

Abstract

A rapid, general route to enantiopure β-fluoroamines and β,β-difluoroamines has been developed employing organocatalysis in both a two-pot and a one-pot procedure. Both chemical yields (64–82%) and enantioselectivity (94–98% ee) were excellent and represent a significant improvement in the art of preparing chemically diverse β-fluoroamines from readily available precursors.
The incorporation of β-fluoroamines into drug candidates has increased dramatically in the past five years, with >150 fluorinated drug candidates in Phase II and Phase III clinical trials.1 The role of the β-fluorine atom is diverse and has been shown to enhance binding interactions, improve metabolic stability, increase CNS penetration and eliminate ancillary ion channel activity by attenuating amine basicity (pKa).1–5 The inductive effects of a β-fluorine atom are pronounced, lowering the pKa of a linear aliphatic amine (pKa ~10.7) to pKa ~9.0, with a single β-flourine to pKa ~7.3 with β,β-difluoro substitution. These effects are general and additive, with a β-CF3 moiety lowering the pKa to ~5.7.1–6
Despite the importance of the β-fluoroamine moiety, there are few synthetic methods in the literature for their preparation.1–7 Two common methods, the ring opening of aziridines with nucleophilic flouride sources8 and the hydrofluorination of olefins,9 deliver β-fluoroamines, but lack generality/substrate scope, require starting materials that are not readily available or, in the latter case, do not provide access to enantiopure β-fluoroamines. The route most utilized involves the treatment of ketones or secondary alcohols with DAST, (diethylamino)sulfur trifluoride, to provide β,β-difluoroamine and β-fluoroamines (with inversion of stereochemistry), respectively.1–10 However, this methodology requires the synthesis of enantiopure secondary alcohols and then suffers from the formation of rearranged and dehydrated products, which, in many published cases, greatly diminished yields of the desired β-fluoroamines.10 In this Letter, we describe general and high-yielding protocols to deliver enantiopure β-fluoroamines and β,β-difluoroamines employing organocatalysis and readily available starting materials.
We were attracted to the classical MacMillan work11 where organocatlysis was employed to deliver enantioselective (91–99% ee) α-fluoroaldehydes 4 by treatment of an aldehyde 1 with NFSI (N-fluorobenzenesulfonamide) 2 and catalytic chiral imidazolidinone ligand 3 (Scheme 1).
Scheme 1.

Organocatalytic α-fluorination of aldehydes
If these chiral α-fluoroaldehydes 4 were subjected to a reductive amination protocol, we surmised that chiral β-fluroamines would result, and depending on the chirality of the imidazolidinone ligand, either the (S)- or (R)-β-fluoroamine would be delivered. Moreover, there are thousands of commercially available amines and aldehydes to employ as reactants, providing improved generality in terms of substrate scope. Surprisingly, this powerful extension of the MacMillan enantioselective α-fluorination of aldehydes has never been described.
We first explored the effect of NFSI equivalents, solvent and temperature on a prototypical α-fluorination reaction and susequent reductive amination sequence employing organocatalyst 3 with phenylpropanal 5 and Bocpiperazine 6 to produce chiral β-fluoroamine 7 (Table 1). Our initial attempt (entry 1) employed the conditions prescribed by MacMillan11 for the α-fluorination, with a quick aqueous work-up prior to the reductive amination step. Conversion and enantioselectivity to the desired β-fluoroamine were excellent, but about 20% of the undesired β,β-difluoroamine was observed. To avoid this side product, we decreased the equivalents for NFSI from 5.0 to 3.0 (entry 2), to 2.0 (entry 3) and finally 1.5 (entry 4). Only in the latter case, was the β,β-difluoroamine side product eliminated; moreover, the success of the α-fluorination was not hindered by decreasing the equivalents of costly NFSI and work-up was greatly improved. Other solvent systems were evaluated, with acetone (entry 6) proving to be generally useful, while CH2Cl2 (entry 7) suffered diminished yields (37%) and low ee (84%). Ultimately, optimal conditions for the two step sequence (entry 10) employed 1.0 equivalent of NFSI in THF at −20 °C for 24 hours, followed by a quick aqueous work-up, suspension of the resulting α-fluoroaldehyde in ClCH2CH2Cl with 6 and NaB(OAc)3H at ambient temeparture to provide enantiopure (>99% ee) 7 with 96% conversion and 80% isolated yield.
Table 1.
Optimization of organocatalytic α-flourination/ reductive amination sequence.
 | ||||||
|---|---|---|---|---|---|---|
| entrya |
2 (equiv) |
solvent | temp (°C) |
time (h) |
convn (%)d |
ee (%)b |
| 1c | 5.0 | THF | −20 | 24 | 99 | >98 |
| 2c | 3.0 | THF | −20 | 24 | 98 | >98 |
| 3c | 2.0 | THF | −20 | 24 | 99 | >99 |
| 4 | 1.5 | THF | 24 | 3 | 97 | >98 |
| 5 | 1.5 | THF | 4 | 12 | 98 | >98 |
| 6 | 1.5 | acetone | 24 | 3 | 91 | >96 |
| 7 | 1.5 | CH2Cl2 | 24 | 3 | 37 | 84 |
| 8 | 1.5 | EtOAc | 24 | 3 | 89 | 95 |
| 9 | 1.2 | THF | −20 | 24 | 99 | >99 |
| 10 | 1.0 | THF | −20 | 24 | 96 | >99 |
All reactions were performed on a 0.05 mmol scale.
Enantiomeric excess were determined using chiral stationary phase HPLC
α,α-difluoro product was observed.
Conversion determined by LC/MS and 1H NMR.
See Supporting Information for complete details.
As shown in Tables 2 and 3, this two-pot protocol is general with respect to the amine component providing yields from 65–82% employing both primary and secondary amines, which include therapeutically relevant GPCR privileged structures.12 Importantly, the (S)- imidazolidinone catalyst 3 provides the corresponding (S)-β-fluoroamines 7a-f in 95–>99% ee (Table 2), while the (R)-imidazolidinone catalyst 3 provides the corresponding (R)-β-fluoroamines 8a–e in 87–>95% ee (Table 3).
Table 2.
Scope of HNR1R2 in enantioselective synthesis of (S)-β-fluoroamines.
 | |||
|---|---|---|---|
| compda | product | yield (%)b | ee (%)c |
| 7a |  |
80 | >99 (S) |
| 7b |  |
70 | >98 (S) |
| 7c |  |
76 | >99 (S) |
| 7d |  |
69 | >98 (S) |
| 7e |  |
65 | >96 (S) |
| 7f | 82 | >95 (S) | |
All reactions were performed on a 0.5 mmol scale and proceeded to complete conversion.
Yield after chromatography.
Enantiomeric excess were determined using chiral stationary phase
Table 3.
Scope of HNR1R2 in enantioselective synthesis of (R)-β-fluoroamines.
 | |||
|---|---|---|---|
| compda | product | yield (%)b | ee (%)c |
| 8a |  |
70 | >98 (R) |
| 8b |  |
80 | >87 (R) |
| 8c |  |
76 | >98 (R) |
| 8d |  |
69 | >94 (R) |
| 8e | 65 | >96 (R) | |
All reactions were performed on a 0.05 mmol scale and proceeded to complete conversion.
Yield after chromatography.
Enantiomeric excess were determinedd using chiral stationary phase HPLC..
See Supporting Information for more details.
At this point, we wanted to explore the scope of this new methodology to produce β-fluoroamines employing alternative aldehydes and to determine if this new strategy would allow access to tertiary β-fluoroamines – a moiety inaccessible through DAST and other known chemistries.8–10 We envisioned that these transformations might require alternative organocatalysts to (S)- and (R)-3, so we assembled a catalyst screening kit comprised of 9–13 for evaluation (Figure 1).
Figure 1.

Organocatalysts examined in determining scope of β-fluoroamine synthesis.
As shown in Table 4, the methdology appears general with respect to both aldehyde and amine component, affording chiral β-fluoramines in good isolated yields (58–92%) and with high enantioselectivities (>96%ee). If the aldehyde bears a β-stereogenic center, as in 15f, the β-fluoroine is still installed with high ee (>95%), but with a 1.2:1 dr. Compounds 15h and 15i represent transformations that all known standard β-fluoroamine methodology can not produce – tertiary β-fluoroamines. 8–10 Standard conditions with (S)-3 provide excellent chemical yield for installation of the tertiary β-fluoroamine, but suffers from low enantioselectivity (15% ee). Catalysts 9, 10 and 13 provided poor conversion (<25%) and low %ee. Like (S)-3, catalysts 11 and 12 installed the tertiary β-fluoroamine in good chemical yields, but the maximum ee observed was 31% with catalyst 11. A similar trend was observed in the synthesis of tertiary β-fluoroamine 15i. Our standard methodolgy with (S)-3 provided good conversion, but only 17% ee. Switching to the tetrazole catalyst 11 provided the desired tertiary β-fluoroamine 15i in comparable yield, but with improved ee (40%). Thus, our new methodology allows access to tertiary β-fluoroamines, and with modest % ee, as opposed to existing methods which are unable to install tertiary β-fluoroamines.
Table 4.
Scope of β-fluoroamine synthesis via organocatalysis.
 | ||||
|---|---|---|---|---|
| compda | product | cat. | yield (%)b | ee (%)c |
| 15a |  |
(S)-3 | 87 | >99 |
| 15b |  |
(S)-3 | 92 | >98 |
| 15c | 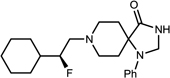 |
(S)-3 | 90 | >96 |
| 15d | 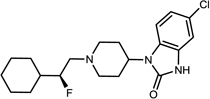 |
(S)-3 | 84 | >99 |
| 15e |  |
(S)-3 | 88 | >99 |
| 15f |  |
(S)-3 | 75 | >95 1.2:1 dre |
| 15g | (S)-3 | 58 | ndd | |
| 15h |  |
(S)-3 | 93 | 15 |
| 9 | 25 | 13 | ||
| 10 | <10 | ndd | ||
| 11 | 94 | 31 | ||
| 12 | 83 | 16 | ||
| 13 | 16 | 12 | ||
| 15i |  |
(S)-3 | 74 | 17 |
| 11 | 75 | 40 | ||
All reactions were performed on a 0.05 mmol scale and proceeded to complete conversion.
Yield after chromatography.
Enantiomeric excess were determined using chiral stationary phase HPLC.
Not determined.
Diastereomer ratios were measured by 19F NMR.
See Supporting Information for complete details.
Our attention now turned to developing a one-pot organocatalytic approach to β-fluoroamines to avoid the aqueous work-up step. As shown in Table 5, this was smoothly accomplished by application of 1.0 equivalent of NFSI, at −20 °C for 3 hours in 10% i-PrOH/THF, followed by direct addition of 1.0 equivalent of mono-Boc-piperazine 6 and 2.2 equivalents of NaB(OAc)3H for 16 hours. The desired β-fluoroamine 7 was delivered in 65% isolated yield and with >96% ee. The Boc protecting groups remained in tact, highlighting the mild conditions of this one-pot procedure.
Table 5.
One-pot β-fluoroamine synthesis via organocatalysis
 | |||||
|---|---|---|---|---|---|
| entrya | solvent | temp (°C) |
time (h) |
yield (%)c |
ee (%)b |
| 1 | THF | 24 | 3 | 45 | >95 |
| 2 | CH3CN | 24 | 3 | 36 | >95 |
| 3 | CH2Cl2 | 24 | 24 | 12 | ndd |
| 4 | DCE | 24 | 24 | 0 | ndd |
| 5 | THF/i-PrOH | −20 | 3 | 65 | >96 |
All reactions were performed on a 0.05 mmol scale.
Enantiomeric excess were determined using chiral stationary phase HPLC.
Yield after chromatography.
Not determined.
Finally, there are many examples in the literature where a β,β-difluoroamine is required to address a specific liability of a candidate molecule.1–6 Based on an earlier observation of β,β-difluoroamine formation (Table 1) when excess NFSI was employed, we quickly developed a two-pot route to rapidly access this valuable moiety. In this instance (Table 6), aldehydes 16 were treated with two equivalents of NFSI, 40 mol% DL-proline in 10% i-PrOH/THF at room tempertaure for 24 hours to provide α,α-difluoroaldehydes 17. After a quick aqueous work-up, the resulting oils were resuspended in DCE, amine and NaB(OAc)3H were added, and the reaction was allowed to stir at room temperature for 16 hours to provide β,β-difluoroamines 18 in isolated yields ranging from 64–77%.
Table 6.
β,β-difluoroamine synthesis via organocatalysis
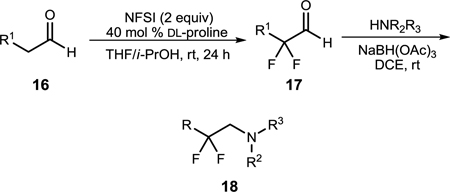




All reactions were performed on a 0.05 mmol scale and proceeded to complete conversion.
Yield after chromatography
In summary, we have developed a powerful extension of the MacMillan enantioselective α-fluorination of aldehydes for the general enantioselective synthesis of β-fluoroamines in yields and % ee far exceeding that of any other reported method, and without rearranged and dehydrated side products. Moreover, our new methodology allows for the first synthesis of tertiary β-fluoroamines with enantioselectivities up to 40%. Furthermore, slight modification of our protocol provides rapid, high yielding access to β,β-difluoroamines. Overall, these novel strategies for the synthesis of β-fluoroamines and β,β-difluoroamines, from readily available precursors, represents a significant improvement in the art to access these therapeutically relevant moieties.
Supplementary Material
Acknowledgment
This work was supported, in part, by the Department of Pharamacology and Vanderbilt University. Funding for the NMR instrumentation was provided in part by a grant from NIH (S10 RR019022).
Footnotes
Supporting Information Available: Experimental procedures, characterization data, chiral LC traces and 1H, 19F and 13C NMR spectra for all new compounds, 7a–f, 15a–i and 18a–d.
References
- 1.Hagmann WK. J. Med. Chem. 2008;51:4359–4369. doi: 10.1021/jm800219f. [DOI] [PubMed] [Google Scholar]
- 2.Muller K, Mainfisch P, Altmann KH, Schlosser M. ChemBioChem. 2004;5:559–572. [Google Scholar]
- 3.Bohm H-J, Banner D, Bendels S, Kansy M, Kuhn B, Muller K, Obst-Sander U, Stahl M. ChemBioChem. 2004;5:637–643. doi: 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]
- 4.Kirk KL. Curr. Top. Med. Chem. 2006;6:1445–1543. doi: 10.2174/156802606777951073. [DOI] [PubMed] [Google Scholar]
- 5.Kirk KL. Curr. Top. Med. Chem. 2006;6:1013–1029. doi: 10.2174/156802606777951073. [DOI] [PubMed] [Google Scholar]
- 6.Morgenthaler M, Schweizer E, Hoffman-Roder F, Benini F, Martin RE, Jaeschke G, Wagner B, Fischer H, Bendels S, Zimmerili D, Schneider J, Hiedrich F, Kansy M, Muller K. ChemMedChem. 2007;2:1100–1115. doi: 10.1002/cmdc.200700059. [DOI] [PubMed] [Google Scholar]
- 7.Percy JM. Sci. Synth. 2005;34:379–416. [Google Scholar]
- 8.Alvenrhe GM, Lacombe S, Laurent AJ. Tetrahderon Lett. 1980;21:289–293. [Google Scholar]
- 9.Thiabaudeau S, Martin-Mingot A, Jouannetaud M-P, Karam O, Zunino F. Chem. Commun. 2007:3198–3200. doi: 10.1039/b703629a. [DOI] [PubMed] [Google Scholar]
- 10.Hsin L-W, Chang L-T, Rothman RB, Dersch CM, Jacobson AE, Rice KC. J. Med. Chem. 2008;51:2795–2806. doi: 10.1021/jm701270n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beeson TD, MacMillan DWC. J. Am.Chem. Soc. 2005;127:8826–8828. doi: 10.1021/ja051805f. [DOI] [PubMed] [Google Scholar]
- 12.Patchett AA, Nargund R. Ann. Reports Med. Chem. 2000;35:289–298. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.