

Abstract
To probe structural changes that occur when a membrane protein is transferred from lipid bilayers to SDS micelles, a fragment of bacteriorhodopsin containing transmembrane helical segments A and B was studied by fluorescence spectroscopy, molecular dynamics (MD) simulation, and stopped flow kinetics. In lipid bilayers, Förster resonance energy transfer (FRET) was observed between tyrosine 57 on helix B and tryptophans 10 and 12 on helix A. FRET efficiency decreased substantially when the peptide was transferred to SDS. MD simulation showed no evidence for significant disruption of helix-helix interactions in SDS micelles. However, a cluster of water molecules was observed to form a hydrogen-bonded network with the phenolic hydroxyl group of tyrosine 57, which probably causes the disappearance of tyrosine-to-tryptophan FRET in SDS. The tryptophan quantum yield decreased in SDS, and the change occurred at nearly the same rate as membrane solubilization. The results provide a clear example of the importance of corroborating distance changes inferred from FRET by using complementary methods.
Keywords: membrane protein folding, FRET, SDS, molecular dynamics simulation
1. Introduction
Most integral membrane proteins contain membrane-spanning α-helices. However, the forces involved in assembling these proteins in lipid bilayers are not well understood. Techniques of reversible unfolding have revealed how water-soluble proteins fold into stable, biologically active structures in aqueous solution, but these techniques are not applicable to helical transmembrane proteins. In various attempts to study the mechanism of membrane protein unfolding, a number of experiments have been reported on the transfer of transmembrane proteins from lipid environments into sodium dodecyl sulfate (SDS) detergent micelles [1, 2]. The premise of these experiments was that SDS causes significant unfolding of helical membrane proteins. However, the structures of membrane proteins in SDS are mostly unknown. MacKenzie et al. [3] used NMR spectroscopy to determine the structure of the glycophorin dimer in SDS. Their results show that glycophorin has intact α-helices and extensive inter-helix side chain interactions in SDS. Recent studies of synthetic peptides in micelles have reached opposite conclusions about the effect of SDS on helix-helix interactions [4, 5]. Many spectroscopic changes have been observed when integral membrane proteins are transferred into SDS micelles. These changes have been variously interpreted as to the extent of unfolding [6–11]. Förster resonance energy transfer (FRET) can give information about proximity between fluorescent groups and near-by chromophores, and a number of studies of integral membrane protein structures have measured distances using FRET (for example [12–16]). To properly apply FRET, the energy donor-acceptor stoichiometries must be matched, but this is often experimentally challenging. One way to avoid this problem, as shown by Eisenhawer et al. [17], is to use tyrosine to tryptophan FRET. Since the donors and acceptors are naturally-occurring parts of the structure, no protein modification is necessary. In the following report, we have used tyrosine to tryptophan FRET, along with molecular dynamics simulations and stopped flow kinetics, to examine the conformation of a two-helix fragment of bacteriorhodopsin in lipid bilayers and in SDS micelles.
2. Materials and methods
2.1 Materials
Purple membrane was obtained from Halobacterium salinarum S9, as previously described [18]. The 1–71 fragment of bacteriorhodopsin, known as C-2 [19] or AB [20], was obtained by chymotrypsin cleavage of purple membrane, followed by gel permeation chromatography [15, 19, 21]. The AB fragment was stored in methanol:chloroform 1:1 containing 0.1 M ammonium acetate in glass vials sealed with teflon-lined caps at −20°C. Chloroform solutions of lipids were obtained from Avanti Polar Lipids, Inc (Alabaster, AL) and stored under argon after opening. Small unilamellar vesicles were prepared by the method of Batzri and Korn [22], as follows. Aliquots of column fractions containing 0.4 nmol of peptide (about 10 μL) were mixed with 20 μL of a 20 mg/mL chloroform solution of DOPC. The samples were dried in a stream of nitrogen and then placed in a vacuum (approx. 1 mtorr) for 5 hrs. The residue was dissolved in 40 μL ethanol, drawn into a syringe, and then slowly delivered through a 25 gauge unbeveled needle into 2.0 mL of rapidly stirred 0.05 M phosphate, pH 7.0, 0.1 M NaCl. For preparation of peptides in SDS micelles, the same drying procedure was used (omitting the lipids), and the residue was directly dissolved in 0.05 M phosphate, pH 7.0, 0.1 M NaCl, 2.0% SDS. Peptides corresponding to the A helix (residues 1–36) and the B helix (residues 38–71) were synthesized by Bio-Synthesis, Inc. (Lewisville, TX) using FMOC chemistry. The peptides were purified by HPLC and the sequences were confirmed by MALDI-TOF mass spectrometry.
2.2 Spectroscopy
Steady-state fluorescence spectra were measured on a Photon Technology International, Inc. (Birmingham, NJ) QM4 fluorometer, with corrected excitation and corrected emission. Fluorescence lifetimes were measured by single photon counting, using a IBH 5000U Jobin Yvon (Glasgow, UK) instrument. Samples were in 0.05 M phosphate buffer, pH 7.0, 0.1 M NaCl. When present, SDS was at a concentration of 2.0 % w/v (i.e. 69 mM). Peptide concentrations were 0.2 μM. Measurements were made at 23°C ± 1°. Lifetime distributions were calculated using the Shannon-Jaynes maximum entropy method, as implemented in Felix32 software from Photon Technology International. Stopped flow kinetics were measured using a Bio-Logic (Claix, FR) SFM-20 instrument.
2.3 Förster resonance energy transfer
Tyrosine to tryptophan FRET was measured by the method of Eisinger [23]. Relative quantum yields for tryptophan, Qo, were measured by comparing fluorescence at 355 nm from excitation at 280 nm and 295 nm, with the fluorescence normalized to constant absorbance. Fractional absorbances of tryptophan and tyrosine, fw and fy, were calculated from published extinction coefficients [24, 25]. The transfer efficiency, E, was calculated from:
| (1) |
For comparison, E was calculated from the atomic coordinates of the AB fragment, either from the crystal structure (Protein Data Bank file 1C3W [26]), or from molecular dynamics simulations (see below), using:
| (2) |
where R is the distance between the energy donor and acceptor and Ro is the Förster critical distance:
| (3) |
For the purposes of this calculation, the fluorescence quantum yield of tyrosine QD was assumed to be 0.14 [23], the spectral overlap of tyrosine emission and tryptophan absorbance, J, was assumed to be 4.8 × 1012 cm3 M−1 nm4 [27], and the index of refraction of the medium, n, was taken to be 1.33. We calculated the dipole orientation factor, κ2, from the atomic coordinates of the AB fragment.
2.4 Molecular dynamics simulations
The AB fragment was inserted into an SDS micelle consisting of 55 SDS molecules. The starting atomic coordinates for the AB fragment were obtained from the bacteriorhodopsin crystal structure [26]. The starting coordinates for the SDS micelle were obtained from Braun et al. [28]. The starting coordinates for a POPC bilayer were obtained from VMD [29]. POPC was used instead of DOPC because of the availability of the atomic coordinates for a lipid bilayer of POPC. For the purposes of this study, the differences between the two lipids should not be significant, because they are known to have similar hydrocarbon thicknesses, volumes, and areas [30]. The AB-SDS complex was explicitly solvated with 11,311 TIP3 waters. Ions (80 sodium and 26 chloride) were added to electrically neutralize the system. The whole system consisted of 37,407 atoms. All simulations of this work were performed using NAMD 2.6. [31]. The starting peptide/membrane and peptide/micelle structures were energy-minimized and equilibrated prior to the simulations. The periodic boundary conditions were employed. The pressure and the temperature were maintained at 1 bar and 300 K respectively. The Langevin damping coefficient was chosen to be 5/ps. The particle-mesh Ewald method was utilized to compute electrostatic interactions. A time step of 1 fs was used for short-range interactions and 4 fs for long-range forces. Covalent bonds of hydrogens were fixed to their equilibrium length. The all-atom CHARMM27 [32, 33] force field was adopted. Equilibrium Langevin dynamics was run for 6 ns. The RMSD of the AB backbone (excluding the two termini) was analyzed to elucidate any structural changes during the 6 ns period of in silico experiments. Similar in silico experiments for 6 ns were implemented for the AB-POPC system. The system consists of AB fragment embedded in a POPC bilayer of 260 POPC molecules.
3. Results and discussion
3.1 Tyrosine to tryptophan Förster resonance energy transfer in bacteriorhodopsin AB fragment
The crystal structure of bacteriorhodopsin (Fig. 1) shows that Tyr 57 is 12.4 Å from Trp 10 and 10.9 Å from Trp 12 (Table 1). We calculated the expected efficiency of Tyr to Trp FRET for each pair (equation 2), using typical values for the spectral properties of Tyr and Trp, along with the particular orientations of donor and acceptor found in the crystal structure. The results (Table 1) suggest that FRET between Tyr 57 and Trp 10 and 12 would be efficient, but little FRET would be expected between the other tyrosines and the two tryptophans. Because Trp 10 and 12 are on helix A, and Tyr 57 is on helix B, the proximity of Tyr 57 to these Trp residues is a measure of the proximity of helices A and B, and therefore a measure of the global folding of the peptide. If the helices become separated in SDS micelles, as has been suggested [34], then the Tyr to Trp FRET should disappear when peptide AB is transferred from a lipid bilayer to SDS.
Figure 1. Structure of bacteriorhodopsin peptide AB.
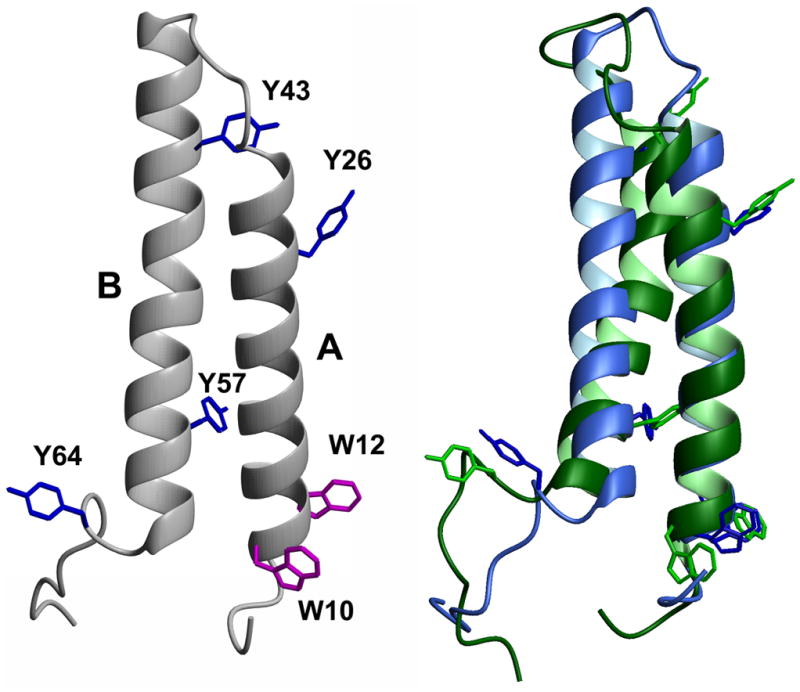
Left side: Amino acids 5–71 from the crystal structure of bacteriorhodopsin (Protein Data Bank 1C3W), comprising transmembrane helices A and B. Tryptophan (W) and tyrosine (Y) residues are indicated. Right side: Superposition of peptide AB structures from final frames of MD simulations in POPC belayed (blue) or SDS micelle (green). Figure generated using Molmol [41].
Table 1.
Crystal structure Tyr to Trp distances and calculated energy transfer efficiencies
| Acceptor | Donor | κ2 | Ro | R | E |
|---|---|---|---|---|---|
| W10 | Y26 | 0.199 | 12.5 | 27.1 | 0.009 |
| Y43 | 0.025 | 8.8 | 33.1 | 0.0004 | |
| Y57 | 0.282 | 13.2 | 12.4 | 0.597 | |
| Y64 | 0.070 | 10.5 | 18.8 | 0.029 | |
| W12 | Y26 | 0.536 | 14.7 | 22.8 | 0.068 |
| Y43 | 2.474 | 19.0 | 29.4 | 0.068 | |
| Y57 | 0.029 | 9.0 | 10.9 | 0.248 | |
| Y64 | 0.084 | 10.8 | 21.9 | 0.014 |
Distances, R, (Å) measured between center of Tyr ring and midpoint of bond between Trp Cδ2 and Cε2 (see Fig. 1). Dipole orientation factor, κ2, Förester critical distance, Ro, and energy transfer efficiency, E, calculated from equations (2) and (3) as described in text.
The steady-state method described by Eisenhawer et al. [17] can detect Tyr to Trp FRET. This method compares emission spectra using 280 nm excitation and emission spectra under the same conditions using 295 nm excitation. The 295 nm spectra are scaled using emission data above 360 nm (where no Tyr emission is expected). Figure 2 shows that tyrosine fluorescence from peptide AB is completely quenched in DOPC vesicles, as indicated by the coincident emission from 280 nm and 295 nm excitation. However, in SDS micelles, peptide AB displays some unquenched tyrosine fluorescence, as indicated by the lack of coincidence of the emission from 280 nm and 295 nm excitation below 340 nm. By contrast, the isolated A helix, which contains one tyrosine and two tryptophans, shows similar levels of tyrosine fluorescence in DOPC vesicles and SDS micelles (Fig. 3), as indicated by the differences between the emission from 280 nm excitation and the scaled emission from 295 nm excitation. Therefore, the quenching of tyrosine fluorescence in DOPC vesicles is likely to be a property of interactions between A and B helices in DOPC vesicles, interactions that are absent in SDS micelles or when the B helix is removed.
Figure 2. Fluorescence emission spectra of peptide AB.
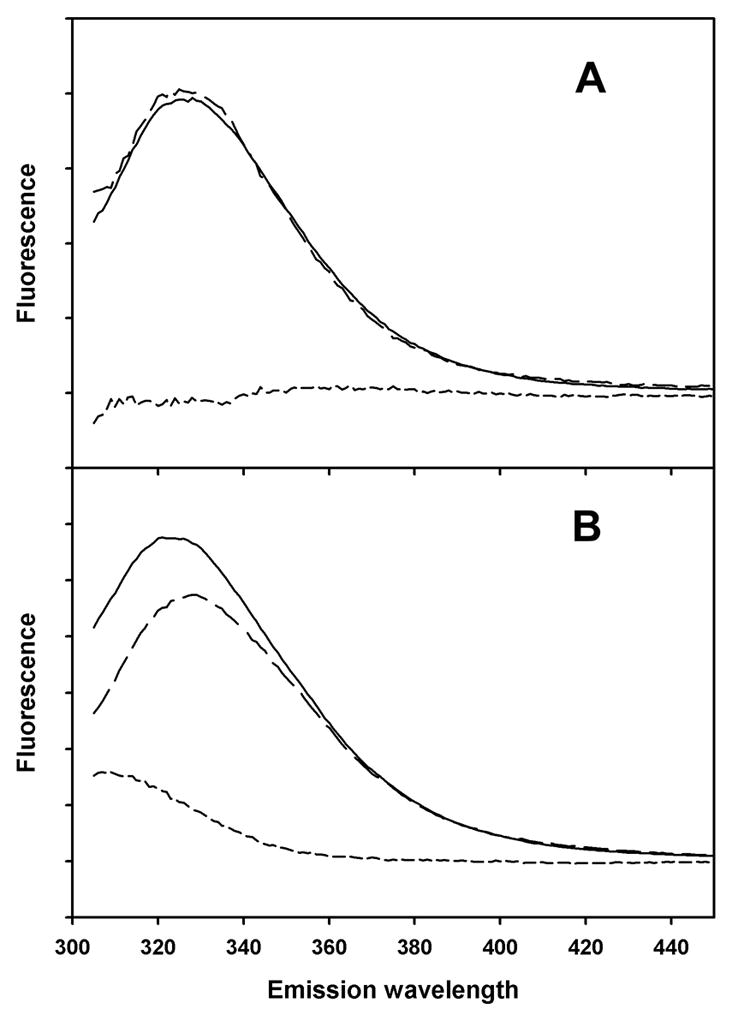
Solid lines: 280 nm excitation. Long dashed lines: 295 nm excitation, with emission scaled to same intensity in 390–400 nm region as from 280 nm excitation. Short dashed lines: difference between emission spectra from 280 nm and 295 nm excitation. A) Peptide AB in small unilamellar vesicles of DOPC. B) Peptide AB in SDS micelles.
Figure 3. Fluorescence emission spectra of peptide A.
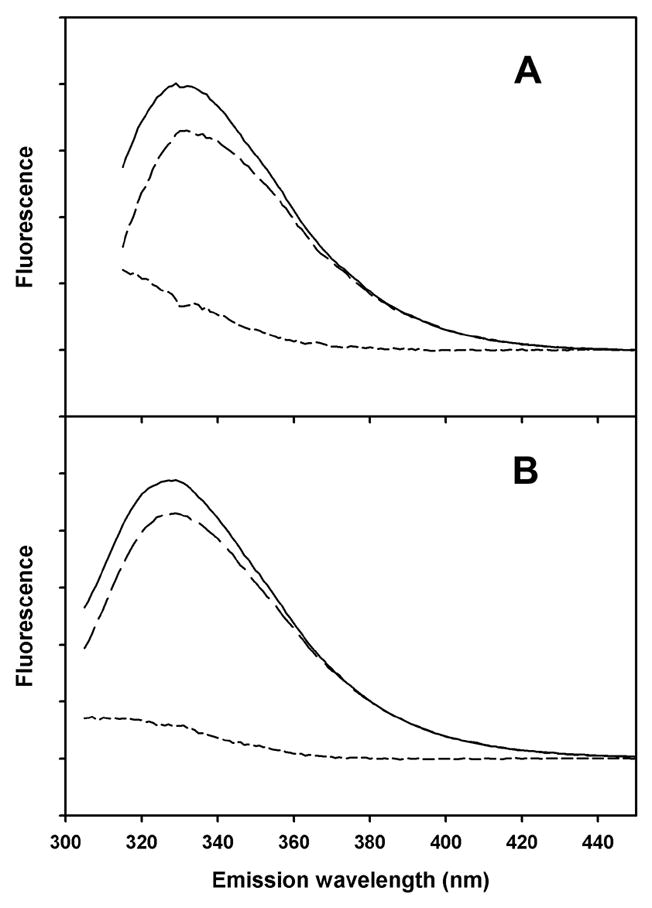
Solid lines: 280 nm excitation. Long dashed lines: 295 nm excitation, with emission scaled to same intensity in 390–400 nm region as from 280 nm excitation. Short dashed lines: difference between emission spectra from 280 nm and 295 nm excitation. A) Peptide A in small unilamellar vesicles of DOPC. B) Peptide A in SDS micelles.
Tyrosine-to-tryptophan energy transfer efficiencies were calculated from equation (1) and from the spectra in Figs. 2 and 3. The results are given in Table 2. The transfer efficiency in DOPC, 0.89 (Table 2), is substantially greater than expected from the maximum E calculated from the crystal structure coordinates, 0.60 (Table 1). This could be due to a difference between the structure of helices A and B in crystals of intact bacteriorhodopsin compared with the structure of the isolated two-helix AB peptide in DOPC bilayers. When the AB peptide is in SDS micelles, there is a dramatic decrease in Tyr-to-Trp FRET. The observed transfer efficiency in SDS is close to what would be expected if the A and B helices separate completely in SDS, eliminating FRET between Tyr 26 and Trp 10/Trp 12. The transfer efficiency observed for peptide A was about the same in DOPC and SDS (Table 2), which is expected, because the only tyrosine in a position to strongly interact with peptide A’s two tryptophans is on peptide B (Tyr 57). However, peptide A’s single tyrosine (Tyr 26) was predicted from the crystal structure (Table 1) to have much lower transfer efficiency in DOPC than what was observed, again raising a question about whether the structures in bilayers and micelles are different from the crystal structure.
Table 2.
Measured relative quantum yields and calculated energy transfer efficiencies
| Peptide | medium | Qo | fw (280 nm) | E |
|---|---|---|---|---|
| AB | DOPC | 0.961 | 0.649 | 0.889 |
| SDS | 0.582 | 0.53 | 0.111 | |
| A | DOPC | 0.916 | 0.881 | 0.29 |
| SDS | 0.844 | 0.772 | 0.32 |
Relative quantum yields of peptide emission (Qo) measured from Figs. 2 & 3. Energy transfer efficiency calculated from equation (1).
3.2 MD Simulation
In order to determine whether the crystal structure coordinates of bacteriorhodopsin are relevant for peptide AB in lipid bilayers and SDS micelles, we computationally modeled the structure of peptide AB in POPC and in SDS. The conformation of the AB fragment in the crystal structure of bacteriorhodopsin (Fig. 1, left) is similar to the conformation in POPC bilayers (Fig. 1, right, blue). The A and B helices do not separate in SDS micelles over a simulation time of 6 ns, and no large fluctuations occur, suggesting that intrusion of SDS monomers between the helices over a longer time period would be unlikely. However, the protein conformation in SDS is different from the crystal and belayed structures (Fig. 1, right, green). The backbone bends at Pro 50, causing the angle between helix A and helix B to increase from 24.6° to 30.2°. Also, the N-terminal end of helix B unwinds one helical turn. Nevertheless, these changes have little effect on the distances (R) between Tyr and Trp side chains, which differ from the crystal structure distances by only 1–2 Å (cf. Tables 1 & 3). We found larger differences in the dipole orientation factors, due to molecular motion. The range of orientation factors was calculated for a sample of frames from the simulations, and the resulting Förester distances (Ro) and energy transfer efficiencies (E) are shown in Table 3. In order to compare the measured and calculated transfer efficiencies, the individual tyrosine quantum yields must be known. It is possible that some of the tyrosine’s are quenched by interactions with near-by polar groups [35]. For example, in the belayed structure of AB, Tyr 43 is hydrogen-bonded to Lees 30, so it is unlikely to emit. Even if Tyr 57 is the only fluorescent Tyr of AB in lipid baitlayers, it is clear from the predicted individual transfer efficiencies (Table 3) that the measured energy transfer is higher than expected. The higher transfer efficiency in baitlayers could be explained by an unusually high quantum yield for Tyr 57 and unusually low quantum yields for the other Tyr groups. The results could also be explained by ant parallel dimidiation of peptide AB, which was previously observed in dimyristoylphosphatidylcholine bicelles [15]. In an ant parallel dimer, Tyr 26 and 43 would be close to Trp 10 and 12 on the opposite protomer, and thus the transfer efficiency would be higher than in a monomer. For peptide A in SDS, the transfer efficiency calculated for Tyr 26 based on MD simulation (Table 3) is closer to the measured result (Table 2) than the transfer efficiency calculated from the crystal structure (Table 1). However, the MD- calculated transfer efficiencies for peptide AB in SDS are even higher than for the crystal structure, and the discrepancy with the measured result is even larger.
Table 3.
Tyr to Trp energy transfer efficiencies calculated from molecular dynamics simulations
| Acceptor | Donor | κ2 | Ro | R | E | |
|---|---|---|---|---|---|---|
| DOPC | W10 | Y26 | 0.092 | 9.2 | 25.8 | 0.006 ± 0.013 |
| Y43 | 0.960 | 15.7 | 32.7 | 0.015 ± 0.011 | ||
| Y57 | 0.237 | 12.4 | 12.8 | 0.465 ± 0.164 | ||
| Y64 | 0.042 | 7.8 | 19.1 | 0.015 ± 0.027 | ||
| W12 | Y26 | 1.436 | 16.0 | 23.3 | 0.140 ± 0.102 | |
| Y43 | 2.370 | 18.8 | 30.0 | 0.058 ± 0.014 | ||
| Y57 | 0.088 | 10.2 | 10.7 | 0.439 ± 0.214 | ||
| Y64 | 0.197 | 11.5 | 21.7 | 0.033 ± 0.025 | ||
| SDS | W10 | Y26 | 0.092 | 9.1 | 27.1 | 0.004 ± 0.006 |
| Y43 | 0.136 | 10.0 | 35.6 | 0.001 ± 0.002 | ||
| Y57 | 0.394 | 13.6 | 13.9 | 0.470 ± 0.140 | ||
| Y64 | 0.148 | 10.2 | 20.7 | 0.040 ± 0.049 | ||
| W12 | Y26 | 1.7063 | 17.6 | 23.2 | 0.169 ± 0.065 | |
| Y43 | 2.515 | 19.0 | 31.5 | 0.047 ± 0.009 | ||
| Y57 | 0.240 | 12.0 | 9.8 | 0.700 ± 0.268 | ||
| Y64 | 0.843 | 15.8 | 24.4 | 0.070 ± 0.012 |
3.3 Tryptophan and tyrosine lifetimes
The emission kinetics of peptides AB, A and B were measured in an attempt to detect the fluorescence lifetimes of the tyrosine donors of peptide AB in the presence and absence of the tryptophan acceptors. Tyrosine emission, if detectable, would be present in 305 nm emission from 280 nm excitation, but absent in 360 nm emission from 280 nm excitation and absent in 305 nm emission from 295 nm excitation. The results are presented in Figure 4, as lifetime distributions calculated using the maximum entropy method. Due to instrument and sample properties, we could not reliably calculate lifetimes below 1 ns, preventing measurement of transfer efficiencies from fluorescence lifetimes. For peptide AB in DOPC baitlayers (Fig. 4A, solid line), the emission has broad lifetimes at about 3 ns and 5.5 ns. These are clearly due to tryptophan, as demonstrated by the two sharp groups of lifetimes observed when the emission is measured from 295 nm excitation (Fig. 4A, short dashed line), which only excites tryptophan. The narrowing of the lifetimes may involve the difference between excitation of both the 1La and 1Lb transitions at 280 nm and the exclusive excitation of the 1La transition at 295 nm. The lifetimes from 280 nm excitation are very well fit with two Gaussian curves (Fig. 4B, curves 2 and 3). Emission at 360 nm (Fig. 4C, solid line) shows two lifetimes similar to those observed at 305 nm, with the 5.5 ns component contributing more at 360 nm than at 305 nm. It is not clear whether the each lifetimes corresponds to a separate tryptophan in a particular environment or whether both tryptophans have mixtures of lifetimes. In the MD simulation, the structure of peptide AB shows Trp 10 in close contact with Thr 5, whereas Trp 12 is in a non-polar environment. Peptide B is useful for analyzing tyrosine lifetimes in the absence of tryptophan acceptors. Three of the four tyrosine groups of peptide AB are on peptide B and none of the tryptophans. Thus, the emission from peptide B (Fig. 4D) is exclusively from tyrosine, and no energy transfer to tryptophan is possible. Two broad groups of lifetimes are observed: a large amplitude group at about 4.5 ns, and a smaller amplitude group at about 2.5 ns. This distribution of lifetimes is similar to curve 5 in Fig. 4B, which is the small difference between the Gaussian curves representing the tyrptophan lifetimes (Fig. 4B, curve 4) and the observed lifetime distribution (Fig. 4B, curve 5). This result provides some insight into the unexpectedly large amount of tyrosine-to-tryptophan FRET observed (Fig. 2A, Table 2). If the larger-than-expected FRET is caused by ant parallel dimers, then curve 5 might represent monomers, having unquenched tyrosine’s, in equilibrium with the dimer. On the other hand, the absence of tyrosine emission in Fig. 2A would be explained if all tyrosine’s were quenched through interactions with polar groups, instead of transfering their excitation to tryptophans via FRET. However, if this were the case, then the unquenched form would not be detectable (formation and breaking of polar interactions would be faster than the fluorescence lifetime). Thus, curve 5 can be interpreted as additional evidence for ant parallel dimer formation. In SDS micelles, the main component of peptide B lifetimes shifts to about 3 ns (dashed line, Fig. 4D). This is also a major component of the emission kinetics of peptide AB in SDS micelles (long dashed line, Fig. 4A). In SDS, peptide AB excited at 295 has one main lifetime component at 4.5 ns (not shown), due to tryptophan. Thus, the large component of lifetime amplitudes observed in SDS in the 2 to 5 ns range must correspond to a mixture of tryptophan and unquenched tyrosine emission. This suggests that, in SDS, the quenching that occurs in DOPC baitlayers has disappeared, presumably due to dissociation of dimers. The transfer of peptide AB from a belayed environment to SDS micelles was previously shown to cause a decrease in the protein absorbance [25] and also a decrease in the fluorescence quantum yield [36]. The fluorescence lifetime differences between DOPC baitlayers and SDS micelles (Fig. 4A,C) are consistent with this, showing changes to shorter lifetimes in SDS.
Figure 4. Fluorescence lifetimes.

Lifetimes calculated from emission kinetics using maximum entropy method. A) Emission at 305 nm from peptide AB: solid line, in DOPC baitlayers excited at 280 nm; long dashed line, in SDS micelles, excited at 280 nm; short dashed line, in DOPC baitlayers excited at 295 nm. B) Gaussian curves (lines 2 and 3) fit to lifetimes of peptide AB in DOPC baitlayers excited at 280 nm (line 1). Line 4: sum of lines 2 and 3. Line 5: difference between lines 1 and 4. C) Emission at 360 nm from peptide AB: solid line, in DOPC baitlayers excited at 280 nm; long dashed line, in SDS micelles, excited at 280 nm. D) Emission at 305 nm from peptide B: solid line, in DOPC baitlayers excited at 280 nm; long dashed line, in SDS micelles, excited at 280 nm.
3.4 Tyrosine environments
The results in Fig. 4 do not explain why so little tyrosine-to-tryptophan FRET is observed in SDS micelles. We looked closely at the tyrosine environments in the MD simulations and found that in lipid baitlayers, Tyr 57 is surrounded by hydrocarbon chains from the lipids (Fig. 5A). By contrast, in SDS the phenol OH of Tyr 57 is tightly hydrogen-bonded to water molecules as part of a hydrogen-bonded chain (Fig. 5B). Therefore, the most likely explanation of the low FRET in SDS is that Tyr 57 is quenched by a chemical interaction with water.
Figure 5. Tyrosine 57 environments in baitlayers and SDS micelles.

Peptide AB backbone helices shown in red; Tyr 57 van der Waals surface shown in gray (carbon), white (hydrogen) and red (oxygen). Solvent accessible surface shown in tan. A) Last frame from MD simulation of peptide AB in POPC belayed. No water molecules are within 3.5 Å of Tyr 57. B) Last frame from MD simulation of peptide AB in SDS micelle. Four water molecules (cyan) are within 3.5 Å of Tyr 57, forming a H-bonded chain that includes the phenol oxygen. Images prepared using Jmol (http://www.jmol.org/).
3.5 Tryptophan environments
The fluorescence lifetime differences between DOPC baitlayers and SDS micelles (Fig. 4A,C) could be due, in part, to the peptide AB conformational change that was indicated by the MD simulation. This change includes distortion of the backbone and a change in interhelix angle, altering the environment of Trp 12. In baitlayers, a string of aliphatic side chains (Ile 11, Leu 15, 19 and 22) interact, locking the two tryptophans into a roughly parallel arrangement, sandwiching Ile 11 between them. In SDS, the bending of the backbone moves the aliphatic chains further apart, releasing Ilea 11 from between the tryptophans and allowing Trp 12 to rotate 90°. This puts Trp 12 more in contact with SDS molecules than its conformation in baitlayers. If the conformational change lags behind the dissolution of the vesicles, the tryptophan fluorescence may report both the change of environment and also the peptide AB conformational change. Membrane protein conformational changes were previously detected primarily on a time scale of seconds when intact bacteriorhodopsin was transferred from a lipid environment to SDS micelles [2]. In contrast, micelle formation typically occurs in microseconds. Therefore, we used stopped flow fluorometry to compare the rate of vesicle dissolution to the rate of protein adjustment after transfer to SDS.
The results of stopped flow mixing of vesicles with SDS are shown in Fig. 6. The rate of vesicle dissolution in SDS was measured by the change in FRET between NBD-PE and lissamine rhodamine B-PE [37] after the vesicles dissolve (Fig. 6B). The kinetics were fit with two exponentials with rate constants of 110±7 sec−1 and 12.4±1.7 sec−1 in approximately a 2:1 amplitude ratio. The kinetics of the change in peptide AB fluorescence (Fig. 6A) were fit with two exponentials with rate constants of 141±21 sec−1 and 11.8±1.7 sec−1 in approximately a 1:1 amplitude ratio. Thus, both rate processes that are observed when the vesicles dissolve are reflected in changes in the peptide AB fluorescence that occur with essentially the same kinetics. Therefore, the quantum yield changes we observe in peptide AB are most likely due to changes the SDS environment, rather than large conformational changes of the peptide.
Figure 6. Kinetics of DOPC belayed dissolution in SDS micelles.

A) Change in peptide AB fluorescence (excitation, 285 nm; emission, >290 nm) after mixing equal volumes of DOPC baitlayers with 0.4% SDS. B) Change in NBD-PE fluorescence (excitation, 470 nm; emission 520 nm). Dark current gives a signal of -0.07 V. Vesicles alone under the conditions in A) give a constant signal at 0.14 V. Data in B) is offset by 0.25 V for clarity.
4. Conclusions
In conclusion, we find from MD simulation no evidence for SDS-induced separation of the two transmembrane helices of the AB peptide. Instead, there is a small rearrangement of the protein backbone in SDS micelles. Because the SDS micelle structure is more open than a lipid belayed, water molecules are able to penetrate into the micelle, and several waters form a strong H-bonded interaction with Tyr 57. This interaction probably decreases the quantum yield of Tyr 57 in SDS, which results in the disappearance of tyrosine to tryptophan FRET. The presence of water molecules interacting with Tyr 57 might be detected by resonance Raman spectroscopy [38].
This study highlights a limitation of FRET. When only one type of donor-acceptor pair is used to probe a structural change, environmental effects could be misinterpreted as changes in proximity. A recent report on the effects of SDS on helix-helix interactions of transmembrane peptides may be an example of this problem [5]. FRET was measured between a fluorescein donor and a tetramethylrhodamine acceptor on dimers of glycophorin transmembrane helices in dodecyl maltoside micelles. With increasing amounts of added SDS, the observed level of FRET declined, which was interpreted as evidence for SDS-induced helix separation. However, the observed change might be an environmental effect caused by the increased surface charge density in the mixed micelles containing higher mole fractions of negatively charged SDS. Both the FRET donor and acceptor are known to be sensitive to environmental effects. The quantum yield of fluorescein decreases dramatically below pH 7 [39], and tetramethylrhodamine self-quenches by dimidiation [40]. In studies of structural differences induced by environmental changes, it would be a good practice to corroborate FRET changes either by using several donor-acceptor pairs having different chemical properties, or by measuring additional physical properties.
Acknowledgments
This work was supported by grants from NIH (GM008194 and GM084834) and the University of Texas at San Antonio Collaborative Research Seed Grant Program. We thank Erica Burns for help with some of the experiments.
Abbreviations
- AB
peptide containing amino acids 1–71 of bacteriorhodopsin
- DOPC
1,2-dioleoyl-sn-glycero-3-phosphocholine
- FRET
Förster resonance energy transfer
- MD
molecular dynamics
- POPC
1-palmitoyl–2-oleoyl-sn-glycero-3-phosphocholine
- SDS
sodium dodecyl sulfate
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Faham S, Yang D, Bare E, Yohannan S, Whitelegge JP, Bowie JU. Side-chain contributions to membrane protein structure and stability. J Mol Biol. 2004;335:297–305. doi: 10.1016/j.jmb.2003.10.041. [DOI] [PubMed] [Google Scholar]
- 2.Curnow P, Booth PJ. Combined kinetic and thermodynamic analysis of alpha-helical membrane protein unfolding. Proc Natl Acad Sci U S A. 2007;104:18970–18975. doi: 10.1073/pnas.0705067104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.MacKenzie KR, Prestegard JH, Engelman DM. A transmembrane helix dimer: structure and implications. Science. 1997;276:131–133. doi: 10.1126/science.276.5309.131. [DOI] [PubMed] [Google Scholar]
- 4.Tulumello DV, Deber CM. SDS micelles as a membrane-mimetic environment for transmembrane segments. Biochemistry. 2009;48:12096–12103. doi: 10.1021/bi9013819. [DOI] [PubMed] [Google Scholar]
- 5.Anbazhagan V, Cymer F, Schneider D. Unfolding a transmembrane helix dimer: A FRET study in mixed micelles. Arch Biochem Biophys. 2010;495:159–164. doi: 10.1016/j.abb.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 6.Riley ML, Wallace BA, Flitsch SL, Booth PJ. Slow alpha helix formation during folding of a membrane protein. Biochemistry. 1997;36:192–196. doi: 10.1021/bi962199r. [DOI] [PubMed] [Google Scholar]
- 7.Lau FW, Bowie JU. A method for assessing the stability of a membrane protein. Biochemistry. 1997;36:5884–5892. doi: 10.1021/bi963095j. [DOI] [PubMed] [Google Scholar]
- 8.Chen GQ, Gouaux E. Probing the folding and unfolding of wild-type and mutant forms of bacteriorhodopsin in micellar solutions: evaluation of reversible unfolding conditions. Biochemistry. 1999;38:15380–15387. doi: 10.1021/bi9909039. [DOI] [PubMed] [Google Scholar]
- 9.Otzen DE. Folding of DsbB in mixed micelles: a kinetic analysis of the stability of a bacterial membrane protein. J Mol Biol. 2003;330:641–649. doi: 10.1016/s0022-2836(03)00624-7. [DOI] [PubMed] [Google Scholar]
- 10.Renthal R. An unfolding story of helical transmembrane proteins. Biochemistry. 2006;45:14559–14566. doi: 10.1021/bi0620454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pan Y, Brown L, Konermann L. Mapping the structure of an integral membrane protein under semi-denaturing conditions by laser-induced oxidative labeling and mass spectrometry. J Mol Biol. 2009;394:968–981. doi: 10.1016/j.jmb.2009.09.063. [DOI] [PubMed] [Google Scholar]
- 12.Adair BD, Engelman DM. Glycophorin A helical transmembrane domains dimerize in phospholipid baitlayers: a resonance energy transfer study. Biochemistry. 1994;33:5539–5544. doi: 10.1021/bi00184a024. [DOI] [PubMed] [Google Scholar]
- 13.Choma C, Gratkowski H, Lear JD, DeGrado WF. Asparagine-mediated self-association of a model transmembrane helix. Nat Struct Biol. 2000;7:161–166. doi: 10.1038/72440. [DOI] [PubMed] [Google Scholar]
- 14.Yano Y, Takemoto T, Kobayashi S, Yasui H, Sakurai H, Ohashi W, Niwa M, Futaki S, Sugiura Y, Matsuzaki K. Topological stability and self-association of a completely hydrophobic model transmembrane helix in lipid baitlayers. Biochemistry. 2002;41:3073–3080. doi: 10.1021/bi011161y. [DOI] [PubMed] [Google Scholar]
- 15.Nannepaga SJ, Gawalapu R, Velasquez D, Renthal R. Estimation of helix-helix association free energy from partial unfolding of bacterioopsin. Biochemistry. 2004;43:550–559. doi: 10.1021/bi034875c. [DOI] [PubMed] [Google Scholar]
- 16.You M, Li E, Wimley WC, Hristova K. Forster resonance energy transfer in liposomes: measurements of transmembrane helix dimidiation in the native belayed environment. Anal Biochem. 2005;340:154–164. doi: 10.1016/j.ab.2005.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Eisenhawer M, Cattarinussi S, Kuhn A, Vogel H. Fluorescence resonance energy transfer shows a close helix-helix distance in the transmembrane M13 procoat protein. Biochemistry. 2001;40:12321–12328. doi: 10.1021/bi0107694. [DOI] [PubMed] [Google Scholar]
- 18.Oesterhelt D, Stoeckenius W. Isolation of the cell membrane of Halobacterium halobium and its fractionation into red and purple membrane. Methods Enzymol. 1974;31:667–678. doi: 10.1016/0076-6879(74)31072-5. [DOI] [PubMed] [Google Scholar]
- 19.Gerber GE, Anderegg RJ, Herlihy WC, Gray CP, Biemann K, Khorana HG. Partial primary structure of bacteriorhodopsin: sequencing methods for membrane proteins. Proc Natl Acad Sci U S A. 1979;76:227–231. doi: 10.1073/pnas.76.1.227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marti T. Refolding of bacteriorhodopsin from expressed polypeptide fragments. J Biol Chem. 1998;273:9312–9322. doi: 10.1074/jbc.273.15.9312. [DOI] [PubMed] [Google Scholar]
- 21.Sigrist H, Wenger RH, Kislig E, Wuthrich M. Refolding of bacteriorhodopsin. Protease V8 fragmentation and chromophore reconstitution from proteolytic V8 fragments. Eur J Biochem. 1988;177:125–133. doi: 10.1111/j.1432-1033.1988.tb14352.x. [DOI] [PubMed] [Google Scholar]
- 22.Batzri S, Korn ED. Single belayed liposomes prepared without sonication. Biochim Biophys Acta. 1973;298:1015–1019. doi: 10.1016/0005-2736(73)90408-2. [DOI] [PubMed] [Google Scholar]
- 23.Eisinger J. Intramolecular energy transfer in adrenocorticotropin. Biochemistry. 1969;8:3902–3908. doi: 10.1021/bi00838a004. [DOI] [PubMed] [Google Scholar]
- 24.Pace CN, Vajdos F, Fee L, Grimsley G, Gray T. How to measure and predict the molar absorption coefficient of a protein. Protein Sci. 1995;4:2411–2423. doi: 10.1002/pro.5560041120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Renthal R, Haas P. Effect of transmembrane helix packing on tryptophan and tyrosine environments in detergent-solubilized bacterio-opsin. J Protein Chem. 1996;15:281–289. doi: 10.1007/BF01887117. [DOI] [PubMed] [Google Scholar]
- 26.Luecke H, Schobert B, Richter HT, Cartailler JP, Lanyi JK. Structure of bacteriorhodopsin at 1.55 A resolution. J Mol Biol. 1999;291:899–911. doi: 10.1006/jmbi.1999.3027. [DOI] [PubMed] [Google Scholar]
- 27.Kaylor J, Bodner N, Edridge S, Yamin G, Hong DP, Fink AL. Characterization of oligomeric intermediates in alpha-synuclein fibrillation: FRET studies of Y125W/Y133F/Y136F alpha-synuclein. J Mol Biol. 2005;353:357–372. doi: 10.1016/j.jmb.2005.08.046. [DOI] [PubMed] [Google Scholar]
- 28.Braun R, Engelman DM, Schulten K. Molecular Dynamics Simulations of Micelle Formation around Dimeric Glycophorin A Transmembrane Helices. Biophys J. 2004;87:754–763. doi: 10.1529/biophysj.104.040279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–38. 27–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 30.Kucerka N, Tristram-Nagle S, Nagle JF. Structure of fully hydrated fluid phase lipid baitlayers with monounsaturated chains. J Membr Biol. 2005;208:193–202. doi: 10.1007/s00232-005-7006-8. [DOI] [PubMed] [Google Scholar]
- 31.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacKerell AD, Bashford D, Bellott, Dunbrack RL, Evanseck JD, Field MJ, Fischer S, Gao J, Guo H, Ha S, Joseph-McCarthy D, Kuchnir L, Kuczera K, Lau FTK, Mattos C, Michnick S, Ngo T, Nguyen DT, Prodhom B, Reiher WE, Roux B, Schlenkrich M, Smith JC, Stote R, Straub J, Watanabe M, Wiorkiewicz-Kuczera J, Yin D, Karplus M. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 33.Feller SE, MacKerell AD. An improved empirical potential energy function for molecular simulations of phospholipids. J Phys Chem B. 2000;104:7510–7515. [Google Scholar]
- 34.Pervushin KV, Orekhov V, Popov AI, Musina L, Arseniev AS. Three-dimensional structure of (1–71)bacterioopsin solubilized in methanol/chloroform and SDS micelles determined by 15N-1H heteronuclear NMR spectroscopy. Eur J Biochem. 1994;219:571–583. doi: 10.1111/j.1432-1033.1994.tb19973.x. [DOI] [PubMed] [Google Scholar]
- 35.Noronha M, Lima JC, Lamosa P, Santos H, Maycock C, Ventura R, Macanita AL. Intramolecular fluorescence quenching of tyrosine by the peptide alpha-carbonyl group revisited. J Phys Chem A. 2004;108:2155–2166. [Google Scholar]
- 36.Luneberg J, Widmann M, Dathe M, Marti T. Secondary structure of bacteriorhodopsin fragments. External sequence constraints specify the conformation of transmembrane helices. J Biol Chem. 1998;273:28822–28830. doi: 10.1074/jbc.273.44.28822. [DOI] [PubMed] [Google Scholar]
- 37.Struck DK, Hoekstra D, Pagano RE. Use of resonance energy transfer to monitor membrane fusion. Biochemistry. 1981;20:4093–4099. doi: 10.1021/bi00517a023. [DOI] [PubMed] [Google Scholar]
- 38.Asher SA, Chi ZH. UV Raman determination of the environment and solvent exposure of Tyr and Trp residues. Journal of Physical Chemistry B. 1998;102:9595–9602. [Google Scholar]
- 39.Whitaker JE, Haugland RP, Ryan D, Hewitt PC, Prendergast FG. Fluorescent rhodol derivatives: versatile, photostable labels and tracers. Anal Biochem. 1992;207:267–279. doi: 10.1016/0003-2697(92)90011-u. [DOI] [PubMed] [Google Scholar]
- 40.Packard BZ, Toptygin DD, Komoriya A, Brand L. Profluorescent protease substrates: intramolecular dimers described by the exciton model. Proc Natl Acad Sci U S A. 1996;93:11640–11645. doi: 10.1073/pnas.93.21.11640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Koradi R, Billeter M, Wuthrich K. MOLMOL: a program for display and analysis of macromolecular structures. J Mol Graph. 1996;14:51–55. 29–32. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]