

Abstract
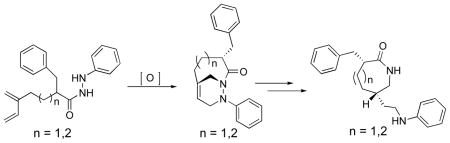
The type 2 intramolecular N-acylazo Diels-Alder reaction provides a regio- and stereoselective synthesis of bicyclic 1,2-diazine systems. A new method for the generation of N-acylazo dienophiles with tetra-n-butylammonium periodate is reported. X-ray crystallographic analysis allowed the quantification of structural distortions of the non-planar bridgehead olefin and lactam functionalities in 1,2-diazine cycloadducts 11 and 15. Synthesis of caprolactams and enantholactams were formed by stereoselective bridgehead alkene reduction, a process which transfers stereochemistry from the bridgehead lactam nitrogen to the bridgehead carbon. The sequence of transformations offers a convenient route for the diastereoselective synthesis of medium-ring nitrogen heterocycles and 1,4-diamines.
Introduction
Nitrogen-containing heterocycles are ubiquitious in nature. Their importance has led to an ongoing search for selective and efficient methods for their preparation.1,2 The type 2 intramolecular Diels-Alder (T2IMDA) reaction has served as a useful reaction to assemble polycyclic compounds in a single step from acyclic precursors.3 In many cases, the reaction offers complete regio- and stereochemical control in the cycloaddition step. More recently, the heteroatom variants of the T2IMDA reaction with N-acylimine and N-acylnitroso dienophiles was employed for the synthesis of bridgehead bicyclic lactams and oxazinolactams (Scheme 1, eq 1,2).3–5 As part of our ongoing interest in the synthesis of nitrogen-containing heterocyclic ring systems, we report T2IMDA reaction with N-acylazo dienophiles (Scheme 1, eq 3).
Scheme 1.

Examples of the hetero type 2 intramolecualr Diels-Alder reaction.
Despite numerous reports utilizing acyclic or cyclic azodicarboxylates as dienophiles in Diels-Alder6 reactions, there are relatively few examples of intramolecular variants7 of this reaction (Scheme 1). The development of the T2IMDA reaction with N-acylazo dienophiles would allow the rapid assembly of bridgehead bicyclic 1,2-diazines with regio- and stereochemical control. The intermediates would offer the potential for the synthesis of seven and eight membered nitrogen-containing heterocyclic ring systems as well as the stereoselective synthesis of 1,4-diamines.
Results and Discussion
Synthesis of the Diels-Alder precursors
The synthesis of T2IMDA reaction precursors began from the commercially available ethyl 4-bromobutyrate (1) (Scheme 2). The corresponding iodoester 38 was prepared by halide exchange with NaI in acetone. In the presence of 3 mol% Li2CuCl4, the coupling reaction of iodoester 38 with chloroprene Grignard (5)9 afforded ester 6.5 This synthetic sequence was subsequently applied to the synthesis of diene ester 75 from commercially available ethyl 5-bromovalerate (2) in 66% overall yield. The acylation reaction of ester 6 or 7 with phenyl hydrazine and Al(CH3)310 afforded hydrazides 8 and 9 in 75% yield and 84% yield, respectively (Scheme 2).
Scheme 2.

Synthesis of Hydrazides 8 and 9.
Type 2 Intramolecular N-acylazo Diels-Alder Reaction
Having established a viable route to the Diels-Alder precursors, we next examined oxidation conditions to form the N-acylazo dienophiles. The reactivity of the N-acylazo functional group towards thermal decomposition and cycloaddition was not known; therefore a search for mild reaction conditions was undertaken. Typically, N-acylazo dienophiles are generated by oxidation of N-acylhydrazides with tert-butylhypochlorite11, lead tetraacetate12, or potassium ferricyanide13. Oxidation of hydrazide 8 with Pb(OAc)4 resulted in a complex mixture of products. The heterogenous oxidation of hydrazide 8 with K3Fe(CN)6 and catalytic 2,4,6-triphenylphenol (1 mol %) in 2N NaOH gave cycloadduct 11 in 65% yield. The oxidation presumably produced N-acylazo dienophile 10, which underwent intramolecular Diels-Alder cycloaddition under the reaction conditions. Despite the acceptable result, the harsh basic conditions limited the general utility of this method. Parallels in structure between hydroxamic acids and hydrazides suggested that n-Bu4NIO4, a reagent used to oxidize hydroxamic acids to the N-acyl nitroso5 intermediate, could be employed for the synthesis of N-acylazo derivatives (Scheme 1). Indeed, oxidation of N-acyl hydrazide 8 proceeded smoothly with 1.3 equiv of n-Bu4NIO4 in CH2Cl2, to form the N-acylazo Diels-Alder precursor 10. Subsequently, compound 10 underwent cycloaddition under these reaction conditions to afford bicyclic 1,2-diazine 11 in 90% yield (Scheme 3). Diels-Alder reactions carried out in water have displayed a significant rate acceleration.14 In the presence of 20 mol% of water in THF, cycloadduct 11 was obtained in 63% yield after 40 h. Interestingly, the oxidation of hydrazide 8 was completed after 5 h and was not inhibit by water; however, the slow rate of the cycloaddition allowed the decomposition of the N-acylazo intermediate 10.
Scheme 3.

Type 2 intramolecular Diels-Alder reaction with N-acylazo dienophiles 10.
To the best of our knowledge the oxidation reaction of hydrazides by this method is unprecedented. Intrigued by the oxidation of hydrazide 8 with n-Bu4NIO4, the generality of this reagent with other hydrazides was examined. Representative examples for this transformation are shown in Scheme 4.15 Subjecting hydrazides to 1.3 equiv n-Bu4NIO4 in CH2Cl2 afforded N-acylazo substrates in high yield. This method provides an alternative procedure for the oxidation of hydrazides and hydrazines.15 n-Bu4NIO4 exhibits functional group tolerance that is lacking in other reagents.
Scheme 4.

Oxidation reaction of hydrazides with n-Bu4NIO4.
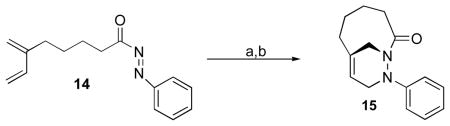
Although oxidation and subsequent intramolecular cycloaddition reaction of hydrazide 8 was complete after 24 h (Scheme 3), the oxidation of hydrazide 9 followed by intramolecular Diels-Alder reaction of N-acylazo dienophile 14 proceeded slowly (Scheme 5). It was established by the 1H-NMR spectrum of the reaction mixture that the periodate oxidation reaction of hydrazide 9 generated the N-acylazo dienophile species after 3 h. However, the cyclization step proceeded relatively slowly affording bicyclic 1,2-diazine 15 in only 55% yield after 60 h. Attempts to isolate the N-acylazo derivative 14 by chromatographic techniques (SiO2, Al2O3 and deactivated SiO2) were unsuccessful. This was attributed to instability of the N-acylazo dienophile 14. Efforts to accelerate the cycloaddition by heating the reaction mixture to 50 °C resulted in decomposition of the remaining N-acylazo intermediate. The presence of both 14 and 15 in the reaction mixture and the instability of intermediate 14 resulted in low isolated yields. These results suggested that the problem was not due to the oxidation step but rather the relative low stability and slow rate of cycloaddition of the N-acylazo dienophile 14.
Scheme 5.

Type 2 intramolecular Diels-Alder reaction of N-acylazo dienophile 14.
To overcome this problem a different set of conditions was required for the generation and isolation of N-acylazo derivative 14. Using a protocol described by Evans and coworkers,18 we found that treating hydrazide 9 with NBS and pyridine in CH2Cl2 for 2 h at 0–23°C resulted in N-acylazo dienophile 14 in 96% yield (Scheme 6). Under these reaction conditions cycloadduct 15 was not observed.
Scheme 6.

Oxidation reaction of hydrazide 9.
This result provided an opportunity to examine the cycloaddition reaction of N-acylazo dienophile 14 under both thermal and Lewis acid catalyzed conditions. Efforts to thermally induce cycloaddition are summarized in Table 1. N-acylazo dienophile 14 was found to be unstable at temperatures > 40 °C in benzene. At these elevated temperatures, the reaction generated a complex mixture of products; cycloadduct 15 was not observed. The best results were obtained at 40 °C, producing cycloadduct 15 in 58% yield.
Table 1.
T2IMDA reaction of N-acylazo dienophile 14.
 | |||
|---|---|---|---|
| entry | temp (°C) | time (h) | yield (%) |
| 1 | 23 | 60 | 45–55 |
| 2 | 40 | 10 | 58 |
| 3 | 60 | 4 | mixture |
| 4 | 80 | 4 | mixture |
Benzene solvent
Concentration 0.010 M
The thermal route did not offer any improvement to the cycloaddition reaction of N-acylazo dienophile 14. We next turned our attention to Lewis acid catalysis. It was found that the cycloaddition of N-acylazo dienophile 14 proceeded smoothly in the presence of 10 mol % ZnCl2 in CH2Cl2 after 5 h to afford cycloadduct 15 in 78% yield (Scheme 7). The two step protocol of oxidation and subsequent Lewis acid catalyzed cycloaddition proved to be the most efficient method for the synthesis of cycloadduct 15. Significantly, this result provides a new method for the Lewis acid-catalyzed Diels-Alder reaction of azo compounds, as the examples of Lewis acid catalyzed Diels-Alder reaction of azo compounds are limited in the literature.6l,6m
Scheme 7.

Lewis acid catalyzed T2IMDA reaction of N-acylazo dienophile 14.
X-ray Crystallography of the Cycloadducts
X-ray crystallographic studies of cycloadducts diamines 11 and 15 reveal structural distortions from the optimal planar olefin and lactam geometry. These distortions are expressed as torsional deformation and are quantified by the angle τ, a value determined from the calculated projection of the two p-orbitals.3–5 The p-orbital overlap in the π bond is presumed to be optimal with τ = 0.0° and lowest at τ = 90.0°. The torsion angle τ is not directly measured but can be calculated from the X-ray crystallographic data.19 Torsional distortions (τ) calculated for bridgehead olefins 11 and 15 are 5.48° and 3.65° respectively with a slightly larger value of τ for the smaller bridgehead alkene 11. Interestingly, the torsional distortion quantified in bridgehead olefins 11 and 15 has little effect on the observed C=C bond lengths. The double bond distances for bridgehead olefins 11 and 15 are 1.3339(15) Å and 1.3327(16) Å respectively, and are within error of the value for cyclohexene (1.335 (3) Å).5
Analysis of the amide linkage of 11 and 15 show significant differences in torsional deformation. For bridgehead lactam 11 the torsional distortion is τ = 0.745(10)° and for 15 τ = 17.56(13)° respectively. It is likely that the somewhat surprising inverse relationship between ring size and τ results from compression in accomodating the five atom bridge in cycloadduct 15. The absence of correlations between bridge size and torsional distortions was previously observed in a series of bridgehead lactams.4
The C-N bond length for bridgehead lactam 15 is slightly longer (1.4013(14) Å) than bridgehead lactam 11 (1.3941(13)Å). In contrast, the C=O bond distance of bridgehead lactam 11 (C=O = 1.2163(12) Å) and bridgehead lactam 15 (C=O = 1.2198(13)Å) are not sensitive to the difference in τ values.
Functionalization of Bicyclic 1,2-diazines
To examine the chemical behavior of the bicyclic 1,2-diazines, a series of transformations were carried out that include reduction of the bridgehead double bond and hydrogenolysis of the N-N bond. When carried out in this order, this sequence transfers stereochemistry from the bridgehead nitrogen to the sp3 bridgehead carbon. The reduction of the bridgehead double bond in cycloadducts 11 and 15 was achieved by catalytic hydrogenation in the presence of 10% Pd/C in EtOH to give the saturated cycloadduct 16 in 95% yield and 18 in 89% (Scheme 8). Several methods have been reported for the N-N bond cleavage including reduction by zinc in acetic acid,20 SmI2,21 and Raney/Ni.22 The most effective method for the cleavage of N-N bond resulted from the treatment of compounds 14 and 18 with Raney/Nickel in ethanol to afford 6-substituted caprolactam 17 in 80% and 7-substituted enantholactam 19 in 87% yield, respectively. This method provides a convenient route for the synthesis of seven and eight member nitrogen-containing heterocyclic ring systems.
Scheme 8.

Synthesis of lactams 17 and 19.
π–facial selectivity in T2IMDA reaction
Analysis of the X-ray crystal structure of cycloadduct 11 revealed a distance of 2.18 Å between the endo hydrogen at C10 and the exo hydrogen at C3 (Figure 1). Based on previous studies5 with N-acylnitroso dienophiles, we anticipated that π–facial selectivity of the T2IMDA reaction with N-acylazo dienophiles would be influenced by the introduction of substituents on the tether at α-position of Diels-Alder precursor. To evaluate the π–facial selectivity in cycloaddition precursors that incorporate substituents at α-position, two derivatives were synthesized (Scheme 9). The synthesis of the α-benzylated esters 205 and 22 was achieved by deprotonation of ester 6 or 7 with LDA, followed by alkylation with benzyl bromide to afford the α-substituted ester derivative 205 in 65% yield and ester 22 in 74% yield. Coupling reaction of ester 20 or 22 with phenylhydrazine and Al(CH3)3 provided hydrazide 21 and 23 in 76% yield and 85% yield, respectively.
Figure 1.

ORTEP plots of cycloadducts 11 and 15 at the 50% probability level.
Scheme 9.

Synthesis of hydrazide 21 and 23.
Under optimized reaction conditions using n-Bu4NIO4, the oxidation of hydrazide 21 generated the N-acylazo dienophile in situ, which underwent intramolecular cycloaddition to afford cycloadduct 24 after 24 h in 91% yield. The product consisted of a single diastereomer as determined by 1H NMR analysis of the crude reaction mixture. The endo diastereomer was established by NOE analysis (Scheme 10).
Scheme 10.

Diastereoselective T2IMDA reaction of hydrazide 21.
Addition of hydrogen to the bridgehead double bond, which occurs in a syn-exo matter, transfers the stereochemistry of the bridgehead nitrogen to the bridgehead carbon.3 However, hydrogenation of cycloadduct 24 in the presence of 10% Pd/C and H2 in resulted a mixture of products that included saturated cycloadduct 25 (68%), 26 (23%), and 27 (2%) (Scheme 11). Bridgehead alkene isomerization competes with hydrogenation resulting in formation of alkenes with less strain than the starting material.
Scheme 11.

Catalytic hydrogenation of cycloadduct 24.
Complete hydrogenation of the bridgehead alkene 22 was achieved in the presence of 10% Pd/C under high pressure (50 psi) to afford saturated cycloadduct 25 in 86% yield. Following hydrogenation the cis-3,6-disubstituted caprolactam 28 was prepared by a reductive N-N bond cleavage using Ra/Ni and 1N NaOH under H2 in 72% yield.
The synthesis of N-acylazo dienophile 29 was achieved using NBS and pyridine in 92% yield. Subsequently, the intramolecular cycloadditon of N-acylazo dienophile 29 proceeded smoothly in the presence of ZnCl2 to afford cycloadduct 30. Only a single endo diastereomer was observed in the 1H NMR spectrum of the crude reaction mixture and the stereochemistry of cycloadduct 30 was established by NOE experiments. Cycloadduct 30 was subjected to a catalytic hydrogenation at 50 psi in the presence of 10% Pd/C to produce saturated bicyclic 1,2-diazine 31 in 90% yield. The stage was now set for the synthesis of cis-3,7-disubstituted enantholactam 32, which was achieved in 77% yield by reductive N-N bond cleavage using Raney/Nickel.
Conclusion
In summary, we have developed the type 2 intramolecular Diels-Alder reaction with N-acylazo dienophiles for the regio- and stereoselective synthesis of bicyclic 1,2-diazines. In the course of our investigation, a new reagent was identified for the oxidation of hydrazides. X-ray crystallographic analysis allowed the quantification of structural distortions of the non-planar bridgehead olefin and lactam functionalities in cycloadducts 11 and 15. The T2IMDA reaction with N-acylazo dienophiles, incorporating substituents at the α-position, underwent stereoselective cycloaddition. These cycloadducts were subsequently elaborated to caprolactams and enantholactams derivatives.
Experimental Section
General Procedure for preparation of the hydrazides.10
To a solution of phenylhydrazine (2.0 equiv) in CHCl3 was added Al(CH3)3 (2.0 equiv, 2.0 M solution in toluene) dropwise. The reaction mixture was stirred at room temperature for 1 h and diene ester (1 equiv) was added dropwise. After 10 h (TLC monitoring), the reaction mixture was cooled to 0 °C and then carefully poured into a solution of HCl (2N) and was allowed to stir for 30 min. The aqueous layer was separated and extracted with 3 portions CHCl3. The combined organic extracts were washed with H2O dried Na2SO4 and concentrated to give a pale yellow oil.
5-Methylene-hept-6-enoic acid N′-phenyl-hydrazide (8)
Diene ester 65 (1.08 g, 6.42 mmol) was added dropwise to a solution of phenylhydrazine (1.39 g, 12.8 mmol) in CHCl3 (50 mL) and Al(CH3)3 (6.4 mL in toluene, 2.0 M). The crude product was purified by flash column chromatography (1:2 EtOAc: Hexanes) to afford hydrazide 8 (1.12 g, 75% yield): 1H NMR (500 MHz, CDCl3) for major rotamer δ 7.28 (d, J = 7.6 Hz, 1H), 7.26 (app t, J = 8.2 Hz, 2H), 6.93 (app t, J = 7.4 Hz, 1H), 6.84 (app d, J = 7.8 Hz, 2H), 6.38 (dd, J = 17.6, 10.8 Hz, 1H), 6.15 (d, J = 3.7 Hz, 1H), 5.26 (d, J = 17.8 Hz, 1H), 5.11 (d, J = 10.9 Hz, 1H), 5.07 (s, 1H), 5.02 (s, 1H), 2.32-2.28 (m overlapped, 4H), 1.93(m, 2H); IR (thin film) νmax : 3258, 1654, 1598, 1498; 13C NMR (125 MHz, CD3OD) δ175.9, 150.1, 147.3, 139.9, 130.3, 121.2, 116.7, 114.2, 114.1, 34.7, 32.2, 25.7. HRMS (ES) m/z calcd. for C14H18N2O [M + Na]+ 253.1317, found 253.1307.
6-Methylene-oct-7-enoic acid N′-phenyl-hydrazide (9)
Diene ester 75 (3.26 g, 17.9 mmol) was added dropwise to a solution of phenylhydrazine (3.87 g, 35.8 mmol) in CHCl3 (75 mL) and Al(CH3)3 (17.9 mL in toluene, 2.0 M). The crude product was purified by flash chromatography (1:2 EtOAc: Hexanes) to give 3.63 g hydrazine 9 (84%): 1H NMR (500 MHz, Tol-d8) for major rotamer δ 7.55 (d, J = 3.4 Hz, 1H), 7.12 (app t, J = 7.3 Hz, 2H), 7.00 (s, 1H), 6.80 (app t, J = 7.3 Hz, 1H), 6.75 (app d, J = 7.8 Hz, 2H), 6.33 (dd, J = 10.8, 17.6 Hz, 1H), 5.19 (d, J = 17.1 Hz, 1H), 5.1 (d, J = 10.8 Hz, 1H), 4.98 (s, 1H), 4.95 (s, 1H), 2.10 (t, J = 7.3 Hz, 2H), 1.86 (t, J = 7.3 Hz, 2H), 1.55 (m, 2H), 1.42 (m, 2H); IR (thin film) νmax: 3265, 3087, 2933, 1655, 1602, 1495 cm−1; 13C NMR (125 MHz, CD3OD) δ176.0, 150.1, 147.8, 140.1, 130.3, 121.2, 116.3, 114.24, 113,47, 34.9, 32.2, 29.2, 26.88; HRMS (ES) m/z calcd. for C15H20N2O [M + Na]+ 267.1473 found, 267.1480.
2-Benzyl-5-methylene-hept-6-enoic acid N′-phenyl-hydrazide (21)
Diene ester 205 (1.98 g, 7.66 mmol) was added to a solution of phenylhydrazine (1.67 g, 15.4 mmol) in CHCl3 (50 mL) and Al(CH3)3 (7.70 mL in toluene, 2.0 M). The crude product was purified by flash chromatography (1:5 EtOAc: Hexanes) to give 1.87 g hydrazide 21 (76 %): 1H NMR (500 MHz, CDCl3) for major rotamer δ 7.2–7.3 (m overlapped, 3H), 7.15 (app d, J = 6.5 Hz, 2H), 7.11 (s, 1H), 7.09 (app d, J = 8.0 Hz, 2H), 6.84 (app t, J = 7.2 Hz, 1H), 6.43 (app d, J = 7.8 Hz, 2H), 6.37 (dd, J = 17.6, 10.8 Hz, 1H), 5.98 (d, J = 3.6 Hz, 1H), 5.22 (d, J = 17.6 Hz, 1H), 5.09 (d, J = 10.9 Hz, 1H), 5.06 (s, 1H), 4.99 (s, 1H), 2.92 (dd, J = 13.4, 10.2 Hz, 1H), 2.81 (dd, J = 13.6, 5.0 Hz, 1H), 2.46 (m, 1H), 2.31 (m, 1H), 2.23 (m, 1H), 2.00 (m, 1H), 1.76 (m, 1H); IR (thin film) νmax: 3248, 3027, 2928, 1667, 1601, 1495 cm−1; 13C NMR (125 MHz, CDCl3) δ 174.9, 147.8, 145.8, 139.5, 138.7, 129.1, 128.6, 126.6, 121.2, 116.3, 113.9, 113.6, 112.6, 47.7, 39.3, 31.1, 29.4. HRMS (ES) m/z calcd. for C21H24N2O [M + H]+ 321.1967 found, 321.1972.
2-Benzyl-6-methylene-oct-7-enoic acid N-phenyl-hydrazide (23)
Diene ester 225 (0.716 g, 2.14 mmol) was added to a solution of Al(CH3)3 (2.14 mL in toluene, 2.0 M) and phenylhydrazine (0.463 g, 4.28 mmol). The crude product was purified by flash chromatography (1:5 EtOAc: Hexanes) to give 1.73 g hydrazine 23 (85 %): 1H NMR (500 MHz, CDCl3) for major rotamer δ 7.31-7.26 (m overlapped, 3H), 7.19 (app d, J = 6.0 Hz, 2H), 7.11 (app t, J = 8.2 Hz, 2H), 6.98 (s, 1H), 6.84 (app t, J = 7.4 Hz, 1H), 6.45 (app d, J = 8.2 Hz, 2H), 6.38 (dd, J = 17.7, 10.8 Hz, 1H), 5.98 (d, J = 3.6 Hz, 1H), 5.23 (d, J = 17.6 Hz, 1H), 5.09 (d, J = 10.8 Hz, 1H), 5.05 (s, 1H), 5.00 (s, 1H), 2.92 (dd, J = 13.4, 10.3 Hz, 1H), 2.79 (dd, J = 13.4, 4.9, 1H), 2.42 (m, 1H), 2.24 (t, J = 6.3 Hz, 2H), 1.83 (m, 1H), 1.65-1.50 (m, 3H); IR (thin film) 3027, 2939, 1661, 1602, 1495 cm−1; 13C NMR (125 MHz, CDCl3) δ 174.9, 147.7, 145.8, 139.4, 138.8, 129.4, 129.04, 128.7, 126.6, 121.1, 116.0, 113.4, 112.5, 48.4, 39.2, 32.9, 31.3, 26.2; HRMS (ES) m/z calcd. for C22H26N2O [M + H]+ 335.2123 found, 335.2122.
General Procedure for the oxidation reaction of hydrazides with n-Bu4NIO4 followed by cycloaddition
To a cooled (0 °C) solution of a hydrazide in dry CH2Cl2 was added n-Bu4NIO4 (1.3 equiv). The reaction mixture was stirred at room temperature for 24h (TLC monitoring) and washed with 2 portions of sat Na2SO3. The organic layers were combined, dried over Na2SO4, filtered, and concentrated in vacuo.
9-Phenyl-1,9-diaza-bicyclo[4.3.1]dec-6-en-2-one (11)
To a solution of hydrazide 8 (0.20 g, 0.87 mmol) in dry CH2Cl2 was added n-Bu4NIO4 (1.3 equiv, 0.49 g, 1.13 mmol) and stirred at room temperature for 24 h. Flash column chromatography (1:2 EtOAc: Hexanes) of the crude product yielded 0.18 g (91%) of cycloadduct 11 as a pale yellow solid. 1H NMR (500 MHz, CDCl3) δ 7.27 (m, 2H), 7.04 (app d, J = 8.7 Hz, 2H), 6.91 (app t, J = 7.3 Hz, 1H), 5.86 (br s, 1H), 4.37 (dd, J =13.8, 7.4 Hz, 1H), 4.15 (d, J = 14.8 Hz, 1H), 3.49 (d, J = 12.7 Hz, 1H), 3.23 (d, J = 14.4 Hz, 1H), 3.12 (td, J = 13.8, 3.2 Hz, 1H), 2.64 (dd, J = 12.1, 6.8 Hz, 1H), 2.55 (dt, J = 13.2, 3.1 Hz, 1H), 2.50 (td, J = 12.1, 6.1 Hz, 1H), 2.29 (m, 1H), 1.95 (m, 1H); IR (thin film) νmax: 1702, 1597, 1492, 1342, 1163 cm−1; 13C NMR (125 MHz, CD2Cl2) δ183.6, 150.9, 150.8, 129.4, 120.1, 119.2, 114.4, 51.6, 48.9, 36.8, 35.0, 33.4. HRMS (ES) m/z calcd. for C14H16N2O [M + Na]+ 251.1160, found 251.1155.
Acetylazobenzene (13a)
To a solution of hydrazide 12a (0.21 g, 1.40 mmol) in dry CH2Cl2 was added n-Bu4NIO4 (1.3 equiv, 0.788 g, 1.81 mmol) and stirred at room temperature for 5 h (TLC monitoring). Flash column chromatography (1:2 EtOAc: Hexanes) of the crude product yielded 0.15 g (72%) of N-acyl azo 13a as a red oil. 1H NMR (500 MHz, CDCl3) δ 7.9 (m, 2H), 7.56 (m, 3H), 2.44 (s, 3H); IR (thin film) νmax: 1743, 1565, 1479 cm−1; 13C NMR (125 MHz, CDCl3) δ 188.7, 151.7, 133.7, 129.5, 123.8, 21.4. HRMS (ES) m/z calcd. For C8H8N2O [M + Na]+ 171.0534, found 171.0540.
Isobutyrylazobenzene (13b)
To a solution of hydrazide 12b14 (0.34 g, 1.91 mmol) in dry CH2Cl2 was added n-Bu4NIO4 (1.3 equiv, 1.07 g, 2.47 mmol) and stirred at room temperature for 5 h (TLC monitoring). Flash column chromatography (1:2 EtOAc: Hexanes) of the crude product yielded 0.31 g (91%) of N-acyl azo 13b as a red oil.1H NMR (500 MHz, CDCl3) δ 7.89 (m, 2H), 7.56 (m, 3H), 3.14 (m, 1H), 1.31 (d, J = 7.3 Hz, 6H); IR (thin film) νmax: 1736, 1501, 1453 cm−1; 13C NMR (500 MHz, CD2Cl2) δ 195.4, 152.4, 133.7, 133.7, 129.9, 123.7, 34.9, 18.3; HRMS (ES) m/z calcd. for C10H12N2O [M + H]+ 177.0950 found, 177.9038.
Ethyl(phenyl)azocarboxylate (13c)
To a solution of hydrazide 12c15 (0.15 g, 0.832 mmol) in dryCH2Cl2 was added n-Bu4NIO4 (1.3 equiv, 0.469 g, 1.08 mmol) and stirred at room temperature for 5h (TLC monitoring). Flash column chromatography (1:2 EtOAc: Hexanes) of the crude product yielded 0.14 g (95%) of N-acyl azo 13c as a red oil.1H NMR (400 MHz, CDCl3) δ 7.94 (m, 2H), 7.56 (m, 3H), 4.53 (q, 2H), 1.48 (t, J = 7.1 Hz, 3H); IR (thin film) 2986, 1755, 1503; 13C NMR (500 MHz, CDCl3) δ 162.4, 151.8, 134.0, 129.5, 123.9, 64.7, 14.4; HRMS (ES) m/z calcd. For C9H10N2O2 [M + Na] 201.0640, found 201.0639.
3-Benzyl-9-phenyl-1,9-diaza-bicyclo[4.3.1]dec-6-en-2-one (24)
To a cooled (0 °C) solution of hydrazide 21 (0.24 g, 0.75 mmol) in CH2Cl2 was added n-Bu4NIO4 (1.3 equiv, 0.42 g, 97 mmol) and stirred at 25 °C. After 24h (TLC monitoring), the reaction mixture was diluted with CH2Cl2 (10 mL) and washed with sat Na2SO3 (2 × 10 mL). The organic layers were combined, dried over Na2SO4, filtered, and concentrated in vacuo. Flash column chromatography (1:3 EtOAc: Hexanes) of the crude product afforded cycloadduct 24 (0.22 g, 91%) as a pale yellow solid. 1H NMR (500 MHz, CDCl3) δ 7.31-7.24 (m, 7H), 6.95 (app d, J = 7.9 Hz, 2H), 6.87 (app t, J = 7.3 Hz, 1H), 5.83 (br s, 1H), 4.33 (dd, J =13.8, 7.3 Hz, 1H), 4.20 (d, J = 14.7, 1H), 3.49 (d, J = 14.5 Hz, 1H), 3.40 (m, 1H), 3.22-3.17 (m, 2H), 2.61 (dd, J = 14.1, 7.7 Hz, 1H), 2.53 (dd, J = 12.3, 6.9 Hz, 1H), 2.39 (m, 1H), 2.17 (d, J = 11.7 Hz, 1H), 1.74 (m, 1H); IR (thin film) 2930, 1694, 1598,1495 cm−1; 13C NMR (125 MHz, CDCl3) δ 184.7, 150.7, 149.9, 140.5, 129.7, 129.4, 128.8, 126.7, 120.48, 119.7, 114.9, 51.0, 49.3, 46.6, 39.5, 38.9, 34.6. HRMS (ES) m/z calcd. for C21H22N2O [M + H]+ 319.1810 found 319.1810.
General Procedure for the preparation of N-acyl azo dienophiles with NBS.16
To a solution of a hydrazide in CH2Cl2 was added pyridine (1 equiv). The reaction mixture was cooled to 0° C and N-bromosuccinimide (1 equiv) was added to the solution. After 2h, the orange reaction mixture was poured into H2O. The layers were separated and the aqueous layer was extracted with 3 portions of CH2Cl2 The combined organic layers were washed with 5% HCl, 10% K2CO3, brine, and dried over Na2SO4. The organic layer was concentrated in vacuo.
6-Methylene-oct-7-enoic acid azobenzene (14)
To a solution of hydrazide 9 (0.101 g, 0.413 mmol) in CH2Cl2 (5 mL) was added pyridine (0.033 g, 0.417 mmol). The reaction mixture was cooled to 0° C and N-bromosuccinimide (0.074g, 0.416 mmoles) was added to the solution. The organic layer was concentrated in vacuo to give 0.096 g of N-azo dienophile 14 in 96% yield and was used without further purification: 1H NMR (500 MHz, CDCl3) δ 7.89 (app d, J = 7.1 Hz, 2H), 7.60-7.51 (m overlapped, 3H), 6.37 (dd, J = 17.6, 10.8 Hz, 1H), 5.23 (d, J = 17.6 Hz, 1H), 5.08 (d, J = 10.8 Hz, 1H), 5.4 (s, 1H), 5.2 (s, 1H) 2.77 (t, J = 7.3 Hz, 2H), 2.27 (t, J = 7.7 Hz, 2H), 1.82 (m, 2H), 1.63 (m, 2H); IR (thin film) νmax: 2941, 1743, 1499 cm−1; 13C NMR (125 MHz, CDCl3) δ191.5, 151.7, 145.9, 138.9, 133.5, 129.4, 123.6, 116.1, 113.4, 34.2, 31.2, 27.7, 23.4. HRMS (ES) m/z calcd. for C15H18N2O [M + Na]+ 265.1317 found, 265.1324.
2-Benzyl-6-methylene-oct-7-enoic acid azobenzene (29)
To a solution of hydrazide 23 (0.023 g, 0.069 mmol) in CH2Cl2 (2 mL) was added pyridine (0.0054 g, 0.0687 mmol). The reaction mixture was cooled to 0 °C and N-bromosuccinimide (0.0122g, 0.0688 mmoles) was added to the solution. The organic layer was concentrated under vacuo to give N-acylazo dienophile 29 (0.0215 g) in 94% yield and used without further purification: 1H NMR (500 MHz, CD2Cl2) δ 7.82 (app d, J = 7.5 Hz, 2H), 7.58-7.55 (m overlapped, 3H), 7.28-7.20 (m, 5H), 6.35 (dd, J = 17.6, 10.8 Hz, 1H), 5.20 (d, J = 17.6 Hz, 1H), 5.05 (d, J = 10.9 Hz, 1H), 4.98 (s, 1H), 4.95 (s, 1H), 3.31 (m, 1H), 3.12 (dd, J = 13.8, 7.7 Hz, 1H), 2.88 (dd, J = 13.8, 6.7, 1H), 2.18 (m, 2H), 1.83 (m, 1H), 1.65-1.53 (m, 3H); IR (thin film) νmax: 2941, 1735, 1594, 1498; 13C NMR (125 MHz, CD2Cl2) δ 193.0, 151.7, 145.9, 139.0, 138.7, 133.3, 129.3, 129.0, 128.3, 126.4, 123.3, 115.7, 113.1, 47.1, 37.1, 31.1, 30.6, 25.5; HRMS (ES) m/z calcd. for C22H24N2O [M + Na]+ 355.1786 found, 355.1785.
General Procedure for the T2IMDA reaction with N-acylazo dienophiles catalyzed by ZnCl2
To a cooled solution (− 78 °C) of N-acylazo dienophile (0.01M) in CH2Cl2 was added ZnCl2 (10 mol %) as a solid in one portion. After 2h, the reaction mixture gradually allowed to warm to 25 °C and was completed after 3h (monitored by TLC). The solution was diluted with CH2Cl2 and poured in H2O. The layers were separated and the aqueous layer was extracted with 3 portions of CH2Cl2. The combined organic layers were washed with NaHCO3, brine and dried over Na2SO4. The organic layer was concentrated in vacuo.
10-Phenyl-1,10-diaza-bicyclo[5.3.1]undec-7-en-2-one (15)
To a solution of N-acylazo dienophile 14 (0.0960 g, 0.396 mmol) in CH2Cl2 (10 mL) was added ZnCl2 (0.0054 g, 0.0396 mmol). Purification of the crude product by column chromatrography (1:3 EtOAc: Hexanes) afforded cycloadduct 15 (0.075 g, 78% yield) as a pale yellow solid: 1H NMR (500 MHz, CDCl3) δ 7.26 (app t, J = 7.8 Hz, 2H), 6.90 (app d, J = 8.7 Hz, 2H), 6.81 (app t, J = 7.3 Hz, 1H), 5.55 (br s, 1H), 4.20 (d, J = 5.2 1H), 4.15 (d, J = 15.5 Hz, 1H), 3.96 (br d, J = 15.7, 1H), 3.56 (br d, J = 15.8 Hz, 1H), 2.62 (dd, J = 13.1, 9.3, 1H), 2.52 (m, 1H), 2.41 (t, J = 11.5, 1H), 2.15-2.10 (m, 3H), 1.90 (m, 1H), 1.44 (m, 1H); IR (thin film) νmax: 2932, 1656, 1599, 1497 cm−1; 13C NMR (125 MHz, CDCl3) δ 180.5, 148.7, 141.7, 129.2, 121.9, 118.7, 112.0, 47.5, 47.1, 37.6, 33.7, 27.2, 24.4. HRMS (ES) m/z calcd. for C15H18N2O [M + Na]+ 265.1317 found, 265.1308.
3-Benzyl-10-phenyl-1,10-diaza-bicyclo[5.3.1]undec-7-en-2-one (30)
To a solution of N-acylazo dienophile 29 (0.087 g, 0.26 mmol) in CH2Cl2 (7 mL) was added ZnCl2 (0.0036 g, 0.011 mmol). Purification of the crude product by column chromatrography (1:4 EtOAc: Hexanes) to afford cycloadduct 30 (0.062 g, 71% yield) as a pale yellow oil: 1H NMR (500 MHz, CDCl3) δ. δ 7.31-7.18 (m overlapped, 7H), 6.76 (app t, J = 7.3 Hz, 1H), 6.64 (app d, J = 8.8 Hz, 2H), 5.66 (br s, 1H), 4.24 (d, J =15.6, 1H), 4.13 (dd, J = 15.5, 5.1, 1H), 3.98 (dt, J = 15.6, 2.1 Hz, 1H), 3.55 (br d, J = 15.6, 1H), 3.20 (dd, J = 13.5, 8.5, 1H), 2.76 (m, 1H), 2.66 (dd, J = 13.5, 6.4 Hz, 1H), 2.49 (br m, 1H), 2.17-2.05 (m, 2H), 1.87 (m, 2H), 1.4 (m, 1H); IR (thin film) νmax: 2930, 1698, 1598, 1497 cm−1; 13C NMR (125 MHz, CDCl3) δ181.9, 148.3, 140.8, 140.1, 129.4, 128.9, 128.3, 126.2, 122.4, 118.3, 111.6, 48.5, 47.6, 46.7, 40.6, 33.9, 31.4, 27.0; HRMS (ES) m/z calcd. for C22H24N2O [M + H]+ 333.1967 found, 333.1963.
General Procedure for the hydrogenation of the bridgehead alkene
To a solution of a cycloadduct in EtOH was added 10% Pd/C. The reaction mixture was stirred under 1 atmosphere or 50 psi of H2 for 5 h. The catalyst was filtered through celite and the filtrate was concentrated in vacuo.
9-Phenyl-1,9-diaza-bicyclo[4.3.1]decan-2-one (16)
To a solution of cycloadduct 11 (0.025, 0.011 mmol) in EtOH (5 mL) was added 10% Pd/C (0.003 g). The reaction mixture was stirred under 1 atmosphere of H2 for 5 h. The clear oil was purified by column chromatography (1:1 EtOAc: Hexanes) to afford 16 (0.023 g, 92% yield). 1H NMR (500 MHz, CDCl3) δ 7.26 (m, 2H), 6.91-6.89 (m, 3H), 3.8 (ddd, J = 9.7, 9.7, 5.3 Hz, 1H), 3.7 (d, J = 14.9 Hz, 1H), 3.24-3.18 (m, 2H), 2.92 (m, 1H), 2.59 (dt, J = 13.7, 4.4 Hz, 1H), 2.18 (m, 1H), 1.98-1.80 (m, 5H), 1.69-1.64 (m, 1H), 13C NMR (125 MHz, CDCl3) δ178.7, 149.3, 129.2, 120.3, 114.1, 49.2, 45.7, 35.4, 31.0, 30.6, 22.9, 19.7; IR (thin film) νmax : 2933, 1682, 1599, 1495 cm−1; HRMS (ES) m/z calcd. for C14H18N2O [M + H]+ 231.1497, found 231. 1498.
10-Phenyl-1,10-diaza-bicyclo[5.3.1]undecan-2-one (18)
To a solution of cycloadduct 15 (0.041, 0.17 mmol) in EtOH (5 mL) was added 10% Pd/C (0.004 g). The reaction mixture was stirred under 1 atmosphere of H2 for 5 h. The clear oil was purified by column chromatography (1:1 EtOAc: Hexanes) to afford 18 (0.036 g, 89% yield): 1H NMR (500 MHz, CDCl3) δ 7.246 (m, 2H), 6.88-6.82 (m, 3H), 3.97 (d, J = 14.4 Hz, 1H), 3.79-3.74 (m, 2H), 3.39 (d, J = 14.3 Hz, 1H), 2.63 (td, J = 13.1, 3.2 Hz, 1H), 2.55 (m, 1H), 2.15-2.05 (m, 2H), 1.99-1.94 (m, 4H), 1.57-1.47 (m, 2H), 1.33-1.25 (m, 1H) 13C NMR (125 MHz, CD3OD) δ176.1, 146.9, 129.5, 119.7, 113.9, 46.4, 40.6, 35.3, 34.7, 30.7, 28.0, 26.0, 25.1. HRMS (ES) m/z calcd. for C15H20N2O [M + Na]+ 267.1473, found 267.1468.
3-Benzyl-9-phenyl-1,9-diaza-bicyclo[4.3.1]decan-2-one (25)
To a solution of cycloadduct 24 (0.037, 0.12 mmol) in EtOH (5 mL) was added 10% Pd/C (0.004 g). The reaction mixture was stirred under high pressure H2 (50 psi) for 5 h. The clear oil was purified by column chromatography (1:2 EtOAc: Hexanes) to afford 25 (0.032 g, 86% yield): 1H NMR (500 MHz, CDCl3) δ 7.33-7.25 (m, 7H), 6.89-6.85 (m, 3H), 3.83-3.78 (m, 2H), 3.32 (dd, J = 14.1, 5.6 Hz, 1H), 3.23-3.17 (m, 3H), 2.67 (dd, J = 14.1, 8.3 Hz, 1H), 2.14 (br s, 1H), 1.92 (m, 1H), 1.83-1.65 (m, 5H); IR (thin film) 2922, 1686, 1599, 1497. 13C NMR (125 MHz, CDCl3) δ 179.8, 149.4, 140.6, 129.6, 129.1, 128.5, 126.3, 120.1, 113.9, 48.7, 45.7, 44.9, 38.2, 31.5, 30.1, 26.2, 22.9. HRMS (ES) m/z calcd. for C21H24N2O [M + H]+ 321.1967, found 321.1960.
3-Benzyl-10-phenyl-1,10-diaza-bicyclo[5.3.1]undecan-2-one (31)
To a solution of cycloadduct 30 (0.040, 0.12 mmol) in EtOH (5 mL) was added 10% Pd/C (0.004 g). The reaction mixture was stirred under high pressure H2 (psi) for 5 h. The clear oil was purified by column chromatography (1:2 EtOAc: Hexanes) to aff ord 31 (0.036 g, 86% yield): 1H NMR (500 MHz, CDCl3) δ 7.28-7.25 (m overlapped, 5H), 7.13 (app t, J = 7.3 Hz, 2H), 6.75 (app t, J = 7.3 Hz, 1H), 6.54 (app d, J = 7.8 Hz, 2H), 4.07 (br d, J = 14.4 Hz, 1H), 3.77-3.70 (m, 2H), 3.27-3.20 (m, 2H), 2.91 (m, 1H), 2.66 (dd, J = 13.4, 5.3 Hz, 1H), 2.08-2.03 (m, 2H), 1.94-1.90 (m, 2H), 185-1.75 (m, 2H), 1.54-1.45 (m, 2H), 1.32 (br d, J = 13.4 Hz, 1H); IR (thin film) νmax: 2921, 1677, 1597, 1497 cm−1; 13C NMR (125 MHz, CDCl3) δ 177.8, 147.1, 140.5, 129.6, 129.4, 128.5, 126.3, 119.4, 113.4, 46.9, 46.2, 39.8, 39.6, 37.6, 35.3, 27.7, 26.9, 24.6. HRMS (ES) m/z calcd. for C22H26N2O [M + H]+ 335.2123 found, 335.2121.
General Procedure for the reductive N-N bond cleavage with Raney-Ni
A solution of saturated cycloadducts in EtOH and Raney nickel was stirred under 1 atmosphere of H2. After 6 h, the reaction mixture was stirred at r.t or refluxed over night. The catalyst was filtered through a pad of celite. The filtrate was concentrate under vacuo and chromatographed.
6-(2-Phenylamino-ethyl)-azepan-2-one (17)
To a solution of 16 (0.020g, 0.087 mmol) in EtOH (5 mL) was added Raney nickel and stirred under 1 atmosphere of H2. After 6 h, the reaction mixture was refluxed over night. Purification of the crude product by column chromatography (1:1 EtOAc: CHCl3) to afford 6-substituted caprolactam 17 (0.016 g, 80%) as a white solid: 1H NMR (500 MHz, CDCl3) δ 7.19 (app t, J = 7.4 Hz, 2H), 6.72 (app t, J = 7.3 Hz, 1H), 6.61 (app d, J = 8.6 Hz, 2H), 6.02 (br s, 1H), 3.59 (br s, 1H), 3.20-310 (m, 4H), 2.48 (dd, J = 6.9, 4.8 Hz, 2H), 1.97 (m, 1H), 1.85 (m, 1H), 1.75 (br m, 1H), 1.69-1.47 (m, 4H and H2O); IR (thin film) νmax: 3369, 2920, 1654 cm−1 ; 13C NMR (125 MHz, CDCl3) δ 178.7, 148.3, 129.5, 117.7, 112.9, 47.5, 41.8, 36.9, 36.6, 36.5, 32.8, 21.7; HRMS (ES) m/z calcd. for C14H20N2O [M + Na]+ 255.1473 found, 255.1473.
7-(2-Phenylamino-ethyl)-azocan-2-one (19)
To a solution of 18 (0.025g, 0.102 mmol) in EtOH (5 mL) was added Raney nickel and stirred under 1 atmosphere of H2. After 6 h, the reaction mixture was refluxed over night. The catalyst was filtered through a pad of celite. The filtrate was concentrate in vacuo and purified by column chromatography (1:1 EtOAc: CHCl3) to afford enantholactam 19 (0.022 g, 87%) as a white solid: 1H NMR (500 MHz, CDCl3) δ 7.18 (app t, J = 7.4 Hz, 2H), 6.73 (app t, J = 7.3 Hz, 1H), 6.61 (app d, J = 7.6 Hz, 2H), 5.92 (br s, 1H), 3.70 (br s, 1H), 3.43 (ddd, J = 9.3, 7.1, 3.8 Hz, 1H), 3.20-3.11 (m, 3H), 2.43 (ddd, J= 8.1, 5.5, 2.9, 2H), 1.85-1.30 (m, 9H); IR (thin film) νmax: 3346, 2925, 1661 cm−1; 13C NMR (125 MHz, CDCl3) δ 177.8, 148.3, 129.5, 117.7, 112.9, 45.7, 42.0, 39.4, 33.1, 32.4, 29.9, 28.5, 23.7; HRMS (ES) m/z calcd. for C15H22N2O [M + H]+ 247.1810, found 247.1811.
3-Benzyl-6-(2-phenylamino-ethyl)-azepan-2-one (28)
To a solution of 25 (0.018 g, 0.056 mmol) in EtOH (5 mL) was added Raney nickel and NaOH (0.2 mL, 1N) stirred under 1 atmosphere of H2. After 6 h, the H2 balloon was removed and stirred for 48 h. Purification by column chromatography (1:1 EtOAc: Hexanes) afforded cis-3,6-substituted caprolactam 28 (0.013 g, 72%) as a white solid: 1H NMR (500 MHz, CDCl3) δ 7.29 -7.16 (m overlapped, 7H), 6.71 (app t, J = 7.3 Hz, 1H), 6.60 (app d, J = 7.9 Hz, 2H), 5.94 (br s, 1H), 3.49 (dd, J = 15.1, 5.6 Hz, 1H), 3.24 (dd, J = 14.1, 5.1 Hz, 1H), 3.11 (m, 3H), 2.71 (m, 1H), 2.57 (dd, J = 14.1, 9.3 Hz, 1H), 1.90-1.50 (m, 8H); IR (thin film) νmax: 3326, 3046, 1643 cm−1; 13C NMR (125 MHz, CDCl3) δ 179.2, 148.2, 140.6, 129.5, 129.4, 128.5, 126.2, 117.8, 113.0, 45.5, 45.3, 42.1, 37.3, 33.8, 33.5, 29.9, 29.3; HRMS (ES) m/z calcd. for C21H26N2O [M + H]+ 323.2123, found 323.2129.
3-Benzyl-7-(2-phenylamino-ethyl)-azocan-2-one (32)
To a solution of 31 (0.021 g, 0.063 mmol) in EtOH (5 mL) was added Raney nickel and NaOH (0.2 mL, 1N) stirred under 1 atmosphere of H2. After 6 h, the H2 balloon was removed and stirred for 48 h. Purification by column chromatography (2:1 EtOAc: Hexanes) afforded cis-3,7-disubstituted enantholactam 32 (0.016 g, 77%) as a clear oil: 1H NMR (500 MHz, CDCl3) δ 7.25-7.15 (m overlapped, 7H), 6.73-6.63 (m overlapped, 3H), 5.75 (br s, 1H), 3.70 (br m, 2H), 3.15-3.05 (m, 4H), 2.91 (m, 1H), 2.66 (dd, J = 13.8, 6.7 Hz, 1H), 1.78 (m, 2H), 1.65 (m, 2H), 1.51 (br s, 2H), 1.36 (m, 2H); IR (thin film) νmax: 3312, 2932, 1651 cm−1; 13C NMR (125 MHz, CDCl3) δ 178.9, 148.3, 140.7, 129.6,129.4, 128.6, 126.3, 117.8, 113.0, 44.5, 42.1, 38.9, 38.6, 35.6, 31.7, 30.2, 29.9, 22.9. HRMS (ES) m/z calcd. for C22H28N2O [M + H]+ 337.2280 found, 337.2280.
Supplementary Material
Scheme 12.

Catalytic Hydrogenation and N-N bond cleavage of cycloadduct 24.
Scheme 13.

Diastereoselective T2IMDA reaction of hydrazide 23.
Acknowledgments
This research was supported by the National Institutes of Health (GM-#). C. L. M. thanks NIH for a research supplement graduate fellowship (GM #71492). We thank Dr. Joseph Ziller for assistance with X-ray crystallographic studies, Dr. John Greaves for mass spectrometric data, and Dr. Phil Dennison for assistance with NMR studies.
Footnotes
Supporting Information Available. Characterization of compounds 26 and 27 are provided in this section. Stereochemical proofs, X-ray crystallographic data, and spectral data for selected compounds are available free or charge via the internet at http://pubs.acs.org.
References
- 1.a Evans PA, Holmes B. Tetrahedron. 1991;47:9131–9166. [Google Scholar]; b Miller ED, Kauffman CA, Jensen PR, Fenical W. J Org Chem. 2007;72:323–330. doi: 10.1021/jo061064g. [DOI] [PubMed] [Google Scholar]
- 2.Thorsett ED, Harris EE, Aster SD, Peterson ER, Snyder JP, Springer JP, Hirshfield J, Tristram EW, Patchett AA, Ulm EH, Vassil TC. J Med Chem. 1986;29:251–260. doi: 10.1021/jm00152a014. [DOI] [PubMed] [Google Scholar]
- 3.Bear BR, Sparks SM, Shea KJ. Angew Chem Int Ed. 2001;40:820–849. [PubMed] [Google Scholar]
- 4.Lease TG, Shea KJ. J Am Chem Soc. 1993;115:2248–2260. [Google Scholar]
- 5.a Sparks SM, Vargas JD, Shea KJ. J Org Lett. 2000;2:1473–1475. doi: 10.1021/ol005811m. [DOI] [PubMed] [Google Scholar]; b Chow CP, Shea KJ, Sparks SM. J Org Lett. 2002;4:2637–2640. doi: 10.1021/ol026075k. [DOI] [PubMed] [Google Scholar]; c Sparks SM, Chow CP, Zhu L, Shea KJ. J Org Chem. 2004;69:3025–3035. doi: 10.1021/jo049897z. [DOI] [PubMed] [Google Scholar]
- 6.a Weinreb SM, Staib RR. Tetrahedron. 1982;38:3087–3128. [Google Scholar]; b Franzus B, Surridge JH. J Org Chem. 1962;27:1951–1957. [Google Scholar]; c Gillis BT, Weinkam R. J Org Chem. 1967;32:3321–3325. [Google Scholar]; d Kealy TJ. J Am Chem Soc. 1962;84:966–973. [Google Scholar]; f Gillis BT, Beck PE. J Org Chem. 1962;27:1947–1951. [Google Scholar]; g Johnson MP, Moody CJ. J Chem Soc Perkin Trans I. 1985:71–74. [Google Scholar]; h Ahern MF, Leopold A, Beadle JR, Gokel GW. J Am Chem Soc. 1982;104:548–554. [Google Scholar]; i Pindur U, Kim M, Rogge M, Massa W, Molinier M. J Org Chem. 1992;57:910–915. [Google Scholar]; j Nugiel DA, Decicco CP, Nelson DJ, Copeland RA, Hardman KD. Bioorg & Med Chem Lett. 1995;5:3053–3056. [Google Scholar]; k Cohen SG, Zand R, Steel C. J Am Chem Soc. 1961;83:2895–2899. [Google Scholar]; l Aburel PS, Zhuang W, Hazell RG, Jorgensen KA. Org Biomol Chem. 2005;3:2344–2349. doi: 10.1039/b503744a. [DOI] [PubMed] [Google Scholar]; m Boger DL, Weinreb SN. Hetero Diels-Alder Methodology in Organic Synthesis. San Diego: Academic Press; 1987. [Google Scholar]; n Kawasaki M, Yamamoto H. J Am Chem Soc. 2006;128:16482–16483. doi: 10.1021/ja066726y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a Kahn M, Wilke S, Chen B, Fujita K. J Am Chem Soc. 1988;110:1638–1639. [Google Scholar]; b Sheradsky T, Milvitskaya J, Pollak IE. Tetrahedron Lett. 1991;32:133–136. [Google Scholar]
- 8.a Tok JBH, Cho J, Rando RR. Tetrahedron. 1999;55:5741–5758. [Google Scholar]; b Frick JA, Thompson CM. J Org Chem. 1989;54:890–896. [Google Scholar]
- 9.a Shea KJ, Pham PQ. Tetrahedron Lett. 1983;24:1003–1006. [Google Scholar]; b Tamura M, Kochi J. Synthesis. 1971:303–305. [Google Scholar]
- 10.a Stavchansky S, Benderly A. Tetrahedron Letters. 1988;29:739–740. [Google Scholar]; b Licandro E, Perdicchia D. Eur J Org Chem. 2004:665–675. [Google Scholar]
- 11.a Cookson RC, Gilani SSH, Stevens IDR. Tetrahedron Lett. 1962;14:614–618. [Google Scholar]; b Amezua MG, Lora-Tamayo M, Soto JL. Tetrahedron Lett. 1970;27:2407–2408. [Google Scholar]
- 12.a Gillis BT, Hagarty JD. J Org Chem. 1967;32:330–333. [Google Scholar]; b Tawil BF, Guggisberg A, Hesse M. Tetrahedron. 1992;48:3375–3780. [Google Scholar]; c Amarasekara A, Chandrasekara S. Org Lett. 2002;4:773–775. doi: 10.1021/ol017256+. [DOI] [PubMed] [Google Scholar]; d Clement RAJ. Org Chem. 1960;25:1724–1727. [Google Scholar]; e Criegge R. Oxidation with lead tetracetate. In: Wiberg KB, editor. Oxidation in Organic Chemistry. Academic Press; New York: 1965. [Google Scholar]
- 13.Dimroth K, Tuncher W. Synthesis. 1977;5:339–340. [Google Scholar]
- 14.a Grieco PA, Yoshida K, Garner P. J Org Chem. 1983;48:3137–3139. [Google Scholar]; b Lindstrom UM. Chem Rev. 2002;102:2751–2772. doi: 10.1021/cr010122p. [DOI] [PubMed] [Google Scholar]
- 15.To further demonstrate the scope and efficiency of tetra-n-butylammonium periodate, we examined the oxidation reaction of 1,2-diphenylhydrazine, 4-phenylurazole, 1,2- dibenzoyhydrazine, and sym-dicarbethoxyhydrazine. Azobenzene was isolated in 88% yield. The oxidation of 4-phenylurazole with tetra-n- butylammonium proceeded smoothly to provide 4-phenyl-1,2,4-triazoline-3,5-dione which was trapped with cyclohexadiene to afford the cycloadduct in 93% yield. The oxidation reaction of 1,2-dibenzoyhydrazine, and sym-dicarbethoxyhydrazine was not successful. See the supporting information section for experimental procedures and spectroscopic data.
- 16.Hearn MJ, Grimwade JE. Dep Chem Organic Preparations and Procedures International. 1980;12:249–51. [Google Scholar]
- 17.Bausch MJ, David B, Dobrowolski P, Guadalupe-Fasano C, Gostowski R, Selmarten D, Prasad V, Vaughn A, Wang LH. J Org Chem. 1991;56:5643–51. [Google Scholar]
- 18.Evans DA, Johnson DS. Org Lett. 1999;1:595–598. doi: 10.1021/ol990113r. [DOI] [PubMed] [Google Scholar]
- 19.The torsional angle (τ) was determined by summing the four atom torsion angles YC1C2W (Φ1) and ZC1C2 (Φ2) and dividing the result by 2 (τ = (Φ1 + Φ2/2).
- 20.Mellor JM, Smith NM. J Chem Soc Perkin Trans I. 1984:2927–2931. [Google Scholar]
- 21.a Ding H, Friestad GK. Org Lett. 2004;6:637–640. doi: 10.1021/ol036480r. [DOI] [PubMed] [Google Scholar]; b Burk MJ, Feaster JE. J Am Chem. 1992;114:6266–6267. [Google Scholar]
- 22.a Grabowski S, Armbruster J, Prinzback H. Tetrahedron Lett. 1997;38:5485–5488. [Google Scholar]; b Denmark SE, Nicaise O, Edwards JP. J Org Chem. 1990;55:6219–6223. [Google Scholar]; c Ghelfi FJ. Org Chem. 2000;65:6249–6253. doi: 10.1021/jo0004153. [DOI] [PubMed] [Google Scholar]; d Hsieh Y, Lee G, Luh T. J Org Chem. 1998;63:1484–1490. [Google Scholar]; e Martin S, Hom RK. Tetrahedron Lett. 1999;40:2887–2890. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.