M3 muscarinic receptors and the PDE4 isozyme PDE4D5 in the lung. In addition, SS markedly intensifies lung inflammation, Th2 cytokine production, and atopy induced through allergic sensitization. While the PDE4-selective inhibitor rolipram (RP) decreased muscarinic receptor expression and AHR, it essentially failed to affect the allergen-induced atopy and lung inflammation.
Materials and Methods
Animals
Pathogen-free BALB/c mice were obtained from the Frederick Cancer Research Facility (Frederick, MD). Animals were housed in shoebox-type plastic cages with hardwood chip bedding and conditioned to whole-body exposure in exposure chambers (H1000; Hazleton Systems, Inc., Aberdeen, MD) for 2 wk before exposure to SS. The chamber temperature was maintained at 26 ± 2°C, and lights were set to a 12-h on/off cycle. Food and water were provided ad libitum. All animal protocols used in this study were approved by Lovelace Respiratory Research Institute’s Institutional Animal Care and Use Committee.
Antibodies and Reagents
Antibody to PDE4 (ab14628) was obtained from Abcam, Inc. MA. All the other reagents, unless otherwise stated, were bought from Sigma Chemical Co. (St. Louis, MO).
Cigarette smoke generation and exposure
Mice were exposed to whole-body SS (the smoke released from the burning end of a cigarette) or filtered air (FA) for 6 h/day, 7 days/wk as described (26). Briefly, a smoking machine (Type 1300; AMESA Electronics, Geneva, Switzerland) generated two 70-cm3 puffs/min from a research cigarette (Type 2R1; Tobacco Health Research Institute, Lexington, KY), and the smoke was captured from the lit end of the cigarette with a plastic manifold placed above it. Mice were placed in whole body chambers and exposed to either FA or SS (total particulate matter: 1.52 ± 0.41 mg/m3). This levels of SS exposure simulates the conditions to which fetuses are likely to be exposed through paternal smoking, is slightly lower than the average SS concentration of 2 mg/m3 found in most smoking bars, and is approximately 70-fold less than the amount of CS inhaled by an average two-pack a day smoker (26, 28). Adult (3–4 month old) male and female mice were separately acclimatized either to SS or FA for 2 wk and then paired for mating under the same exposure conditions. After ascertaining pregnancy by vaginal smear, pregnant mice were separated, housed singly in plastic cages, and continued to receive SS or FA until the pups were born. The mothers and pups continued to be exposed to FA or SS until the pups were weaned at 3 wk of age.
Allergen (Af) sensitization and challenge
A lyophilized culture filtrate preparation (29, 30) of Af was used as the allergic sensitization as described previously (27). Briefly, mice were sensitized intratracheal (i.t) with Af (50 μg/0.1 ml sterile endotoxin-free saline or saline alone) and subsequently challenged i.t. with 100 μg/0.1 ml of Af-extract three times at 5-day intervals. Where indicated, RP (250 μg/kg) was injected intraperitoneal (i.p.) into animals at the time of Af sensitization. Control animals received sterile saline alone.
Pulmonary function test
At 48 h after the last Af/saline challenge, airway resistance (RL) was measured by the Flexivent system (SCIREQ, Montreal, Quebec, Canada). Briefly, mice were anesthetized by i.p. injection of avertin (250 mg/kg). A small incision was made in the trachea through which a 20-gauge needle hub was inserted; a saline-filled catheter was placed in the thoracic esophagus via the mouth to obtain transthoracic pressure. The mouse was placed on the Flexivent apparatus and ventilated through the tracheal cannula. Then the paralytic doxacurium (0.5 mg/kg) was administered i.p. Heart rate and electrocardiogram were monitored (via a Grass Instruments Recorder with Tachograph). AHR was assessed using increasing doses of aerosolized methacholine (MCh). RL and elastance were measured by manipulation of ventilatory patterns and measurement of upstream and downstream pressures. The peak responses at each MCh concentration were used for data analysis.
Bronchoalveolar lavage (BAL) and cell Collection
Mice were anesthetized and euthanized by exsanguination 24 h after the last allergen (Af) challenge. Before excision of the lungs, the trachea was surgically exposed and cannulated. The left lung lobe was tied off with a silk thread suture, and the right lobe was lavaged with 1 ml PBS (2x) and pooled for each animal. BAL cells were collected by centrifugation and resuspended in PBS. Approximately 5 × 104 cells from each sample were cytospun on duplicate slides and stained with Diff Quik (Baxter Healthcare, Miami, FL) to score eosinophils (EOS), macrophages (MACS), neutrophils, and lymphocytes using standard hemocytologic criteria. At least 200 cells from each slide were counted to obtain the differential cell count.
Analysis of cytokines/chemokines in the BAL fluid (BALF)
BALF was analyzed for cytokines and chemokines by the commercially available Mouse Cytokine Ten-Plex ELISA kit (Biosource, Invitrogen, Camarillo, CA) and used according to the manufacturer’s directions. Cytokine concentrations were determined utilizing Bioplex manager software with four-parameter data analysis. The sensitivity of the assay was < 10 pg/ml.
Quantitative RT- PCR (qPCR) analysis
Total RNA was extracted from the lung samples using TRI reagent (Molecular Res. Center, Cincinnati, OH) and quantified following the manufacturer’s instructions. The lung expression of cytokines (IL-4, IL-5, IL-6, and IL-13), muscarinic receptors (M1, M2, and M3), 18S RNA, and GAPDH was determined by qPCR using the One-Step RT-PCR Master Mix and specific labeled primers/probes set (Applied Biosystems, Foster City, CA). The relative expression of each mRNA was calculated as previously described (13).
Total serum IgE
Blood from the mice were collected on the day of sacrifice, centrifuged in a serum separator, and stored at −80° C until analysis. Total serum IgE titers were determined by the ELISA method as previously described (30).
Western Blot Analysis
Lung tissues were homogenized in RIPA buffer (20 mM Tris, 150 mM NaCl, 20 mM β-glycerylphosphate, 1% Triton-X, 10 mM NaF, 5 mM EDTA, 1 mM Na3VO4, and protease inhibitors, 1 mM PMSF, 1 μg/ml each of aprotinin, antipain, and leupeptin) at 4°C. Protein content of the extracts was determined by the BCA Protein Assay Kit (Pierce, Rockford, IL) according to the manufacturer’s directions. The samples were electrophoresed on 10% SDS-PAGE and transferred on to nitrocellulose membrane (Bio-Rad, Hercules, CA). Membranes were blocked using 5% non-fat dry milk in Tris-buffered saline with 0.05% Tween-20 (TBST) for 1 h at room temperature and probed with primary antibody to PDE4 (rabbit polyclonal ab14628: Abcam, Inc.) overnight at 4°C. The blots were washed three times with TBST, incubated for 1 h at room temperature with HRP-conjugated anti-rabbit serum (Santa Cruz Biotechnology, Santa Cruz, CA) at room temperature, and developed by ECL (Amersham Biosciences, UK) on an X-ray film.
Statistical analysis
All data were analyzed using Graph Pad Prism software 3.0 using student’s t test or by two-way ANOVA. Results are presented as the means ± SD of the combined experiments. The differences with p value of ≤0.05 were considered statistically significant.
Results
PDE4D5 expression is upregulated in the lungs of mice exposed to maternal SS
We have previously shown that the activity of PDE4 is higher in animals exposed in utero to mainstream CS (26). However, the identity of the PDE4 isozyme(s) affected by gestational exposure to CS was not known. Also, it was not clear whether the increased enzymatic activity reflected increased PDE4 protein and whether the increase was independent of allergen sensitization. Although there are nine isoforms of PDE4, including alternative spliced products (31), under these conditions the Western blot analysis of the lung homogenates showed the presence of only three immunoreactive forms of PDE4 isozymes in the mouse lung that correspond to 105 kDa (PDE4D5), 95 kDa (PDE4D3), and 79 kDa (PDE4A/B) (Fig. 1). Results indicate that prenatal exposure to SS or Af sensitization of control animals alone does not significantly alter their content in the lung. However, after Af sensitization, the lungs from SS-exposed animals (SS/Af) exhibit increased PDE4 proteins, particularly the PDE4D5 isoform of the enzyme. Treatment of SS/Af animals with the PDE4 selective inhibitor RP completely blocked the rise in PDE4 content, including that of the PDE4D5 isoform. These data suggest that prenatal exposure to CS per se does not elevate PDE4, but primes the lung to make more PDE4, particularly the PDE4D5 isoform, after an allergic challenge. Moreover, essentially all the increase in PDE4D5 is blocked by RP treatment.
FIGURE 1. Maternal exposure to SS increases allergen-induced lung expression of PDE4D5.
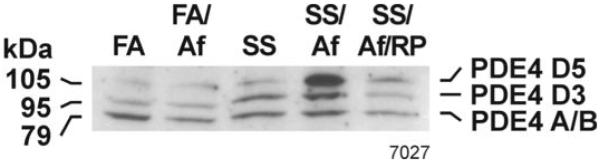
Lung homogenate (75 μg) were fractionated using SDS-PAGE, transferred on to nitrocellulose membrane, and probed with anti-PDE4 antibody. The figure represents the response of three animals from each group: FA, FA/Af, SS, SS/Af, and SS/Af treated with rolipram (RP).
Increased RL in Af-sensitized mice exposed to maternal CS
Children exposed to tobacco smoke either through maternal smoking or exposure of the mother to SS are at higher risk of developing asthma (32). We have demonstrated that prenatal exposure to mainstream CS increases AHR detected by Baxco plethysmography (26). However, the validity of the Baxco system to measure bronchoconstriction has been strongly questioned (33). Therefore, to determine whether maternal exposure to CS affected AHR, we used the Flexivent system to measure RL in response to MCh in animals exposed prenatally to SS. Results presented in Fig. 2A (inset) show that maternal exposure to SS slightly but significantly increased AHR at the baseline. Af sensitization of FA-exposed animals significantly increased the AHR response; however, compared to animals exposed to FA or FA+Af, the increase in the RL in response to MCh of prenatally SS-exposed animals sensitized with Af was extremely dramatic (Fig. 2A). Moreover, as seen by changes in the ED50 for MCh, treatment of animals with the PDE4 selective inhibitor RP significantly attenuated the MCh-induced RL in Af-sensitized SS-treated animals (Fig. 2B). Thus, gestational exposure to SS primes the lung for increased allergen-induced AHR, and the increase is at least partly related to the increased lung phosphodiesterase expression after the allergic sensitization.
FIGURE 2. Maternal SS exposure strongly increases MCh-induced and rolipram (RP)-insensitive RL in Af-treated animals.

Indicated groups of animals were examined for RL by the Flexivent System as described in Materials and Methods. A; Peak values for RL are plotted against each MCh concentration, and the values represent mean ± SD of 5-7 mice/group in one experiment. The difference in the p value between SS + Af group with other groups was ≤0.001. Inset: Because the response of SS/Af to MCh was much higher than other animals, the y-axis scale was expanded to visualize differences in the response between other groups (n = 5-7/group). B. Doses of MCh needed to elicit 50% (ED50) of the peak response in SS-exposed animals before and after Af and Af + RP treatment. Bars are mean ± SD. *** represents p ≤ 0.001.
Maternal SS exposure increases lung leukocytic infiltration after allergic sensitization
Allergic asthma is associated with elevated numbers of EOS and neutrophils in the lung (34, 35). To determine whether maternal exposure to SS promoted leukocytic infiltration in the lung before and after an allergen challenge, prenatally FA- and SS-exposed animals were challenged through i.t.( instillation with Af extracts. BAL cells were collected and stained for differential cell count. The total number of BAL cells in FA- and SS-exposed animals without Af sensitization were similar (6-8 × 104); however, after Af challenge, the number of cells in FA/Af rose to 44 ± 7× 104, and the number was further increased to 96 ± 14 × 104 in SS/Af animals. Moreover, as shown in Fig. 3, prior to Af sensitization, MACS represented the predominant cell population in FA- and SS-exposed animals; following Af sensitization, however, the composition of BAL cells changed. The percentage of EOS and polymorphs (PMN) relative to MACS increased significantly, and the increase was markedly higher in Af-sensitized animals exposed maternally to SS. Thus, in the 200 cells counted, PNM and EOS were over three fold higher in SS/Af mice than in the FA/Af mice (35 ± 12, and 31 ± 11 vs 9 ± 5 and 12 ± 3, respectively). Interestingly, RP did not significantly alter the total number or distribution of leukocyte subtypes in the SS/Af BAL. These results suggest that maternal exposure to SS primes the lung for increased inflammatory responses, and the increase is not significantly related to changes in the lung PDE4 activity.
FIGURE 3. Changes in leukocyte subtypes in the BAL cells after maternal SS exposure, Af treatment, and Af + rolipram (RP) treatment.

Differential cell count was accessed by staining cytospin slides using standard hemocytologic criteria (n = 4-6/group). Two hundred cells were counted per slide (MACS, macrophage; PMN, neutrophils; LYMPH, lymphocytes; EOS, eosinophils). Bars represents mean ± SD. Differences in the p values between SS and SS+Af and SS+ Af +RP are ≤ 0.01.
Maternal exposure to SS increases the propensity of Th2 cytokine production in the lung
To ascertain whether the increased lung inflammation in maternally SS-exposed animals is associated with increased production of proinflammatory cytokines, we determined the expression of various proinflammatory cytokines by ELISA in the BALF and by qPCR of the lung mRNA in animals exposed prenatally to FA or SS before and after sensitization with Af. Compared to FA/Af, levels of the Th2 cytokines (IL-4, IL-5, IL-6, and IL-13) were significantly higher in the BALF from SS/Af animals (Fig. 4A). Similarly, the lung mRNA expression for IL-4, IL-5, IL-6, and IL-13 was significantly higher in the SS/Af than the FA/Af group (Fig. 4B). There was no detectable level of IFN-γ or IL-2 in any BALF sample, and the lung mRNA level of IFN-γ was low and unaltered by any treatment (not shown). Moreover, RP had no significant effect on the expression of Th2 cytokines. Thus, it is likely that the maternal exposure to SS induces epigenetic changes that primarily increase the propensity of the lung to upregulate the expression of Th2 cytokines after Af sensitization, and the SS-induced changes in Th2 cytokine expression are not significantly related to the increase in PDE4 activity.
FIGURE 4. Maternal exposure to SS strongly upregulates cytokine expression in the lung after Af sensitization that is insensitive to rolipram (RP).

Cytokines levels in BALF (A) and expression in the lung (B) were determined by ELISA (n = 4-6/group) and qPCR (n = 3/group), respectively. * indicates a significant change at p ≤ 0.05; ** p ≤ 0.01, and *** p ≤ 0.001.
Maternal exposure to SS increases serum IgE levels
Atopy is the hallmark of allergic asthma, and the Th2 cytokines IL-4/IL-13 are critical in the production of IgE (36). Moreover, Af sensitization stimulates IgE production in the murine model of aspergillosis (37). To determine the effect of maternal exposure to SS on IgE, total IgE was assayed in the serum from the FA- and maternal SS-exposed mice before and after Af sensitization. Sensitization with Af significantly increased the serum IgE concentration of FA and SS animals; however, compared to FA-exposed mice, the serum level of IgE was nearly 3-fold higher in the in SS-exposed animals after Af sensitization. Moreover, RP treatment failed to significantly alter the serum concentration of IgE (Fig. 5). These results suggest that maternal SS exposure increases serum IgE in response to allergic sensitization, but the increased IgE levels are not linked to changes in the lung PDE4 activity.
FIGURE 5. Maternal exposure to SS exacerbates atopy following Af sensitization that is insensitive to rolipram (RP).

Total serum IgE was determined by a commercially developed kit as described in Materials and Methods (n = 4-6/group). ** p ≤ 0.01.
Maternal exposure to SS upregulates the muscarinic acetylcholine receptor (mAChR) expression in the lung
Lungs have an abundant expression of mAChRs. These receptors modulate airway smooth muscle contraction and affect AHR and asthma (38). To investigate whether increased AHR in animals maternally exposed to SS and sensitized with Af was related to the effects of SS exposure on the mAChR expression in the lung, we determined the expression of M1, M2, and M3 mAChRs in the lung by qPCR. These three mAChR subtypes have been detected in murine and human airways (39). It is clear from the results presented in Fig. 6 that compared to Af-sensitized FA animals, the mRNA expression of M1, M2, and M3 receptors is markedly upregulated in Af-sensitized, SS-exposed animals by approximately 43-, 13-, and 6-fold, respectively. Moreover, RP treatment completely blocked the increase in expression of these mAChR subtypes, indicating a crucial role for PDE4 in the maternal smoke-induced elevated expression of mAChRs.
FIGURE 6. Maternal exposure to SS upregulates the expression of M1, M2, and M3 muscarinic receptors in the lung.

The lung expression of muscarinic receptors was determined by qPCR as detailed in Materials and Methods (n = 3/group). A: M1, B: M2, and C: M3. Bars represents mean ± SD of triplicate values.
Discussion
In utero exposure to parental smoking is associated with an increased risk for development of childhood atopy and asthma (40), indicating that direct maternal smoking as well as exposure of mothers to SS may be significant factors in eliciting childhood asthma. To study the role of maternal SS exposure in childhood allergic asthma, we used the murine model of allergic bronchopulmonary aspergillosis, where Aspergillus proteins act as allergens and increase the total and Aspergillus-reactive IgE in adult animals (27, 37). Allergic bronchopulmonary aspergillosis is also seen in humans, and IgE from these patients reacts with Aspergillus proteins (29). In the Af-sensitized animals, the allergic response is further aggravated after the administration of recombinant IL-4, indicating a significant role of this cytokine in the allergic response (29). In addition, BAL cells from Af-sensitized animals produce increased levels of proinflammatory cytokines after in vitro stimulation (42). Thus, this model simulates typical characteristics of allergic asthma, including increased AHR, inflammation, and atopy. We have used this model to demonstrate that maternal exposure to mainstream CS increases AHR that is causally related to increased PDE4 activity (26). However, the experiment represented a single exposure to Af, which did not induce significant lung inflammation or atopy (26). Because of these concerns, in the current studies we measured RL by the Flexivent system and after Af sensitization conditions that produced significant increases in lung inflammation and IgE levels.
It is clear from our results that, similar to mainstream CS exposure (26), compared to controls, gestational exposure to SS slightly increases the baseline RL, and the difference becomes highly exaggerated when the animals are sensitized with Af. These results suggest that the potential for higher AHR is established during in utero SS exposure, but requires allergic sensitization for the full expression. Therefore, it is likely that CS exposure during fetal development produced conditions that lead to an increased propensity for AHR. Moreover, the fact that the concentration of SS to which the animals were exposed during gestation (1.52 ± 0.41 mg/m3 total particulate matter) is nearly 70-fold lower than the level of exposure seen in a two-pack a day human smoker (43) or the mice exposed to mainstream CS in our earlier studies (26) suggests that the fetus is extraordinarily sensitive to CS. Af sensitization of prenatally SS-exposed animals also led to increased expression of the 105 kD isoform of PDE4–PDE4D5, whereas the expression of PDE4D3 (95 kDa) was not significantly affected by the allergen and SS exposure. Other variants of the PDE4D isoenzyme were not detectable under our assay conditions. The PDE4 selective inhibitor RP blocked the rise in PDE4D5 expression and the increased AHR observed in Af-sensitized SS animals, suggesting that the two responses are interrelated.
In recent years the pharmaceutical industry has actively followed the development of subtype-specific PDE4 inhibitors for the treatment of asthma and several compounds such as roflumilast, filaminast (44), and cilomilast are in clinical trials (45). Results with novel PDE4 inhibitors BAY 19-8004 (46) and roflumilast (47) have shown only marginal clinical benefits in asthmatics and COPD patients. Similarly, despite the initial optimism, the clinical trials of many other PDE4 inhibitors have been discontinued primarily due to the low therapeutic ratio that severely limits the dose that can be administered to humans (44). Our data clearly show that in spite of ameliorating AHR and blocking increased expression of PDE4D5, RP has no significant effect on lung inflammatory parameters, including leukocytic infiltration and Th2 cytokine/chemokine expression. These data are similar to those obtained in the PDE4D−/− mice, where the disruption of the PDE4D gene attenuated AHR without affecting the inflammatory response in the lung (48). However, some PDE4 inhibitors have decreased lung inflammation in the murine ovalbumin model (49). While it is conceivable that some PDE4 inhibitors may have beneficial effects on lung inflammation (49, 50), at present it is not clear whether the anti-inflammatory properties of these inhibitors are related to PDE4 activity. Indeed, the anti-inflammatory property of theophylline is manifested at drug levels well below the Ki for phosphodiesterase inhibition and might relate to its histone modulating activity (51, 52). In general, the problem is the complexity of PDE4, because it includes four gene families and 20 splice variants (53), and the expression of these isozymes in multiple cell types (45, 54). Given the critical role of cAMP as the second messenger in numerous biological functions, a global PDE4 inhibitor is likely to have undesirable side effects. The identification of PDE4D5 in these studies as the RP-sensitive PDE4D isozyme suggests the possibility of targeting this isoform for therapeutic interventions to reduce AHR. With anti-inflammatory (particularly Th2-specific) drugs, it could be useful in controlling allergic asthma in children. An agent without PDE4D5 inhibition may lack sufficient therapeutic efficacy to control AHR in children exposed to maternal CS.
While, the precise mechanism by which PDE4D5 controls AHR is not clear, cAMP, the substrate for PDE4, is a bronchodilator (55), and decreased levels of cAMP in the lung (26) is likely to increase bronchoconstriction. However, the major effect of SS-exposure on AHR is attained only after Af sensitization, which is associated with increased lung expression of muscarinic receptors M1, M2, and M3. Five muscarinic receptor subtypes (M1–M5) have been identified (56); however, the lung mainly expresses the M1, M2, and M3 subtypes (39, 57). While M1, M2, and M3 receptors participate in bronchoconstriction, M3 accounts for most of the bronchoconstriction in the normal lung (39). Our data show that compared to controls, the expression of all three muscarinic receptors is strongly upregulated in SS-exposed lungs. Therefore, it is likely that the increased muscarinic receptor expression accounts for the increased sensitivity of these animals to MCh-induced RL. RP attenuates AHR and the increased muscarinic receptor expression in SS-exposed lungs, indicating a causal relationship between the PDE4D5 activity and muscarinic receptor expression in allergic asthma. However, the mechanistic basis of this relationship is not clear at present. Although RP essentially blocked the allergen-induced rise in PDE4D5 expression and muscarinic receptor expression, it did not completely eliminate the allergen-induced AHR. It is possible that other mediators, such as mast cell-derived serotonin and histamine that cause bronchial hyperresponsiveness through receptors distinct from muscarinic receptors (58), are not affected by phosphodiesterase inhibitors.
In addition to AHR and inflammation, the clinical signature of allergic asthma is atopy (25), and the serum IgE levels are significantly higher in Af-sensitized SS animals than in FA-exposed animals. However, as with lung inflammation, RP treatment did not decrease the allergen-induced IgE response, indicating that atopy in these animals is essentially independent of PDE4 activity. The inability of RP to control atopy is understandable, because the RP treatment failed to significantly suppress the production of IL-4/IL-13 and IL-6 in SS-exposed, Af-sensitized animals. These cytokines are critical in IgE isotype switching and B cell proliferation/differentiation, respectively (36, 59). Moreover, the development of marked lung eosinophilia persisted in RP-treated, SS-exposed animals, suggesting a lack of effect on IL-5 and eotaxin, the main EOS proliferation/differentiation cytokine (60) and the potent eosinophilic chemoattractant (61), respectively. Thus, it is likely that the inability of RP to control the inflammation and atopy in our allergic asthma model is its failure to control the allergen-induced Th2 cytokine/chemokine production.
Mechanistically, the connection between maternal CS exposure during gestation and the increased susceptibility to allergic asthma remains somewhat undefined. Although these animals were exposed to CS both prenatally and perinatally to simulate the potential maternal exposure, we have observed that neonatal exposure to CS does not affect AHR (26) and, in fact, neonatal or adult exposure to CS or nicotine causes immunosuppression (1, 12, 13). Interestingly, the major difference in AHR between FA- and SS-exposed animals is manifested after exposure to Af, indicating that gestational CS creates the conditions that allow allergens to induce excessive AHR and lung inflammation. This is likely through epigenetic changes associated with gestational exposure to CS that encourages higher expression of Th2 cytokine/chemokine genes, PDE4D5, and muscarinic receptors. Because changes in methylation alter gene transcription (51, 52), we examined the methylation status of PDE4 and M1 muscarinic receptor genes in FA- and SS-exposed animals. Our preliminary results indicate that PDE4 essentially lacked methyl islands and the M1 gene is methylated to only about 5% (unpublished observations). Therefore, it is unlikely that methylation of these genes contributes to potential changes in the gene expression during allergic asthma in animals exposed prenatally to CS. It is possible that changes in the methylation status of other genes such as cytokines and chemokines or mechanisms other than gene methylation such as covalent modifications of the histone tails, nucleosome occupancy and turnover, and higher-order chromatin folding (62) are involved in the epigenetic modulation by gestational CS.
Acknowledgement
We express our deep appreciation to Dr. Ted Barrett for his assistance in developing animal protocols for these studies, Mr. Steve Randock for help with graphics, and Ms. Paula Bradley for editorial help.
These studies were supported in part by grants from FAMRI (Flight Attendant Medical Research Institute and the NIH (R01 DAO17003 and RO1 DAO4208-17).
Abbreviations used in this paper
- CS
Cigarette smoke
- PDE4D5
phosphodiesterase-4D5
- Af
Apergillus fumigatus extraxt
- AHR
airway hyperreactivity
- RP
rolipram
- SS
secondhand cigarette smoke
- FA
filtered air
- BAL
bronchoalveolar lavage
- BALF
bronchoalveolar lavage fluid
- qPCR
real-time PCR
- RL
airway resistance
- i.p.
intraperitoneal
- MCh
methacholine
- mAChR
muscarinic acetylcholine receptor
- i.t.
intratracheal
References
- 1.Sopori M. Effects of cigarette smoke on the immune system. Nat. Rev. Immunol. 2002;2:372–377. doi: 10.1038/nri803. [DOI] [PubMed] [Google Scholar]
- 2.DiFranza JF, Aligne A, Weitzman M. Prenatal and postnatal environmental tobacco smoke exposure and children’s health. Pediatrics. 2004;113:1007–1015. [PubMed] [Google Scholar]
- 3.Bradley JP, Bacharier LB, Bonfiglio J, Schechtman KB, Strunk R, Storch G, Castro M. Severity of respiratory syncytial virus bronchiolitis is affected by cigarette smoke exposure and atopy. Pediatrics. 2005;115:e7–e14. doi: 10.1542/peds.2004-0059. [DOI] [PubMed] [Google Scholar]
- 4.Environmental Protection Agency . Respiratory health effects of passive smoking: lung cancer and other disorders. US EPA Office Research and Development; Washington, DC: 1992. [Google Scholar]
- 5.Cliver SP, Goldenberg RL, Cutter GR, Hoffman HJ, Davis RO, Nelson KG. The effect of cigarette smoking on neonatal anthropometric measurements. Obstet. Gynecol. 1995;85:625–630. doi: 10.1016/0029-7844(94)00437-I. [DOI] [PubMed] [Google Scholar]
- 6.Wen SW, Goldenberg RL, Cutter GR, Hoffman HJ, Cliver SP, Davis RO, DuBard MB. Smoking, maternal age, fetal growth, and gestational age at delivery. Am. J. Obstet. Gynecol. 1990;162:53–58. doi: 10.1016/0002-9378(90)90819-s. [DOI] [PubMed] [Google Scholar]
- 7.Taylor B, Wadsworth J. Maternal smoking during pregnancy and lower respiratory tract illness in early life. Arch. Dis. Child. 1987;62:786–91. doi: 10.1136/adc.62.8.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sekhon HS, Proskocil BJ, Clark JA, Spindel ER. Prenatal nicotine exposure increases connective tissue expression in foetal monkey pulmonary vessels. Eur. Respir. J. 2004;23:906–915. doi: 10.1183/09031936.04.00069604. [DOI] [PubMed] [Google Scholar]
- 9.Tager IB, Hanrahan JP, Tosteson TD, Castile RG, Brown RW, Weiss ST, Speizer FE. Lung function, pre- and post-natal smoke exposure, and wheezing in the first year of life. Am. Rev. Respir. Dis. 1993;147:811–817. doi: 10.1164/ajrccm/147.4.811. [DOI] [PubMed] [Google Scholar]
- 10.Cunningham J, Dockery DW, Speizer FE. Maternal smoking during pregnancy as a predictor of lung function in children. Am. J. Epidemiol. 1994;139:1139–1152. doi: 10.1093/oxfordjournals.aje.a116961. [DOI] [PubMed] [Google Scholar]
- 11.Mathews TJ. Smoking during pregnancy in the 1990s. Nat. Vital Stat. Rep. 2001;49:1–14. [PubMed] [Google Scholar]
- 12.Singh SP, Razani-Boroujerdi S, Peña-Philippides JC, Langley RJ, Mishra NC, Sopori ML. Early postnatal exposure to cigarette smoke impairs the antigen-specific T-cell responses in the spleen. Toxicol. Lett. 2006;167:231–237. doi: 10.1016/j.toxlet.2006.10.001. [DOI] [PubMed] [Google Scholar]
- 13.Mishra NC, Rir-Sima-ah J, Langley RJ, Singh SP, Peña-Philippides JC, Koga T, Razani-Boroujerdi S, Hutt J, Campen M, Kim KC, Tesfaigzi Y, Sopori ML. Nicotine primarily suppresses lung Th2 but not goblet cell and muscle cell responses to allergens. J. Immunol. 2008;180:7655–7663. doi: 10.4049/jimmunol.180.11.7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warren CP. Extrinsic allergic alveolitis: a disease commoner in non-smokers. Thorax. 1977;32:567–569. doi: 10.1136/thx.32.5.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Calkins BM. A meta-analysis of the role of smoking in inflammatory bowel disease. Dig. Dis. Sci. 1989;34:1841–1854. doi: 10.1007/BF01536701. [DOI] [PubMed] [Google Scholar]
- 16.Baron JA. Beneficial effects of nicotine and cigarette smoking: the real, the possible, and the spurious. Br. Med. Bull. 1996;52:58–73. doi: 10.1093/oxfordjournals.bmb.a011533. [DOI] [PubMed] [Google Scholar]
- 17.Netscher DT, Clamon J. Smoking: adverse effects on outcome for plastic surgical patients. Plast. Surg. Nurs. 1994;14:205–210. doi: 10.1097/00006527-199401440-00003. [DOI] [PubMed] [Google Scholar]
- 18.Eskenazi B, Warner ML. Epidemiology of endometriosis. Obstet. Gynecol. Clin. North Am. 1997;24:235–258. doi: 10.1016/s0889-8545(05)70302-8. [DOI] [PubMed] [Google Scholar]
- 19.Baldacci S, Modena P, Carrozi L, Pedreschi M, Vellutini M, Biavati P, Simoni M, Sapigni T, Viegi G, Paoletti P, Giuntini C. Skin prick test reactivity to common aeroallergens in relation to total IgE, respiratory symptoms, and smoking in a general population sample of northern Italy. Allergy. 1996;51:149–156. doi: 10.1111/j.1398-9995.1996.tb04579.x. [DOI] [PubMed] [Google Scholar]
- 20.Troisi RJ, Speizer FE, Rosner B, Trichopoulos D, Willett WC. Cigarette smoking and incidence of chronic bronchitis and asthma in women. Chest. 1995;108:1557–1561. doi: 10.1378/chest.108.6.1557. [DOI] [PubMed] [Google Scholar]
- 21.Kalra R, Singh SP, Peña-Philippides JC, Langley RJ, Razani-Boroujerdi S, Sopori ML. Immunosuppressive and anti-inflammatory effects of nicotine administered by patch in an animal model. Clin. Diag. Lab. Immunol. 2004;11:563–568. doi: 10.1128/CDLI.11.3.563-568.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Razani-Boroujerdi S, Singh SP, Knall C, Hahn FF, Peña-Philippides JC, Kalra R, Langley RJ, Sopori ML. Chronic nicotine inhibits inflammation and promotes influenza infection. Cell. Immunol. 2004;230:1–9. doi: 10.1016/j.cellimm.2004.07.007. [DOI] [PubMed] [Google Scholar]
- 23.Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421(6921):384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- 24.Saeed RW, Varma S, Peng-Nemeroff T, Sherry B, Balakhaneh D, Huston J, Tracey KJ, Al-Abed Y, Metz CN. Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J. Exp. Med. 2005;201(7):1113–1123. doi: 10.1084/jem.20040463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thomson NC, Chaudhari R, Livingston E. Asthma and cigarette smoking. Eur. Respir. J. 2004;24:822–833. doi: 10.1183/09031936.04.00039004. [DOI] [PubMed] [Google Scholar]
- 26.Singh SP, Barrett EG, Kalra R, Razani-Boroujerdi S, Langley RJ, Kurup V, Tesfaigzi Y, Sopori ML. Prenatal cigarette smoke decreases lung cAMP and increases airway hyperresponsiveness. Am. J. Respir. Crit. Care Med. 2003;168:342–47. doi: 10.1164/rccm.200211-1262OC. [DOI] [PubMed] [Google Scholar]
- 27.Kurup VP, Mauze S, Choi H, Seymour BW, Coffman RL. A murine model of allergic bronchopulmonary aspergillosis with elevated eosinophils and IgE. J. Immunol. 1992;148:3783–3788. [PubMed] [Google Scholar]
- 28.Barrett EG, Wilder J, March TH, Espindola T, Bice DE. Cigarette smoke-induced airway hyperresponsiveness is not dependent on elevated immunoglobulin and eosinophilic inflammation in a mouse model of allergic airway disease. Am. J. Respir. Crit. Care Med. 2002;165:1410–1418. doi: 10.1164/rccm.2106029. [DOI] [PubMed] [Google Scholar]
- 29.Kurup VP. Aspergillus antigens: which are important? Med. Mycol. 2005;43(Suppl 1):S189–196. doi: 10.1080/13693780500064763. [DOI] [PubMed] [Google Scholar]
- 30.Brummund W, Resnick A, Fink JN, Kurup VP. Aspergillus fumigatus-specific antibodies in allergic bronchopulmonary aspergillosis and aspergilloma: evidence for a polyclonal antibody response. J. Clin. Microbiol. 1987;25:5–9. doi: 10.1128/jcm.25.1.5-9.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Levallet G, Levallet J, Bouraima-Lelong H, Bonnamy PJ. Expression of cAMP-phosphodiesterase PDE4D isoforms and age-related changes in follicle-stimulating hormone-stimulated activities in immature rat sertoli cells. Biol. Reprod. 2007;76:794–803. doi: 10.1095/biolreprod.106.055343. [DOI] [PubMed] [Google Scholar]
- 32.Weitzman M, Gortmaker S, Sobol A. Racial, social, and environmental risks for childhood asthma. Am. J. Dis. Child. 1990;144(11):1189–1194. doi: 10.1001/archpedi.1990.02150350021016. [DOI] [PubMed] [Google Scholar]
- 33.Lundblad LK, Irvin CG, Adler A, Bates JH. A reevaluation of the validity of unrestrained plethysmography in mice. J. Appl. Physiol. 2002;93:1198–1207. doi: 10.1152/japplphysiol.00080.2002. [DOI] [PubMed] [Google Scholar]
- 34.Hauk PJ, Krawiec M, Murphy J, Boguniewicz J, Schiltz A, Goleva E, Liu AH, Leung DY. Neutrophilic airway inflammation and association with bacterial lipopolysaccharide in children with asthma and wheezing. Pediatr. Pulmonol. 2008;43:916–23. doi: 10.1002/ppul.20880. [DOI] [PubMed] [Google Scholar]
- 35.Jones CA, Holt PG. Immunopathology of allergy and asthma in childhood. Am. J. Respir. Crit. Care Med. 2000;162:S36–S39. doi: 10.1164/ajrccm.162.supplement_1.maic-10. [DOI] [PubMed] [Google Scholar]
- 36.Poulsen LK, Hummelshoj L. Triggers of IgE class switching and allergy development. Ann. Med. 2007;39:440–456. doi: 10.1080/07853890701449354. [DOI] [PubMed] [Google Scholar]
- 37.Kurup VP, Grunig G. Animal models of allergic bronchopulmonary aspergillosis. Mycopathologia. 2002;153:165–77. doi: 10.1023/a:1014963600314. [DOI] [PubMed] [Google Scholar]
- 38.Gosens R, Zaagsma J, Meurs H, Halayko AJ. Muscarinic receptor signaling in the pathophysiology of asthma and COPD. Respir. Res. 2006;7:73–88. doi: 10.1186/1465-9921-7-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Struckmann N, Schwering S, Wiegand S, Gschnell A, Yamada M, Kummer W, Wess J, Haberberger RV. Role of muscarinic receptor subtypes in the constriction of peripheral airways: studies on receptor-deficient mice. Mol. Pharmacol. 2003;64:1444–1451. doi: 10.1124/mol.64.6.1444. [DOI] [PubMed] [Google Scholar]
- 40.Floreani AA, Rennard SI. The role of cigarette smoke in the pathogenesis of asthma and as a trigger for acute symptoms. Curr. Opin. Pulm. Med. 5:38–46. doi: 10.1097/00063198-199901000-00007. [DOI] [PubMed] [Google Scholar]
- 41.Kurup VP. Aspergillus antigens: which are important? Med. Mycol. 2005;43(Suppl 1):S189–196. doi: 10.1080/13693780500064763. [DOI] [PubMed] [Google Scholar]
- 42.Seymour BW, Peake JL, Pinkerton KE, Kurup VP, Gershwin LJ. Second-hand smoke increases nitric oxide and alters the IgE response in a murine model of allergic aspergillosis. Clin. Dev. Immunol. 2005;12:113–124. doi: 10.1080/17402520500116806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Finch GL, Nikula KJ, Chen BT, Barr EB, Chang IY, Hobbs CH. Effect of chronic cigarette smoke exposure on lung clearance of tracer particles inhaled by rats. Fundam. Appl. Toxicol. 1995;24:76–85. doi: 10.1006/faat.1995.1009. [DOI] [PubMed] [Google Scholar]
- 44.Giembycz MA. Can the anti-inflammatory potential of PDE4 inhibitors be realized: guarded optimism or wishful thinking? Br. J. Pharmacol. 2008;155:288–290. doi: 10.1038/bjp.2008.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spina D. PDE4 inhibitors: current status. Br. J. Pharmacol. 2008;155:308–15. doi: 10.1038/bjp.2008.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grootendorst DC, Gauw SA, Benschop N, Sterk PJ, Hiemstra PS, Rabe KF. Efficacy of the novel phosphodiesterase-4 inhibitor BAY 19-8004 on lung function and airway inflammation in asthma and chronic obstructive pulmonary disease (COPD) Pulm. Pharmacol. Ther. 16:341–347. doi: 10.1016/S1094-5539(03)00090-7. [DOI] [PubMed] [Google Scholar]
- 47.Field SK. Roflumilast: an oral, once-daily selective PDE-4 inhibitor for the management of COPD and asthma. Expert Opin. Investig. Drugs. 2008;17:811–818. doi: 10.1517/13543784.17.5.811. [DOI] [PubMed] [Google Scholar]
- 48.Hansen G, Jin S, Umetsu DT, Conti M. Absence of muscarinic cholinergic airway responses in mice deficient in the cyclic nucleotide phosphodiesterase PDE4D. Proc. Natl. Acad. Sci. USA. 2000;97:6751–6756. doi: 10.1073/pnas.97.12.6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deng YM, Xie QM, Tang HF, Sun JG, Deng JF, Chen JQ, Yang SY. Effects of ciclamilast, a new PDE 4 PDE4 inhibitor, on airway hyperresponsiveness, PDE4D expression and airway inflammation in a murine model of asthma. Eur. J. Pharmacol. 2006;547:125–135. doi: 10.1016/j.ejphar.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 50.Wollin L, Bundschuh DS, Wohlsen A, Marx D, Beume R. Inhibition of airway hyperresponsiveness and pulmonary inflammation by roflumilast and other PDE4 inhibitors. Pulm. Pharmacol. Ther. 2006;19:343–352. doi: 10.1016/j.pupt.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 51.Barnes PJ, Adcock IM, Ito K. Histone acetylation and deacetylation: importance in inflammatory lung diseases. Eur. Respir. J. 2005;25:552–563. doi: 10.1183/09031936.05.00117504. [DOI] [PubMed] [Google Scholar]
- 52.Adcock IM, Ito K, Barnes PJ. Histone deacetylation: an important mechanism in inflammatory lung diseases. COPD. 2005;2:445–455. doi: 10.1080/15412550500346683. [DOI] [PubMed] [Google Scholar]
- 53.Lynch MJ, Baillie GS, Houslay MD. cAMP-specific phosphodiesterase-4D5 (PDE4D5) provides a paradigm for understanding the unique non-redundant roles that PDE4 isoforms play in shaping compartmentalized cAMP cell signalling. Biochem. Soc. Trans. 2007;35:938–941. doi: 10.1042/BST0350938. [DOI] [PubMed] [Google Scholar]
- 54.Houslay MD, Schafer P, Zhang KY. Keynote review: phosphodiesterase-4 as a therapeutic target. Drug Discov. Today. 2005;10:1503–1519. doi: 10.1016/S1359-6446(05)03622-6. [DOI] [PubMed] [Google Scholar]
- 55.Mehats C, Jin S-LC, Wahlstrom J, Law E, Umetsu DL, Conti M. PDE4D plays a critical role in the control of airway smooth muscle contraction. FASEB J. 2003;17:1831–1841. doi: 10.1096/fj.03-0274com. [DOI] [PubMed] [Google Scholar]
- 56.Caulfield MP, Birdsall NJM. International Union of Pharmacology. XVII, Classification of muscaranic acetylcholine receptors. Pharmacol. Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- 57.Hislop AA, Mak JC, Reader JA, Barnes PJ, Haworth SG. Muscaranic receptor subtype in the porcine lung during postnatal development. Eur. J. Pharmacol. 1998;359:211–221. doi: 10.1016/s0014-2999(98)00585-8. [DOI] [PubMed] [Google Scholar]
- 58.Nagai T, Kim DJ, Delay RJ, Roper SD. Neuromodulation of transduction and signal processing in the end organs of taste. Chem. Senses. 1996;21:353–365. doi: 10.1093/chemse/21.3.353. [DOI] [PubMed] [Google Scholar]
- 59.Kishimoto T. Interleukin-6: discovery of a pleiotropic cytokine. Arthritis Res. Ther. 2006;8(Suppl 2):S2. doi: 10.1186/ar1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Foster PS, Rosenberg HF, Asquith KL, Kumar RK. Targeting eosinophils in asthma. Curr. Mol. Med. 2008;8:585–590. doi: 10.2174/156652408785748013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Conroy DM, Williams TJ. Eotaxin and the attraction of eosinophils to the asthmatic lung. Respir. Res. 2001;2:150–156. doi: 10.1186/rr52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Van der Maarel SM. Epigenetic mechanisms in health and disease. Ann. Rheum. Dis. 2008;67(Suppl 3):iii, 97–100. doi: 10.1136/ard.2008.098392. [DOI] [PubMed] [Google Scholar]