

Abstract
Cyclic AMP, generated by adenylyl cyclase (AC), serves as a second messenger in signaling pathways regulating many aspects of cardiac physiology including contraction rate and action potential duration, and in the pathophysiology of hypertrophy and heart failure. A kinase-anchoring proteins (AKAPs) localize the effect of cAMP in space and time by organizing receptors, adenylyl cyclase, protein kinase A and other components of the cAMP cascade into multiprotein complexes. In this review we discuss how interaction of AKAPs with distinct AC isoforms affects cardiovascular physiology.
Keywords: adenylyl cyclase, A-kinase anchoring protein, protein kinase A, G proteincoupled receptor, adrenergic receptor, phosphodiesterase
3′–5′-cyclic adenosine monophosphate (cAMP) is a small diffusible intracellular second messenger generated by a family of adenylyl cyclase (AC) enzymes. Catalytic activity of AC is stimulated in response to activation of Gs protein-coupled receptors (GPCRs) by a number of hormones and neurotransmitters.1 In heart, cAMP activates protein kinase A (PKA), in addition to cAMP-activated exchange proteins (EPAC), cAMP-gated channels (HCN), and a subset of phosphodiesterases (PDEs). In myocytes the most well studied of these downstream cAMP targets is PKA, whose activation leads to the increase in phosphorylation of multiple cellular targets, including regulators of contractility.2,3 The regulation of HCN channels by cAMP in pacemaker cells is important for basal heart rate,4 while EPAC activation can have effects on cardiac calcium handling and hypertrophy.5 The subsequent degradation of cAMP to 5′-AMP by cyclic nucleotide PDEs, sets up a finely tuned temporal balance between cAMP production and breakdown and ultimately controls cellular responses.
Traditionally, cAMP signaling has been pharmacologically targeted through GPCR modulators and PDE inhibitors. Abnormal regulation of cAMP contributes to the progressive deterioration of cardiac function leading to heart failure. The benefits of lowered cAMP are the basis for the use of β-adrenergic receptor blockers (β-blockers) in treating congestive heart failure, cardiac arrhythmias, and angina pectoris. However, recently AC has emerged as a limiting component in GPCR downstream signaling. AC levels may limit maximal β-adrenergic receptor (βAR) response and potency in cardiac myocytes or whole hearts.6
AC Isoforms and Cardiac Function
The AC family comprises nine membrane-bound and one soluble isoforms. All isoforms except AC8 are expressed in the heart.1,7 AC1 is expressed only in sino-atrial mode where it modulates the I(f) pacemaker current.8 Although ACs 2, 3, 4, 5/6, and 7 are readily detected in cardiac fibroblasts,9 the major isoforms present in myocytes are AC5 and AC6.10,11 Much lower levels of AC2 and AC9 can also be detected in myocytes.12,13 The roles for these latter ACs are unknown. AC5 is dominantly expressed in adult cardiac myocytes,10 while AC6 is expressed at higher levels in fetal cardiac myocytes and adult cardiac fibroblasts. These ACs may exert opposite effects on the heart, since cardiac overexpression of AC6 appears to be protective,14,15 whereas disruption of type 5 AC prolongs longevity and protects against cardiac stress.10,16,17 The deletion of AC5 results in ~40% decreased isoproterenol- and forskolin-stimulated activity in cardiac membranes and isolated myocytes.11 Although differences in AC5 deletion strains exist, the decrease in cAMP results in decreased isoproterenol-stimulated left ventricular (LV) ejection fraction.11 Effects of AC5 deletion are not limited to sympathetic regulation, as loss of AC5 also eliminates parasympathetic control of cAMP levels and attenuates baroreflexes.11
Much of the interest in AC inhibitors stems from the protective effects of limiting cAMP in the heart, particularly in terms of AC5. Chronic activation of cAMP signaling by prolonged βAR stimulation or the overexpression of βAR, Gsα, or PKA results in cardiac myopathy.2,18,19 The use of β-blockers for treatment of congestive heart failure has been employed to disrupt this pathway. AC5 deletion protects the heart against chronic βAR stimulation and chronic pressure overload by attenuating the decline in cardiac function and defending against increased apoptosis. This is consistent with the increase in AC5 levels in mice with pressure overload.20 AC5 disruption is also protective against age-related cardiac myopathy and gives rise to an increased lifespan as compared to wild type animals.17 Thus direct pharmacological inhibitors of AC5 might serve as an alternative to the common βAR blockade therapy in the treatment of cardiovascular diseases, with the potential added benefit of preventing age-related heart disease to prolong the human lifespan.10
By contrast, overexpression of AC6 also has cardioprotective effects under numerous stress settings,15,21 although results for pressure overload models are mixed.14,22 Deletion of AC6 reveals unique biological functions not duplicated by AC5.23 Disruption of AC6 reduces PKA and Akt activity, phospholamban (PLN) phosphorylation, and βAR-stimulated LV contractile function, in addition to displaying abnormalities in calcium transients.23 Curiously, female AC6 knockout animals show decreased LV hypertrophy in response to pressure overload, although their male counterparts show significantly elevated mortality rates.24 A possible correlation between protective effects for AC6 upregulation and AC5 deletion is unknown.
AC Compartmentalization by Lipid Rafts
The differences in phenotypes between AC5 and AC6 deletions suggest that distinct pools of cAMP must be generated within cardiac myocytes. Although the mechanism for generation of different cAMP pools is still controversial, the spatial distribution of ACs must be a significant aspect underlying their physiological functions. Spatial distribution of cAMP signaling components can be accomplished by either localization to distinct membrane structures or association with protein scaffolds. Cardiac myocytes and fibroblasts contain membrane lipid raft domains that have decreased fluidity compared to other portions of the plasma membrane based upon their high cholesterol and sphingolipid content.25,26 A growing number of proteins involved in GPCR signal transduction are enriched in lipid raft regions of cardiac membranes, including a subset of GPCRs, G proteins, PKA-RII, PKC, ACs, and numerous K+ and Ca2+ channels.25,27 Cardiac tissue is protected from hypoxia-reperfusion when preconditioned with opioids or short periods of ischemic conditions. Disruption of caveolae in adult cardiac myocytes with cholesterol-depleting agents in vitro eliminates this protection from damage by hypoxiareperfusion.28 Similar effects have been observed with intact hearts,29 suggesting that the spatial distribution of signaling components is an important aspect of cellular functions.
Different AC isoforms localize to distinct membrane compartments. Of the nine isoforms of the AC family,7 the calcium-sensitive ACs (AC 1, 3, 5, 6, and 8) but not the Ca2+-insensitive ACs (AC 2, 4, 7, and 9) are localized to lipid raft structures, independent of caveolin expression. Extraction of cholesterol destroys lipid rafts and disrupts regulation of AC6 and AC8 by capacitative calcium entry,30 suggesting that these structures are required for at least regulation by calcium. This may be due in part to the co-localization of the sodium-hydrogen exchanger type 1 and/or L-type calcium channels (LTCC) in lipid rafts which can modulate Ca2+ sensitivity of ACs.31,32
AKAP Organization of Adenylyl Cyclase/cAMP Signaling
Organization of upstream- and downstream- components of the cAMP cascade by scaffolding proteins has been recognized for some time.33–35 A kinase-anchoring proteins (AKAPs) can tether PKA, downstream effectors, and a subset of GPCRs to specific subcellular compartments. In addition, they often bring together opposing regulatory molecules such as kinases and phosphatases or PKA and phosphodiesterases to set up localized temporal regulation of signal transduction pathways. A number of AKAPs have been identified in heart including mAKAP, Yotiao, AKAP15/18 splice variants, AKAP-Lbc, AKAP79/150, Gravin, and synemin, to name a few.35,36 The functional importance for overall AKAP anchoring of PKA in heart has been demonstrated using the expression of AKAP-PKA disruptors (Ht31). Disruption of PKA anchoring in cardiac myocytes results in impaired cAMP regulation of cystic fibrosis transmembrane conductance regulator chloride channels,37 LTCC,38 and the phosphorylation of a number of PKA targets,3 resulting in altered cardiac contraction in both myocytes and in heart.39 More recently, a number of AKAPs have been shown to also anchor ACs, providing a potential mechanism for localized signaling by individual AC isoforms.40 The roles for unique AKAP-AC complexes in heart are discussed below (Fig. 1).
Figure 1.
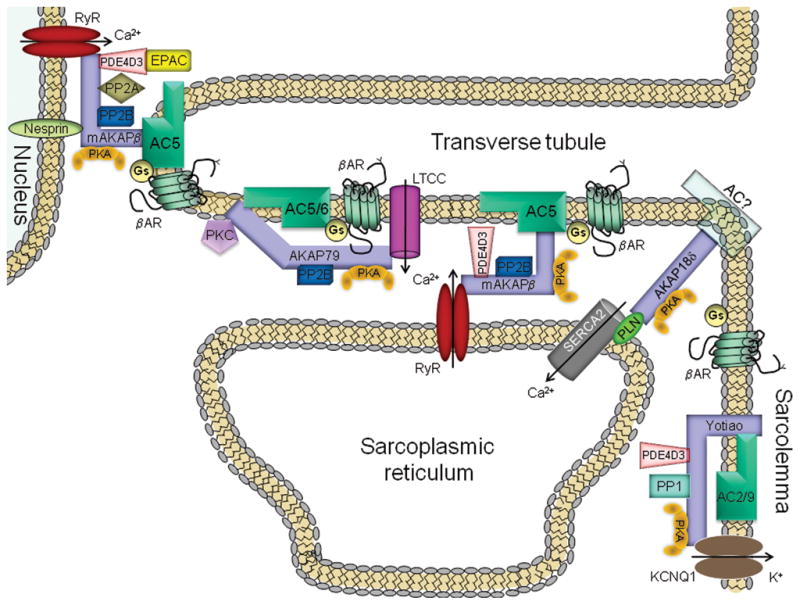
AC-AKAP complexes in cardiac myocytes. Abbreviations are: AC, adenylyl cyclase; AKAP, A kinase-anchoring protein; βAR, β-adrenergic receptor; EPAC, exchange protein directly activated by cAMP; Gs, stimulatory G protein; KCNQ1, α subunit of delayed rectifier K+ channel; LTCC, L-type calcium channel; PDE4D3, phosphodiesterase; PK, protein kinase; PLN, phospholamban; PP, protein phosphatase; RyR, ryanodine receptor; SERCA2, sarcoplasmic reticulum type 2 Ca2+-ATPase.
AKAP79/150 (AKAP5)
The AKAP79/150 family consists of human AKAP79, mouse AKAP150, and bovine AKAP75 and has multiple roles in the heart and vascular system. AKAP79/150 can associate with AC isoforms 2, 3, 5, 6, 8, and 9 in cell culture systems,41,42 and specifically with AC 5 and 6 in heart.43 This is a direct interaction mediated by the N-termini of AC 5/6 and a polybasic region of AKAP79 (aa 77–153). The interaction with AKAP79/150 inhibits Gsα-stimulation of a subset of AC isoforms (2, 5 and 6) in isolated plasma membranes. The formation of an AKAP79/150-AC-PKA complex facilitates preferential phosphorylation of AC 5/6 by anchored PKA to inhibit AC activity.41 This sets up an important negative feed-back loop to temporally regulate cAMP production and downstream signaling.
The physiological relevance of AKAP79/150 complexes in the sympathetic response has been examined in cardiac myocytes isolated from wild type and AKAP150 knockout animals. Deletion of AKAP150 results in loss of β-adrenergic stimulated calcium transients and the phosphorylation of substrates involved in calcium handling, reminiscent of AC6 deletion phenotypes.23,43 The scaffolding protein AKAP79/150 targets AC5/6, PKA, and type 2B protein phosphatase (PP2B or calcineurin) to a caveolin 3–associated complex in ventricular myocytes that also binds a unique subpopulation of Cav1.2 LTCC. However, in the AKAP150 knockout heart, the organization of this signaling complex is disrupted and AC 5/6 no longer associates with caveolin 3 in the transverse tubules. The signaling domain created by AKAP150 is also essential for the PKA-dependent phosphorylation of ryanodine receptors and phospholamban. Therefore, one role of AKAP79/150 is to recruit AC5/6 to a membrane-associated complex of signaling molecules which directs the PKA phosphorylation of PLN, ryanodine receptor (RyR), and a subpopulation of LTCC.
AKAP79/150 also has PKA-independent scaffolding effects on LTCC in cardiac myocytes and arterial myocytes. AKAP150 is required for calcium sparking in arterial myocytes, anchoring PKC to LTCC.44 Deletion of AKAP150 results in loss of angiotensin-induced hypertension. AKAP150 is also involved in the coupled gating of LTCC, which can result in cardiac arrhythmias in patients with Timothy syndrome.45
AKAP79/150 complexes may also be important in the cardioprotective effects of the hormone relaxin but due to scaffolding of a different AC isoform. In cardiovascular disease models, activation of the relaxin family peptide receptor 1 (RXFP1) in cardiac fibroblasts results in the inhibition of hypertrophy and fibrosis.46 Sub-picomolar levels of relaxin drive the assembly of a signalosome complex in HEK293 cells, which includes Gsα, Gβγ, AC2, and RXFP1.46 AKAP79/150 binds directly to helix 8 of the relaxin receptor to facilitate coupling between the receptor and AC2. The generation of cAMP is opposed by PDE4D3 activity anchored to the receptor via β-arrestin. The assembly of this elaborate signalosome may explain the high sensitivity to relaxin in rat cardiac fibroblasts.
Yotiao (AKAP9)
Yotiao is a plasma membrane-associated protein, a product of the smallest (250 kDa) splicing variant of AKAP9 gene. Alternative splicing of this gene results in several other isoforms that localize to the centrosome and the Golgi apparatus. In heart, Yotiao has been shown to anchor PKA,47 PP1,48 PDE4D3,49 and slowly activating delayed rectifier K+ current (IKs) α subunit (KCNQ1).50
In heart, sympathetic nervous system regulation of cardiac action potential duration, mediated by the βAR receptor, requires assembly of Yotiao with KCNQ1.50 The control of the sympathetic nervous system over the duration of cardiac action potential requires PKAmediated phosphorylation of the KCNQ1 subunit of the IKs channel with the consequent increase in IKs current, accelerated repolarization and increased heart rate. KCNQ1 mutations that disrupt this complex cause type 1 long-QT syndrome (LQT1), one of the potentially lethal heritable arrhythmia syndromes. A single mutation S1570L in Yotiao, localized to the C-terminal KCNQ1 binding domain, is found in 2% subjects with a clinically robust phenotype for LQT syndrome.51 The inherited S1570L mutation reduces the interaction between KCNQ1 and Yotiao, reduces the cAMP-induced phosphorylation of the channel, eliminates the functional response of the IKs channel to cAMP, and prolongs the action potential in a computational model of the ventricular cardiac myocytes. Therefore, Yotiao is responsible for restoration of potential after contractility.
Yotiao interacts and regulates several AC isoforms with unique specificity.52 Yotiao anchors AC 1, 2, 3 and 9, but surprisingly not with the main cardiac isoforms AC 5/6. Thus in cardiac myocytes, Yotiao likely forms complexes with either AC2 or AC9. Yotiao/AC2 interaction is mediated by the N-terminus of AC2, which binds directly to amino acids 808–957 of Yotiao. The association with the Yotiao scaffold inhibits AC 2 and 3. Peptides that disrupt Yotiao-AC2 interactions reverse the inhibition of AC2 in membranes, but have no effect on AC activity alone. The mechanism for AC inhibition by Yotiao is unknown; however we hypothesize that it may result from interaction with regulatory proteins recruited to the scaffolding protein rather than with Yotiao per se. The IKs response to cAMP is also regulated by Yotiao-anchored PDE4D3, but not PDE4D5.49 Based on these results we suggest that Yotiao/AC/PDE4D3/PP1 complex in heart40 provides a feedback loop for tight control IKs-dependent heart rate and repolarization.
mAKAP
The cardiac splice variant of muscle AKAP (mAKAPβ) is anchored to the nuclear envelope by the membrane-spanning protein, nesprin, and is also present in the sarcoplasmic reticulum (SR) of cardiac myocytes.53–55 In spite of the intracellular localization of mAKAPβ, it associates with AC5 and AC2, but not with AC6.56 This association may be possible when AC is located on transverse tubules of cardiac myocytes.57 These invaginations protrude from the plasma membrane, bringing AC in close contact with the SR and the outer nuclear membrane.58
The mAKAPβ-associated AC activity was completely absent in hearts deleted of AC5, demonstrating that mAKAPβ-AC5 is the predominant complex in heart. AC5 directly interacts with amino acid residues 275–340 of mAKAPβ, a region that does not overlap with binding sites for other known mAKAP-associated proteins.56 Similar to the regulation of ACs by other AKAPs, mAKAPβ inhibits AC5 in a PKA-dependent manner, generating several feed-back forms of regulation within the mAKAP complex.
Many molecules implicated in the regulation of cardiac hypertrophy are anchored by mAKAP, including protein phosphatases PP2A and PP2B, PDE4D3, hypoxia-inducible factor 1α, and indirectly EPAC1, and the ERK5 and MEK5 mitogen-activated protein kinases.53,59–66 At the SR, mAKAP directly interacts with the RyR2, and facilitates PKA-mediated phosphorylation of the receptor in response to βAR stimulation to allow for Ca2+ release from SR.55,67 Association of AC5 with this complex may help to facilitate localized cAMP signaling at the Ttubule/SR junction. In addition, AC5 would be negatively regulated by both anchored PKA and calcium levels generated during excitation-contraction coupling by L-type Ca2+ channels.7
At the nuclear envelope of cardiac myocytes, mAKAP appears to facilitate hypertrophic signaling induced by isoproterenol, phenylephrine and the leukemia inhibitor factor.62,66 Expression of a peptide to disruption of mAKAPβ-AC5 complexes in cultured rat neonatal ventricular myocytes resulted in an increase in total cellular cAMP levels.56 Additionally, disruption of mAKAPβ-AC5 gave rise to myocyte hypertrophy with an increase in cell size, greater myofibrillar organization, and increased protein synthesis. The increased cAMP levels and subsequent hypertrophy may be due to the loss of multiple feed-back loops assembled on mAKAP to constrain cAMP levels, including PKA-mediated increases in PDE4D3 activity, regulation of PDE4D3 activity through EPAC and the MAPK cascade, and PKA-phosphorylation of AC5 activity to decrease cAMP generation.
The proposed function of an mAKAPβ-AC5 complex in cardiac stress responses is consistent with evidence that knockdown of mAKAPβ expression in myocytes blocks hypertrophic signaling,62,66 and that disruption of the AC5 gene prevents the development of heart failure16 and protects against age-induced cardiac hypertrophy, apoptosis, and fibrosis.17
Conclusions
Cardiac myocytes contain at least four distinct AC-AKAP complexes at various locations throughout the cell, however, other complexes may also exist. For example, mAKAP has been shown to anchor the sodium-calcium exchanger (NCX1).68 A complex containing mAKAP/NCX1/AC5 could facilitate calcium extrusion from myocytes. Another possibility may be AKAP18, which anchors well-documented PKA targets including the L-type Ca2+ channel and phospholamban.
AKAP15/18 (AKAP7) has four splice variants with molecular weights ranging from 15 to 50 kDa. The smaller α, β and larger δ isoforms are expressed in the heart.69–71 AKAP18α (AKAP15) has been shown to target PKA to the LTCC. Disruption of PKA anchoring to the LTCC via AKAP18α significantly inhibits the βAR mediated regulation of the channel.72,73 Although clearly an AC isoform must be present in the general vicinity of this complex, the small size of AKAP18α may not be sufficient to allow for direct anchoring of AC.
On the other hand, AKAP18δ acts as a scaffold that coordinates PKA phosphorylation of PLN and the β-adrenergic effect on Ca2+ re-uptake by the SR Ca2+-ATPase (SERCA2).71 Although there is no evidence for an interaction between AC and AKAP18δ, such a complex could facilitate regulation of Ca2+ re-uptake, counter-balancing the effect of Ca2+ release from SR by AC5/mAKAPβ. In this case different AC/AKAP complexes may control not only cAMP, but also Ca2+, which has a direct physiological relevance to cardiac muscle contraction and relaxation.
Certainly one challenge for the future will be to determine the effect of AC anchoring on temporal and spatial regulation of targeted effectors. AC binding sites on AKAPs are not well conserved and each appears to be unique. For example, an AC5-AKAP79 complex cannot be disrupted by an AC5-mAKAP disruption peptide.42 This may be an advantage for future studies aimed at selectively targeting individual complexes to downstream physiological processes in heart.
Acknowledgments
Acknowledgement of Support:
This work was supported by NIH grant GM060419 and AHA grant 09GRNT2200034
References
- 1.Sadana R, Dessauer CW. Physiological Roles for G Protein-Regulated Adenylyl Cyclase Isoforms: Insights from Knockout and Overexpression Studies. NeuroSignals. 2009;17:5–22. doi: 10.1159/000166277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Antos CL, Frey N, Marx SO, Reiken S, Gaburjakova M, Richardson JA, Marks AR, Olson EN. Dilated cardiomyopathy and sudden death resulting from constitutive activation of protein kinase a. Circ Res. 2001;89:997–1004. doi: 10.1161/hh2301.100003. [DOI] [PubMed] [Google Scholar]
- 3.Fink MA, Zakhary DR, Mackey JA, Desnoyer RW, Apperson-Hansen C, Damron DS, Bond M. AKAP-Mediated Targeting of Protein Kinase A Regulates Contractility in Cardiac Myocytes. Circ Res. 2001;88:291–297. doi: 10.1161/01.res.88.3.291. [DOI] [PubMed] [Google Scholar]
- 4.Alig J, Marger L, Mesirca P, Ehmke H, Mangoni ME, Isbrandt D. Control of heart rate by cAMP sensitivity of HCN channels. Proc Natl Acad Sci U S A. 2009;106:12189–12194. doi: 10.1073/pnas.0810332106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Metrich M, Berthouze M, Morel E, Crozatier B, Gomez AM, Lezoualc’h F. Role of the cAMP-binding protein Epac in cardiovascular physiology and pathophysiology. Pflugers Arch - Eur J Physiol. 2010;459:535–546. doi: 10.1007/s00424-009-0747-y. [DOI] [PubMed] [Google Scholar]
- 6.Gao M, Ping P, Post S, Insel PA, Tang R, Hammond HK. Increased expression of adenylylcyclase type VI proportionately increases beta-adrenergic receptor-stimulated production of cAMP in neonatal rat cardiac myocytes. Proc Natl Acad Sci U S A. 1998;95:1038–1043. doi: 10.1073/pnas.95.3.1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Willoughby D, Cooper DM. Organization and Ca2+ regulation of adenylyl cyclases in cAMP microdomains. Physiol Rev. 2007;87:965–1010. doi: 10.1152/physrev.00049.2006. [DOI] [PubMed] [Google Scholar]
- 8.Mattick P, Parrington J, Odia E, Simpson A, Collins T, Terrar D. Ca2+-stimulated adenylyl cyclase isoform AC1 is preferentially expressed in guinea-pig sino-atrial node cells and modulates the I(f) pacemaker current. J Physiol (Lond) 2007;582:1195–1203. doi: 10.1113/jphysiol.2007.133439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ostrom RS, Naugle JE, Hase M, Gregorian C, Swaney JS, Insel PA, Brunton LL, Meszaros JG. Angiotensin II enhances adenylyl cyclase signaling via Ca2+/calmodulin. Gq-Gs cross-talk regulates collagen production in cardiac fibroblasts. J Biol Chem. 2003;278:24461–24468. doi: 10.1074/jbc.M212659200. [DOI] [PubMed] [Google Scholar]
- 10.Iwatsubo K, Minamisawa S, Tsunematsu T, Nakagome M, Toya Y, Tomlinson JE, Umemura S, Scarborough RM, Levy DE, Ishikawa Y. Direct inhibition of type 5 adenylyl cyclase prevents myocardial apoptosis without functional deterioration. J Biol Chem. 2004;279:40938–40945. doi: 10.1074/jbc.M314238200. [DOI] [PubMed] [Google Scholar]
- 11.Okumura S, Kawabe J, Yatani A, Takagi G, Lee MC, Hong C, Liu J, Takagi I, Sadoshima J, Vatner DE, Vatner SF, Ishikawa Y. Type 5 adenylyl cyclase disruption alters not only sympathetic but also parasympathetic and calcium-mediated cardiac regulation. Circ Res. 2003;93:364–371. doi: 10.1161/01.RES.0000086986.35568.63. [DOI] [PubMed] [Google Scholar]
- 12.Boivin B, Lavoie C, Vaniotis G, Baragli A, Villeneuve LR, Ethier N, Trieu P, Allen BG, Hébert TE. Functional beta-adrenergic receptor signalling on nuclear membranes in adult rat and mouse ventricular cardiomyocytes. Cardiovasc Res. 2006;71:69–78. doi: 10.1016/j.cardiores.2006.03.015. [DOI] [PubMed] [Google Scholar]
- 13.Ping P, Anzai T, Gao M, Hammond HK. Adenylyl cyclase and G protein receptor kinase expression during development of heart failure. Am J Physiol Heart Circ Physiol. 1997;273:H707–717. doi: 10.1152/ajpheart.1997.273.2.H707. [DOI] [PubMed] [Google Scholar]
- 14.Guellich A, Gao S, Hong C, Yan L, Wagner TE, Dhar SK, Ghaleh B, Hittinger L, Iwatsubo K, Ishikawa Y, Vatner SF, Vatner DE. Effects of cardiac overexpression of type 6 adenylyl cyclase affects on the response to chronic pressure overload. Am J Physiol Heart Circ Physiol. 2010;299:H707–712. doi: 10.1152/ajpheart.00148.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Takahashi T, Tang T, Lai NC, Roth DM, Rebolledo B, Saito M, Lew WY, Clopton P, Hammond HK. Increased cardiac adenylyl cyclase expression is associated with increased survival after myocardial infarction. Circulation. 2006;114:388–396. doi: 10.1161/CIRCULATIONAHA.106.632513. [DOI] [PubMed] [Google Scholar]
- 16.Okumura S, Takagi G, Kawabe J, Yang G, Lee MC, Hong C, Liu J, Vatner DE, Sadoshima J, Vatner SF, Ishikawa Y. Disruption of type 5 adenylyl cyclase gene preserves cardiac function against pressure overload. Proc Natl Acad Sci U S A. 2003;100:9986–9990. doi: 10.1073/pnas.1733772100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yan L, Vatner DE, O’Connor JP, Ivessa A, Ge H, Chen W, Hirotani S, Ishikawa Y, Sadoshima J, Vatner SF. Type 5 adenylyl cyclase disruption increases longevity and protects against stress. Cell. 2007;130:247–258. doi: 10.1016/j.cell.2007.05.038. [DOI] [PubMed] [Google Scholar]
- 18.Engelhardt S, Hein L, Wiesmann F, Lohse MJ. Progressive hypertrophy and heart failure in beta1-adrenergic receptor transgenic mice. Proc Natl Acad Sci U S A. 1999;96:7059–7064. doi: 10.1073/pnas.96.12.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwase M, Uechi M, Vatner DE, Asai K, Shannon RP, Kudej RK, Wagner TE, Wight DC, Patrick TA, Ishikawa Y, Homcy CJ, Vatner SF. Cardiomyopathy induced by cardiac Gs alpha overexpression. Am J Physiol Heart Circ Physiol. 1997;272:H585–589. doi: 10.1152/ajpheart.1997.272.1.H585. [DOI] [PubMed] [Google Scholar]
- 20.Hu CL, Chandra R, Ge H, Pain J, Yan L, Babu G, Depre C, Iwatsubo K, Ishikawa Y, Sadoshima J, Vatner SF, Vatner DE. Adenylyl cyclase type 5 protein expression during cardiac development and stress. Am J Physiol Heart Circ Physiol. 2009;297:H1776–1782. doi: 10.1152/ajpheart.00050.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao MH, Tang T, Guo T, Miyanohara A, Yajima T, Pestonjamasp K, Feramisco JR, Hammond HK. Adenylyl cyclase type VI increases Akt activity and phospholamban phosphorylation in cardiac myocytes. J Biol Chem. 2008;283:33527–33535. doi: 10.1074/jbc.M805825200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sugano Y, Lai NC, Gao MH, Firth AL, Yuan JX, Lew WY, Hammond HK. Activated expression of cardiac adenylyl cyclase 6 reduces dilation and dysfunction of the pressure-overloaded heart. Biochem Biophys Res Commun. 2011;405:349–355. doi: 10.1016/j.bbrc.2010.12.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tang T, Gao MH, Lai NC, Firth AL, Takahashi T, Guo T, Yuan JX, Roth DM, Hammond HK. Adenylyl cyclase type 6 deletion decreases left ventricular function via impaired calcium handling. Circulation. 2008;117:61–69. doi: 10.1161/CIRCULATIONAHA.107.730069. [DOI] [PubMed] [Google Scholar]
- 24.Tang T, Lai NC, Hammond HK, Roth DM, Yang Y, Guo T, Gao MH. Adenylyl cyclase 6 deletion reduces left ventricular hypertrophy, dilation, dysfunction, and fibrosis in pressure-overloaded female mice. J Am Coll Cardiol. 2010;55:1476–1486. doi: 10.1016/j.jacc.2009.11.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Das M, Das DK. Lipid raft in cardiac health and disease. Curr Cardiol Rev. 2009;5:105–111. doi: 10.2174/157340309788166660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Insel PA, Patel HH. Membrane rafts and caveolae in cardiovascular signaling. Curr Opin Nephrol Hypertens. 2009;18:50–56. doi: 10.1097/MNH.0b013e3283186f82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ostrom RS, Liu X, Head BP, Gregorian C, Seasholtz TM, Insel PA. Localization of adenylyl cyclase isoforms and G protein-coupled receptors in vascular smooth muscle cells: expression in caveolin-rich and noncaveolin domains. Mol Pharmacol. 2002;62:983. doi: 10.1124/mol.62.5.983. [DOI] [PubMed] [Google Scholar]
- 28.Patel HH, Tsutsumi YM, Head BP, Niesman IR, Jennings M, Horikawa Y, Huang D, Moreno AL, Patel PM, Insel PA, Roth DM. Mechanisms of cardiac protection from ischemia/reperfusion injury: a role for caveolae and caveolin-1. FASEB J. 2007;21:1565–1574. doi: 10.1096/fj.06-7719com. [DOI] [PubMed] [Google Scholar]
- 29.Das M, Gherghiceanu M, Lekli I, Mukherjee S, Popescu LM, Das DK. Essential role of lipid raft in ischemic preconditioning. Cell Physiol Biochem. 2008;21:325–334. doi: 10.1159/000129391. [DOI] [PubMed] [Google Scholar]
- 30.Fagan KA, Graf RA, Tolman S, Schaack J, Cooper DMF. Regulation of a Ca2+-sensitive adenylyl cyclase in an excitable cell - Role of voltage-gated versus capacitative Ca2+ entry. J Biol Chem. 2000;275:40187–40194. doi: 10.1074/jbc.M006606200. [DOI] [PubMed] [Google Scholar]
- 31.Willoughby D, Masada N, Crossthwaite AJ, Ciruela A, Cooper DM. Localized Na+/H+ exchanger 1 expression protects Ca2+-regulated adenylyl cyclases from changes in intracellular pH. J Biol Chem. 2005;280:30864–30872. doi: 10.1074/jbc.M414355200. [DOI] [PubMed] [Google Scholar]
- 32.Yu HJ, Ma H, Green RD. Calcium entry via L-type calcium channels acts as a negative regulator of adenylyl cyclase activity and cyclic AMP levels in cardiac myocytes. Mol Pharmacol. 1993;44:689. [PubMed] [Google Scholar]
- 33.Dodge-Kafka KL, Langeberg L, Scott JD. Compartmentation of cyclic nucleotide signaling in the heart - The role of A-kinase anchoring proteins. Circ Res. 2006;98:993–1001. doi: 10.1161/01.RES.0000218273.91741.30. [DOI] [PubMed] [Google Scholar]
- 34.Logue JS, Scott JD. Organizing signal transduction through A-kinase anchoring proteins (AKAPs) FEBS J. 2010;277:4370–4375. doi: 10.1111/j.1742-4658.2010.07866.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scott JD, Santana LF. A-kinase anchoring proteins: getting to the heart of the matter. Circulation. 2010;121:1264–1271. doi: 10.1161/CIRCULATIONAHA.109.896357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mauban JR, O’Donnell M, Warrier S, Manni S, Bond M. AKAP-scaffolding proteins and regulation of cardiac physiology. Physiology (Bethesda) 2009;24:78–87. doi: 10.1152/physiol.00041.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kockskamper J, Sendhoff K, Erlenkamp S, Bordusa F, Cerovsky V, Glitsch HG. Differences in the protein-kinase-A-dependent regulation of CFTR Cl- channels and Na+-K+ pumps in guinea-pig ventricular myocytes. Pflugers Arch. 2001;441:807–815. doi: 10.1007/s004240000485. [DOI] [PubMed] [Google Scholar]
- 38.Gao TY, Yatani A, DellAcqua ML, Sako H, Green SA, Dascal N, Scott JD, Hosey MM. CAMP-dependent regulation of cardiac L-type Ca2+ channels requires membrane targeting of PKA and phosphorylation of channel subunits. Neuron. 1997;19:185–196. doi: 10.1016/s0896-6273(00)80358-x. [DOI] [PubMed] [Google Scholar]
- 39.McConnell BK, Popovic Z, Mal N, Lee K, Bautista J, Forudi F, Schwartzman R, Jin JP, Penn M, Bond M. Disruption of protein kinase A interaction with A-kinase-anchoring proteins in the heart in vivo: effects on cardiac contractility, protein kinase A phosphorylation, and troponin I proteolysis. J Biol Chem. 2009;284:1583–1592. doi: 10.1074/jbc.M806321200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dessauer CW. Adenylyl cyclase--A-kinase anchoring protein complexes: the next dimension in cAMP signaling. Mol Pharmacol. 2009;76:935–941. doi: 10.1124/mol.109.059345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bauman AL, Soughayer J, Nguyen BT, Willoughby D, Carnegie GK, Wong W, Hoshi N, Langeberg LK, Cooper DM, Dessauer CW, Scott JD. Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Mol Cell. 2006;23:925–931. doi: 10.1016/j.molcel.2006.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Efendiev R, Samelson BK, Nguyen BT, Phatarpekar PV, Baameur F, Scott JD, Dessauer CW. AKAP79 interacts with multiple adenylyl cyclase (AC) isoforms and scaffolds AC5 and -6 to alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptors. J Biol Chem. 2010;285:14450–14458. doi: 10.1074/jbc.M110.109769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nichols CB, Rossow CF, Navedo MF, Westenbroek RE, Catterall WA, Santana LF, McKnight GS. Sympathetic stimulation of adult cardiomyocytes requires association of AKAP5 with a subpopulation of L-type calcium channels. Circ Res. 2010;107:747–756. doi: 10.1161/CIRCRESAHA.109.216127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Navedo MF, Nieves-Cintron M, Amberg GC, Yuan C, Votaw VS, Lederer WJ, McKnight GS, Santana LF. AKAP150 Is Required for Stuttering Persistent Ca2+ Sparklets and Angiotensin II-Induced Hypertension. Circ Res. 2008;102:e1–11. doi: 10.1161/CIRCRESAHA.107.167809. [DOI] [PubMed] [Google Scholar]
- 45.Navedo MF, Cheng EP, Yuan C, Votaw S, Molkentin JD, Scott JD, Santana LF. Increased coupled gating of L-type Ca2+ channels during hypertension and Timothy syndrome. Circ Res. 2010;106:748–756. doi: 10.1161/CIRCRESAHA.109.213363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Halls ML, Cooper DM. Sub-picomolar relaxin signalling by a pre-assembled RXFP1, AKAP79, AC2, beta-arrestin 2, PDE4D3 complex. EMBO J. 2010;29:2772–2787. doi: 10.1038/emboj.2010.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin JW, Wyszynski M, Madhavan R, Sealock R, Kim JU, Sheng M. Yotiao, a novel protein of neuromuscular junction and brain that interacts with specific splice variants of NMDA receptor subunit NR1. J Neurosci. 1998;18:2017–2027. doi: 10.1523/JNEUROSCI.18-06-02017.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Westphal RS, Tavalin SJ, Lin JW, Alto NM, Fraser ID, Langeberg LK, Sheng M, Scott JD. Regulation of NMDA receptors by an associated phosphatase-kinase signaling complex. Science. 1999;285:93–96. doi: 10.1126/science.285.5424.93. [DOI] [PubMed] [Google Scholar]
- 49.Terrenoire C, Houslay MD, Baillie GS, Kass RS. The cardiac IKs potassium channel macromolecular complex includes the phosphodiesterase PDE4D3. J Biol Chem. 2009;284:9140–9146. doi: 10.1074/jbc.M805366200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Marx SO, Kurokawa J, Reiken S, Motoike H, D’Armiento J, Marks AR, Kass RS. Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science. 2002;295:496–499. doi: 10.1126/science.1066843. [DOI] [PubMed] [Google Scholar]
- 51.Chen L, Marquardt ML, Tester DJ, Sampson KJ, Ackerman MJ, Kass RS. Mutation of an A-kinase-anchoring protein causes long-QT syndrome. Proc Natl Acad Sci U S A. 2007;104:20990–20995. doi: 10.1073/pnas.0710527105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Piggott LA, Bauman AL, Scott JD, Dessauer CW. The A-kinase anchoring protein Yotiao binds and regulates adenylyl cyclase in brain. Proc Natl Acad Sci U S A. 2008;105:13835–13840. doi: 10.1073/pnas.0712100105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kapiloff MS, Schillace RV, Westphal AM, Scott JD. mAKAP: an A-kinase anchoring protein targeted to the nuclear membrane of differentiated myocytes. J Cell Sci. 1999;112:2725–2736. doi: 10.1242/jcs.112.16.2725. [DOI] [PubMed] [Google Scholar]
- 54.Pare GC, Easlick JL, Mislow JM, McNally EM, Kapiloff MS. Nesprin-1alpha contributes to the targeting of mAKAP to the cardiac myocyte nuclear envelope. Exp Cell Res. 2005;303:388–399. doi: 10.1016/j.yexcr.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 55.Ruehr ML, Russell MA, Ferguson DG, Bhat M, Ma JJ, Damron DS, Scott JD, Bond M. Targeting of protein kinase A by muscle a kinase-anchoring protein (mAKAP) regulates phosphorylation and function of the skeletal muscle ryanodine receptor. J Biol Chem. 2003;278:24831–24836. doi: 10.1074/jbc.M213279200. [DOI] [PubMed] [Google Scholar]
- 56.Kapiloff MS, Piggott LA, Sadana R, Li J, Heredia LA, Henson E, Efendiev R, Dessauer CW. An adenylyl cyclase-mAKAPbeta signaling complex regulates cAMP levels in cardiac myocytes. J Biol Chem. 2009;284:23540–23546. doi: 10.1074/jbc.M109.030072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gao T, Puri TS, Gerhardstein BL, Chien AJ, Green RD, Hosey MM. Identification and subcellular localization of the subunits of L-type calcium channels and adenylyl cyclase in cardiac myocytes. J Biol Chem. 1997;272:19401–19407. doi: 10.1074/jbc.272.31.19401. [DOI] [PubMed] [Google Scholar]
- 58.Escobar M, Cardenas C, Colavita K, Petrenko NB, Franzini-Armstrong C. Structural evidence for perinuclear calcium microdomains in cardiac myocytes. J Mol Cell Cardiol. 50:451–459. doi: 10.1016/j.yjmcc.2010.11.021. [DOI] [PubMed] [Google Scholar]
- 59.Dodge-Kafka KL, Bauman A, Mayer N, Henson E, Heredia L, Ahn J, McAvoy T, Nairn AC, Kapiloff MS. cAMP-stimulated protein phosphatase 2A activity associated with muscle A kinase-anchoring protein (mAKAP) signaling complexes inhibits the phosphorylation and activity of the cAMP-specific phosphodiesterase PDE4D3. J Biol Chem. 2010;285:11078–11086. doi: 10.1074/jbc.M109.034868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wong W, Goehring AS, Kapiloff MS, Langeberg LK, Scott JD. mAKAP compartmentalizes oxygen-dependent control of HIF-1alpha. Sci Signal. 2008;1:ra18. doi: 10.1126/scisignal.2000026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dodge KL, Khouangsathiene S, Kapiloff MS, Mouton R, Hill EV, Houslay MD, Langeberg LK, Scott JD. mAKAP assembles a protein kinase A/PDE4 phosphodiesterase cAMP signaling module. EMBO J. 2001;20:1921–1930. doi: 10.1093/emboj/20.8.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dodge-Kafka KL, Soughayer J, Pare GC, Carlisle Michel JJ, Langeberg LK, Kapiloff MS, Scott JD. The protein kinase A anchoring protein mAKAP coordinates two integrated cAMP effector pathways. Nature. 2005;437:574–578. doi: 10.1038/nature03966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kapiloff MS, Jackson N, Airhart N. mAKAP and the ryanodine receptor are part of a multi-component signaling complex on the cardiomyocyte nuclear envelope. J Cell Sci. 2001;114:3167–3176. doi: 10.1242/jcs.114.17.3167. [DOI] [PubMed] [Google Scholar]
- 64.Michel JJ, Townley IK, Dodge-Kafka KL, Zhang F, Kapiloff MS, Scott JD. Spatial restriction of PDK1 activation cascades by anchoring to mAKAPalpha. Mol Cell. 2005;20:661–672. doi: 10.1016/j.molcel.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 65.Michel JJC, Dodge KL, Wong W, Mayer NC, Langeberg LK, Scott JD. PKAphosphorylation of PDE4D3 facilitates recruitment of the mAKAP signalling complex. Biochem J. 2004;381:587–592. doi: 10.1042/BJ20040846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pare GC, Bauman AL, McHenry M, Michel JJ, Dodge-Kafka KL, Kapiloff MS. The mAKAP complex participates in the induction of cardiac myocyte hypertrophy by adrenergic receptor signaling. J Cell Sci. 2005;118:5637–5646. doi: 10.1242/jcs.02675. [DOI] [PubMed] [Google Scholar]
- 67.Marx SO, Reiken S, Hisamatsu Y, Jayaraman T, Burkhoff D, Rosemblit N, Marks AR. PKA phosphorylation dissociates FKBP12.6 from the calcium release channel (ryanodine receptor): defective regulation in failing hearts. Cell. 2000;101:365–376. doi: 10.1016/s0092-8674(00)80847-8. [DOI] [PubMed] [Google Scholar]
- 68.Schulze DH, Muqhal M, Lederer WJ, Ruknudin AM. Sodium/calcium exchanger (NCX1) macromolecular complex. J Biol Chem. 2003;278:28849–28855. doi: 10.1074/jbc.M300754200. [DOI] [PubMed] [Google Scholar]
- 69.Fraser ID, Tavalin SJ, Lester LB, Langeberg LK, Westphal AM, Dean RA, Marrion NV, Scott JD. A novel lipid-anchored A-kinase Anchoring Protein facilitates cAMP-responsive membrane events. EMBO J. 1998;17:2261–2272. doi: 10.1093/emboj/17.8.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gray PC, Johnson BD, Westenbroek RE, Hays LG, Yates JR, 3rd, Scheuer T, Catterall WA, Murphy BJ. Primary structure and function of an A kinase anchoring protein associated with calcium channels. Neuron. 1998;20:1017–1026. doi: 10.1016/s0896-6273(00)80482-1. [DOI] [PubMed] [Google Scholar]
- 71.Lygren B, Carlson CR, Santamaria K, Lissandron V, McSorley T, Litzenberg J, Lorenz D, Wiesner B, Rosenthal W, Zaccolo M, Tasken K, Klussmann E. AKAP complex regulates Ca(2+) re-uptake into heart sarcoplasmic reticulum. EMBO Rep. 2007;8:1061–1067. doi: 10.1038/sj.embor.7401081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hulme JT, Lin TW, Westenbroek RE, Scheuer T, Catterall WA. Beta-adrenergic regulation requires direct anchoring of PKA to cardiac CaV1.2 channels via a leucine zipper interaction with A kinase-anchoring protein 15. Proc Natl Acad Sci U S A. 2003;100:13093–13098. doi: 10.1073/pnas.2135335100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hulme JT, Westenbroek RE, Scheuer T, Catterall WA. Phosphorylation of serine 1928 in the distal C-terminal domain of cardiac CaV1.2 channels during beta1-adrenergic regulation. Proc Natl Acad Sci U S A. 2006;103:16574–16579. doi: 10.1073/pnas.0607294103. [DOI] [PMC free article] [PubMed] [Google Scholar]