

Abstract
Acute and chronic Plasmodium falciparum infections alter the immune competence of the host possibly through changes in dendritic cell (DC) functionality. DCs are the most potent activators of T cells, and migration is integral to their function. Mature DCs express lymphoid chemokine receptors (CCRs), expression of which enables them to migrate to the lymph nodes, where they encounter naïve T cells. The present study aimed to investigate the impact of the synthetic analog to malaria parasite pigment hemozoin, i.e., β-hematin, or infected erythrocytes (iRBCs) on the activation status of human monocyte-derived DCs and on their expression of CCRs. Human monocyte-derived DCs partially matured upon incubation with β-hematin as indicated by an increased expression of CD80 and CD83. Both β-hematin and iRBCs provoked the release of proinflammatory and anti-inflammatory cytokines, such as interleukin-6 (IL-6), IL-10, and tumor necrosis factor alpha, but not IL-12, and induced upregulation of the lymphoid chemokine receptor CXCR4, which was coupled to an increased migration to lymphoid ligands. Taken together, these results suggest that the partial and transient maturation of human myeloid DCs upon stimulation with malaria parasite-derived products and the increased IL-10 but lack of IL-12 secretion may lead to suboptimal activation of T cells. This may in turn lead to impaired adaptive immune responses and therefore insufficient clearance of the parasites.
INTRODUCTION
Plasmodium falciparum malaria is one of the most severe infectious diseases in the world, where as many as 3.3 billion people live at risk of infection and 247 million cases were reported in 2006. The vast majority of victims are children below 5 years of age (49). Immunity to malaria is developed only after repeated exposure and is not long lasting (18). Several studies have demonstrated impairment of immune responses in P. falciparum malaria infection (46).
Previous studies indicate that an early proinflammatory cytokine-mediated mechanism is crucial to control parasitemia and for the clearance of parasites. Excessive proinflammatory responses, however, can cause severe disease (31). The rapid production of cytokines implies release from either preexisting memory T cells or cells of the innate immune system, such as antigen-presenting cells (APCs) or natural killer (NK) cells.
Among APCs, dendritic cells (DCs) are the most potent in initiating, controlling, and regulating functionally distinct T-cell responses (2, 3). Three signals are required from a DC to potently activate naive T cells, and they consist of high levels of peptide-loaded human leukocyte antigen (HLA) class II molecules, costimulatory molecules, and cytokine secretion.
For proper stimulation of T cells, upon encountering a pathogen in the periphery, DCs have to migrate to secondary lymphoid organs in order to encounter T cells (24). The migratory capacities of DCs are guided by the expression of chemokine receptors (CCRs), which in turn is regulated by DC maturation. In the periphery, the expression of CCR1 and CCR5 enables DCs to detect inflammation and migrate toward the causative agent. Upon maturation, those inflammatory CCRs are downregulated and, instead, lymphoid CCRs such as CXCR4 and CCR7, whose ligands are expressed in lymphatic tissues, are upregulated (2).
The P. falciparum parasite has been shown to affect DC responses through the action of the malaria parasite pigment hemozoin (Hz) or the synthetic analog β-hematin or through adhesion of P. falciparum-infected red blood cells (iRBCs). However, the data obtained so far are somewhat contradictory. Coban et al. reported activation of a proportion of human monocyte-derived DCs (MoDCs) upon Hz stimulation, as indicated by elevated expression of maturation markers and IL-12 secretion (5). On the other hand, Hz-loaded human monocytes do not differentiate into MoDCs, and Hz-loaded MoDCs exhibit an impaired maturation in response to lipopolysaccharide (LPS), suggesting that Hz could have an inhibitory role in the development of inflammatory DCs (36).
It has been shown that high doses of mature iRBCs inhibit LPS-induced maturation of human MoDCs and alter their immunostimulatory abilities (44). It was later suggested that this effect could have been induced by apoptosis of DCs upon contact with high doses of mature iRBCs (10). Another study has demonstrated that low doses of iRBCs induce semimaturation of human MoDCs and release of tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), and IL-10, but not IL-12, whereas free merozoites inhibited CD40L-induced maturation of MoDCs (27). Furthermore, P. falciparum induces incomplete maturation of other DC subsets, such as plasmacytoid DCs (pDCs). Parasite schizont extracts and iRBCs stimulate human pDCs to induce release of alpha interferon (IFN-α) but not TNF-α and to upregulate CCR7 and costimulatory molecules. Moreover, this effect was reproduced in murine pDCs and shown to be dependent on Toll-like receptor 9 (TLR9) (29).
In vivo, the proportion of circulating DCs is reduced in children with P. falciparum malaria (45). Recent evidence shows that a minor blood-myeloid-DC population, the blood-dendritic cell antigen (BDCA)-3+ DCs, is expanded in children with severe malaria compared to healthy controls and was associated with augmented IL-10 plasma levels and impaired DC allostimulatory ability, indicating an immunomodulatory function for this APC subset (43). Furthermore, analysis of the distribution of DCs in spleens of fatal P. falciparum malaria cases showed that interdigitating DCs are reduced in the white pulp of the spleen, whereas prion protein-expressing DCs are enriched in the red pulp and the marginal zone compared to findings for fatal cases of sepsis or controls (20). Therefore, despite contradictory in vitro results, analysis of DCs during natural P. falciparum infection suggests that a proportion of DCs are activated during infection and, importantly, exhibit an immunomodulatory phenotype compared to classical type 1 DC activation, which leads to a Th1-skewed T-cell response.
In the present study, the phenotype and function of human MoDCs were evaluated upon exposure to β-hematin, parasite lysates, and iRBCs. As a measure of function, the expression of CCRs and maturation markers, cytokine release, and the capacity of human MoDCs to migrate toward chemoattractants and to activate allogeneic T cells were investigated.
MATERIALS AND METHODS
Dendritic cell cultures.
Peripheral blood mononuclear cells (PBMCs) from anonymous blood donors (Karolinska Hospital, Stockholm, Sweden) were isolated on a density gradient by centrifugation using Ficoll-Paque (GE Healthcare, Uppsala, Sweden) and used for further cell purification. MoDCs were generated by modification of a described method (32). Briefly, monocytes were isolated by negative selection using MACS microbeads according to the manufacturer's instructions (monocyte isolation kit II; Miltenyi Biotech, Bergisch Gladbach, Germany). Purified monocytes were resuspended in complete RPMI 1640 supplemented with 10% fetal calf serum (FCS), 35 ng/ml IL-4, and 50 ng/ml granulocyte-macrophage colony-stimulating factor (R&D Systems, Minneapolis, MN). Cell differentiation was monitored by light microscopy and flow cytometry. At day 5, cells were harvested and 89% ± 1% of the cells exhibited a typical MoDC phenotype (CD14− CD1a+). Parasite lysate, β-hematin, or iRBCs were then added to differentiated MoDC cultures. In order to induce maturation of MoDCs, 1 μM prostaglandin E2 (PGE2) (R&D Systems) and 50 ng/ml of TNF-α (R&D Systems) were added to the culture medium at day 5, if not stated otherwise.
Preparation of β-hematin.
β-Hematin, also called synthetic Hz, is structurally similar to Hz formed naturally by parasites (37, 40). It has been suggested that the manner of preparation of β-hematin from hemin could affect the size and therefore the effect of the crystals. Coban et al. as well as other groups showed recently that β-hematin prepared from an acid-catalyzed reaction had an optimal adjuvant effect compared to that of a preparation based on an organic solution, and we used this protocol with minor modifications (4, 9, 41). Briefly, 75 mg of porcine hemin (Sigma-Aldrich, Steinheim, Netherlands) was dissolved in 14.5 ml NaOH (0.1 M) and incubated at 60°C. Within 10 min, 1.45 ml HCl (1 M) and 8.825 ml sodium acetate (12.8 M) were added and the solution was incubated for 2 h at 60°C with constant stirring at a rate of ∼150 rpm. The solution was then centrifuged at 15,000 × g for 13 min, and the pellet was resuspended in Tris-HCl buffer (100 mM, pH 8) containing 2.5% SDS (KEBO Lab, Stockholm, Sweden) and incubated for 30 min 37°C to dissolve heme aggregates. The solution was centrifuged at 15,000 × g for 13 min, and the pellet was resuspended in prewarmed NaHCO3 (100 mM, pH 9.2). After the suspension was washed twice, Hz was dried at 37°C. The resulting pellet was weighed and resuspended to 10 mg/ml in sterile water. In agreement with previous findings (8), the infrared (IR) spectrum of Hz showed intense bands at 1,710, 1,660, 1,299, 1,279, and 1,209 cm−1 (see Fig. S1 in the supplemental material). Two intense bands at 1,660 and 1,209 cm−1 were present in our preparation, indicating a high grade of purity of the sample (9). The endotoxin level in the preparation was below 0.05 endotoxin units (EU)/ml (Bacteriology Lab, Sahlgrenska Hospital, Göteborg, Sweden) in our highest working concentration. The highest working concentration used in this study was 20 μg of β-hematin/ml, which corresponds to 30 μM and has been calculated to be equivalent to the amount of hemozoin released into the bloodstream at the burst of schizonts when parasitemia is 0.5% (12).
Parasite cultures.
P. falciparum parasite strains were cultured using human O+ RBCs (Karolinska Hospital, Stockholm, Sweden) in RPMI 1640 medium containing 25 mM HEPES, 2 mM l-glutamine, 50 μg/ml gentamicin, and 0.2% sodium bicarbonate and supplemented with 0.5% Albumax (Gibco, Paisley, United Kingdom). The F32 Tanzanian laboratory strain used in this study adheres to CD36 but not to CD54 or chondroitin sulfate A (unpublished data). The parasites were maintained in continuous cultures in candle jars as previously described (14). Parasite cultures were set aside once a month and cultured separately without antibiotics for regular testing for Mycoplasma contamination by use of the mycoplasma detection kit Mycoalert (Lonza, Rockland ME). The parasite cultures were kept synchronous by treatment with 5% d-sorbitol in distilled water as previously described (17). In order to produce parasite lysates, late-stage parasites of the F32 strain were purified on a 60% Percoll gradient by centrifugation, sonicated as previously described (42), and stored at −80°C until used.
Intact iRBCs were harvested from cultures of late parasite stages (schizonts and late trophozoites) when the parasitemia reached approximately 10% and purified by using magnetic separation as described previously (48).
Briefly, parasite cultures were pelleted and resuspended in phosphate-buffered saline (PBS) supplemented with 2% bovine serum albumin (BSA) and 2 mM EDTA. Parasites were added to CS columns (Miltenyi Biotech) and incubated for 10 min before the column was extensively washed. The column was then removed from the magnet and parasites were rinsed out with PBS. The percentage of late-stage iRBCs was determined by staining an aliquot of purified late-stage parasites with acridine orange and counting under a UV microscope. Uninfected RBCs run in parallel under the same conditions were used as negative controls. Uninfected red blood cells and iRBCs were obtained from the same donors in each experiment.
MoDC-parasite cocultures.
In order to prevent burst of the infected erythrocytes during coculture, iRBCs were incubated in aphidicolin, which arrests the parasite cell cycle in the trophozoite stage (13). Briefly, synchronous parasite cultures at ring stage (8 to 10% parasitemia) were cultured in 15 μg/ml of aphidicolin (Sigma-Aldrich, St. Louis, MO) for a maximum of 24 h. When the parasites had reached the trophozoite stage, the cultures were purified, including extensive washing to remove any aphidicolin remnants, using magnetic separation as previously described (48). Immature MoDCs were harvested and resuspended at 106 cells/ml in 24-well plates. Purified trophozoites (90% ± 1% purity) were added at ratios of iRBCs/DCs of 10:1 and 100:1, or uninfected RBCs were added at the high ratio. On the basis of the assumption that each iRBC contains 47 femtograms of Hz (15), DCs were in contact with 4.7 μg/ml of Hz when incubated with iRBCs at the ratio of 100:1 (iRBC/DCs). MoDCs and uninfected or infected RBCs were cocultured for 10 h. TNF-α and PGE2 were added to separate DC cultures as a maturation control.
Immunophenotyping of MoDCs.
All antibodies used were mouse monoclonal antibodies either unconjugated or conjugated with fluorescein isothiocyanate (FITC) or phycoerythrin (PE). Conjugated antibodies used for cell surface staining included those recognizing CD1a, CD14, CD80, CD83, CD86, and HLA-DR (Pharmingen, San Diego, CA). Unconjugated antibodies were CCR7 and CXCR4 (R&D Systems), which were made detectable using a secondary polyclonal goat antimouse-PE conjugated antibody (Dako Cytomation, Dako A/S, Denmark). Data acquisition and analysis were performed on a FACSCalibur flow cytometer (Becton Dickinson, Mountain View, CA) using BD CellQuestPro (version 5.2.1) software. Fluorescence-activated cell sorter (FACS) analysis data are presented here as mean fluorescence intensity (MFI).
Migration assays.
Migration of MoDCs was evaluated using a 48-well Boyden chamber (Neuroprobe, Pleasanton, CA) with 5-μm-pore-size polycarbonate filters. Medium without FCS was used as a control of background migration. Cells were allowed to migrate for 1.5 h in response to either CXCL12–stromal cell-derived factor 1α (SDF-1α) or CCL19–macrophage inflammatory protein 3β (MIP-3β) (PeproTech Inc., Rocky Hill, NJ) at a final concentration of 100 ng/ml in RPMI 1640 medium containing 1% FCS. Thereafter, MoDCs adhering to the membrane were stained and counted. All samples were assayed in duplicate, and the number of cells that migrated in five visual fields (original magnification, ×100) was determined for each well, as previously described (38). The net number of cells that migrated was calculated by subtracting the number of cells that migrated in response to the medium alone from the number of cells that migrated in response to chemokines.
Cytokine measurement in supernatants of cell cultures.
Supernatants from MoDCs that were cultured in the presence or absence of parasite-derived products were collected and subsequently stored at −80°C. Supernatants were analyzed for the concentrations of IFN-γ, IL-1β, IL-2, IL-5, IL-6, IL-8, IL-10, IL-12p70, TNF-α, and tumor necrosis factor beta (TNF-β) using a human Th1/Th2 11-plex bead assay (Bender MedSystems, Vienna, Austria) according to the manufacturer's instructions. Data acquisition and analysis were performed by FACSCalibur and the FCAP Array software (Soft Flow, Hungary Ltd. Hungary).
MLR.
The ability of MoDCs stimulated with either β-hematin or iRBCs to induce T-cell proliferation was assessed in an allogeneic mixed leukocyte reaction (MLR). As responder cells, we used allogeneic PBMCs that were depleted of monocytes by CD14 negative selection (MACS microbeads; Miltenyi Biotech) and then stained with carboxyfluorescein diacetate succinimidyl ester (CFSE) according to the manufacturer's instructions (Invitrogen, OR). Cells were then resuspended in culture medium, and 105 cells/well were added to a U-bottom 96-well plate. MoDCs that were cocultured with infected or uninfected erythrocytes for 10 h as described above were isolated on a density gradient centrifugation using Ficoll-Paque (GE Healthcare, Uppsala, Sweden), washed, and resuspended in culture medium. MoDCs stimulated with 20 μg β-hematin were washed and resuspended in culture medium. Differently stimulated MoDCs were then added to responder cells at MoDC-to-PBMC ratios of 1:10, 1:30, 1:100, and 1:300. Cells were cocultured for 5 days before being harvested and stained for subsequent analysis. Monoclonal antibodies directed against CD3 and CD4 (Pharmingen, San Diego, CA) that were conjugated with peridinin chlorophyll protein (PerCP) and allophycocyanin (APC), respectively, were used to stain the cells for subsequent FACS analysis. Data acquisition and analysis were performed on a FACSCalibur flow cytometer (Becton Dickinson, Mountain View, CA) using BD CellQuestPro (version 5.2.1) software. MoDC-stimulated CD4+ CD3+ cells that expressed less CFSE than CD4+ CD3+ cells in unstimulated PBMCs were gated as proliferating lymphocytes.
Statistical analysis.
Data were analyzed using the Wilcoxon rank-sum test. Page's trend test is a variant of the nonparametric Friedman's test and was used to analyze the dose-response effects of β-hematin on MoDCs by employing increasing doses of this compound. Comparisons were considered statistically significant at P values of ≤0.05.
RESULTS
β-Hematin induces partial maturation of MoDCs and increases lymphoid CCR expression.
In order to evaluate the effect of β-hematin on the phenotype of MoDCs, cells were stimulated with β-hematin or TNF-α–PGE2 overnight. MoDCs cultured in medium alone were used as a negative control. As expected, cells that were stimulated with TNF-α–PGE2 showed a statistically significant increased expression of CD80, CD83, HLA-DR, CCR7, and CXCR4 (Fig. 1).
Fig. 1.

β-Hematin induces partial maturation of MoDCs and causes upregulation of lymphoid CCR expression. MoDCs were incubated with or without β-hematin (10 or 20 μg/ml), and TNF-α–PGE2 was added or not as a maturation stimulus in designated wells. After 18 h of incubation, cells were harvested, stained for surface markers, and analyzed by FACS analysis. The y axis reports MFIs for CD80, CD83, HLA-DR, CXCR4, and CCR7. The box plots illustrate the medians and the 25th and 75th quartiles, and the whiskers represent minimum and maximum values for 5 donors. Data were analyzed using Wilcoxon's rank sum test. *, P < 0.05; **; P < 0.01.
In order to examine what amount of β-hematin could affect MoDCs, we incubated MoDCs with increasing doses of this compound. The results indicate a clear dose-response relation for the activation markers CD80, CD83, and HLA-DR (see Fig. S2 in the supplemental material), and the most potent effect was observed for the highest dose of 20 μg/ml.
After stimulation with β-hematin, we observed increased expression of CD80 on MoDCs (P = 0.046 and P = 0.018 for 10 and 20 μg/ml β-hematin, respectively). Unstimulated cells expressed low levels of CD83, and expression of CD83 on MoDCs increased significantly in the presence of 10 μg/ml β-hematin (P = 0.043) but not 20 μg/ml β-hematin. TNF-α–PGE2 strongly increased the expression of HLA-DR and CCR7 molecules, whereas the expression levels of these markers were only slightly, but not significantly, increased upon β-hematin stimulation (Fig. 1). Conversely, β-hematin induced significantly increased expression of CXCR4 compared to unstimulated MoDCs when it was added at the concentration of 20 μg/ml (P = 0.028), and this increased expression was comparable to the upregulation of CXCR4 induced by TNF-α–PGE2 (Fig. 1). A similar increase in CXCR4 expression on MoDCs was observed in the presence of 10 μg/ml β-hematin, although this was not significant (P = 0.079). Thus, β-hematin induces partial maturation of MoDCs and increases the expression of the lymphoid chemokine receptor CXCR4 on the cell surface.
To evaluate the kinetics of MoDC activation upon β-hematin or TNF-α–PGE2 stimulation, we performed a phenotypic analysis at 12, 36, and 60 h after incubation with these stimuli. Given our initial results, the kinetics of expression of activation markers on MoDCs was analyzed only after stimulation with 20 μg/ml of β-hematin. At first, we evaluated the data without taking into consideration the time points, thus confirming the partial activation described above. The baseline expression of CD80 was 56 ± 3 MFI (mean value ± standard error), and expression increased to 85 ± 4 MFI and to 96 ± 4 MFI when cells were stimulated with β-hematin and TNF-α–PGE2, respectively. For CD83, the background expression was 21 ± 4 MFI, and expression increased to 35 ± 6 MFI and to 68 ± 6 MFI when cells were stimulated with β-hematin and TNF-α–PGE2, respectively. HLA-DR expression increased from 75 ± 3 to 91 ± 2 MFI upon β-hematin stimulation and to 100 ± 1 MFI using TNF-α–PGE2.
When we examined the kinetics of cell activation, increased expression of CD80 and HLA-DR on MoDCs was maintained after 60 h of stimulation regardless of the type of stimulus (data not shown). For CD83, maximum expression was observed 36 h after addition of the stimulus, but expression levels dropped close to baseline over the 60-h period in response to β-hematin (Fig. 2). Statistical analysis showed that the decrease in CD83 expression on MoDCs was significant for unstimulated cells (P = 0.021) and cells stimulated with β-hematin between the 12- and 60-h time points (P = 0.043) (Fig. 2). When the cells were stimulated with TNF-α–PGE2, however, no statistically significant difference was observed for CD83 expression levels (P = 0.19) between the same time points. Thus, the partial activation of MoDCs by β-hematin resulted in stably increased expression of CD80 and HLA-DR, while expression of CD83 was transient, as the levels were not maintained after 60 h of incubation.
Fig. 2.

Kinetics of CD83 expression levels on MoDCs upon β-hematin stimulation. MoDCs (1 × 106 cells/ml) were left unstimulated or were stimulated with β-hematin (20 μg/ml) or the maturation stimulus (TNF-α–PGE2) and harvested after 12, 36, or 60 h of incubation. Cells were stained for the maturation marker CD83 (n = 4 donors) and analyzed by FACS analysis. Data were analyzed using Wilcoxon's rank sum test. *, P < 0.05; NS, not significant.
β-Hematin impairs the upregulation of CCR7 induced by maturation stimuli on MoDCs.
In addition to the transient maturation of MoDCs with β-hematin, we investigated whether β-hematin could interfere with their maturation induced by TNF-α–PGE2. As shown in Fig. 3, addition of β-hematin to the cultures prior to TNF-α–PGE2 stimulation significantly impaired the upregulation of CCR7 (P = 0.042 and P = 0.017 for 10 μg and 20 μg of β-hematin, respectively). No differences were observed for the expression levels of CD80, CD83, HLA-DR, and CXCR4 on MoDCs matured with TNF-α–PGE2 in the presence or absence of β-hematin (data not shown). Thus, β-hematin selectively blocks CCR7 upregulation induced by TNF-α–PGE2 on MoDCs.
Fig. 3.
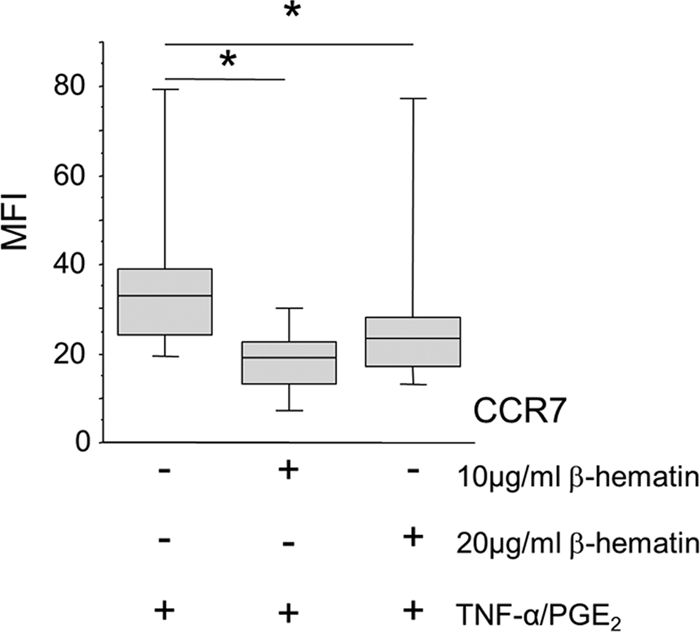
β-Hematin impairs upregulation of CCR7 upon DC maturation. MoDCs were incubated with or without β-hematin (10 or 20 μg/ml; n = 5 and n = 8 donors examined, respectively) for 4 h and then treated with maturation stimuli (TNF-α–PGE2). Cells were incubated for an additional 18 h, harvested, stained for surface markers, and analyzed by FACS analysis. The y axis indicates MFI for CCR7. The box plots illustrate the medians and the 25th and 75th quartiles, and the whiskers represent the minimum and maximum values. Data were analyzed using Wilcoxon's rank sum test. *, P < 0.05.
Contact with iRBCs induces upregulation of the lymphoid chemokine receptor CXCR4.
Next, we examined whether parasite lysates or intact iRBCs could induce transient maturation of MoDCs. MoDCs from four donors were incubated for 18 h with 15 or 75 μg/ml of parasite lysate. The cells were subsequently harvested and stained for surface expression of CD80, CD83, HLA-DR, CXCR4, and CCR7 and analyzed by FACS analysis. No significant differences in the investigated markers were observed upon contact with parasite lysates compared to medium (data not shown).
To evaluate whether P. falciparum antigens expressed on iRBCs could have an effect on MoDCs, cells were incubated with aphidicolin-treated iRBCs, (F32 laboratory strain) or uninfected RBCs for 10 h and then stained for CD83, HLA-DR, CCR7, and CXCR4. Aphidicolin arrests parasite development at the trophozoite stage, and therefore, we were able to analyze the effect of intact iRBCs on MoDC maturation independent of the effect of free merozoites (12).
The expression levels of the investigated markers did not differ significantly among MoDCs cultured in medium alone or with uninfected RBCs. In addition, no signs of maturation were observed by incubating MoDCs with low ratios of infected RBCs (1:1, 5:1, or 10:1 iRBC/MoDCs; data not shown). Nevertheless, we observed marginal upregulation of CD83, HLA-DR, and CCR7 from MFIs of 17 ± 4 (mean value ± standard error), 81 ± 5, and 49 ± 6, respectively, in the unstimulated cultures to MFIs of 24 ± 5, 91 ± 6, and 60 ± 11, respectively, upon coincubation with a high ratio of iRBCs to MoDCs (100:1), although the differences were not statistically significant (Fig. 4). Interestingly, the expression of CXCR4 was significantly increased from 78 ± 7 MFI in unstimulated cells to 93 ± 7 MFI when MoDCs were stimulated with high doses of iRBCs (P = 0.043) and to 106 ± 11 MFI when they were stimulated with TNF-α–PGE2 (P = 0.028).
Fig. 4.

Incubation with iRBCs induces upregulation of CXCR4 expression. MoDCs were cocultured with uninfected RBCs (Ctrl RBC) or iRBCs at a ratio of 100:1 (RBCs/DCs). Maturation stimulus (TNF-α–PGE2) was added in separate wells as a positive control for maturation. Cells were incubated for 10 h, harvested, stained for surface markers, and analyzed by FACS analysis. The MFIs of HLA-DR, CD83, CXCR4, and CCR7 are presented. The box plots illustrate the medians and the 25th and 75th quartiles, and the whiskers represent the minimum and maximum values of five donors. Data were analyzed using Wilcoxon's rank sum test. *, P < 0.05.
A separate experiment was performed to assess the impact of aphidicolin-treated RBCs on MoDCs. No significant difference in the expression of the maturation markers CD80, CD83, and HLA-DR was observed in MoDCs incubated with either untreated RBCs and aphidicolin-treated RBCs (data not shown), indicating that aphidicolin does not affect maturation of MoDCs.
Thus, high doses of iRBCs seem to induce marginal upregulation of HLA-DR and costimulatory molecules on MoDCs but a significant increase in expression of the lymphoid-chemokine receptor CXCR4. Importantly, intact parasitized erythrocytes are required to induce this effect, since parasite lysates did not induce any maturation of DCs.
β-Hematin-induced maturation of MoDCs is coupled to increased migratory capacity in response to lymphoid chemokines.
Upon maturation, MoDCs acquire the ability to migrate in response to lymphoid chemokines by upregulating the expression of CXCR4 and CCR7 molecules (47). Since we observed that β-hematin induced significant upregulation of CXCR4 and marginally increased levels of CCR7, the capacity of β-hematin-treated MoDCs to migrate in response to lymphoid chemokines was assessed.
MoDCs treated with TNF-α–PGE2 exhibited a significantly increased migratory capacity in response to the CXCR4 ligand CXCL12/SDF-1α (P = 0.03) and the CCR7 ligand CCL19/MIP-3β (P = 0.04) compared to untreated cells (Fig. 5). Significantly increased migration toward CXCL12/SDF-1α (P = 0.03) as well as CCL19/MIP-3β (P = 0.04) was also observed in β-hematin-treated MoDCs compared to untreated cells, indicating that the increased expression of CXCR4 and CCR7 on β-hematin-treated MoDCs is functional.
Fig. 5.
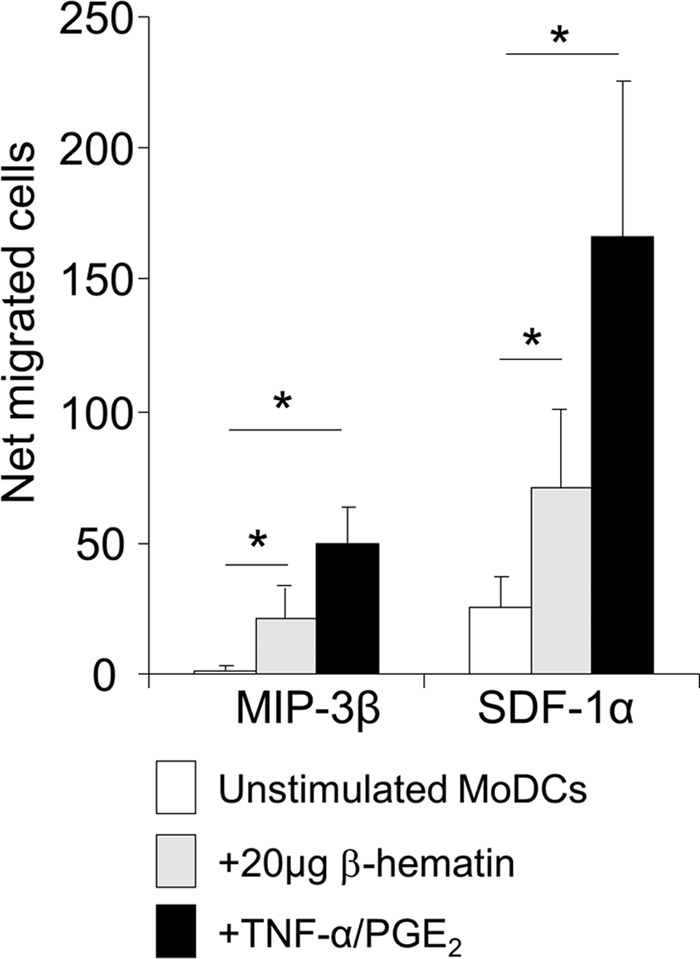
Partial maturation of MoDCs induced by β-hematin is coupled to increased migration in response to lymphoid chemokines. MoDCs were stimulated with β-hematin (20 μg/ml) or the maturation stimulus (TNF-α–PGE2) and incubated for 18 h. Cells were then harvested and seeded in the upper compartments of a Boyden chamber, and 100 ng/ml of CXCL12/SDF-1α or CCL19/MIP-3β was added to the lower compartments. Culture medium alone was used as a control for background migration. Cells were incubated for 90 min to allow migration, after which the cells that had migrated to the lower compartment were counted in 5 representative fields for each well. The net number of cells that migrated was calculated by subtracting the number of cells that migrated in response to medium alone from the number of cells that migrated in response to chemokines. The diagram represents mean values ± standard error of the mean of cells that migrated in response to CXCL12/SDF-1α (n = 6 donors) and to CCL19/MIP-3β (n = 5 donors). Data were analyzed using Wilcoxon's rank sum test. *, P < 0.05.
MoDCs exhibit increased secretion of pro- and anti-inflammatory cytokines after stimulation with β-hematin or high concentrations of iRBCs.
To investigate whether malaria parasite-derived stimuli could induce the production of soluble mediators in MoDCs, the concentration of selected cytokines was measured in the cell culture supernatant obtained from β-hematin-treated, iRBC-treated, and untreated MoDCs. Baseline production of all inflammatory cytokines except IL-8 in unstimulated MoDCs was negligible, as shown in Fig. 6 A and B. In agreement with published data (21), the concentration of all examined cytokines was unaffected upon stimulation with TNF-α–PGE2 (data not shown). In contrast, MoDCs stimulated with 20 μg/ml of β-hematin released significantly higher levels of IL-6 (P = 0.009) and IL-10 (P = 0.021) than unstimulated cells (Fig. 6A). The secretion of IL-6 was also increased when MoDCs were stimulated with 10 μg/ml of β-hematin (P = 0.05). We noted a slight increase in TNF-α secretion in β-hematin-stimulated cultures compared to unstimulated controls, but this did not reach statistical significance (P = 0.174). No differences in IL-1β, IL-8, or IL-12p70 secretion were found between unstimulated cultures and β-hematin-stimulated cultures.
Fig. 6.

Increased secretion of pro- and anti- inflammatory cytokines by MoDCs upon stimulation with high concentrations of β-hematin and iRBCs. MoDCs were left unstimulated or incubated with 10 or 20 μg/ml of β-hematin, and cell culture supernatants were subsequently analyzed for cytokine levels (A). MoDCs were incubated with uninfected RBCs (Ctrl RBCs) or with a high (MoDCs + iRBCs, 1:100) or a low (MoDCs + iRBCs, 1:10) concentration of iRBCs (B). Cell culture supernatants were collected and subsequently analyzed for cytokine levels. Each graph illustrates the mean values and standard error of the mean of cytokine concentration (n = 4 donors). Data were analyzed using Wilcoxon's rank sum test. *, P < 0.05.
Next, we analyzed cytokine release by MoDCs after stimulation with iRBCs. At the low iRBC/MoDC (10:1) ratio, no significant increment in the release of IL-1β, IL-6, IL-8, IL-10, IL-12p70, or TNF-α was observed (Fig. 6B). However, using a high iRBC/MoDC (100:1) ratio, there was a significantly increased secretion of IL-1β (P = 0.021), IL-6 (P = 0.021), IL-10 (P = 0.021), and TNF-α (P = 0.043) by MoDCs. Of note, the secretion of IL-12p70 was very low upon iRBC stimulation in all donors examined.
When the levels of cytokines secreted by DCs upon stimulation with β-hematin and iRBCs were compared, the same cytokines except for IL-1β were elevated with both stimuli; IL-1β was not increased upon β-hematin stimulation. The levels of IL-6, IL-10, and TNF-α were considerably higher when MoDCs were stimulated with iRBCs than with β-hematin. Thus, both β-hematin and iRBCs induce secretion of a number of pro- and anti-inflammatory cytokines in MoDCs but not IL-12 secretion.
iRBC-treated but not β-hematin-treated MoDCs induce increased T-cell proliferation.
To investigate whether P. falciparum-stimulated MoDCs could induce T-cell proliferation at early time points, we measured proliferation of allogeneic T cells in response to different doses of DCs cocultured with either 20 μg of β-hematin or a high number of iRBCs (1:100). MoDCs cocultured with β-hematin induced T-cell proliferation comparable to that induced by MoDCs that were unstimulated or cocultured with uninfected RBCs, in line with their only partial phenotypic maturation (Fig. 7). In contrast, MoDCs that had been cocultured with a high dose of intact iRBCs activated allogeneic T cells to a similar degree as TNF-α–PGE2-treated MoDCs, despite their partial phenotypic maturation, suggesting a difference in the mode of action between MoDCs cocultured with β-hematin and iRBCs.
Fig. 7.

iRBC- but not β-hematin-treated MoDCs induce increased T-cell proliferation. MoDCs were cocultured with medium alone (Ctrl DCs), 100:1 uninfected RBCs (Ctrl RBCs), 20 μg β-hematin (β-hematin), 100:1 iRBCs, or maturation stimulus (TNF-α–PGE2). MoDCs where then put in cocultures with CFSE-stained, allogeneic PBMCs for 5 days. After incubation, the percentage of CD4+ CD3+ cells that had proliferated were gated and are presented on the y axis, while the different ratios of MoDC/T cells are presented on the x axis. The graph illustrates mean values of separate experiments from a minimum of 6 donors. Error bars depict the standard error of the mean.
DISCUSSION
In this study, we show that β-hematin treatment of human MoDCs induces a significant upregulation of CXCR4 as well as a slight upregulation of CCR7. The increased levels of expression of lymphoid CCRs on MoDCs upon β-hematin stimulation were coupled with significantly increased migration toward the ligands for CXCR4 and CCR7, CXCL12/SDF-1α, and CCL19/MIP-3β. In addition, exposure of MoDCs to high doses of iRBCs resulted in a similar pattern of upregulation of these lymphoid CCRs.
Both CXCR4 and CCR7 have been found to be upregulated in association with maturation of MoDCs (33). Upregulation of these receptors enables DCs to migrate to T-cell-rich areas in secondary lymphoid organs and screen massive numbers of naive T cells (31). Therefore, our findings suggest that β-hematin increases migration of DCs to lymphoid organs. In contrast, current evidence from rodent models of malaria shows that the time of interaction between T cells and DCs is shortened, both in vitro and in vivo, when DCs had been exposed to Hz (26). Together these results suggest that increased migration of DCs toward lymphoid-chemokine gradients upon stimulation with Hz may be coupled to an improper DC-T cell interaction.
We also detected a significant upregulation of some but not all costimulatory markers when MoDCs were stimulated with β-hematin. It is unlikely that this effect is mediated by TLR9 since recent evidence suggests that neither β-hematin (28) nor parasite-derived Hz (50) activates murine myeloid DCs via TLR9 and TLR9 is not expressed on human myeloid DCs (35). Conversely, β-hematin activation of myeloid DCs may act through the Nalp3 inflammasome, in accordance with previous findings in murine macrophages (7, 11). In this context, it should be noted that our study was not designed to assess such aspects.
A kinetic study revealed that increased expression of CD80 and HLA-DR was maintained on MoDCs after 60 h of stimulation with β-hematin or TNF-α–PGE2. However, although the expression of CD83 was maintained for 60 h upon stimulation with TNF-α–PGE2, this was not true for MoDCs activated by β-hematin. Other studies have shown that CD83 has a profound effect on DC maturation (16) and that modulation of CD83 can interfere with DC-T cell interactions (19). Thus, our data indicate that β-hematin-induced DC maturation, reflected by CD83 expression, appears to be transient and not as potent as the one induced by TNF-α–PGE2.
In contrast, exposure to iRBCs resulted only in upregulation of chemokine receptor expression, whereas upregulation of HLA-DR and costimulatory molecules was only marginal and did not reach statistical significance. We (B. C. Urban and colleagues) and others have shown that iRBCs alone did not induce upregulation of HLA-DR and costimulatory molecules on MoDCs (10, 44), but the expression levels of chemokine receptors were not investigated. These results are therefore in agreement, particularly when taking into account the fact that the marginal upregulation of HLA-DR and CD83 that we report here was observed after 10 h of coculture, as opposed to the 48 to 68 h coculture of MoDCs and iRBCs in earlier studies (10, 44).
In addition, we observed increased secretion of pro- and anti-inflammatory cytokines by MoDCs incubated with P. falciparum-derived stimuli within 24 h of incubation and increased immunostimulatory abilities of MoDCs after 10 h coculture with iRBCs, whereas others found reduced allostimulation and no cytokine release by MoDCs upon stimulation with iRBCs for a longer time (10). However, consistent in all reports is the lack of IL-12 secretion by MoDCs stimulated with any P. falciparum-derived product.
Of note, production of proinflammatory cytokines was much higher in MoDCs cocultured with iRBCs than those cocultured with β-hematin, suggesting that iRBCs contain additional factors that drive secretion of inflammatory cytokines such as P. falciparum glycosylphosphatidylinositol (6). The high concentrations of IL-1β, IL-6, and TNF-α secreted within 10 h by MoDCs cocultured with iRBCs could contribute to their ability to induce proliferation of allogeneic T cells.
Together, our results suggest that parasite products can induce partial and transient phenotypic maturation of DCs at early time points but not IL-12 release. This is coupled to increased DC immunostimulatory ability upon contact with intact iRBCs but not with β-hematin. How these phenomena can influence adaptive immune responses to the parasite can only be speculated. Partial and possibly transient DC maturation associated with a lack of IL-12 secretion and high levels of IL-10 is a phenomenon that could support a blunted adaptive immune response.
Importantly, we report here the sustained upregulation of chemokine receptors resulting in migration of MoDCs toward lymphokine gradients. These observations are in line with findings by Millington et al. (25, 26), who observed a significant decrease of mature DCs in vivo by day 12 in mice infected with the parasite P. chabaudi chabaudi and no or only marginal maturation of rodent DCs in response to iRBCs or Hz. Nevertheless, an increased migration of myeloid DCs into the spleen was observed during P. chabaudii chabaudi infection (39), and in particular, migration from the marginal zone of the spleen into the CD4+ T-cell area was observed within 5 days after parasites entered the bloodstream (20). In this context, DCs induce activation of T cells but fail to sustain the prolonged contact necessary for T-cell proliferation and differentiation (26).
Our in vitro observations of the effect of β-hematin and to a lesser extent iRBCs on human DCs suggest that a similar mechanism may apply in humans; accumulation of malaria parasite pigment in myeloid DCs could induce migration of DCs into T-cell areas of secondary lymphoid organs due to increased expression of lymphoid chemokine receptors, followed by increased stimulation of T-cell proliferation at early time points. However, these phenomena are coupled with a lack of sustained upregulation of CD83 and a lack of IL-12 secretion but high levels of IL-10, which may result in altered T-cell differentiation.
We also observed that β-hematin selectively impaired the upregulation of CCR7 induced by TNF-α–PGE2 stimulation. Recent evidence indicates that, apart from chemotaxis, CCR7 controls the cytoarchitecture, the migratory speed, and the maturation rate of DCs (34). Therefore, impairing the CCR7 upregulation on maturing MoDCs may be an effective tactic for the malaria parasite to block DC functionality, thus hampering the initiation of the T-cell responses, as seen in other parasitic infections (1).
It is well established that malaria parasites induce a large inflammatory response during acute infection, and a correlation between high levels of certain cytokines and severe complications during malaria parasite infection has been found (22, 23, 30). Our findings of rapid release of pro- and anti-inflammatory mediators upon short-term stimulation with P. falciparum-derived stimuli imply that myeloid DCs may make a significant contribution to the large inflammatory response that is observed during acute P. falciparum infection.
In conclusion, we found that contact of MoDCs with β-hematin or iRBCs induced partial and transient maturation of these cells, provoked the release of pro- and anti-inflammatory cytokines other than IL-12, and upregulated lymphoid CCR on the cell surface. This was coupled to increased migration to lymphoid ligands when cells were stimulated with β-hematin and increased proliferation of allogeneic T cells upon stimulation with iRBCs but not β-hematin.
We therefore believe that myeloid DCs become activated at least early on during P. falciparum infection. However, the complex nature of the parasite may lead to a transient and suboptimal maturation process but increased migration to secondary lymphoid organs and subsequently impaired activation of lymphocytes. Once careful studies with different parasite strains and their products have established how DCs are affected by the parasite, it may become feasible to manipulate DCs during malaria parasite infection to induce more potent adaptive immunity.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the Swedish Agency for Research and Development with Developing Countries (SIDA/SAREC; to M.T.-B. and S.V.), as well as a grant within the BioMalPar European Network of Excellence (LSHP-CT-2004-503578; to M.T.-B.) and the European Community's Seventh Framework Programme (FP7/2007-2013) under grant agreement 242095. B.C.U. is a Wellcome Trust Senior Fellow in Biomedical Science (grant number 079082).
Footnotes
Supplemental material for this article may be found at http://iai.asm.org/.
Published ahead of print on 4 April 2011.
REFERENCES
- 1. Ato M., Stager S., Engwerda C. R., Kaye P. M. 2002. Defective CCR7 expression on dendritic cells contributes to the development of visceral leishmaniasis. Nat. Immunol. 3:1185–1191 [DOI] [PubMed] [Google Scholar]
- 2. Banchereau J., et al. 2000. Immunobiology of dendritic cells. Annu. Rev. Immunol. 18:767–811 [DOI] [PubMed] [Google Scholar]
- 3. Banchereau J., Steinman R. M. 1998. Dendritic cells and the control of immunity. Nature 392:245–252 [DOI] [PubMed] [Google Scholar]
- 4. Coban C., et al. 2010. Immunogenicity of whole-parasite vaccines against Plasmodium falciparum involves malarial hemozoin and host TLR9. Cell Host Microbe 7:50–61 [DOI] [PubMed] [Google Scholar]
- 5. Coban C., Ishii K. J., Sullivan D. J., Kumar N. 2002. Purified malaria pigment (hemozoin) enhances dendritic cell maturation and modulates the isotype of antibodies induced by a DNA vaccine. Infect. Immun. 70:3939–3943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Corrigan R. A., Rowe J. A. 2010. Strain variation in early innate cytokine induction by Plasmodium falciparum. Parasite Immunol. 32:512–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dostert C., et al. 2009. Malarial hemozoin is a Nalp3 inflammasome activating danger signal. PLoS One 4:e6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Egan T. J., Hempelmann E., Mavuso W. W. 1999. Characterisation of synthetic beta-haematin and effects of the antimalarial drugs quinidine, halofantrine, desbutylhalofantrine and mefloquine on its formation. J. Inorg. Biochem. 73:101–107 [DOI] [PubMed] [Google Scholar]
- 9. Egan T. J., Mavuso W. W., Ncokazi K. K. 2001. The mechanism of beta-hematin formation in acetate solution. Parallels between hemozoin formation and biomineralization processes. Biochemistry 40:204–213 [DOI] [PubMed] [Google Scholar]
- 10. Elliott S. R., et al. 2007. Inhibition of dendritic cell maturation by malaria is dose dependent and does not require Plasmodium falciparum erythrocyte membrane protein 1. Infect. Immun. 75:3621–3632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Griffith J. W., Sun T., McIntosh M. T., Bucala R. 2009. Pure hemozoin is inflammatory in vivo and activates the NALP3 inflammasome via release of uric acid. J. Immunol. 183:5208–5220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hanscheid T., Egan T. J., Grobusch M. P. 2007. Haemozoin: from melatonin pigment to drug target, diagnostic tool, and immune modulator. Lancet Infect. Dis. 7:675–685 [DOI] [PubMed] [Google Scholar]
- 13. Inselburg J., Banyal H. S. 1984. Plasmodium falciparum: synchronization of asexual development with aphidicolin, a DNA synthesis inhibitor. Exp. Parasitol. 57:48–54 [DOI] [PubMed] [Google Scholar]
- 14. Jensen J. B., Trager W. 1978. Plasmodium falciparum in culture: establishment of additional strains. Am. J. Trop. Med. Hyg. 27:743–746 [DOI] [PubMed] [Google Scholar]
- 15. Keller C. C., et al. 2004. Elevated nitric oxide production in children with malarial anemia: hemozoin-induced nitric oxide synthase type 2 transcripts and nitric oxide in blood mononuclear cells. Infect. Immun. 72:4868–4873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kotzor N., Lechmann M., Zinser E., Steinkasserer A. 2004. The soluble form of CD83 dramatically changes the cytoskeleton of dendritic cells. Immunobiology 209:129–140 [DOI] [PubMed] [Google Scholar]
- 17. Lambros C., Vanderberg J. P. 1979. Synchronization of Plasmodium falciparum erythrocytic stages in culture. J. Parasitol. 65:418–420 [PubMed] [Google Scholar]
- 18. Langhorne J., Ndungu F. M., Sponaas A. M., Marsh K. 2008. Immunity to malaria: more questions than answers. Nat. Immunol. 9:725–732 [DOI] [PubMed] [Google Scholar]
- 19. Lechmann M., et al. 2001. The extracellular domain of CD83 inhibits dendritic cell-mediated T cell stimulation and binds to a ligand on dendritic cells. J. Exp. Med. 194:1813–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Leisewitz A. L., et al. 2004. Response of the splenic dendritic cell population to malaria infection. Infect. Immun. 72:4233–4239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Luft T., et al. 2002. Functionally distinct dendritic cell (DC) populations induced by physiologic stimuli: prostaglandin E(2) regulates the migratory capacity of specific DC subsets. Blood 100:1362–1372 [DOI] [PubMed] [Google Scholar]
- 22. Luty A. J., et al. 2000. Low interleukin-12 activity in severe Plasmodium falciparum malaria. Infect. Immun. 68:3909–3915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lyke K. E., et al. 2004. Serum levels of the proinflammatory cytokines interleukin-1 beta (IL-1beta), IL-6, IL-8, IL-10, tumor necrosis factor alpha, and IL-12(p70) in Malian children with severe Plasmodium falciparum malaria and matched uncomplicated malaria or healthy controls. Infect. Immun. 72:5630–5637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mellman I., Steinman R. M. 2001. Dendritic cells: specialized and regulated antigen processing machines. Cell 106:255–258 [DOI] [PubMed] [Google Scholar]
- 25. Millington O. R., Di Lorenzo C., Phillips R. S., Garside P., Brewer J. M. 2006. Suppression of adaptive immunity to heterologous antigens during Plasmodium infection through hemozoin-induced failure of dendritic cell function. J. Biol. 5:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Millington O. R., et al. 2007. Malaria impairs T cell clustering and immune priming despite normal signal 1 from dendritic cells. PLoS Pathog. 3:1380–1387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mukherjee P., Chauhan V. S. 2008. Plasmodium falciparum-free merozoites and infected RBCs distinctly affect soluble CD40 ligand-mediated maturation of immature monocyte-derived dendritic cells. J. Leukoc. Biol. 84:244–254 [DOI] [PubMed] [Google Scholar]
- 28. Parroche P., et al. 2007. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc. Natl. Acad. Sci. U. S. A. 104:1919–1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pichyangkul S., et al. 2004. Malaria blood stage parasites activate human plasmacytoid dendritic cells and murine dendritic cells through a Toll-like receptor 9-dependent pathway. J. Immunol. 172:4926–4933 [DOI] [PubMed] [Google Scholar]
- 30. Prakash D., et al. 2006. Clusters of cytokines determine malaria severity in Plasmodium falciparum-infected patients from endemic areas of Central India. J. Infect. Dis. 194:198–207 [DOI] [PubMed] [Google Scholar]
- 31. Riley E. M., Wahl S., Perkins D. J., Schofield L. 2006. Regulating immunity to malaria. Parasite Immunol. 28:35–49 [DOI] [PubMed] [Google Scholar]
- 32. Sallusto F., Lanzavecchia A. 1994. Efficient presentation of soluble antigen by cultured human dendritic cells is maintained by granulocyte/macrophage colony-stimulating factor plus interleukin 4 and downregulated by tumor necrosis factor alpha. J. Exp. Med. 179:1109–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sallusto F., et al. 1998. Rapid and coordinated switch in chemokine receptor expression during dendritic cell maturation. Eur. J. Immunol. 28:2760–2769 [DOI] [PubMed] [Google Scholar]
- 34. Sanchez-Sanchez N., Riol-Blanco L., Rodriguez-Fernandez J. L. 2006. The multiple personalities of the chemokine receptor CCR7 in dendritic cells. J. Immunol. 176:5153–5159 [DOI] [PubMed] [Google Scholar]
- 35. Schreibelt G., et al. 2010. Toll-like receptor expression and function in human dendritic cell subsets: implications for dendritic cell-based anti-cancer immunotherapy. Cancer Immunol. Immunother. 59:1573–1582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Skorokhod O. A., Alessio M., Mordmuller B., Arese P., Schwarzer E. 2004. Hemozoin (malarial pigment) inhibits differentiation and maturation of human monocyte-derived dendritic cells: a peroxisome proliferator-activated receptor-gamma-mediated effect. J. Immunol. 173:4066–4074 [DOI] [PubMed] [Google Scholar]
- 37. Slater A. F., et al. 1991. An iron-carboxylate bond links the heme units of malaria pigment. Proc. Natl. Acad. Sci. U. S. A. 88:325–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sozzani S., et al. 1997. Receptor expression and responsiveness of human dendritic cells to a defined set of CC and CXC chemokines. J. Immunol. 159:1993–2000 [PubMed] [Google Scholar]
- 39. Sponaas A. M., et al. 2006. Malaria infection changes the ability of splenic dendritic cell populations to stimulate antigen-specific T cells. J. Exp. Med. 203:1427–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sullivan D. J., Jr., Gluzman I. Y., Goldberg D. E. 1996. Plasmodium hemozoin formation mediated by histidine-rich proteins. Science 271:219–222 [DOI] [PubMed] [Google Scholar]
- 41. Tripathi A. K., Khan S. I., Walker L. A., Tekwani B. L. 2004. Spectrophotometric determination of de novo hemozoin/beta-hematin formation in an in vitro assay. Anal. Biochem. 325:85–91 [DOI] [PubMed] [Google Scholar]
- 42. Troye-Blomberg M., Perlmann H., Patarroyo M. E., Perlmann P. 1983. Regulation of the immune response in Plasmodium falciparum malaria. II. Antigen specific proliferative responses in vitro. Clin. Exp. Immunol. 53:345–353 [PMC free article] [PubMed] [Google Scholar]
- 43. Urban B. C., et al. 2006. The frequency of BDCA3-positive dendritic cells is increased in the peripheral circulation of Kenyan children with severe malaria. Infect. Immun. 74:6700–6706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Urban B. C., et al. 1999. Plasmodium falciparum-infected erythrocytes modulate the maturation of dendritic cells. Nature 400:73–77 [DOI] [PubMed] [Google Scholar]
- 45. Urban B. C., et al. 2001. Peripheral blood dendritic cells in children with acute Plasmodium falciparum malaria. Blood 98:2859–2861 [DOI] [PubMed] [Google Scholar]
- 46. Urban B. C., Roberts D. J. 2003. Inhibition of T cell function during malaria: implications for immunology and vaccinology. J. Exp. Med. 197:137–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. van Helden S. F., et al. 2006. A critical role for prostaglandin E2 in podosome dissolution and induction of high-speed migration during dendritic cell maturation. J. Immunol. 177:1567–1574 [DOI] [PubMed] [Google Scholar]
- 48. Vogt A. 2008. Selection of trophozoites by using magnetic cell sorting (MACS), p. 31–33In Moll K., Ljungström I., Perlmann H., Scherf A., Wahlgren M.(ed.), Methods in malaria research, 5th ed. MR4/ATCC, Manassas, VA [Google Scholar]
- 49. World Health Organization 2008. World malaria report 2008. World Health Organization, Geneva, Switzerland [Google Scholar]
- 50. Wu X., Gowda N. M., Kumar S., Gowda D. C. 2010. Protein-DNA complex is the exclusive malaria parasite component that activates dendritic cells and triggers innate immune responses. J. Immunol. 184:4338–4348 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.