

Abstract
Neutrophil accumulation is a critical event to clear bacteria. Since uncontrolled neutrophil recruitment can cause severe lung damage, understanding neutrophil trafficking mechanisms is important to attenuate neutrophil-mediated damage. While monocyte chemoattractant protein 1 (MCP-1) is known to be a monocyte chemoattractant, its role in pulmonary neutrophil-mediated host defense against Gram-negative bacterial infection is not understood. We hypothesized that MCP-1/chemokine (C-C motif) ligand 2 is important for neutrophil-mediated host defense. Reduced bacterial clearance in the lungs was observed in MCP-1−/− mice following Escherichia coli infection. Neutrophil influx, along with cytokines/chemokines, leukotriene B4 (LTB4), and vascular cell adhesion molecule 1 levels in the lungs, was reduced in MCP-1−/− mice after infection. E. coli-induced activation of NF-κB and mitogen-activated protein kinases in the lung was also reduced in MCP-1−/− mice. Administration of intratracheal recombinant MCP-1 (rMCP-1) to MCP-1−/− mice induced pulmonary neutrophil influx and cytokine/chemokine responses in the presence or absence of E. coli infection. Our in vitro migration experiment demonstrates MCP-1-mediated neutrophil chemotaxis. Notably, chemokine receptor 2 is expressed on lung and blood neutrophils, which are increased upon E. coli infection. Furthermore, our findings show that neutrophil depletion impairs E. coli clearance and that exogenous rMCP-1 after infection improves bacterial clearance in the lungs. Overall, these new findings demonstrate that E. coli-induced MCP-1 causes neutrophil recruitment directly via chemotaxis as well as indirectly via modulation of keratinocyte cell-derived chemokine, macrophage inflammatory protein 2, and LTB4.
INTRODUCTION
Bacterial pneumonia is a leading cause of death in both immunocompromised and immunocompetent individuals (12, 25, 33). Successful host defense against bacterial infection in the lung is dependent on effective recruitment of phagocytes into the alveolar space. During cellular recruitment, neutrophil infiltration is the first event, followed by monocyte/macrophage accumulation at the site of infection. Several lines of evidence suggest that cytokines/chemokines regulate leukocyte migration in concert with cell adhesion molecules, including intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1) (1, 11, 32). In this context, monocyte chemoattractant protein 1 (MCP-1/chemokine [C-C motif] ligand 2 [CCL2]) has been shown to be a potent monocyte/macrophage and T cell chemoattractant against bacteria in lung infection (10, 31, 34, 35, 39, 49). MCP-1 is produced by a variety of cells, including epithelial cells (41), endothelial cells, fibroblasts (42), and monocytes/macrophages (48), and it binds to the G protein-coupled transmembrane receptor chemokine receptor 2 (CCR2).
Prior studies have unequivocally demonstrated the important role of MCP-1 in monocyte/macrophage-mediated host defense against bacterial infection. During Streptococcus pneumoniae infection, overexpression of MCP-1 was shown to improve bacterial clearance (46). Using MCP-1−/− mice, Winter et al. (45) have demonstrated that MCP-1-mediated macrophage recruitment is important to prevent bacterial dissemination following pulmonary Streptococcus pneumoniae infection. During Pseudomonas aeruginosa infection, MCP-1 promoted resolution and repair of the lung by enhancing the uptake of apoptotic neutrophils by alveolar macrophages (2). In addition, mice deficient in CCR2 showed impairment in macrophage migration and clearance of bacteria from the lungs and extrapulmonary organs after intravenous challenge with Listeria monocytogenes (24). MCP-1 is also shown to be important for the survival of mice during P. aeruginosa and Salmonella enterica serotype Typhimurium infections, and MCP-1 promotes bacterial killing by macrophages (35).
MCP-1 is further implicated in cellular trafficking to the inflamed lung via multiple mechanisms. MCP-1 modulates the expression of β2 integrin and thereby triggers firm monocyte adhesion to the inflamed endothelium (28). Studies have shown that CCR2 plays an essential role in the induction of adaptive arms of the immune system via the induction of Th1 cytokines (10). However, the role of MCP-1 in neutrophil recruitment is debatable, and the role of MCP-1 in neutrophil recruitment during Escherichia coli infection of the lungs has not been studied. In one study, it has been shown that administration of exogenous MCP-1 alone did not cause neutrophil influx into the lungs, whereas administration of MCP-1 along with lipopolysaccharide (LPS) did cause excessive neutrophil recruitment (29). In another investigation, it has been demonstrated that neutrophils express CCR2 and show chemotaxis toward MCP-1 (21). However, the role of MCP-1 in neutrophil trafficking and activation in the lung in acute bacterial pneumonia has not been studied.
The objective of the current investigation was to determine the role of MCP-1 in neutrophil-mediated host defense following E. coli infection. In this regard, we used MCP-1 gene-deficient (MCP-1−/−) mice. Our findings demonstrate that MCP-1 is induced upon E. coli infection and can be produced by both myeloid and nonmyeloid cells. MCP-1 contributes to bacterial clearance in the lungs via neutrophil recruitment in a murine model. Our findings indicate that MCP-1 causes neutrophil recruitment to the lungs directly via chemotaxis as well as indirectly via modulation of the levels of keratinocyte cell-derived chemokine (KC) and macrophage inflammatory protein 2 (MIP-2) following E. coli infection. Furthermore, we show that neutrophils from blood and bronchoalveolar fluid (BALF) express CCR2 and its level is increased upon E. coli infection.
MATERIALS AND METHODS
Mice.
Eight- to 10-week-old female mice genetically deficient in MCP-1 (16) or myeloid differentiation protein-2 (MD-2) (9) were used, while age- and gender-matched C57BL/6 mice were used as controls. Animal studies were approved by the Louisiana State University and National Jewish Health Animal Care and Use Committees. The mice ranged from 19 to 25 g in weight.
Infection model.
Bacteria were prepared for mouse inoculation, as described in previous studies (8, 22). E. coli (American Type Culture Collection [ATCC] 25922) was grown in Trypticase soy broth (TSB) at 37°C overnight under constant agitation. Bacteria were harvested, washed, and resuspended in sterile 0.9% saline at a concentration of 20 × 106 CFU/ml. Mouse strains were anesthetized with intraperitoneal ketamine-xylazine (250 mg/kg), followed by intratracheal (i.t.) inoculation of 50 μl of bacteria (106 CFU/mouse), whereas control mice were i.t. inoculated with 50 μl of saline. The initial mouse inocula were confirmed by plating serial 10-fold dilutions on MacConkey agar and tryptic soy agar (TSA) plates. For enumerating bacterial CFU in the lung and spleen, whole lungs and spleens were homogenized in 2 ml sterile saline for 30 s, and 20 μl of the resulting homogenates was plated by serial 10-fold dilutions on MacConkey agar and TSA plates. In a similar manner, spleens were homogenized for 15 s for bacterial culture. Bacterial colonies were counted after incubation overnight at 37°C. For CFU studies, we used a higher dose of E. coli (5 × 106 CFU/50 μl/mouse) since a low dose (106 CFU/mouse) did not induce substantial mortality either in MCP-1−/− or in wild-type (WT) mice.
Lung pathology.
The lungs of WT and MCP-1−/− mice were perfused from the right ventricle of the heart with 10 ml isotonic saline at 24 h postinfection. Lungs were then removed and fixed in 4% phosphate-buffered formalin. Fixed tissue samples were processed in paraffin blocks, and 5-μm sections were cut with a microtome and stained with hematoxylin-eosin (H&E). Analysis of histopathology was performed in blinded fashion by a veterinary pathologist according to the following scoring scale: 0, no inflammatory cells (macrophages or neutrophils) present in section; 1, <5% of section is infiltrated by inflammatory cells; 2, 5 to 10% of section is infiltrated by inflammatory cells; and 3, >10% of section is infiltrated by inflammatory cells.
BALF collection.
BALF was collected, and total and differential cell counts and cytokine/chemokine levels were determined. Approximately 3 ml of lavage fluid was retrieved per mouse. Total leukocytes in BALF were determined using a hemocytometer. Cytospin samples were subsequently prepared from BALF cells and stained with Diff-Quick (Fisher). Differential cell counts were determined by direct counting of stained slides. For examination of cytokines/chemokines, the remainder (2 ml) of the undiluted cell-free BALF was passed via a 0.22-μm-pore-size filter and used immediately or stored at −80°C.
Bone marrow transplantation.
Bone marrow chimeras were generated as described in our earlier publications (7, 9). Recipient mice were irradiated with gamma rays from a cesium source in two 525-rad doses 3 h apart. Bone marrow cells (8 × 106/mouse) from donor mice were injected into the tail vein of the irradiated recipients. Mice that had undergone transplantation were maintained on 0.2% neomycin sulfate for the first 2 weeks. The reconstituted mice were used 2 months after the transplantation. In parallel experiments, we used green fluorescent protein-expressing donor cells. Sample blood was collected from these recipients at between 6 and 8 weeks after transplantation, and hematological parameters (differential counts) were assessed. Greater than 90% of blood leukocytes were derived from donor mice at the time that the mice were used for experiments (8 weeks after transplantation; data not shown). Irradiated mice that were not transplanted with donor cells died between days 20 and 22 after transplantation (data not shown).
Cytokine, chemokine, and LTB4 determination.
We used BALF and lungs that were obtained from animals after E. coli infection or saline instillation. Enzyme-linked immunosorbent assay (ELISA) kits for tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6), leukotriene B4 (LTB4), and MCP-1 were obtained from eBiosciences, PA, whereas kits for KC and MIP-2 were obtained from R&D Systems, MN. The minimum detection limit is 8 pg/ml cytokine protein, whereas the detection limit for LTB4 is 13.7 pg/mg/ml.
Western blotting.
The lungs were collected at the designated time points and used for immunoblotting as described in our previous publications (8). The primary antibodies (Abs) to phospho-NF-κB/p65(Ser536), NF-κB/p65, phospho-IKKα/β (Ser176/180), IKKβ, phospho-IκBα (Ser32), IκBα, VCAM-1, ICAM-1, phospho-p44/42 mitogen-activated protein kinase (MAPK; extracellular signal-regulated kinase 1 [ERK1]/ERK2) (Thr202/Tyr204), phospho-p38 MAPK (Thr180/Tyr182), phospho-Jun N-terminal protein kinase (phospho-JNK; Thr183/Tyr185), p47phox, p67phox, and inhalational nitric oxides were obtained from Cell Signaling, Boston, MA, and were added at a 1:1,000 dilution. The primary Abs to total p38 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; Santa Cruz Biotechnology, CA) were added at a 1:5,000 dilution. Immunostaining was performed using appropriate secondary Ab at a dilution of 1:2,000 and developed with an enhanced chemiluminescence plus Western blot detection system (Amersham Pharmacia Biotech, Piscataway, NJ). To demonstrate equal protein loading on gels, the blots were stripped and reprobed with Ab specific for total p38, pan-cadherin, or GAPDH.
NF-κB DNA binding assay.
Nuclear proteins were extracted from 50 to 80 mg lung tissue collected at 6 and 24 h after E. coli or saline administration. A total of 7.5 μg nuclear extract was mixed with binding buffer, and the mixture was added to the precoated plate (with the DNA binding motif of NF-κB) and incubated for 1 h at room temperature according to the manufacturer's protocol (TransAM ELISA kit; Active Motiff, Carlsbad, CA). Wells were then washed, and plates were incubated with NF-κB/p65 antibody for 1 h. Plates were then washed three times, horseradish peroxidase-conjugated anti-rabbit IgG was added to each well, and plates were incubated for 1 h. Plates were read at 450 nm after addition of the developing reagent. This method has been described in our earlier publications (8, 9, 22).
Neutrophil transmigration assay.
Chemotaxis assays in a Transwell system were performed using 24-well tissue culture plates (polycarbonate membrane polystyrene plates; 3415; Costar; Corning Inc., Corning, NY) with a pore size of 3.0 μm. Chemoattractants, either KC (2 μg/ml, 1 μg/ml) or MCP-1 (2 μg/ml) (R&D, MN), and phosphate-buffered saline (PBS) supplemented with bovine serum albumin (BSA; 2 μg/ml) was added to each of the lower wells (final volume, 500 μl). A 100-μl suspension of 1 × 106 LPS-activated polymorphonuclear leukocytes (PMNs) in Dulbecco modified Eagle medium (DMEM)-0.1% BSA was added to each well of the upper filter plate. After incubation at 5% CO2 at 37°C for 3 h, the upper plate was removed and cells from 20 nonoverlapping fields in the lower plate were counted by using an inverted microscope (18, 19).
Examination of CCR2 expression on neutrophils.
A total of 50 μl of whole blood or BALF of WT and MCP-1−/− mice treated with saline or E. coli was aliquoted into flow cytometry tubes, and 0.10 μl of Fc-blocked mouse-conjugated anti-mouse Gr-1, CCR2, and CXCR-2 (R&D, MN) antibodies was added to appropriate tubes. Samples were vortexed and incubated for 30 min at room temperature in the dark. Then, cells were washed by adding 2 ml of 1× PBS, and the mixture was centrifuged at 1,000 rpm (200 × g) for 8 min. Erythrocytes were lysed by adding 2 ml of NH4Cl lysing buffer to each sample tube, the contents were mixed well, and the tube was incubated at room temperature for 10 min. Then, samples were centrifuged immediately at 1,000 rpm (200 × g) for 8 min, and the supernatant was removed. Cells were washed twice with PBS. Cells were fixed by adding 200 μl of cold 1% formaldehyde-PBS and stored at 1 to 6°C for fluorescence-activated cell sorter analysis.
Neutrophil depletion.
The protocol used to deplete neutrophils (Gr1 positive) in mice has been described in our earlier reports, with minor modifications (7). A total of 50 μg anti-Gr1/Ly6G monoclonal antibody (MAb; sodium azide free and low LPS content; 1A8; BD Pharmingen, San Diego, CA) in 250 μl was administered intraperitoneally at 12 and 2 h before bacterial infection. In control experiments, 50 μg of isotype control MAb in equal volumes was administered at the same time points prior to bacterial infection. To validate the efficiency of neutrophil depletion by anti-Gr1/Ly6G MAb, we obtained neutrophil counts in blood every 12 h up to 3 days, and ≤3% neutrophils were found up to 3 days after depletion.
AM culture.
Alveolar macrophages (AMs) were isolated as described in earlier publications (7). Positive selection by CD11b MicroBeads (Stem Cell Technologies, Vancouver, British Columbia, Canada) was applied to isolate AMs from BALF cells. Pooled BALF cells were resuspended in RoboSep buffer, and CD11b phycoerythrin (PE) labeling reagent was mixed with cell suspension, followed by incubation at room temperature for 15 min. EasySep PE selection mixture was thereafter added to the cell suspension, and the suspension was further incubated for 15 min at room temperature. Lastly, EasySep magnetic nanoparticles were mixed with the cell suspension, followed by magnetic separation of CD11b+ cells. Cells were washed with PBS, centrifuged at 1,200 rpm for 10 min, and resuspended in DMEM containing 5% fetal bovine serum. Cells were infected with E. coli (multiplicity of infection, 1) and harvested at designated time points using urea-3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate-Tris buffer. Samples were centrifuged, and supernatants were used for Western blotting. Finally, the total number of live cells and the percentage of purified AMs were determined by trypan blue exclusion and Wright- Giemsa staining of cytospin preparations, respectively. Percent recovery of AMs was calculated as the ratio of the number of AMs to the total number of macrophages isolated from the BALF. Using this technique, we obtained 86 to 92% AMs (data not shown).
Statistical analysis.
Data are expressed as means ± standard errors. The intensity of immunoreactive bands was determined using gel digitizing software (UN-SCAN-IT gel) from Silk Scientific, Inc., UT. Data were analyzed by analysis of variance, followed by Bonferroni's post hoc analysis for multiple comparisons. All statistical calculations were performed using InStat software and GraphPad Prism (version 4.0) software. Differences were considered statistically significant at P values of <0.05 compared with the control.
RESULTS
MCP-1 is essential for bacterial clearance following E. coli infection.
We used an experimental model of pulmonary E. coli infection to demonstrate the MCP-1 protein expression in BALF and lung homogenates. MCP-1 levels in the BALF (Fig. 1A) and the lungs (Fig. 1B) of WT (C57BL/6) mice were increased at 6 and 24 h compared to those in the saline-challenged animals after i.t. E. coli (106/mouse) infection. In order to determine the cell type that can produce MCP-1 during E. coli infection, we used chimeric MD-2−/− mice infected with E. coli at 24 h postinfection. We found that both myeloid and nonmyeloid cell-derived MD-2 is important to produce MCP-1 following E. coli infection (Fig. 1C). To determine whether deficiency in MCP-1 compromised host defense against E. coli in the lung, mice were infected i.t. with E. coli (1 × 106 CFU/mouse) and killed at 6 and 24 h postinfection. The lungs and spleens were isolated to determine the numbers of bacterial CFU. MCP-1−/− mice had greater numbers of CFU in the lung at 6 and 24 h postinfection (Fig. 2A). However, we did not observe bacterial dissemination in the bloodstream in either MCP-1−/− mice or their controls at 6 or 24 h (data not shown). Since we observed higher bacterial burdens in MCP-1−/− mice both at 6 and at 24 h following the regular dose of E. coli (1 × 106/mouse) infection (Fig. 2A), we chose to use the regular dose in the rest of our experiments.
Fig. 1.
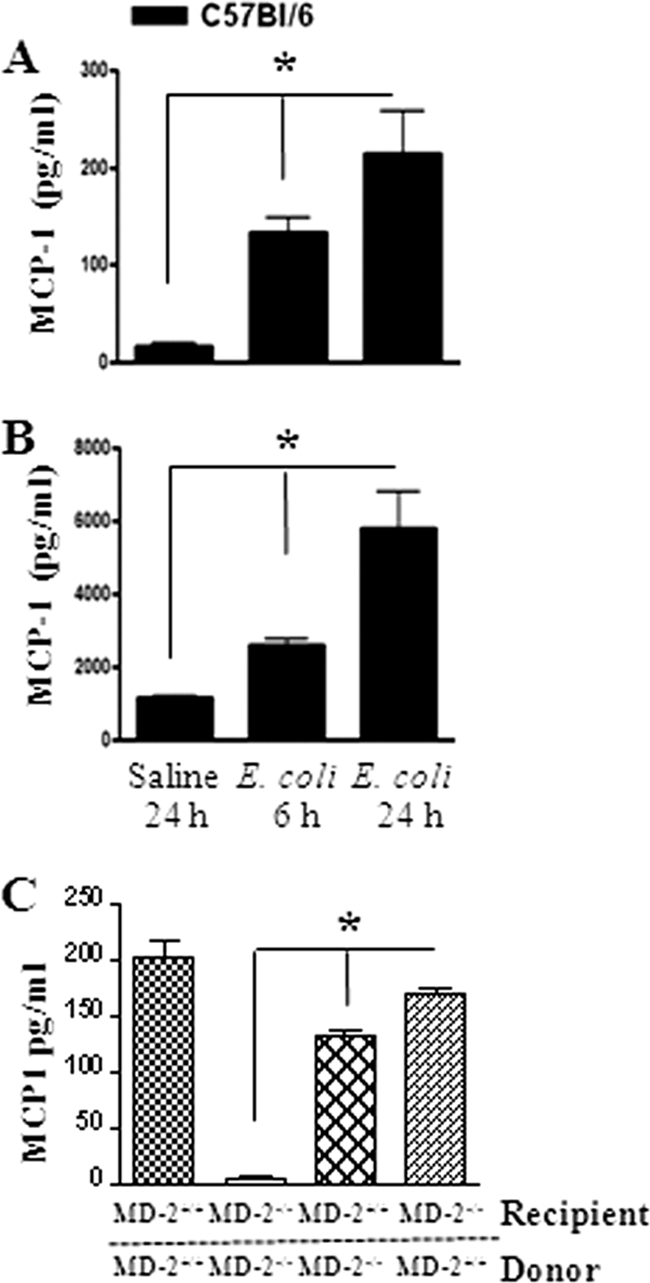
(A and B) Kinetics of MCP-1 production. The MCP-1 concentration was measured in BALF (A) and lung homogenate (B) from WT mice by ELISA after i.t. infection with E. coli (106/mouse). *, significant difference between MCP-1−/− and WT mice (P < 0.005; n = 4 to 5 mice/group/time point; the figure is a representation of 3 individual experiments). (C) MCP-1 expression in mice transplanted with bone marrow following E. coli infection. *, significant difference between knockout and WT mice (P < 0.005). Data shown are a representation of 3 individual experiments.
Fig. 2.

(A) Bacterial burden in the lungs of MCP-1−/− mice following E. coli infection (106/mouse). Lungs were collected from control and infected groups of mice at the designated times and homogenized, and the bacteria were enumerated (n = 5 to 6 mice/group). *, significant difference between knockout and WT mice (P < 0.005). Data shown are a representation of 3 individual experiments. (B and C) Cellular infiltration in the lung in MCP-1−/− mice against E. coli. Mice were inoculated with E. coli (106 CFU/mouse), BALF was obtained at 6 and 24 h postinfection, and cell enumeration was performed to determine neutrophil and macrophage infiltration to the lung (n = 4 to 5 mice/group; P < 0.005; data are a representation of 3 individual experiments). (D) Lung histology in MCP-1−/− mice following E. coli infection. Mice were inoculated with E. coli (106 CFU/mouse), and lungs were obtained at 24 h postinfection. This picture is representative of 3 separate mice with identical results.
MCP-1−/− mice show attenuated cellular recruitment to the lung following E. coli infection.
Leukocyte recruitment to the site of infection is a critical step to clear bacteria. Therefore, we determined the total as well as the differential leukocyte count in BALF at 6 and 24 h postinfection with the regular dose (1 × 106 CFU/mice) (Fig. 2B and C). We observed reduced leukocyte influx at 24 h postinfection in MCP-1−/− mice, and this reduction was primarily due to reduced neutrophil influx, although macrophage numbers were reduced at this time (Fig. 2B and C). Furthermore, attenuated leukocyte numbers and alveolar edema were noted in lung histology of MCP-1−/− mice at 24 h; i.e., lungs from WT mice infected with E. coli show a score of 2 (average of 3 mice), whereas lungs from MCP-1−/− mice infected with bacteria show a score of 1 (average of 3 mice) (Fig. 2D). These findings show that MCP-1 is important for the neutrophil accumulation, in addition to macrophage influx, during E. coli infection.
MCP-1 regulates the production of cytokines/chemokines and expression of cell adhesion molecules.
Leukocyte recruitment during infection is a multistep process in which cytokine/chemokine production along with upregulation of cell adhesion molecules plays an important role. Hence, we used BALF to determine cytokine/chemokine levels following E. coli infection. Our data demonstrate that levels of cytokines, such as TNF-α and IL-6 (Fig. 3A and B), and neutrophil chemoattractants, such as KC and MIP-2 (Fig. 3C and D), were reduced at 24 h following E. coli infection in MCP-1−/− mice. We also examined the expression of the neutrophil chemotactic lipid LTB4 in the lungs of MCP-1−/− mice following E. coli administration and found that the levels of LTB4 were decreased in MCP-1−/− mice at 6 h but not at 24 h (Fig. 3E). Although ICAM-1 expression in the lung remained unchanged, VCAM-1 expression was substantially reduced in MCP-1−/− mice at 24 h after infection with E. coli (Fig. 3F to H). Our data suggest that MCP-1 controls the expression of cytokines, neutrophil chemoattractants, and cellular adhesion molecules.
Fig. 3.

(A to E) Cytokine, chemokine, and chemotactic lipid levels in the lung following E. coli infection. Mice were infected by intratracheal instillation of E. coli (106 CFU/mouse), and BALF was collected from the lungs at designated time points. Concentrations (pg/ml) of TNF-α (A), IL-6 (B), KC (C), and MIP-2 (D) in BALF were quantified by sandwich ELISA. *, significant difference between MCP-1−/− and WT mice (P < 0.005; n = 4 to 6 mice in each group at each time point; data are a representation of 3 separate experiments). (E) Expression of LTB4 in the lung following E. coli infection. Lung homogenates were prepared after E. coli infection, and the levels of LTB4 were measured in homogenates and were normalized against total protein concentration. (F) Expression of ICAM-1 and VCAM-1 in the lung in response to E. coli challenge. Infected lungs were homogenized, and total proteins were isolated, resolved on an SDS-polyacrylamide gel, and transferred onto a nitrocellulose membrane. The membrane was blotted with Abs for ICAM-1, VCAM-1, and GAPDH. This is a representative blot of 3 independent experiments with identical results. (G and H) Densitometric analysis was performed in 3 blots to demonstrate the expression of ICAM-1 and VCAM-1 in the lung following E. coli infection.
MCP-1 activates NF-κB and MAPKs in the lung following E. coli infection.
NF-κB and MAPKs regulate the expression of multiple proinflammatory genes in the inflammatory setting. We therefore investigated the activation of NF-κB and MAPKs in the lungs after E. coli infection. Our findings demonstrate that NF-κB activation was reduced in MCP-1−/− mice at 24 h postinfection (Fig. 4A to G). In addition, MCP-1−/− mice infected with E. coli showed reduced activation of p38 kinase and ERK at 24 h (Fig. 5A and B), whereas JNK activation was reduced at an earlier time point (6 h) in MCP-1−/− mice (Fig. 5A and B).
Fig. 4.

(A and B) Activation of NF-κB in the lung following infection with E. coli. Lung homogenates and nuclear lysates from MCP-1−/− mice and their controls were prepared at 6 and 24 h after infection with E. coli. NF-κB binding assay was performed in nuclear lysates from lung (A), and expression and phosphorylation of NF-κB pathway members were determined using Western blots of lung homogenates (B). The blots are representative of 3 independent experiments with identical results. (C to G) Relative densities normalized against GAPDH are representatives of 3 independent experiments. OD, optical density; encircled P, phosphorylated form; *, P < 0.005.
Fig. 5.
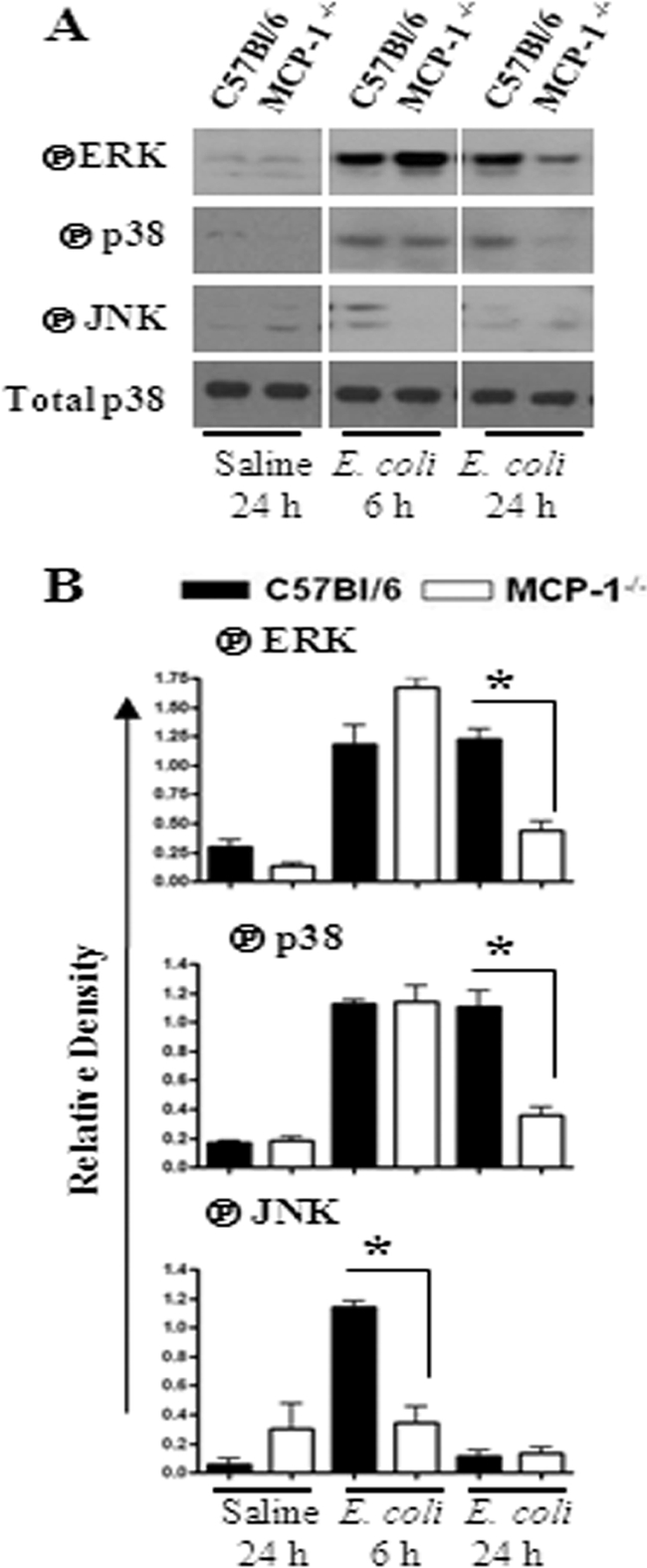
(A) Activation of MAPKs in the lung following E. coli infection. Total proteins in the lung were isolated from MCP-1−/− and control mice at 6 and 24 h after infection with E. coli and resolved on an SDS-polyacrylamide gel, and the membrane was blotted with the Abs against the activated/phosphorylated (encircled P) form of MAPKs as described in Materials and Methods. This is a representative of 3 separate experiments with identical results. (B) Densitometric analysis of MAPK activation was performed from 3 separate blots. *, P < 0.05 for difference between MCP-1−/− mice and their WT controls.
rMCP-1 induces neutrophil influx and cytokine/chemokine expression in the lungs of MCP-1−/− mice.
Since MCP-1 deficiency causes attenuated neutrophil influx, we determined whether reconstituting MCP-1−/− mice with exogenous rMCP-1 prior to E. coli infection can cause neutrophil accumulation and cytokine/chemokine expression in the lungs. In this regard, MCP-1−/− and WT mice were i.t. infected with E. coli at 1 h after i.t. rMCP-1 (10 μg/mouse) administration. At 24 h after infection, the neutrophil count was increased in BALF of MCP-1−/− mice which received rMCP-1 (Fig. 6A and B). Furthermore, MCP-1−/− mice reconstituted with rMCP-1 showed increased expression of cytokines (TNF-α and IL-6; Fig. 6C and D) and chemokines (KC and MIP-2; Fig. 6E and F) in BALF after E. coli infection. Moreover, activation of NF-κB and MAPKs and expression of VCAM-1 were increased in the lungs of MCP−/− mice after reconstitution with rMCP-1 (Fig. 7A to C).
Fig. 6.

(A and B) Cellular infiltration in BALF at 24 h after i.t. treatment with rMCP-1 (50 μg) and E. coli infection. Fifty micrograms of rMCP-1 was i.t. administered at 1 h after E. coli infection, and BALF was collected 24 h after infection. (C to F) Cytokine (C and D) and chemokine (E and F) production in the lungs of infected animals after rMCP-1 treatment. For the experiments indicated in panels A to F, n = 4 to 6 mice/group and data are representative of 3 experimental repeats. WBC, white blood cells; SAL, saline. (G) Numbers of CFU in lungs of WT and MCP-1−/− mice administered rMCP-1 following i.t. E. coli infection. The controls were treated with PBS. Each group had 5 to 6 mice, and the result shown is representative of 3 independent experiments. (H) Numbers of CFU in lungs of neutrophil-depleted mice at 24 h after E. coli infection. Neutrophils were depleted by using anti-Ly6G Ab intraperitoneally at 12 and 2 h prior to infection, and control mice were treated with isotype Ab prior to infection. Lung samples were homogenized, diluted, and plated to enumerate bacterial CFU (n = 5 to 6 mice/group/time point, and data are from 3 separate experiments). *, P < 0.05.
Fig. 7.

(A) Activation of NF-κB and MAPKs, and expression of cellular adhesion molecules in lungs of mice administered rMCP-1 (10 μg) at 24 h after E. coli infection. (B and C) Densitometric analysis of expression/phosphorylation levels of identified proteins normalized with GAPDH. The results are representative of 3 independent experiments with identical results. *, P < 0.05; encircled P, phosphorylated form.
Since our findings show that rMCP-1 induced cytokine/chemokine secretion and neutrophil influx into the lungs, we wanted to determine whether rMCP-1 treatment affects bacterial clearance from the lungs. We reconstituted MCP-1−/− mice with rMCP-1 prior to infection, and the lung CFU were enumerated at 24 h postinfection. MCP-1−/− mice treated with rMCP-1 show reduced numbers of CFU in the lung that were comparable to those in wild-type mice (Fig. 6G). These observations indicate that rMCP-1 improves E. coli clearance in the lungs. In order to determine whether neutrophils are important for bacterial clearance, we depleted neutrophils in C57BL/6 (WT) mice and challenged them with E. coli. We observed substantial increases in bacterial burden in the lungs of neutrophil-depleted mice compared to control mice (Fig. 6H). These findings illustrate the importance of neutrophils in E. coli clearance from the lungs.
As we found that rMCP-1 causes neutrophil recruitment to the lungs of MCP-1−/− mice following infection, we determined whether administration of exogenous rMCP-1 in MCP-1−/− mice can cause neutrophil recruitment in the absence of E. coli infection. In this regard, it has been shown that i.t. administration of 50 μg rMCP-1 in BALB/c mice did not cause any neutrophil influx (29). In our experiments, 2 doses (10 μg or 50 μg/mouse) of rMCP-1 via the i.t. route were used in C57BL/6 mice, and we found that the neutrophil count was increased in BALF and lungs (Fig. 8A) of the rMCP-1-treated mice in a concentration-dependent manner.
Fig. 8.

(A) Cellular recruitment into the alveoli after i.t. treatment with rMCP-1 (10 μg and 50 μg) alone. After 12 h, BALF was processed for cellular count (n = 5 to 6 mice/group; *, P < 0.005; data are a representation of 3 individual experiments). MPO, myeloperoxidase. (B) CCR2 expression in neutrophils. Flow cytometric analysis of BALF and blood from WT mice at 24 h after i.t. E. coli (1 × 106 CFU/mouse) infection. Data are representative of 3 independent experiments with identical results. (D) Picture demonstrating the number of PMNs in the lower chamber of a transwell plate after incubation with chemoattractants rMCP-1 and KC. The figure is representative of 20 random fields from 3 separate experiments. (E) Chemotaxis of neutrophils toward MCP-1. PMN numbers in the lower chamber of a transwell after 3 h of incubation with rMCP-1 and KC. Data shown here are a representation of 3 individual experiments (n = 3 to 5; *, P < 0.05). (F) Bone marrow neutrophils produce MCP-1 at 2 h after E. coli infection (n = 4 to 6 mice/group from 3 separate experiments).
Neutrophils migrate toward rMCP-1 in vitro.
Since rMCP-1 can rescue neutrophil accumulation in MCP-1−/− mice in the presence or absence of E. coli infection, we examined whether this is due to the direct chemotactic effect of MCP-1. In this regard, we first used flow cytometry to determine whether neutrophils express CCR2. For these experiments, BALF and blood obtained from WT mice after E. coli infection were used, since chemokines produced in the lungs after infection can induce neutrophil release from the bone marrow to blood. In this regard, we used dual staining of Gr-1-Ly6G-fluorescein isothiocyanate-CCR2-allophycocyanin to enumerate CCR2 expression on neutrophils. We first gated neutrophils on the basis of their size and granularity and then determined the double-positive cell population from the gated population of neutrophils. Notably, neutrophils in BALF and blood express CCR2, and this expression is enhanced during infection (Fig. 8B). In addition, bone marrow neutrophils also express CCR2, and it is induced upon E. coli infection (Fig. 8C).
Since we observed CCR2 expression on neutrophils, we examined whether these neutrophils show chemotaxis toward MCP-1. In this context, a transwell transmigration assay was used in which we used 1 × 106 LPS-activated neutrophils in DMEM-0.1% BSA. KC was used as a positive control at two concentrations (500 ng and 1,000 ng), whereas MCP-1 was used at 1,000 ng. Neutrophils show chemotaxis toward MCP-1 (Fig. 8D and E), although the number of cells that show chemotaxis toward MCP-1 is much less than the number of cells that show chemotaxis toward KC at the same concentration. Interestingly, neutrophils that are challenged with E. coli produce substantial MCP-1 at 2 h postinfection (Fig. 8F). These findings demonstrate that neutrophils produce MCP-1 and respond to MCP-1.
Macrophages obtained from MCP-1−/− mice show reduced NF-κB and MAPK activation.
In order to confirm the activation of NF-κB and MAPKs in isolated lung cells, we used alveolar macrophages obtained from WT and MCP-1−/− mice. Macrophages were infected with E. coli, and samples were collected at 2 and 6 h for Western blotting. We found reduced NF-κB and MAPK (JNK, p38, and ERK) activation in MCP-1−/− mice following infection (Fig. 9A and B).
Fig. 9.

(A and B) Activation of NF-κB and MAPKs in alveolar macrophages obtained from WT and MCP-1−/− mice following infection with E. coli. Representative Western blots from 3 separate experiments are shown. (B) Relative densities normalized against GAPDH are representatives of 3 independent experiments (n = 4 to 5 mice/group; *, P < 0.05). Encircled P, phosphorylated form.
DISCUSSION
In the present investigation, we sought to determine the importance of MCP-1 in an experimental model of bacterial pneumonia and in the human system. We found that MCP-1 expression is substantially increased following intratracheal E. coli infection and that MCP-1 is important for bacterial clearance during E. coli infection of the lungs. In addition, we also show that MCP-1 is being produced by both myeloid and nonmyeloid cells following E. coli infection. Collectively, our study demonstrates that MCP-1 directly (via chemotaxis) and indirectly (via modulation of KC and MIP-2) contributes to neutrophil recruitment during E. coli pneumonia (Fig. 10).
Fig. 10.
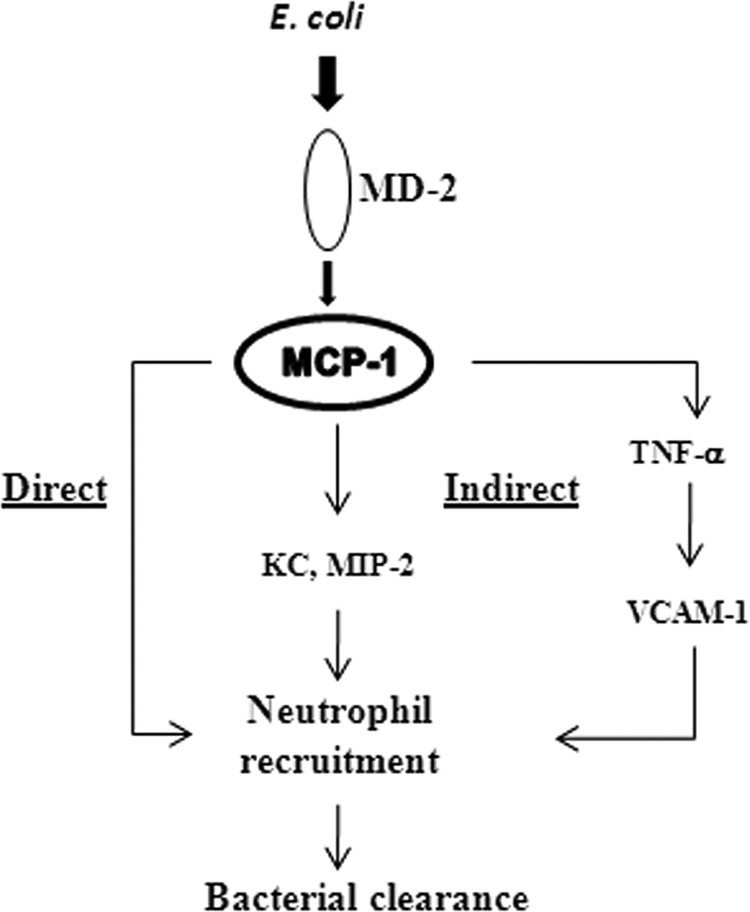
Proposed model of MCP-1-mediated neutrophil recruitment to the lung during E. coli infection. E. coli induces MCP-1 through the MD-2 signaling pathway, which involves Toll-like receptors. MCP-1 in turn regulates neutrophil recruitment directly via chemotaxis and indirectly through neutrophil chemokines, such as KC, MIP-2, as well as the cytokine TNF-α.
In previous studies using mouse models, MCP-1 has been shown to play a protective role against pulmonary pathogens, including Streptococcus pneumoniae (13, 45, 46), Pseudomonas aeruginosa (2, 35), and Cryptococcus neoformans (20). In studies of pneumococcal and Pseudomonas pneumonia, inhibition of MCP-1 by antibodies reduced monocyte but not neutrophil recruitment (2, 13). MCP-1 has also been shown to be associated with the influx of T lymphocytes to inflammatory sites (10). Our results demonstrate that rMCP-1 in the presence or absence of E. coli infection causes neutrophil and macrophage influx into the lungs. Our findings also demonstrate attenuated macrophage and neutrophil counts in the lungs of the gene-deficient mice after E. coli challenge. These findings are consistent with an earlier report demonstrating that blocking MCP-1/CCR2 reduced neutrophil accumulation in the lung following i.t. treatment with MCP-1/LPS (30) or Cryptococcus neoformans infection (20). The Maus group (29) showed that administering MCP-1 alone did not bring neutrophils to the lungs but that MCP-1 plus LPS synergistically cause neutrophil trafficking to the lungs of BALB/c mice. However, we found that rMCP-1 alone causes greater neutrophil influx into the lungs of the WT (C57BL/6) mice than in the PBS controls. Further, we show that neutrophils from blood, BALF, and bone marrow express CCR2, which is enhanced upon infection, and bone marrow neutrophils migrate toward rMCP-1, suggesting that MCP-1 can act as a direct neutrophil chemoattractant. In addition, the Beck-Schimmer group (4) has shown that LPS-induced neutrophil recruitment depends on MCP-1 production. A study by Iida et al. has shown that murine bone marrow neutrophils express functional CCR2 and migrate toward MCP-1 (21). Additional studies have shown that normal neutrophils are not responsive to MCP-1, although neutrophils become responsive to MCP-1 when animals become septic after cecal ligation and puncture (CLP) (40). A study by the Johnston group (23) shows that chronic inflammation induces neutrophils to express CCR2 in order to cause migration toward MCP-1. Furthermore, in other acute inflammatory models, such as models of acute pancreatitis and colitis, MCP-1 has been shown to be critical for neutrophil recruitment and blocking of MCP-1 with bindarit reduced neutrophil numbers and the inflammation (5, 6). These observations strongly suggest that neutrophils are directly responsive to MCP-1, and our findings show the importance of MCP-1 in neutrophil recruitment to the lungs.
Our findings show that MCP-1 can indirectly regulate neutrophil influx by regulating the expression of CXC chemokines. We observed reduced expression of cytokines (TNF-α and IL-6) and chemokines (KC and MIP-2) in MCP-1−/− mice compared to the controls after E. coli infection, and when MCP-1−/− mice were reconstituted with rMCP-1, the cytokine and chemokines levels returned to the level of WT mice during E. coli infection. In line with this finding, a previous study in a rat gastric ulcer model suggests that MCP-1 can regulate the expression of CXC chemokines and cause neutrophil recruitment (44). Moreover, Gouwy et al. showed that extracellular signaling by CCL2 significantly enhances CXCL-8 expression on monocytes (15), suggesting that CCL2/CCR2 signaling can regulate the expression of CXC chemokines. Further, the Herbold group (17) demonstrates that CXCR-2 signaling can lead to macrophage recruitment during S. pneumoniae infection. These observations suggest that there is a complex interplay between CC and CXC chemokines.
LTB4 is a known neutrophil chemotactic lipid which can be generated by leukocytes from arachidonic acid (19) and act via G protein-coupled receptors to cause leukocyte accumulation, microbial killing, and generation of proinflammatory mediators (26, 36). In an earlier study using the CLP model, it has been shown that MCP-1 inhibition attenuated neutrophil influx via the downregulation of LTB4 production (37). From our results, it is also possible that the attenuated level of LTB4 was the result of the reduced number of migrating neutrophils in MCP-1−/− mice. However, the attenuation was significant at 6 h and not significant at 24 h after E. coli infection. Since LTB4 itself can lead to generation of other proinflammatory mediators and neutrophil chemoattractants (36), reduction of the neutrophil chemoattractant lipid LTB4 at 6 h would have led to reduced neutrophil influx at 24 h.
We observed reduced NF-κB activation and activation of MAPKs in MCP-1 gene-deficient mice during E. coli infection. These observations suggest that the MCP-1/CCR2 axis regulates NF-κB and MAPK activation. Consistent with this finding, Viedt and Orth (43) show that MCP-1 can activate NF-κB and MAPKs during renal inflammation. It is also possible that other transcription factors, such as activator protein 1 (AP-1) and signal transducer and activator of transcription 1 (STAT-1), can be activated via MAPKs by MCP-1 (14). Therefore, our results suggest that MCP-1/CCR2 signaling regulates the expression of CXC chemokines (MIP-2 and KC) through NF-κB and MAPKs. Regulation of CXC chemokines and cytokines through NF-κB and MAPKs by MCP-1 can be attributed to initial alveolar macrophage activation. We used alveolar macrophages from wild-type and MCP-1−/− mice to determine whether MCP-1 deficiency has any effect on activation of NF-κB and MAPKs. We observed reduced activation of transcription factor NF-κB and MAPKs in the MCP-1−/− macrophages following E. coli infection, confirming our in vivo data that MCP-1 could regulate NF-κB- and MAPK-mediated macrophage activation through an autocrine and paracrine manner in the lungs.
Recruitment of leukocytes to the tissues involves upregulation of cell adhesion molecules (3). In this regard, we found that VCAM-1 upregulation was dependent on MCP-1. This was most pronounced at 24 h following E. coli infection, when neutrophil influx in the lungs was at peak. These observations suggest that VCAM-1 plays an important role in the transmigration of both neutrophils and monocytes to the lungs during E. coli infection in a MCP-1-dependent manner.
Previous studies have shown that MCP-1 is important for bacterial killing by macrophages (35, 38). Furthermore, earlier studies have shown that neutrophils play an important role in bacterial clearance during Gram-negative bacterial infections (27, 47). Since we found reduced neutrophil numbers and enhanced bacterial burden in MCP-1−/− mice following E. coli infection, we depleted neutrophils in wild-type mice to determine the role of neutrophils in augmenting clearance of E. coli. Our results show that neutrophils are indeed important for the clearance of E. coli in the lungs. Further experiments involving MCP-1 restoration in MCP-1−/− mice demonstrate the importance of MCP-1 in bacterial clearance from the lungs. Although there is a higher bacterial burden in the lungs of MCP-1−/− mice, both WT and MCP-1−/− mice cleared infection after 24 h, and no mortality was observed after infection.
Overall, our study reveals a new role for MCP-1, in that MCP-1 is directly as well as indirectly involved in the neutrophil recruitment during E. coli pneumonia. Herein we show evidence that MCP-1 is a direct neutrophil chemoattractant and indirect evidence that it regulates the expression of other chemokines (KC and MIP-2) and cytokines (TNF-α and IL-6) to cause neutrophil trafficking (Fig. 10). Identification of these novel molecular MCP-1-mediated mechanisms triggered by E. coli could be useful to design better therapeutic strategies during infections by Gram-negative bacterial pathogens.
ACKNOWLEDGMENTS
This study was supported by a scientist award from the Flight Attendant Medical Research Institute (YCSA-062466) and grants from the NIH (R01 HL-091958 and R01 HL-091958S1 via ARRA) to S.J.
We thank Barrett Rollins at Harvard Medical School for providing MCP-1−/− mice. We thank Rachel Zemans and Ken Malcolm at National Jewish Health and Dan Chisenhall and Pete Mottram at LSU for critical reading of the manuscript. We also thank Laboratory of Lung Biology members Malaya Sahoo, Himanshu Raje, Jin Liliang, and Kanapathipillai Jeyagowri for helpful discussions and critical reading of the manuscript.
Footnotes
Published ahead of print on 25 April 2011.
REFERENCES
- 1. Abraham E. 2003. Neutrophils and acute lung injury. Crit. Care Med. 31:S195–S199 [DOI] [PubMed] [Google Scholar]
- 2. Amano H., et al. 2004. Essential contribution of monocyte chemoattractant protein-1/C-C chemokine ligand-2 to resolution and repair processes in acute bacterial pneumonia. J. Immunol. 172:398–409 [DOI] [PubMed] [Google Scholar]
- 3. Bazan-Socha S., Bukiej A., Marcinkiewicz C., Musial J. 2005. Integrins in pulmonary inflammatory diseases. Curr. Pharm. Des. 11:893–901 [DOI] [PubMed] [Google Scholar]
- 4. Beck-Schimmer B., et al. 2005. Alveolar macrophages regulate neutrophil recruitment in endotoxin-induced lung injury. Respir. Res. 6:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bhatia M., et al. 2008. Treatment with bindarit, an inhibitor of MCP-1 synthesis, protects mice against trinitrobenzene sulfonic acid-induced colitis. Inflamm. Res. 57:464–471 [DOI] [PubMed] [Google Scholar]
- 6. Bhatia M., Ramnath R. D., Chevali L., Guglielmotti A. 2005. Treatment with bindarit, a blocker of MCP-1 synthesis, protects mice against acute pancreatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 288:G1259–G1265 [DOI] [PubMed] [Google Scholar]
- 7. Cai S., Batra S., Lira S., Kolls J. K., Jeyaseelan S. Keratinocyte cell-derived chemokine (KC) induces pulmonary host defense against Klebsiella pneumoniae via multiple mechanisms. Am. J. Respir. Crit. Care Med. 181:A1796 [Google Scholar]
- 8. Cai S., Batra S., Lira S. A., Kolls J. K., Jeyaseelan S. 2010. CXCL1 regulates pulmonary host defense to Klebsiella infection via CXCL2, CXCL5, NF-κB and MAPKs. J. Immunol. 185:6214–6225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cai S., Zemans R. L., Young S. K., Worthen G. S., Jeyaseelan S. 2009. Myeloid differentiation protein-2-dependent and -independent neutrophil accumulation during Escherichia coli pneumonia. Am. J. Respir. Cell Mol. Biol. 40:701–709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Carr M. W., Roth S. J., Luther E., Rose S. S., Springer T. A. 1994. Monocyte chemoattractant protein 1 acts as a T-lymphocyte chemoattractant. Proc. Natl. Acad. Sci. U. S. A. 91:3652–3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Craig A., Mai J., Cai S., Jeyaseelan S. 2009. Neutrophil recruitment to the lungs during bacterial pneumonia. Infect. Immun. 77:568–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fein A. M. 1999. Pneumonia in the elderly: overview of diagnostic and therapeutic approaches. Clin. Infect. Dis. 28:726–729 [DOI] [PubMed] [Google Scholar]
- 13. Fillion I., et al. 2001. Role of chemokines and formyl peptides in pneumococcal pneumonia-induced monocyte/macrophage recruitment. J. Immunol. 166:7353–7361 [DOI] [PubMed] [Google Scholar]
- 14. Goh K. C., Haque S. J., Williams B. R. G. 1999. p38 MAP kinase is required for STAT1 serine phosphorylation and transcriptional activation induced by interferons. EMBO J. 18:5601–5608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gouwy M., et al. 2008. Synergy between coproduced CC and CXC chemokines in monocyte chemotaxis through receptor-mediated events. Mol. Pharmacol. 74:485–495 [DOI] [PubMed] [Google Scholar]
- 16. Gu L., et al. 1998. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol. Cell 2:275–281 [DOI] [PubMed] [Google Scholar]
- 17. Herbold W., et al. 2010. Importance of CXC chemokine receptor 2 in alveolar neutrophil and exudate macrophage recruitment in response to pneumococcal lung infection. Infect. Immun. 78:2620–2630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hollingsworth J. W., et al. 2007. CD44 regulates macrophage recruitment to the lung in lipopolysaccharide-induced airway disease. Am. J. Respir. Cell Mol. Biol. 37:248–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang L., et al. 2004. Leukotriene B4 strongly increases monocyte chemoattractant protein-1 in human monocytes. Arterioscler. Thromb. Vasc. Biol. 24:1783–1788 [DOI] [PubMed] [Google Scholar]
- 20. Huffnagle G., et al. 1995. The role of monocyte chemotactic protein-1 (MCP-1) in the recruitment of monocytes and CD4+ T cells during a pulmonary Cryptococcus neoformans infection. J. Immunol. 155:4790–4797 [PubMed] [Google Scholar]
- 21. Iida S., Kohro T., Kodama T., Nagata S., Fukunaga R. 2005. Identification of CCR2, flotillin, and gp49B genes as new G-CSF targets during neutrophilic differentiation. J. Leukoc. Biol. 78:481–490 [DOI] [PubMed] [Google Scholar]
- 22. Jeyaseelan S., et al. 2006. Toll/IL-1R domain-containing adaptor protein (TIRAP) is a critical mediator of antibacterial defense in the lung against Klebsiella pneumoniae but not Pseudomonas aeruginosa. J. Immunol. 177:538–547 [DOI] [PubMed] [Google Scholar]
- 23. Johnston B., et al. 1999. Chronic inflammation upregulates chemokine receptors and induces neutrophil migration to monocyte chemoattractant protein-1. J. Clin. Invest. 103:1269–1276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kurihara T., Warr G., Loy J., Bravo R. 1997. Defects in macrophage recruitment and host defense in mice lacking the CCR2 chemokine receptor. J. Exp. Med. 186:1757–1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lynch J. P., III, Martinez F. J. 1998. Community-acquired pneumonia. Curr. Opin. Pulm. Med. 4:162–172 [PubMed] [Google Scholar]
- 26. Mancuso P., Lewis C., Serezani C. H., Goel D., Peters-Golden M. Intrapulmonary administration of leukotriene B4 enhances pulmonary host defense against pneumococcal pneumonia. Infect. Immun. 78:2264–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mancuso P., Nana-Sinkam P., Peters-Golden M. 2001. Leukotriene B4 augments neutrophil phagocytosis of Klebsiella pneumoniae. Infect. Immun. 69:2011–2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maus U., et al. 2002. Role of endothelial MCP-1 in monocyte adhesion to inflamed human endothelium under physiological flow. Am. J. Physiol. Heart Circ. Physiol. 283:H2584–H2591 [DOI] [PubMed] [Google Scholar]
- 29. Maus U., Huwe J., Maus R., Seeger W., Lohmeyer J. 2001. Alveolar JE/MCP-1 and endotoxin synergize to provoke lung cytokine upregulation, sequential neutrophil and monocyte influx, and vascular leakage in mice. Am. J. Respir. Crit. Care Med. 164:406–411 [DOI] [PubMed] [Google Scholar]
- 30. Maus U., et al. 2002. The role of CC chemokine receptor 2 in alveolar monocyte and neutrophil immigration in intact mice. Am. J. Respir. Crit. Care Med. 166:268–273 [DOI] [PubMed] [Google Scholar]
- 31. Mehrad B., Standiford T. J. 1999. Role of cytokines in pulmonary antimicrobial host defense. Immunol. Res. 20:15–27 [DOI] [PubMed] [Google Scholar]
- 32. Mizgerd J. P. 2008. Acute lower respiratory tract infection. N. Engl. J. Med. 358:716–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mizgerd J. P. 2006. Lung infection—a public health priority. PLoS Med. 3:e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Moore T. A., Standiford T. J. 2001. Cytokine immunotherapy during bacterial pneumonia: from benchtop to bedside. Semin. Respir. Infect. 16:27–37 [DOI] [PubMed] [Google Scholar]
- 35. Nakano Y., et al. 1994. Protection against lethal bacterial infection in mice by monocyte-chemotactic and -activating factor. Infect. Immun. 62:377–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Peters-Golden M., Canetti C., Mancuso P., Coffey M. J. 2005. Leukotrienes: underappreciated mediators of innate immune responses. J. Immunol. 174:589–594 [DOI] [PubMed] [Google Scholar]
- 37. Rios-Santos F., Benjamim C. F., Zavery D., Ferreira S. H., Cunha Fde Q. 2003. A critical role of leukotriene B4 in neutrophil migration to infectious focus in cecal ligaton and puncture sepsis. Shock 19:61–65 [DOI] [PubMed] [Google Scholar]
- 38. Rollins B., Walz A., Baggiolini M. 1991. Recombinant human MCP-1/JE induces chemotaxis, calcium flux, and the respiratory burst in human monocytes. Blood 78:1112–1116 [PubMed] [Google Scholar]
- 39. Scott M. G., et al. 2007. An anti-infective peptide that selectively modulates the innate immune response. Nat. Biotechnol. 25:465–472 [DOI] [PubMed] [Google Scholar]
- 40. Speyer C. L., et al. 2004. Novel chemokine responsiveness and mobilization of neutrophils during sepsis. Am. J. Pathol. 165:2187–2196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Standiford T. J., Kunkel S. L., Phan S. H., Rollins B. J., Strieter R. M. 1991. Alveolar macrophage-derived cytokines induce monocyte chemoattractant protein-1 expression from human pulmonary type II-like epithelial cells. J. Biol. Chem. 266:9912–9918 [PubMed] [Google Scholar]
- 42. Strieter R. M., et al. 1989. Monocyte chemotactic protein gene expression by cytokine-treated human fibroblasts and endothelial cells. Biochem. Biophys. Res. Commun. 162:694–700 [DOI] [PubMed] [Google Scholar]
- 43. Viedt C., Orth S. R. 2002. Monocyte chemoattractant protein-1 (MCP-1) in the kidney: does it more than simply attract monocytes? Nephrol. Dial. Transplant. 17:2043–2047 [DOI] [PubMed] [Google Scholar]
- 44. Watanabe T., et al. 2004. Monocyte chemotactic protein-1 regulates leukocyte recruitment during gastric ulcer recurrence induced by tumor necrosis factor-α. Am. J. Physiol. Gastrointest. Liver Physiol. 287:G919–G928 [DOI] [PubMed] [Google Scholar]
- 45. Winter C., et al. 2009. Important role for CC chemokine ligand 2-dependent lung mononuclear phagocyte recruitment to inhibit sepsis in mice infected with Streptococcus pneumoniae. J. Immunol. 182:4931–4937 [DOI] [PubMed] [Google Scholar]
- 46. Winter C., et al. 2007. Lung-specific overexpression of CC chemokine ligand (CCL) 2 enhances the host defense to Streptococcus pneumoniae infection in mice: role of the CCL2-CCR2 axis. J. Immunol. 178:5828–5838 [DOI] [PubMed] [Google Scholar]
- 47. Ye P., et al. 2001. Interleukin-17 and lung host defense against Klebsiella pneumoniae infection. Am. J. Respir. Cell Mol. Biol. 25:335–340 [DOI] [PubMed] [Google Scholar]
- 48. Yoo J. K., et al. 2005. IL-18 induces monocyte chemotactic protein-1 production in macrophages through the phosphatidylinositol 3-kinase/Akt and MEK/ERK1/2 pathways. J. Immunol. 175:8280–8286 [DOI] [PubMed] [Google Scholar]
- 49. Zisman D. A., et al. 1997. MCP-1 protects mice in lethal endotoxemia. J. Clin. Invest. 99:2832–2836 [DOI] [PMC free article] [PubMed] [Google Scholar]