

Abstract
Gastric epithelial cells (GECs) express the class II major histocompatibility complex (MHC) and costimulatory molecules, enabling them to act as antigen-presenting cells (APCs) and affect local T cell responses. During Helicobacter pylori infection, GECs respond by releasing proinflammatory cytokines and by increasing the surface expression of immunologically relevant receptors, including class II MHC. The CD4+ T cell response during H. pylori infection is skewed toward a Th1 response, but these cells remain hyporesponsive. Activated T cells show decreased proliferation during H. pylori infection, and CD4+ CD25+ FoxP3+ regulatory T cells (Tregs) are present at the site of infection. In this study, we examined the mechanisms surrounding the CD4+ T cell responses during H. pylori infection and found that transforming growth factor β (TGF-β) plays a major role in these responses. GECs produced TGF-β1 and TGF-β2 in response to infection. Activated CD4+ T cells in culture with H. pylori-treated GECs were decreased in proliferation but increased upon neutralization of TGF-β. Naïve CD4+ T cell development into Tregs was also enhanced in the presence of GEC-derived TGF-β. Herein, we demonstrate a role for GEC-produced TGF-β in the inhibition of CD4+ T cell responses seen during H. pylori infection.
INTRODUCTION
Helicobacter pylori is a Gram-negative bacterium that colonizes the gastric mucosa of more than 50% of the world's population. Although many infections are asymptomatic, the clinical magnitude is significant since H. pylori infection is the major cause of chronic gastritis, gastric and duodenal ulcers, and gastric carcinomas throughout the world (10, 21). Additionally, H. pylori is responsible for the development of a non-Hodgkin's-type lymphoma that is the only known malignancy cleared when an infection is cleared. The persistence of infection suggests that the host immune response is inadequate at clearing the infection. Macrophages, neutrophils, B cells, and T cells are present in the infected mucosa (33), but H. pylori evades, subverts, or suppresses the host response. This bacterium may induce immune-regulatory mechanisms that prolong low-level pathogenesis while evading immune-mediated clearance.
The infected gastric mucosa is infiltrated by T cells, most of which are CD4+ cells. The T helper response is generally polarized toward a Th1 response, as interleukin-12 (IL-12) and gamma interferon are the main T cell cytokines seen in the gastric mucosa of infected individuals (3, 13). The Th1 response may be due in part to the H. pylori neutrophil-activating protein (NAP), which has been shown to skew the response toward a Th1 response and is able to shift allergen-specific Th2 cells to become Th1 cells (1, 27). However, during H. pylori infection, Th1 cells do not respond robustly and may be impaired in proliferation (9). Various studies have suggested that H. pylori may inhibit the T cell response by inducing T cell anergy and apoptosis as a method of avoiding immune clearance. T cell inhibition may be in part due to H. pylori virulence factors that play a role in T cell inhibition. One mechanism of T cell inhibition is through the vacuolating toxin (VacA) virulence factor, which is a secreted bacterial toxin that is capable of arresting T cell cycle events (34). One group showed that the proliferation of activated T cells incubated with wild-type H. pylori lysates was reduced in comparison to that of cells incubated with lysates from a cag pathogenicity island A (cagA) isogenic mutant, along with direct inhibition of T cells by a recombinant fragment of CagA, which also suggests a role for this virulence factor in T cell inhibition (25, 30).
In addition to H. pylori having a direct effect on T cell proliferation, it may affect the T cell activation indirectly through the response of other cell types. Gastric epithelial cells (GECs) separate H. pylori from T cells in the lamina propria and represent a cell type that plays a crucial role in the T cell response during infection. GECs have been shown by us and others to express the class II major histocompatibility complex (MHC) as well as CD80 and CD86, which allow them to act as antigen-presenting cells (APCs) (4, 15). We have shown this mechanism not only with epithelial cells but also with subepithelial myofibroblasts of the gastrointestinal tract, which demonstrates an important role for nonprofessional APC phenotypes in the gut immune response (31). On the other hand, we also demonstrated that H. pylori upregulates coinhibitory molecules, such as B7-H1 and B7-DC, that bind PD-1 on activated T cells and inhibit their proliferation and IL-2 production (9). Further, B7-H1 expression by the gastric epithelium induces CD4+ CD25high FoxP3+ regulatory T cells (Tregs) from naïve CD4+ T cells, which in turn inhibit proliferation of CD4+ effector T cells (6). Tregs inhibit activated T cells by direct contact, by competition for APC binding, or by secreted mediators, such as IL-10 and transforming growth factor β (TGF-β) (32). The presence of Tregs in the H. pylori-infected gastric mucosa has been confirmed by multiple groups, some studying infected individuals and others studying mice (11, 16, 23). Additionally, we previously showed that Treg may be locally induced when naïve CD4+ T cells come into contact with GECs that have been exposed to H. pylori, and these Tregs can inhibit adjacent activated T cells (6).
A major determining factor in T cell function and lineage commitment is TGF-β. There are three isoforms of TGF-β, known as TGF-β1, TGF-β2, and TGF-β3. The three isoforms share three receptors, TGF-βRI, TGF-βRII, and TGF-βRIII, with TGF-βRI and TGF-βRII having high affinity for TGF-β1, while TGF-βRIII has high affinity for both TGF-β1 and TGF-β2 (7). Binding of TGF-β1 to TGF-βRII dimers initiates the interaction of two TGF-βRI molecules, allowing signaling to occur (19). The TGF-β1 and TGF-β2 isoforms have a number of documented functions, including the inhibition of T cell proliferation and the development of Tregs. In Treg development, TGF-β induces the expression of the forkhead box P3 (FoxP3) transcription factor, which is essential for their development (8). Tregs also produce TGF-β, which in turn may inhibit adjacent T cells. Although a positive correlation between the increase of FoxP3+ T cells and TGF-β has been reported for the H. pylori-infected gastric mucosa (18), the role of TGF-β isoforms in Treg development during CD4+ T cell interaction with the gastric epithelium is unknown. Therefore, understanding the development of Tregs during H. pylori infection is crucial to understanding the pathogenesis of infection and the multiple outcomes of infection known to be associated with H. pylori.
Previously, we demonstrated that the interaction of H. pylori with GECs leads to the enhanced suppression of the protective immune responses and evaluated the role of cell contact-mediated mechanisms in those interactions (6, 9). Herein, we investigate additional mechanisms and evaluate the role of soluble factors, produced by GECs in response to the interaction with H. pylori, in the above-mentioned suppression (7). In this study, we found that GECs produce TGF-β1 and TGF-β2 in response to H. pylori infection. The GEC response was also associated with an increase of TGF-β receptors on CD4+ T cells. A role for H. pylori-induced GEC production of TGF-β on naïve CD4+ T cell differentiation into Tregs, along with their role in the inhibition of activated CD4+ effector T cell proliferation, was established in this study.
MATERIALS AND METHODS
Cell lines and bacterial cultures.
N87 human gastric carcinoma epithelial cells were obtained from the American Type Culture Collection (ATCC) and maintained in RPMI 1640 with 10% fetal bovine serum (FBS) and 2 mM l-glutamine. HS-738 nontransformed fetal gastric/int cells were obtained from the ATCC and maintained in high-glucose Dulbecco's modified Eagle's medium (DMEM) with 10% FBS and 2 mM l-glutamine. Cells were added to 48-well plates for coculture with CD4+ T cells. H. pylori strains 26695, 51B, 51B cagA−, and 51B vacA− were obtained from Yoshio Yamaoka at Baylor College of Medicine and grown on tryptic soy agar (TSA) plates with 5% sheep's blood (Becton Dickinson, San Jose, CA) and blood agar plates with 2.5 μg/ml of chloramphenicol (Technova, Hollister, CA) to maintain 51B cagA− and 51B vacA− strains at 37°C under microaerophilic conditions. Cells were incubated at a 50:1 bacterium/cell ratio of H. pylori for 24 h.
Cytokine assays.
Supernatants from H. pylori-infected GECs were analyzed by a Luminex bead array assay for TGF-β1, TGF-β2, and TGF-β3 (Millipore, Billerica, MA). Supernatants from T cell coculture experiments were analyzed for IL-2 by a singleplex assay (Bio-Rad, Hercules, CA). Assays were performed according to the manufacturer's instructions.
Naïve CD4+ T cell isolation and incubation with GECs.
Heparinized venous blood samples were obtained from healthy adult volunteers negative for H. pylori (IRB-approved protocol 06-122 at the University of Texas Medical Branch [UTMB] and 09-617 at the University of New Mexico [UNM]). Peripheral blood mononuclear cells (PBMC) were prepared from collected blood by density gradient centrifugation over Ficoll-Paque Plus. Naïve CD4+ T cells were isolated from the PBMC using negative selection (Stem Cell Technologies, Vancouver, WA). Supernatants from H. pylori-treated cultures were filtered to remove bacteria (H. pylori-conditioned medium [Hp-CM]), and cells were washed twice with phosphate-buffered saline (PBS) to remove attached bacteria before addition of T cells. Conditioned medium was incubated with cells in cocultures to measure the effects of TGF-β on T cells. In order to neutralize TGF-β, anti-TGF-β1 and anti-TGF-β2 antibodies (Abs) (Thermo Scientific, R&D Systems, Minneapolis, MN) or an isotype control was added to cells for 30 min before addition of T cells. Some samples were treated with recombinant TGF-β1 and TGF-β2 (Cell Sciences, Canton, MA) to examine their effects on T cells. For coculture with GECs, naive CD4+ T cells were added to each well at a 10:1 T cell/GEC ratio and incubated at 37°C with 5% CO2 for 5 days for proliferation assays and 7 days for Treg experiments.
CFSE.
Freshly isolated CD4+ T cells were preloaded with carboxyfluorescein diacetate succinimidyl ester (CFSE) according to the manufacturer's instructions (Invitrogen, Carlsbad, CA). Cells were activated using T cell activation beads (Miltenyi Biotech, Auburn, CA) coupled with anti-CD3 and anti-CD28 antibodies. Cells were added to cultures with GECs for 5 days and analyzed for proliferation by flow cytometry. Nonactivated cells were used as a control to measure nonproliferating cells.
Flow cytometry.
Freshly isolated naïve CD4+ T cells, resting or activated with T cell activation beads, were incubated with conditioned medium from GECs exposed to H. pylori and filtered to remove bacteria. After 24 h, cells were stained for TGF-βRI (AF3025; R&D Systems) tagged with fluorescein isothiocyanate (FITC) using an Alexa Fluor 488 labeling kit (Invitrogen), TGF-βRII–FITC, or TGF-βRIII-phycoerythrin (PE) (FAB241F and FAB242P; R&D Systems). After being stained, cells were washed and analyzed by flow cytometry. To examine active TGF-β1 and -2, CD4+ T cells were incubated with H. pylori-conditioned medium for 2 h and stained for cell surface TGF-β1 (clone 9016.2; Thermo Scientific) and TGF-β2 (AB-112-NA; R&D systems) labeled with FITC using Zenon antibody labeling kits (Invitrogen). Blocking antibodies for TGF-βRII (AF-241-NA) were from R&D Systems. For examination of isolated CD4+ T cells, cells were stained with anti-human CD4-FITC (OKT-4; eBioscience, San Diego, CA) to assess purity. For Treg experiments, CD4+ T cells were cocultured with GECs for 7 days. After 7 days, cells were harvested, washed, and stained with anti-human CD25–FITC antibodies (clone BC96; Biolegend, San Diego, CA) or isotype control antibodies for 1 h at 4°C. After being washed, cells were permeabilized using a FoxP3 fixation and permeabilization kit from Biolegend according to the manufacturer's instructions. Cells were then incubated with mouse anti-human FoxP3–PE-conjugated antibodies (clone 236A/E7; eBioscience) or an isotype control for 1 h at 4°C. In order to examine IL-10 expression by CD4+ T cells, brefeldin A (Biolegend) was added to cultures according to the manufacturer's instructions for 4 h. T cells were then harvested, permeabilized, and stained with FITC-conjugated anti-IL-10 antibodies (clone JES3-9D7; Biolegend) for flow cytometry. After being washed, cells were analyzed by flow cytometry on a Guava EasyCyte Plus (Millipore) and analyzed using FCS Express software.
Real-time PCR.
Total cellular RNA was isolated from GECs and T cells using an RNeasy RNA isolation kit (Qiagen, Valencia, CA) according to the manufacturer's instructions. The sample concentration was measured by a spectrophotometer at 260 nm, and RNA quality was determined with a 1% agarose gel. Real-time PCR was performed according the Applied Biosystems two-step real-time PCR protocol (Applied Biosystems, Foster City, CA). All reagents were purchased from Applied Biosystems. The reverse transcriptase (RT) reaction mixture included random 2.5 μM hexamers, 500 μM deoxynucleoside triphosphates (dNTPs), 0.4 U/μl of the RNase inhibitors, 5.5 mM MgCl2, MultiScribe reverse transcriptase (3.125 U/μl), and its buffer, 1 μg of cellular RNA. The RT volume mixture was adjusted to a final volume of 50 μl using RNase- and DNase-free H2O. The RT step was performed according to the following protocol: 10 min at 25°C, 60 min at 37°C, and 5 min at 95°C. cDNA samples were stored at −80°C and used for the PCR step. The PCR mixture was prepared using the assays-on-demand gene expression assay mixture (Applied Biosystems) for human 18S, TGF-β1, TGF-β2, TGF-βRI, TGF-βII, TGF-βRIII, and FoxP3 (a 20× mix of unlabeled PCR primers and TaqMan MGB probe, 6-carboxyfluorescein [FAM] dye labeled), and 2 μl of cDNA was added to the PCR mixture for a final volume of 20 μl using Bio-Rad's iQ5 real-time PCR machine. The reaction was carried out according to the following protocol: 2 min at 50°C, 10 min at 95°C (1 cycle), and 15 s 95°C and 1 min at 60°C (45 cycles). The negative controls were included in the RT real-time two-step reaction. The endpoint used in real-time PCR quantification, the threshold cycle (CT), was defined as the PCR cycle number that crosses the signal threshold. Quantification of cytokine gene expression was performed using the comparative CT method (sequence detector user bulletin 2; Applied Biosystems) and reported as the fold difference relative to the level for the human housekeeping gene, 18S mRNA.
Statistical analysis.
Results were expressed as the mean values ± standard errors (SE) of the data. Differences between means were evaluated by analysis of variance (ANOVA) using Student's t test for multiple comparisons. P values of <0.05 were considered statistically significant.
RESULTS
GECs produce TGF-β in response to H. pylori.
Since the gastric epithelium is known to produce an array of cytokines that play a major role in the immune response to H. pylori and are known to affect the T cell response during infection, the ability of GECs to produce TGF-β during exposure to H. pylori was examined. N87 and HS-738 GECs were exposed to H. pylori strains 26695 and 51B (50:1 H. pylori bacterium/cell ratio) for 24 h. Two wild-type strains of H. pylori that are both positive for the major virulence factors of H. pylori, including CagA and VacA, were examined in order to show that similar responses are seen with multiple strains of H. pylori. Supernatants were harvested and analyzed for TGF-β production by a Luminex bead array assay for TGF-β1, TGF-β2, and TGF-β3. GECs were found to produce both TGF-β1 and TGF-β2 (Fig. 1A and B) but not TGF-β3 (data not shown). Both N87 and HS-738 cells produced TGF-β1 and TGF-β2, with approximately 8 times more TGF-β2 produced than TGF-β1. Both H. pylori strains tested induced TGF-β isoforms at similar levels. In order to further examine GEC expression of TGF-β, mRNA levels were measured by quantitative real-time PCR after 16 h of incubation with H. pylori. Figure 1C shows that the H. pylori 51B strain led to HS-738 cell increases of approximately 3.4-fold in TGF-β1 mRNA and approximately 5.0-fold in TGF-β2 mRNA over the level of untreated cells, demonstrating that not only is TGF-β released by GECs, but the amount of transcript is also increased. The cagA knockout induced a 2.1-fold increase in TGF-β1 mRNA, which was decreased from the 3.4-fold increase induced by the wild type, and a 3.7-fold increase in TGF-β2 mRNA, compared to the 5.0-fold increase induced by the wild type. However, there was a more profound difference between the wild type and the vacA knockout, with a 1.9-fold increase in TGF-β1 mRNA and a 2.7-fold increase in TGF-β2 mRNA levels. These results suggest that two of the major virulence factors of H. pylori play a role in GEC production of TGF-β1 and TGF-β2 isoforms in response to infection.
Fig. 1.

GECs exposed to H. pylori produce TGF-β. GECs incubated at a 50:1 H. pylori bacterium/cell ratio for 24 h produce TGF-β1 (A) and TGF-β2 (B), as shown by a Luminex bead array assay. *, P < 0.05 for comparison to untreated cells. (C) HS-738 cells incubated with H. pylori strain 51B for 16 h have increased levels of TGF-β1 and TGF-β2 mRNA compared to untreated cells, and strains lacking cagA or vacA were found to induce reduced amounts compared to the wild type when examined by quantitative real-time PCR. *, P < 0.05 for comparison to untreated cells, and **, P < 0.05 for comparison to 51B. Data shown are means ± standard errors of the means (SEM) of results from 8 experiments.
CD4+ T cells exposed to H. pylori-conditioned medium upregulate TGF-β receptors.
TGF-β1 and TGF-β2 induce signaling by binding to TGF-β receptors, which complex after ligand binding. Here, we examined the expression of these receptors on CD4+ T cell surfaces and examined the effect of H. pylori-conditioned medium (Hp-CM) on expression. CD4+ T cells, activated with anti-CD3, anti-CD28 beads, were exposed to Hp-CM for 24 h, and TGF-β receptor expression was examined by flow cytometry. Upon exposure to H. pylori-conditioned medium, TGF-βRI, TGF-βRII, and TGF-βRIII expression levels were increased on CD4+ T cells at 24 h (Fig. 2 A to C). Similar results were seen with nonactivated CD4+ T cells (not shown). Expression was further examined by quantitative real-time PCR. Figure 2D shows that TGF-βRI, TGF-βRII, and TGF-βRIII mRNA levels are increased by 2- to 3-fold in CD4+ T cells following exposure to H. pylori-conditioned medium from GECs. These results indicate that factors released by GECs in response to H. pylori induce the expression of the TGF-β receptors on T cells. GEC production of TGF-β1 and TGF-β2, along with the production of soluble mediators that increase the expression of TGF-β receptors with high affinity for TGF-β1 and TGF-β2, suggests a mechanism by which the GEC response to H. pylori infection may affect local T cell responses by releasing soluble factors.
Fig. 2.

H. pylori-conditioned medium increases TGF-β receptor expression on CD4+ T cells. Activated CD4+ T cells incubated with Hp-CM have increased TGF-βRI (A), TGF-βRII (B), and TGF-βRIII (C) expression compared to the solid peak isotype controls. Representative figures for 8 experiments are shown here. (D) RNA levels are also increased by quantitative real-time PCR. Data shown are means ± SEM of results from 6 experiments. *, P < 0.05 for comparison to untreated cells.
GECs produce active TGF-β that binds to CD4+ T cells.
Since TGF-β is released in an inactive form with the latency-associated peptide, this peptide must be cleaved in order for TGF-β to be active (2). In assays commonly used to measure TGF-β, such as an enzyme-linked immunosorbent assay (ELISA) or a Luminex bead array assay, samples must be acid activated to measure TGF-β. However, in order to further examine TGF-β in this system, we developed a method to test for active TGF-β1 and TGF-β2. Since TGF-β must be in an active form to bind to a receptor, we incubated activated CD4+ T cells with Hp-CM, which contains TGF-β1 and TGF-β2 in order to examine binding of TGF-β to TGF-βRII on the surface of the CD4+ T cells by flow cytometry. T cells were activated with anti-CD3- and anti-CD28-coated beads to increase expression of TGF-β receptors. Hp-CM consisted of supernatants from GECs incubated with H. pylori for 24 h and then filtered to remove the bacteria to prevent direct effects of bacteria on the T cells, but the soluble mediators, including TGF-β, were still present. After 2 h of incubation with Hp-CM, surface-bound TGF-β1 and TGF-β2 (active TGF-β) were examined by flow cytometry. TGF-βRII was blocked to demonstrate active TGF-β binding to receptor on CD4+ T cells. TGF-βRII was blocked in this assay since both TGF-β1 and TGF-β2 have been shown to bind to it; however, TGF-β2 may require coexpression of TGF-βRIII for binding (29). Fig. 3A and B show that both TGF-β1 and TGF-β2 are bound to the surfaces of activated T cells. Blocking TGF-βRII decreased binding of both TGF-β1 and TGF-β2, indicating that there is active TGF-β released by GECs in response to H. pylori.
Fig. 3.
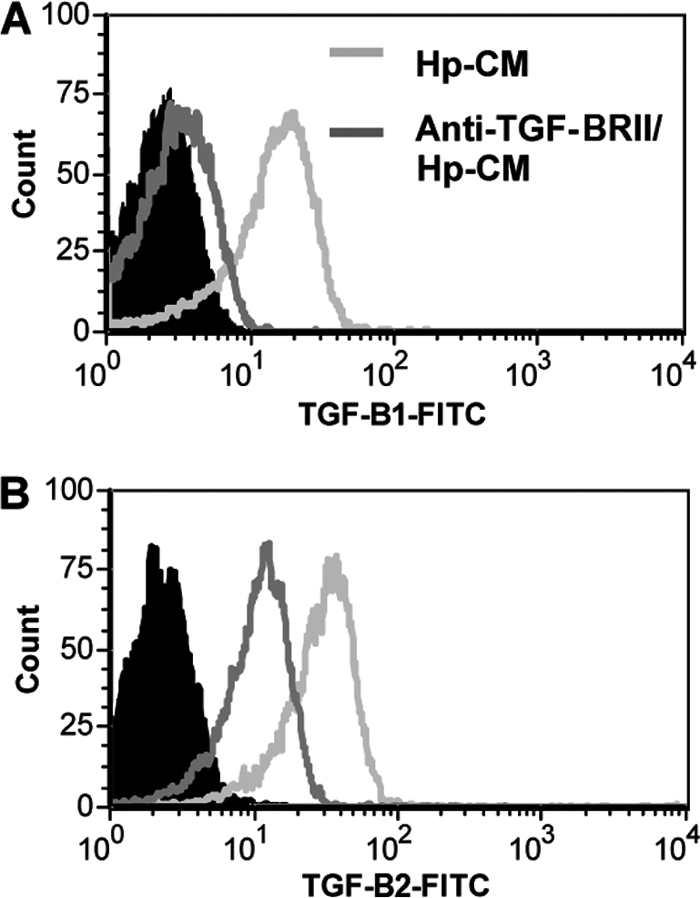
Active TGF-β binds to CD4+ T cells. CD4+ T cells incubated with Hp-CM have TGF-β1 (A) and TGF-β2 (B) bound to the surface compared to the solid peak isotype controls. Representative figures for 6 experiments are shown here.
TGF-β produced by GECs inhibits CD4+ T cell proliferation.
Since TGF-β can inhibit the action of T cells, we examined the effect GEC-derived TGF-β induced by H. pylori on activated CD4+ T cell proliferation. For this purpose, the effects of GEC-conditioned medium (CM), untreated or preexposed to H. pylori (CM or Hp-CM, respectively) for 24 h, on proliferation of CFSE-labeled, anti-CD3- and anti-CD28-activated CD4+ T cells in coculture with GECs were measured. Some cultures were treated with anti-TGF-β1 and TGF-β2 neutralizing antibodies. We observed that the proliferation of activated CD4+ T cells was substantially lower in the presence of Hp-CM than in the presence of the CM control. Proliferating cells were determined by comparison to nonproliferating control cells (Fig. 4A to C). In these experiments, two different H. pylori strains were used, 26695 and 51B. The Hp-CM derived from both strains demonstrated strong inhibitory capacity (up to 30%) on the proliferation of the activated T cells compared to CM from non-H. pylori-treated cells (Fig. 4D). This inhibitory effect of Hp-CM was partially reversed in the presence of anti-TGF-β1 or anti-TGF-β2 neutralizing antibodies. Simultaneous addition of both Abs almost completely abrogated the inhibitory effect of Hp-CM on the proliferation of activated T cells. Importantly, adding the mixture of recombinant TGF-β1 and TGF-β2 at levels similar to those produced by GECs during H. pylori infection to the activated T cell cultures decreases the number of proliferating cells as well, but not to the extent of H. pylori treatments. Thus, our data suggested that TGF-β1 and TGF-β2 isoforms induced by H. pylori in GECs have a suppressive role in activated CD4+ T cell proliferation. Taken together, our data indicate that both TGF-β1 and TGF-β2 play a key role in the suppression of the proliferation of activated T cells. The cagA and vacA knockouts reduced the ability of H. pylori to inhibit T cell proliferation (Fig. 4E). Since these virulence factors induce less TGF-β1 and TGF-β2 production, as shown in Fig. 1C, and we show in Fig. 4D that TGF-β plays a role in this inhibition, it is likely that this mechanism is in part due to the CagA and VacA proteins.
Fig. 4.

H. pylori-induced GEC-derived TGF-β inhibits activated CD4+ T cell proliferation. Proliferation of CD4+ T cells was examined by CFSE by flow cytometry where resting CD4+ T cells fluoresce brightly as nonproliferating cells (A), activated CD4+ T cells in coculture with untreated HS-738 cells proliferate for several generations (B), and activated CD4+ T cells in coculture with H. pylori-exposed HS-738 cells show decreased proliferation compared to activated CD4+ T cells in coculture with untreated HS-738 cells, as shown in example histograms, where the M1 populations are resting cells and the M2 populations are proliferating cells (C). (D) Compiled CFSE results, where M2 populations are shown as percentages of proliferating CD4+ T cells from culture with N87 and HS-738 cells exposed to H. pylori 26695 and 51B, where blocking of TGF-β1 and TGF-β2 led to increased proliferation. (E) CFSE results comparing proliferating CD4+ T cells from culture with HS-738 cells incubated with 51B, 51B cagA−, and 51B vacA−. Data shown are means ± SEM of results from 8 experiments. *, P < 0.05 for comparison to untreated cells; **, P < 0.05 for comparison to H. pylori-treated cells.
In order to further examine the significance of the TGF-β isoforms present in Hp-CM on T cell activity in these cultures, we examined levels of IL-2, a cytokine produced by activated T cells, present in cultures by a Luminex bead assay (Fig. 5). We demonstrated that IL-2 levels in these cultures parallel results seen with CFSE assays. IL-2 production by the activated T cells was reduced in the cocultures with Hp-CM, compared to the level for the untreated CM control. Moreover, the neutralization of TGF-β1 and TGF-β2 in those cultures increased IL-2 production. Taken together, our data suggest that increase in the TGF-β1 and TGF-β2 production by GECs in response to H. pylori infection may contribute to the suppression of the activated CD4+ effector T cell proliferation and, thus, to H. pylori escape from the protective host immune response.
Fig. 5.

H. pylori-induced GEC-derived TGF-β inhibits activated CD4+ T cell production of IL-2. IL-2 production in cocultures of anti-CD3- and anti-CD28-activated CD4+ T cells with H. pylori-exposed GECs was decreased but increased when TGF-β1 and TGF-β2 were blocked. Results are shown as means ± SEM for 8 experiments. *, P < 0.05 for comparison to untreated cells; **, P < 0.05 for comparison to H. pylori-treated cells.
TGF-β produced by GECs in response to H. pylori promotes development of resting naïve CD4+ T cells into CD4+ CD25+ FoxP3+ regulatory T cells.
Another mechanism of T cell inhibition during H. pylori infection may be through recruitment or expansion of the CD4+ FoxP3+ CD25+ regulatory T cells. We recently described that H. pylori-exposed GECs may induce Tregs by a contact-mediated mechanism (6). Here, we extend our previous work and sought to determine the role of TGF-β in the induction of Tregs from naïve CD4+ T cells. In order to do this, naïve CD4+ T cells were isolated from whole blood and incubated with GECs in the presence of Hp-CM or nontreated CM for 7 days since our previous study showed this to be the ideal time for Treg development (6). T cells were harvested from coculture and stained with monoclonal antibodies for CD25 and FoxP3 for analysis by flow cytometry. T cells isolated by magnetic bead separation showed 98% purity in CD4 staining by flow cytometry (Fig. 6 A). As demonstrated in Fig. 6B and C, addition of Hp-CM to cultures resulted in a strong generation of Tregs, with up to 18% of the CD4+ T cells staining positive for the Treg phenotype, CD25+ FoxP3+, compared to the level for the CM control (up to 5%) (Fig. 6B). Compiled data for both N87 and HS-738 are shown in Fig. 6D. Neutralization of TGF-β1 and TGF-β2 in Hp-CM-treated cultures resulted in an approximately 50% decrease in Treg generation with both strains of H. pylori. Similar observations were made for FoxP3 mRNA levels. No significant changes in Treg development were seen when cagA or vacA knockout strains were tested in these experiments (not shown). Figure 6E indicates a 3.3-fold increase in FoxP3 mRNA from cells in culture with 51B-treated GECs over the level for untreated GECs, which was decreased when TGF-β was neutralized. In order to further examine the presence of Tregs in these cultures, some cells were stained for intracellular IL-10, a cytokine associated with the inhibitory activity of Tregs. Figure 7A and B show that addition of Hp-CM to the T cell-GEC cocultures resulted in increased production of IL-10 but that this production was decreased when TGF-β1 and TGF-β2 were neutralized. These results suggest a second mechanism by which TGF-β contributes to T cell inhibition during H. pylori infection, as we previously showed that induced Tregs in coculture could effectively inhibit activated T cells in coculture (6).
Fig. 6.

H. pylori-induced GEC-derived TGF-β promotes resting naïve CD4+ T cell development into CD4+ CD25+ FoxP3+ regulatory T cells. Naïve CD4+ T cells isolated by negative selection were stained with anti-CD4 antibodies to assess purity (A). Isolated cells were cocultured with GECs (B) or GECs (C) exposed to H. pylori and stained for CD25 and FoxP3 in representative dot plots. (D) Compiled data for CD25 and FoxP3 staining of CD4 T cells from cultures with untreated cells, H. pylori-exposed cells, and H. pylori-exposed cells with anti-TGF-β1 and anti-TGF-β2 neutralizing antibodies. (E) FoxP3 mRNA levels were also examined by quantitative real-time PCR. Results indicate fold changes over the level for the isotype control relative to the level for the 18S housekeeping gene. Data shown are means ± SEM of results from 4 experiments, each performed in duplicate (n = 8). *, P < 0.05 for comparison to untreated cells; **, P < 0.05 for comparison to H. pylori-treated cells.
Fig. 7.
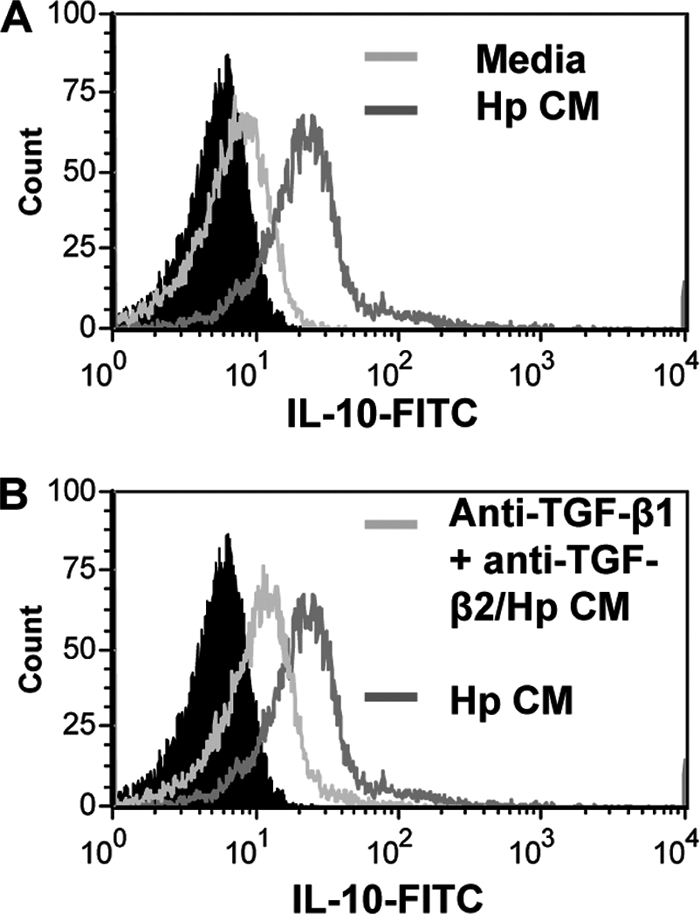
H. pylori-induced GEC-derived TGF-β promotes resting naïve CD4+ T cell expression of IL-10. CD4+ T cells from cultures with untreated cells, H. pylori-exposed cells (A), and H. pylori-exposed cells with anti-TGF-β1 and anti-TGF-β2 neutralizing antibodies (B) were incubated with brefeldin A, stained with anti-IL-10 antibodies for flow cytometry, and compared to the solid peak isotype controls. Representative figures for 8 experiments are shown here.
DISCUSSION
TGF-β has many complex effects on cells. During H. pylori infection, it may be induced as a mechanism by which the bacterium manipulates host responses to enhance its own survival. One study indicates that TGF-β may enhance bacterial colonization and attachment to host cells (17). This group also suggests that the production of TGF-β may depend on bacterial load and decrease when high levels of bacteria are present. Another study demonstrates that increased TGF-β1 in the human cardia and antrum are associated with increased Treg and H. pylori colonization (18). Here, we show that at a moderate level of infection, TGF-β inhibits the CD4+ T cell response by both inhibiting proliferation of activated T cells and inducing Treg development, which also suggests a mechanism for H. pylori persistence.
TGF-β affects both resting and activated T cells to determine phenotypic development and proliferation. TGF-β has multiple isoforms, TGF-β1, TGF-β2, and TGF-β3, which bind with different affinities to receptors. We found TGF-β1 and TGF-β2 to be produced by N87 and HS-738 cells in response to H. pylori infection, with both isoforms having similar effects on T cells in our studies. All three receptors (TGF-βRI, TGF-βRII, and TGF-βRIII) were also upregulated on resting and activated CD4+ T cells when exposed to H. pylori-conditioned medium, allowing increased binding of TGF-β to T cells. Since TGF-β is known to have such a crucial affect on T cell responses, and GECs are known to influence T cell responses during H. pylori infection, GEC-produced TGF-β may be among the key factors that steer the T cell response during infection.
As we have previously shown a role for GEC expression of PD-L1 in T cell inhibition and in Treg development (6), here we show that GEC-produced soluble factors TGF-β1 and TGF-β2 also play a role in decreased activated CD4+ proliferation and in Treg development. The CD4+ T cell response in general during H. pylori infection has been shown to be repressed, with decreased proliferation, anergy, and apoptotic mechanisms in place (9, 30, 34). Tregs likely play an important role in these decreased responses since they have been shown to be present in the infected gastric mucosa in both gastritis and cancer (16). Factors such as TGF-β are upregulated by the infected epithelium and contribute to local development of Tregs. Here, we utilized the N87 cell line, a mucus-producing cell line that not only represents the target of infection in vivo but also has been noted to reproduce many of the features of human primary antral cells based on analysis of adherens junction proteins, the actin cytoskeleton, cell matrix protein expression, and cell migration. Studies to determine what gastric cancer cell lines are appropriate for use as models to study human gastric diseases indicated that N87 is an appropriate cell line (5). We also used the nontransformed cell line HS-738, which, like N87, produces TGF-β1 and TGF-β2 in response to H. pylori. TGF-β and TGF-βRI have been shown to be expressed during H. pylori-associated gastritis and were shown to play a role in the pathogenesis during infection (22). In that study, the focus was on mononuclear cells as a source of TGF-β. Here, we have focused on epithelial cells as a primary target of H. pylori infection and an abundant cell type in the gastric mucosa. These data demonstrate yet another novel mechanism for an H. pylori immunosuppressive mechanism, whereby H. pylori-infected GECs produce TGF-β1 and -2, which affect protective T cell responses during H. pylori infection by decreasing proliferation of CD4+ effector T cells and inducing Tregs. Thus, the upregulation of TGF-β may not only contribute to pathogenesis associated with H. pylori infection but also be a mechanism by which the bacterium manipulates host responses to enhance its own survival and persistent infection. Additionally, we found that knocking out the virulence factor genes vacA and cagA led to the decrease in bacterium-mediated gene expression of TGF-β1 and -2 by GECs and, consequently, to the decrease in immunosuppression of proliferation of activated T cells. The vacA and cagA knockout strains did not, however, affect Treg development. This is consistent with the work of Kao et al., who showed that Treg development from interaction of naïve CD4+ T cells with dendritic cells was independent of these virulence factors but required TGF-β (20). Likely, we see this because although TGF-β1 and TGF-β2 are decreased with the virulence factor knockout, they are still produced at increased levels compared to the basal level, with 2- to 4-fold increases, as we showed here.
A major question that remains for T cell responses in H. pylori infections is whether the outcome of infection is dependent on T cell responses. In a mouse model of infection, Tregs were shown to keep gastritis in check and allow increased colonization of H. pylori by inhibiting the immune response to infection (26). One study suggests that children do not often develop ulcers during H. pylori infection because they develop more Tregs than adults (14). In this study, children were noted to have substantially less gastritis than adults, coupled with increased levels of TGF-β1 mRNA. Interestingly, HS-738 fetal cells produced higher levels of both TGF-β isoforms than N87 cells, although similar amounts of cells and bacteria were used in this study. Although the role of Tregs during infection is not yet clear, one may speculate that those cells contribute to maintaining the balance between chronic infection and the host response. In support of this idea, one study suggests that Tregs allow for H. pylori to persist without harm to the human host, while a lack of a Treg response in some infected people leads to ulceration (28). They further showed that people with ulcers had reduced Treg responses and increased Th1 and Th2 responses compared with those without ulcers. These studies suggest a protective role for TGF-β and Tregs, but there are also some studies indicating that Tregs induced during H. pylori infection may promote prolonged infection, and these studies lend support to the notion that Tregs are important in the chronicity of infection (12). Another group demonstrated that memory T cells from infected individuals did not respond well to H. pylori membrane preparations compared to memory cells from uninfected individuals, but the activity was restored in infected individuals when Tregs were depleted (24). Furthermore, several studies suggest that the presence and influence of the Treg response during infection may promote carcinogenesis (11, 16). These studies point to Tregs being protective against pathogenesis, but in chronic infection and carcinogenesis, Tregs may exacerbate the situation. All of these studies demonstrate a key role for Tregs and TGF-β in the T cell response during H. pylori infection but suggest different effects on different outcomes of infection.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants K22AI68712, R56DK090090-01, and R21CA127022, by the DHHS/NIH/NCRR, by grant 1UL1RR031977-01 from the University of New Mexico Clinical and Translational Science Center, and by American Cancer Society grant RSG-10-159-01-LIB.
The costs of publication of this article were defrayed in part by the payment of page charges. The article must therefore be marked advertisement in accordance with 18 U.S.C. section 1734 solely to indicate this fact.
Footnotes
Published ahead of print on 11 April 2011.
REFERENCES
- 1. Amedei A., et al. 2006. The neutrophil-activating protein of Helicobacter pylori promotes Th1 immune responses. J. Clin. Invest. 116:1092–1101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Annes J. P., Munger J. S., Rifkin D. B. 2003. Making sense of latent TGFbeta activation. J. Cell Sci. 116:217–224 [DOI] [PubMed] [Google Scholar]
- 3. Bamford K. B., et al. 1998. Lymphocytes in the human gastric mucosa during Helicobacter pylori have a T helper cell 1 phenotype. Gastroenterology 114:482–492 [DOI] [PubMed] [Google Scholar]
- 4. Barrera C., et al. 2001. Expression of cathepsins B, L, S, and D by gastric epithelial cells implicates them as antigen presenting cells in local immune responses. Hum. Immunol. 62:1081–1091 [DOI] [PubMed] [Google Scholar]
- 5. Basque J. R., Chenard M., Chailler P., Menard D. 2001. Gastric cancer cell lines as models to study human digestive functions. J. Cell. Biochem. 81:241–251 [PubMed] [Google Scholar]
- 6. Beswick E. J., Pinchuk I. V., Das S., Powell D. W., Reyes V. E. 2007. Expression of the programmed death ligand 1, B7-H1, on gastric epithelial cells after Helicobacter pylori exposure promotes development of CD4+ CD25+ FoxP3+ regulatory T cells. Infect. Immun. 75:4334–4341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cheifetz S., et al. 1987. The transforming growth factor-beta system, a complex pattern of cross-reactive ligands and receptors. Cell 48:409–415 [DOI] [PubMed] [Google Scholar]
- 8. Chen W., Konkel J. E. 2010. TGF-beta and ‘adaptive’ Foxp3(+) regulatory T cells. J. Mol. Cell Biol. 2:30–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Das S., et al. 2006. Expression of B7-H1 on gastric epithelial cells: its potential role in regulating T cells during Helicobacter pylori infection. J. Immunol. 176:3000–3009 [DOI] [PubMed] [Google Scholar]
- 10. Dunn B. E., Cohen H., Blaser M. J. 1997. Helicobacter pylori. Clin. Microbiol. Rev. 10:720–741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Enarsson K., et al. 2006. Function and recruitment of mucosal regulatory T cells in human chronic Helicobacter pylori infection and gastric adenocarcinoma. Clin. Immunol. 121:358–368 [DOI] [PubMed] [Google Scholar]
- 12. Goll R., et al. 2007. Helicobacter pylori stimulates a mixed adaptive immune response with a strong T-regulatory component in human gastric mucosa. Helicobacter 12:185–192 [DOI] [PubMed] [Google Scholar]
- 13. Haeberle H. A., et al. 1997. Differential stimulation of interleukin-12 (IL-12) and IL-10 by live and killed Helicobacter pylori in vitro and association of IL-12 production with gamma interferon-producing T cells in the human gastric mucosa. Infect. Immun. 65:4229–4235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harris P. R., et al. 2008. Helicobacter pylori gastritis in children is associated with a regulatory T-cell response. Gastroenterology 134:491–499 [DOI] [PubMed] [Google Scholar]
- 15. Ishii N., et al. 1992. Expression of MHC class II antigens (HLA-DR, -DP, and -DQ) on human gastric epithelium. Gastroenterol. Jpn. 27:23–28 [DOI] [PubMed] [Google Scholar]
- 16. Jang T. J. 2010. The number of Foxp3-positive regulatory T cells is increased in Helicobacter pylori gastritis and gastric cancer. Pathol. Res. Pract. 206:34–38 [DOI] [PubMed] [Google Scholar]
- 17. Jo Y., et al. 2010. Suppressed gastric mucosal TGF-beta1 increases susceptibility to H. pylori-induced gastric inflammation and ulceration: a stupid host defense response. Gut Liver 4:43–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kandulski A., et al. 2008. Naturally occurring regulatory T cells (CD4+, CD25high, FOXP3+) in the antrum and cardia are associated with higher H. pylori colonization and increased gene expression of TGF-beta1. Helicobacter 13:295–303 [DOI] [PubMed] [Google Scholar]
- 19. Kang J. S., Liu C., Derynck R. 2009. New regulatory mechanisms of TGF-beta receptor function. Trends Cell Biol. 19:385–394 [DOI] [PubMed] [Google Scholar]
- 20. Kao J. Y., et al. 2010. Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterology 138:1046–1054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kuipers E. J. 1997. Helicobacter pylori and the risk and management of associated diseases: gastritis, ulcer disease, atrophic gastritis and gastric cancer. Aliment. Pharmacol. Ther. 11(Suppl. 1):71–88 [DOI] [PubMed] [Google Scholar]
- 22. Li Z., Li J. 2006. Local expressions of TGF-beta1, TGF-beta1RI, CTGF, and Smad-7 in Helicobacter pylori-associated gastritis. Scand. J. Gastroenterol. 41:1007–1012 [DOI] [PubMed] [Google Scholar]
- 23. Lundgren A., et al. 2005. Mucosal FOXP3-expressing CD4+ CD25high regulatory T cells in Helicobacter pylori-infected patients. Infect. Immun. 73:523–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lundgren A., Suri-Payer E., Enarsson K., Svennerholm A. M., Lundin B. S. 2003. Helicobacter pylori-specific CD4+ CD25high regulatory T cells suppress memory T-cell responses to H. pylori in infected individuals. Infect. Immun. 71:1755–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Paziak-Domanska B., Chmiela M., Jarosinska A., Rudnicka W. 2000. Potential role of CagA in the inhibition of T cell reactivity in Helicobacter pylori infections. Cell. Immunol. 202:136–139 [DOI] [PubMed] [Google Scholar]
- 26. Rad R., et al. 2006. CD25+/Foxp3+ T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterology 131:525–537 [DOI] [PubMed] [Google Scholar]
- 27. Reyes V. E., Beswick E. J. 2007. Helicobacter pylori neutrophil activating proteint's potential as tool in therapeutic immune modulation. Expert Opin. Ther. Pat. 17:1315–1320 [Google Scholar]
- 28. Robinson K., et al. 2008. Helicobacter pylori-induced peptic ulcer disease is associated with inadequate regulatory T cell responses. Gut 57:1375–1385 [DOI] [PubMed] [Google Scholar]
- 29. Rodriguez C., Chen F., Weinberg R. A., Lodish H. F. 1995. Cooperative binding of transforming growth factor (TGF)-beta 2 to the types I and II TGF-beta receptors. J. Biol. Chem. 270:15919–15922 [DOI] [PubMed] [Google Scholar]
- 30. Rudnicka W., Covacci A., Wadstrom T., Chmiela M. 1998. A recombinant fragment of Helicobacter pylori CagA affects proliferation of human cells. J. Physiol. Pharmacol. 49:111–119 [PubMed] [Google Scholar]
- 31. Saada J. I., et al. 2006. Subepithelial myofibroblasts are novel nonprofessional APCs in the human colonic mucosa. J. Immunol. 177:5968–5979 [DOI] [PubMed] [Google Scholar]
- 32. Scheffold A., Murphy K. M., Hofer T. 2007. Competition for cytokines: T(reg) cells take all. Nat. Immunol. 8:1285–1287 [DOI] [PubMed] [Google Scholar]
- 33. Suarez G., Reyes V. E., Beswick E. J. 2006. Immune response to H. pylori. World J. Gastroenterol. 12:5593–5598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sundrud M. S., Torres V. J., Unutmaz D., Cover T. L. 2004. Inhibition of primary human T cell proliferation by Helicobacter pylori vacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proc. Natl. Acad. Sci. U. S. A. 101:7727–7732 [DOI] [PMC free article] [PubMed] [Google Scholar]