

Spinal Muscular Atrophy (SMA) is a motor-neuron disease and the leading genetic cause of infant mortality; it is caused by loss-of-function mutations in the Survival motor neuron 1 (SMN1) gene1. Humans have a paralog, SMN2, whose exon 7 is predominantly skipped2; the limited amount of functional, full-length SMN it expresses cannot fully compensate for the lack of SMN1. SMN is important for spliceosomal snRNP biogenesis3, but downstream splicing targets involved in pathogenesis remain elusive. There is no effective treatment for SMA, but SMN restoration in spinal-cord motor neurons is believed to be necessary and sufficient for therapy 4. Non-CNS pathologies, including cardiovascular defects, were recently reported in severe SMA patients and mouse models5–8, reflecting autonomic dysfunction or direct effects in cardiac tissues. Here we compared the effects of systemic versus CNS restoration of SMN in a severe mouse model with 10-day survival9,10. We used an antisense oligonucleotide (ASO-10-27) that effectively corrects SMN2 splicing and restores SMN expression in motor neurons after intracerebroventricular (ICV) injection11,12. Surprisingly, systemic administration to neonates robustly rescued severe SMA mice, much more effectively than ICV administration; subcutaneous injections extended the median lifespan by 25-fold. We further demonstrate decreased expression of hepatic Igfals in neonatal SMA mice, leading to a pronounced reduction of circulating insulin-like growth factor 1 (IGF1); ASO treatment restored IGF1 to normal levels. These results suggest an important role of the liver in SMA pathogenesis, underscoring the importance of SMN in peripheral tissues, and demonstrate the efficacy of a promising drug candidate.
To compare the effectiveness of ASO 10-27 delivered centrally versus systemically, we injected 20 μg of ASO-10-27 ICV at postnatal day 1 (P1) to increase SMN in CNS tissues, or we injected the ASO on two separate days subcutaneously (SC) at 50 μg per g of body weight (μg/g), between P0 and P3 (2 doses). These doses were based on our previous studies with this ASO 11,13. We also evaluated combined ICV and SC injections, and repeated SC injections (Supplementary Table 1). Control heterozygous mice (Smn+/−; SMN2+/0) that received ICV and/or SC ASO injections had normal survival and behavior. Severe SMA mice (Smn−/−; SMN2+/0) that received ICV and/or SC saline- injections survived 1–2 weeks, with a median survival of ~10 days, similar to untreated mice (Fig. 1a; Supplementary Figs. 1a,2a, Video 1). ASO delivered only into the CNS efficiently corrected SMN2 exon 7 splicing in the spinal cord and led to a striking increase in SMN protein levels, but modestly extended the median survival to 16 days, with a single pup surviving for one month (Fig. 1a–c; Supplementary Fig. 2b–d). In marked contrast, systemic treatment with two SC injections resulted in a median survival of 108 days. Combining ICV and SC ASO injections further increased the median survival to 173 days; and additional SC injections at P5-P7, after the initial SC injections at P0-P3, extended the median survival to 137 days (Fig. 1d).
Figure 1.
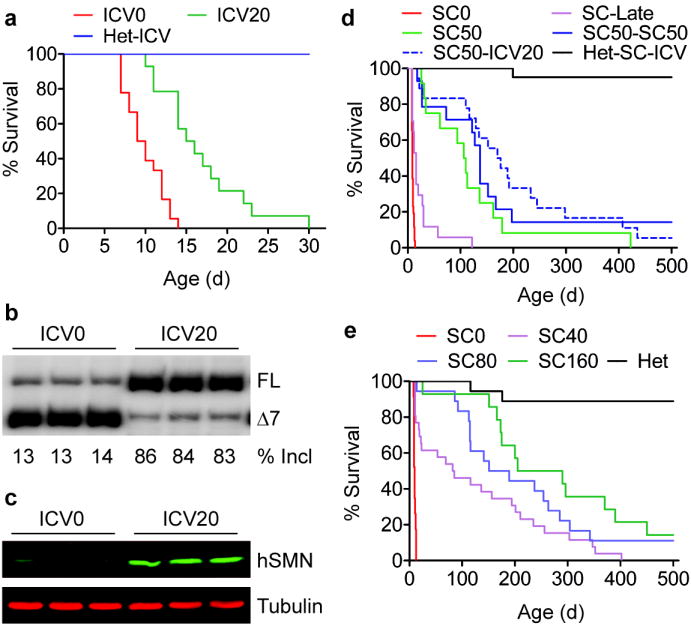
Systemic versus ICV ASO-10-27 injections in SMA mice. a, Survival curves after ICV administration at P1. 20 μg ASO (ICV20, n=14) or saline (ICV0, n=18) gave mean survivals of 17 and 10 d, respectively (P<0.001). ASO-treated heterozygotes (Het-ICV, n=15) served as controls. Spinal-cord RNA and protein samples (n=3) were analyzed at P7 by radioactive RT-PCR (b) or immunoblotting with SMN-KH mAb (c). FL: full-length mRNA; Δ7, exon-7-skipped mRNA; incl, exon 7 inclusion. d, Survival curves after SC administration with saline (SC0, n=26) or ASO (SC50, n=12) twice between P0 and P3. SC50-SC50 (n=14) mice received two additional SC injections at P5-P7. Het-SC-ICV (n=13) and SC50-ICV20 (n=18) were heterozygous and SMA mice, respectively, that received combined P1 ICV and P0-P3 SC injections. Each SC injection dose was 50 μg/g. P<0.0001 for all groups versus SC0. e, Dose-dependent survival after two SC injections at P0-P3 with 40 (n=26), 80 (n=18), or 160 (n=14) μg/g/injection. Saline-treated SMA (SC0, n=23) or heterozygous mice (Het, n=18) served as controls. P<0.0001 for all groups versus SC0.
Treated mice varied in size from comparable to heterozygous littermates to runt; the average weight was low, and the tails were much shorter (Supplementary Figs. 3,4). The surviving runts slowly gained weight, reaching ~18 g at ~3 months. Most rescued SMA mice could run and climb normally; however, their tail and ears developed necrosis, and were gradually lost, resembling the phenotype of type III SMA mice (Supplementary Fig. 3e, f). Additional ASO delivered either by ICV injection at P1 or repeat SC injections at P5-P7 delayed necrosis (Supplementary Fig. 3g).
To further characterize the effects of the ASO administered systemically, we carried out a dose-response study with 0 (SC0), 40 (SC40), 80 (SC80) and 160 (SC160) μg/g/injection, given twice between P0 and P3. Systemic treatment with the ASO resulted in a dose-dependent increase in survival (Fig. 1e), with the median survival increasing from 10 to 84, 170, and 248 days, respectively. At the highest dose tested, ASO-10-27 given systemically resulted in long-term survival comparable to the best results achieved by adeno-associated-virus (AAV) expression of the SMN protein in a slightly less severe mouse model14–16. Remarkably, 2/14 mice in the SC160 group, and 2/18 in the SC80 group are still alive and active after >500 days. A similar survival benefit was achieved by intraperitoneal (IP) dosing (Supplementary Fig. 5). There was no significant difference in weight gain among the three SC-dosing groups; however, SC160 mice had significantly longer tails (Supplementary Fig. 6a–e). We also observed dose-dependent rescue of ear and tail necrosis, and dose-dependent delays in the development of cataracts and rectal prolapse (Supplementary Fig. 6f–h). Administration of the ASO on days P5 and P7 resulted in a modest increase in survival, compared to earlier treatments on P0 and P3, emphasizing the importance of early-postnatal therapeutic intervention (Fig. 1d).
To examine SMN2 splicing changes in various tissues after SC ASO injection, we performed RT-PCR on RNA samples from P7 mice, and detected a dose-dependent increase in exon 7 inclusion in spinal cord, brain, liver, heart, kidney, and skeletal muscle, with the strongest effect occurring in the liver and the weakest in the kidneys. In contrast, ICV administration of the ASO resulted in a much more robust change in exon 7 inclusion in brain and spinal cord tissues, but very limited effects in peripheral tissues (Fig. 2a,b; Supplementary Fig. 7, 8). Immunoblotting of the spinal cord, liver, and heart tissue samples from mice treated by SC administration demonstrated a corresponding increase in full-length SMN protein (Fig. 2c; Supplementary Fig. 7a). Exon 7 inclusion in liver significantly decreased after P30 (Fig. 2d,e; Supplementary Fig. 9), consistent with a measured ASO half-life of 22 days in liver, (data not shown). These data suggest that transiently increasing SMN expression in peripheral tissues during the first few weeks of life has a profound effect on long-term survival.
Figure 2.

SMN2 splicing and protein expression in mouse tissues after SC ASO injection. a, Radioactive RT-PCR of RNA from P7 SMA mice after two SC injections between P0 and P3 at 0, 40, 80, or 160 μg/g/injection. b, Statistics of exon 7 inclusion (n=3). c, Protein samples from P7 SMA mice (n=3) treated with 160 μg/g/injection were analyzed by immunoblotting with SMN-KH mAb (Supplementary Fig. 7a). d, RT-PCR of liver RNA from P10, P30, and P180 SMA mice shows decreasing effect of ASO-10-27 over time. e, Statistics of data in panel d. *P<0.05, **P<0.001, ***P<0.0001 compared to saline controls. Means ± s.d. are shown.
The SMN2-splicing changes were consistent with the ASO distribution assayed by immunohistochemistry, with the apparent exception of the kidney, but in this case most of the ASO was not internalized into cells (Supplementary Fig. 10, 11). We also observed some ASO accumulating in spinal-cord motor neurons (Supplementary Fig. 10, 11). The limited ASO distribution and the moderate SMN2-splicing changes in the CNS after systemic administration likely reflect incomplete closure of the BBB in neonates17, and/or ASO retrograde transport. However, we detected strong cytoplasmic SMN staining and/or a pronounced increase in gem number in spinal-cord motor neurons after ICV injection of 20 μg ASO, but not after two SC injections of ASO at 80 μg/g/injection between P0 and P3, a dosage that markedly rescued the severe SMA mice (Supplementary Fig. 12, 13). Therefore, the effect on splicing in the CNS after systemic administration probably contributes to the extended survival, which is consistent with the combined ICV and SC treatment giving even better survival than SC administration alone (Fig. 1d); yet, the striking effects of systemic administration on survival in this severe mouse model cannot be solely explained by a direct effect on SMN2 splicing in the CNS.
The rescue of severe SMA mice by systemic administration of, e.g., histone deacetylase inhibitors or AAV vectors, has been attributed to their ability to cross the BBB10,15. However, our data indicate that SMN restoration in peripheral tissues, in combination with partial restoration in the CNS, can achieve efficient rescue of severe SMA mice.
Histological examination of tissues/organs associated with SMA in mice treated systemically with 160 μg/g of ASO-10-27 and sacrificed at P9, revealed striking improvements, consistent with the markedly increased survival of mice in the SC160 group. The α-motor-neuron counts in spinal cord were comparable to the control heterozygous littermates, and the mean area of muscle fiber cross-sections was >80% of that in heterozygotes (Fig. 3a,b; Supplementary Fig. 14a). Likewise, the heart weight, and the thickness of the inter-ventricular septum and left ventricular wall were similar in ASO-treated mice and heterozygous littermates (Fig. 3c,d; Supplementary Fig. 14b). Finally, staining of neuromuscular junctions (NMJ) showed that NMJ integrity was similar to that in heterozygous littermates (Fig. 3e).
Figure 3.

Evaluation of affected tissues and motor-function. H&E-stained tissues from P9 ASO-treated SMA mice (SC160, n=6, two SC injections at 160 μg/g/injection at P0-P3) versus saline controls (SC0, n=6) and untreated heterozygotes (Het, n=6) (Supplementary Fig. 14). Saline-treated mice were ambulant at P9, and were expected to live for another 3–5 d. α-motor-neuron counts in each cross-section of L1-L2 spinal cord (a), mean fiber-cross-sectional area (200 fibers) of rectus femoris muscle (b), heart weight (c), and thickness of the heart inter-ventricular septum (IVS) and left ventricular wall (LVW) (d) significantly improved in ASO-treated mice. e, Arborisation complexity of NMJs was restored in ASO-treated mice. P90 SC40 (n=12, 124 trials), SC80 (n=13, 137 trials), SC160 (n=11, 117 trials) and untreated heterozygous mice (Het, n=12, 135 trials) were tested 3–5 times/d for 3 d on a Rotarod, using an acceleration profile. The mean times for staying on the spinning rod (f), and the number of no-fall-trials and of mice with ≥1 no-fall trial (g) are shown. h, Grip strength (grams-force) of SC160 mice (n=6) evaluated at 5 and 9 months (m) reached ~80% of that of heterozygous mice (n=6). *P<0.05; **P<0.01. Means ± s.d. are shown.
Most systemic-ASO-treated mice showed no overt signs of motor dysfunction (Supplementary Video 2, Supplementary Table 2). We employed three tests to evaluate their behavior and motor function. The first was a Rotarod test, which requires limb-muscle strength, as well as balance and coordination. Three-month-old mice in the SC80 and SC160 groups could stay on the rotating rod for ~12 sec, i.e., shorter than heterozygotes, but longer than mice in group SC40. Some treated mice passed a 30-sec acceleration-profile test that many heterozygotes failed (Fig. 3f,g; Supplementary Video 3). Considering that SMA is a neuromuscular disease, this performance represents a remarkable phenotypic improvement. The second test evaluated muscle strength in mice from the SC160 group at 5 and 9 months. At both ages, the forelimb grip strength of treated SMA mice was ~80% that of heterozygous mice (Fig 3h). The final test employed HomeCageScan, a video-based platform for automated high-resolution behavior analysis18. Treated SMA mice performed various behaviors similarly to heterozygous mice, except for rearing, suggesting some hindlimb weakness (Supplementary Fig. 15).
Two reasons prompted us to examine the growth-hormone (GH)/IGF1 axis. First, all severe SMA mice are small9,10,19, reflecting growth retardation. Second, the major effect of SC ASO injection on SMN2 splicing is in the liver, which contributes ~75% of circulating IGF120. Moreover, liver-derived IGF1 is sufficient to support normal postnatal growth in Igf1-null mice20. IGF1 is a potent neurotrophic factor21, and is also involved in cardiac development and function22. ELISA of P6-P9 serum samples showed undetectable or greatly reduced IGF1 levels, compared to heterozygote controls, and SC ASO administration restored IGF1 to normal levels (Fig. 4a).
Figure 4.

IGF1 system is disrupted in SMA mice. Treated mice received two SC ASO injections at 80 μg/g/injection between P0 and P3 (SC80). a, IGF1 serum levels in P6 and P9 SMA mice (SC0), measured by ELISA (mean of three measurements per sample), were strikingly lower than in heterozygous littermates (Het) or treated SMA mice (P<0.001 for all samples). b, Total liver RNA from P1 and P5 SMA mice and heterozygous littermates was analyzed by radioactive RT-PCR to measure Igf1, Igfals, and Igfbp3 expression, with Gapdh as control. c, ASO treatment restored hepatic Igfals expression, assayed at P5. d, Quantitation of hepatic Igfals expression; *P<0.01 versus heterozygous or ASO-treated SMA samples. Means ± s.d. are shown.
RT-PCR showed that hepatic Igf1 mRNA was not reduced in SMA mice, and increased from P1 to P5 in both heterozygote and SMA mice (Fig. 4b). IGF-binding-protein acid labile subunit (IGFALS), which is postnatally stimulated by GH, binds to IGF1 and IGFBP3 to form a stable ternary complex, extending the half-life of IGF1 from 10 min to >12 h23. Inactivation of Igfals results in low circulating IGF1 and IGFBP3, as well as impaired postnatal growth23. RT-PCR revealed a marked reduction in Igfals mRNA in the liver of both P1 and P5 SMA mice; moreover, ASO administration rescued Igfals expression (Fig. 4c–d). We conclude that the striking reduction in serum IGF1 levels in SMA mice is likely caused by decreased Igfals expression, which correlates with SMN deficiency and SMA progression.
Because Igfals expression is decreased at P1, when the pups are still healthy, we propose that the early deficiency in circulating IGF1 may be one of the factors that contribute to the pathogenesis of severe SMA mice (Supplementary Fig. 1b). Though in several mouse mutants, an impaired GH/IGF-1 axis results in increased lifespan24, a severe lack of IGF1 may contribute to SMA progression in concert with other defective factors. Consistent with our hypothesis, two recent studies showed that a local increase of IGF1 in either spinal cord or muscle increases survival of severe SMA mice25,26. Indeed, disruption of the IGF1 system is a common feature of neurodegenerative diseases, including Alzheimer’s and amyotrophic lateral sclerosis (ALS)27. IGF1-null mice also display some phenotypic similarity to SMA mice, such as small size, severe and generalized muscle dystrophy, including of the diaphragm and heart, with most of them dying at birth28. Moreover, dysregulation of the IGF1 receptor and its downstream signaling pathway has been observed in type I SMA patients29. However, in light of the inconsistent results of IGF1 therapy for ALS between humans and mice21,30, it will be crucial to determine to what extent SMA mouse models accurately mimic human SMA. This will further refine our understanding of the mouse models, and influence therapeutics development and clinical treatments for SMA.
METHODS SUMMARY
ASO-10-27 was synthesized as described11 and dissolved in 0.9 % saline. The severe SMA mouse model was generated from an SMA type III mouse model, as described10. All mouse protocols were in accordance with Cold Spring Harbor Laboratory’s Institutional Animal Care and Use Committee guidelines. Treated mice were provided with additional gel food. The procedures for neonatal ICV injection, tissue-sample collection, RT-PCR, and Western blotting, and the human-specific anti-SMN antibody (SMN-KH) were described previously11. Primers for gene expression analysis are shown in Supplementary Table 3. Serum IGF1 was analyzed with a Quantikine Mouse/Rat IGF1 Immunoassay kit (R&D Systems).
Mouse spinal cord, quadriceps, and heart were fixed and H&E stained as described12. α-motor neurons in serial 10–20 μm cross-sections of lumbar L1-L2 spinal-cord were counted. Muscle-fiber cross-sectional area was calculated with Axivision LE velocity. NMJ in toto staining was performed as described12.
For the Rotarod (AccuScan Instruments) test, a four-phase profile was used: phase 1, from 1 to 10 rpm in 7.5 s; phase 2, from 10 to 0 rpm in 7.5 s; phase 3, from 0 to 10 rpm in 7.5 s in the opposite direction; phase 4, from 10 to 0 rpm in 7.5 s. A grip-strength meter (Columbus Instruments) was used for the gripping test. Mice were allowed to grasp a triangular bar with their forelimbs, and were pulled back horizontally. The test was repeated 5 times for each mouse, and the highest value was recorded as the grip force for that animal.
Statistical significance was analyzed by two-tailed Student’s t-tests. Kaplan-Meier survival data were analyzed with Mantel-Cox tests using GraphPad Prism.
Supplementary Material
Acknowledgments
We gratefully acknowledge support from the Muscular Dystrophy Association, the National Institute of General Medical Sciences, and St. Giles Foundation. We thank Jie Bu and Marco Passini for protocols and advice on NMJ staining, and Stephen Hearn for assistance with microscope imaging.
Footnotes
Author Contributions Y.H, A.R.K., and C.F.B. designed the study and wrote the paper. Y.H., K.S., F.R., G.Hung, and G. Horev carried out experiments and data analysis. All authors read the manuscript.
Author Information Reprints and permissions information is available at www.nature.com/reprints. F.R., G. Hung, and C.F.B. may materially benefit either directly or indirectly through stock options. Y.H., K.S., and A.R.K., along with their employer could materially benefit if a therapeutic for SMA results from this work.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature
References
- 1.Lefebvre S, et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 2.Lorson CL, Rindt H, Shababi M. Spinal muscular atrophy: mechanisms and therapeutic strategies. Hum Mol Genet. 2010;19:R111–118. doi: 10.1093/hmg/ddq147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Burghes AH, Beattie CE. Spinal muscular atrophy: why do low levels of survival motor neuron protein make motor neurons sick? Nat Rev Neurosci. 2009;10:597–609. doi: 10.1038/nrn2670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gavrilina TO, et al. Neuronal SMN expression corrects spinal muscular atrophy in severe SMA mice while muscle-specific SMN expression has no phenotypic effect. Hum Mol Genet. 2008;17:1063–1075. doi: 10.1093/hmg/ddm379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rudnik-Schoneborn S, et al. Congenital heart disease is a feature of severe infantile spinal muscular atrophy. J Med Genet. 2008;45:635–638. doi: 10.1136/jmg.2008.057950. [DOI] [PubMed] [Google Scholar]
- 6.Bevan AK, et al. Early heart failure in the SMNDelta7 model of spinal muscular atrophy and correction by postnatal scAAV9-SMN delivery. Hum Mol Genet. 2010;19:3895–3905. doi: 10.1093/hmg/ddq300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heier CR, Satta R, Lutz C, DiDonato CJ. Arrhythmia and cardiac defects are a feature of spinal muscular atrophy model mice. Hum Mol Genet. 2010;19:3906–3918. doi: 10.1093/hmg/ddq330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shababi M, et al. Cardiac defects contribute to the pathology of spinal muscular atrophy models. Hum Mol Genet. 2010;19:4059–4071. doi: 10.1093/hmg/ddq329. [DOI] [PubMed] [Google Scholar]
- 9.Gogliotti RG, Hammond SM, Lutz C, Didonato CJ. Molecular and phenotypic reassessment of an infrequently used mouse model for spinal muscular atrophy. Biochem Biophys Res Commun. 2010;391:517–522. doi: 10.1016/j.bbrc.2009.11.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Riessland M, et al. SAHA ameliorates the SMA phenotype in two mouse models for spinal muscular atrophy. Hum Mol Genet. 2010;19:1492–1506. doi: 10.1093/hmg/ddq023. [DOI] [PubMed] [Google Scholar]
- 11.Hua Y, et al. Antisense correction of SMN2 splicing in the CNS rescues necrosis in a type III SMA mouse model. Genes Dev. 2010;24:1634–1644. doi: 10.1101/gad.1941310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Passini MA, et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci Transl Med. 2011;3:72ra18. doi: 10.1126/scitranslmed.3001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hua Y, Vickers TA, Okunola HL, Bennett CF, Krainer AR. Antisense masking of an hnRNP A1/A2 intronic splicing silencer corrects SMN2 splicing in transgenic mice. Am J Hum Genet. 2008;82:834–848. doi: 10.1016/j.ajhg.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Passini MA, et al. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J Clin Invest. 2010;120:1253–1264. doi: 10.1172/JCI41615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Foust KD, et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat Biotechnol. 2010;28:271–274. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 16.Dominguez E, et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum Mol Genet. 2010 doi: 10.1093/hmg/ddq514. [DOI] [PubMed] [Google Scholar]
- 17.Ek CJ, Habgood MD, Dziegielewska KM, Saunders NR. Structural characteristics and barrier properties of the choroid plexuses in developing brain of the opossum (Monodelphis Domestica) J Comp Neurol. 2003;460:451–464. doi: 10.1002/cne.10661. [DOI] [PubMed] [Google Scholar]
- 18.Steele AD, Jackson WS, King OD, Lindquist S. The power of automated high-resolution behavior analysis revealed by its application to mouse models of Huntington’s and prion diseases. Proc Natl Acad Sci U S A. 2007;104:1983–1988. doi: 10.1073/pnas.0610779104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Park GH, Kariya S, Monani UR. Spinal muscular atrophy: new and emerging insights from model mice. Curr Neurol Neurosci Rep. 2010;10:108–117. doi: 10.1007/s11910-010-0095-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu Y, Sun H, Yakar S, LeRoith D. Elevated levels of insulin-like growth factor (IGF)-I in serum rescue the severe growth retardation of IGF-I null mice. Endocrinology. 2009;150:4395–4403. doi: 10.1210/en.2009-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaspar BK, Llado J, Sherkat N, Rothstein JD, Gage FH. Retrograde viral delivery of IGF-1 prolongs survival in a mouse ALS model. Science. 2003;301:839–842. doi: 10.1126/science.1086137. [DOI] [PubMed] [Google Scholar]
- 22.Colao A. The GH-IGF-I axis and the cardiovascular system: clinical implications. Clin Endocrinol (Oxf) 2008;69:347–358. doi: 10.1111/j.1365-2265.2008.03292.x. [DOI] [PubMed] [Google Scholar]
- 23.Domene HM, et al. Human acid-labile subunit deficiency: clinical, endocrine and metabolic consequences. Horm Res. 2009;72:129–141. doi: 10.1159/000232486. [DOI] [PubMed] [Google Scholar]
- 24.Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120:449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 25.Shababi M, Glascock J, Lorson CL. Combination of SMN trans-splicing and a neurotrophic factor increases the life span and body mass in a severe model of spinal muscular atrophy. Hum Gene Ther. 2011;22:135–144. doi: 10.1089/hum.2010.114. [DOI] [PubMed] [Google Scholar]
- 26.Bosch-Marce M, et al. Increased IGF-1 in muscle modulates the phenotype of severe SMA mice. Hum Mol Genet. 2011;20:1844–1853. doi: 10.1093/hmg/ddr067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trejo JL, Carro E, Garcia-Galloway E, Torres-Aleman I. Role of insulin-like growth factor I signaling in neurodegenerative diseases. J Mol Med. 2004;82:156–162. doi: 10.1007/s00109-003-0499-7. [DOI] [PubMed] [Google Scholar]
- 28.Powell-Braxton L, et al. IGF-I is required for normal embryonic growth in mice. Genes Dev. 1993;7:2609–2617. doi: 10.1101/gad.7.12b.2609. [DOI] [PubMed] [Google Scholar]
- 29.Millino C, et al. Different atrophy-hypertrophy transcription pathways in muscles affected by severe and mild spinal muscular atrophy. BMC Med. 2009;7:14. doi: 10.1186/1741-7015-7-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sorenson EJ, et al. Subcutaneous IGF-1 is not beneficial in 2-year ALS trial. Neurology. 2008;71:1770–1775. doi: 10.1212/01.wnl.0000335970.78664.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.