

Abstract
This Letter describes the synthesis and SAR of a series of analogs of the mGlu5 partial antagonist 5-(phenylethynyl)pyrimidine. New molecular switches are identified that modulate the pharmacological activity of the lead compound. Slight structural modifications around the proximal pyrimidine ring change activity of the partial antagonist lead to that of potent and selective full negative allosteric modulators and positive allosteric modulators, that demonstrate in vivo efficacy in rodent models for anxiolytic activity and antipsychotic, respectively.
Glutamate is the major excitatory neurotransmitter in the mammalian central nervous system and exerts its effects through both ionotropic and metabotropic glutamate receptors. The metabotropic glutamate receptors (mGluRsγ) are members of the G Protein-coupled recpetor (GPCR) family C, which are characterized by a large extracellular amino-terminal agonist-binding domain. To date, eight mGluRs have been cloned, sequenced and assigned to three groups (Group I: mGlu1 and mGlu5; Group II: mGlu2 and mGlu3; Group III: mGlu4,6,7,8) based on their sequence homology, pharmacology, and coupling to effector mechanisms.1-2 In preclinical models, studies with the negative allosteric modulators (NAMs) 1 (MPEP) and 2 (MTEP) have demonstrated that selective antagonism of mGlu5 has therapeutic potential for chronic disorders such as pain, anxiety, depression, addiction and Fragile X syndrome.3-7 Furthermore, there is direct clinical validation of anxiolytic activity by allosteric antagonism of mGlu5 in patients with fenobam 3.8 Alternatively receptor activity can be enhanced through positive allosteric modulators (PAMs) such as 4 (DFB), 5 (CPPHA), 6 (CDPPB) and 7 (ADX-47273), which, with the exception of 5, share the same allosteric binding site as 1.9-13 PAMs 6 and 7, both ago-potentiators, have demonstrated in vivo proof of concept in preclinical schizophrenia models in which other known antipsychotics show similar positive effects.10-13 Recently, pure mGlu5 PAMs have been developed based on 7, by the incorporation of a basic heterocycle in the 3-position of the oxadiazole.14 Based on our experience in the development of allosteric modulators of mGluRs with a broad range of activities including negative allosteric modulators, positive allosteric modulators and neutral allosteric site ligands at the allosteric binding site occupied by 1, together with theoretical models of allosteric function, we postulated that it might be possible to develop ‘partial antagonists’. As envisioned, a ‘partial antagonist’ would fully occupy the binding site of 1 on the mGlu5 receptor but only partially block agonist response, resulting in partial mGlu5 inhibition; moreover, Rodriguez et al. identified several mGlu5 partial antagonists.15 In 2008, Sharma et. al. conducted a limited optimization effort focused on the mGlu5 partial antagonist lead 8. Within two 24-member libraries, SAR elucidated a “molecular switch” to modulate pharmacological activity (Fig. 1).16 Lead 8, with an unsubstituted distal phenyl ring, fully occupied the allosteric binding site of 1, possessed an IC50 of 486 nM, but only afforded partial response (29% response, 71% partial antagonism), that is, allosteric partial antagonism. Incorporation of small chemical moieties in the 3-position of the distal phenyl ring, such as a 3-methyl group, delivered 9, a full noncompetitive mGlu5 antagonist (IC50 = 7.5 nM). When the methyl group was moved from the 3-position to the 4-position as in 10, an efficacious (99% of glutamate max) mGlu5 PAM resulted (EC50 =3.3 μM, 4.2-fold shift), which also represented a new mGlu5 PAM chemotype.16 The observation of a conserved molecular switch, accessed by toggling between 3- and 4-substitution on the distal phenyl ring, within this chemical series was unprecedented. These preliminary data encouraged us to further optimize 8, and survey the impact of incorporating incorporating substituents on the pyrimidine ring, as well as examining regioisomeric pyrimidines in an attempt to develop potent and selective mGlu5 NAMs and PAMs suitable for in vivo studies to confirm the observed in vitro pharmacology.
Figure 1.

Identification of ‘molecular switches’ that converts an mGlu5 partial antagonist 8, to either a full non–competitive antagonist (NAM) 9, or a weak, but fully efficacious mGlu5 positive allosteric modulator (PAM) 10.
For the next round of chemical lead optimization, we relied on an iterative analog library synthesis approach17 to rapidly prepare a 24-member library18 in which 2-substituted-5-bromo-pyrimidines 11 were treated with either phenylacetylene 13, 3-methylphenyl acetylene (the NAM ‘switch’) 14 or 4-methylphenyl acetylene (the PAM ‘switch’) 15 under microwave-assisted Sonogashira conditions (Scheme 1) to provide analogs 16. In parallel, we prepared a small 3-member library employing the regiosiomeric 2-bromopyrimidine 12 and 13-15 to deliver analogs 17.
Scheme 1.
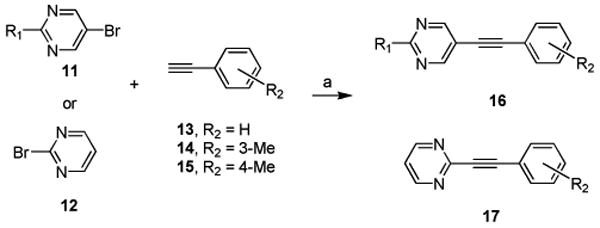
Synthesis of Analogs of 16 and 17a.
aReagents and conditions: (a) 10 mol% Pd(PPh3)4, 20 mol% CuI, 20.0 equiv. diethylamine, DMF, mw, 70 °C, 10 min, 16-95%; all compounds purified by mass-directed HPLC to >98% purity.19
SAR from this library was ‘flat’, with few actives (Table 1); however, unexpected modulation of the mode of mGlu5 pharmacology was observed. All new analogs 16 containing the 4-methyl phenyl moiety were uniformly inactive, save for 16f, a weak mGlu5 PAM. When R1 was an ethoxy group in combination with the NAM ‘switch’ 3-methyl phenyl, 16a resulted, a potent mGlu5 NAM (IC50 = 21 nM). The remaining analogs 16 were either inactive, or more surprisingly, potent mGlu5 PAMs. When an aminomethyl group was incorporated at the 2-position of the pyrimidine, in conjunction with an unsubstituted phenyl ring, 16b resulted, which represents the most potent (EC50 = 14.3 nM, 15-fold shift) rat mGlu5 PAM reported to date (10-15-fold more potent than 6 and 7). Addition of the NAM ‘switch’ 3-methyl phenyl moiety with the 2-aminomethyl group 16c unexpectedly afforded a similarly potent mGlu5 PAM (EC50 = 21.1 nM, 5.9-fold shift) – suggesting the 3-methylphenyl moiety is not a conserved molecular switch for engendering NAM activity. Interestingly, the NAM 16a differs from the PAM 16c by substitution at the 2-position of the pyrimidine, OEt versus NHMe, respectively, with equal potency (IC50 = 21 nM and EC50 = 21 nM, respectively), but opposite mode of pharmacology. Other groups were tolerated in the 2-position of the pyrimidine such as SMe 16d and t-Bu 16e, and found to engender mGlu5 PAM activity (EC50 = 120 nM, 11-fold shift and EC50= 247 nM, 6-fold shift, respectively), but were inactive in the presence of the 3- or 4-Me phenyl moieties. Overall, 16b represents a 235-fold improvement in potency over mGlu5 PAM 10, was selective for mGlu5 (>10 μM vs. mGlu1,2,3,4,7,8), and warranted further evaluation.
Table 1.
Structures, Activity and Mode of Pharmacology of Analogs 16 and 17.
 | ||||||
|---|---|---|---|---|---|---|
| R1 | R2 | Allosteric Activity | IC50/EC50 (nM)a | % Antagonisma | Fold Shifta | |
| 8 | H | H | PA | 486±28 | 71 | N/A |
| 9 | H | 3-Me | NAM | 7.5±1.2 | 100 | N/A |
| 10 | H | 4-Me | PAM | 3,300±290 | N/A | 3.3 |
| 16a | OEt | 3-Me | NAM | 21.1±2.8 | 100 | N/A |
| 16b | NHMe | H | PAM | 14.3±2.3 | N/A | 15 |
| 16c | NHMe | 3-Me | PAM | 21.1±1.8 | N/A | 5.9 |
| 16d | SMe | H | PAM | 120±25 | N/A | 11 |
| 16e | t-Bu | H | PAM | 247±24 | N/A | 6.0 |
| 16f | NHMe | 4-Me | PAM | 704±86 | N/A | 5.7 |
| 17a | N/A | H | NAM | 195±65 | 100 | N/A |
| 17b | N/A | 3-Me | NAM | 10.8±2.1 | 100 | N/A |
| 17c | N/A | 4-Me | N/A | >10,000 | N/A | N/A |
IC50s, EC50s, % Antagonism and Fold Shift are the average of at least three independent determinations. N/A = not applicable PA = Partial Antagonist NAM = Negative Allosteric Modualtor PAM = positive Allosteric Modulator. Fold-shift at 10 μM fixed concentration of cmpd
The PAMs reported here demonstrated no activity in the absence of glutamate, but in the presence of a sub-threshold concentration of glutamate (EC20), a concentration dependent potentiation of mGlu5 response was observed (Fig. 2). Importantly, 16b is a pure mGlu5 PAM, not an ago-potentiator like 6 and 7. In addition, compound 16b demonstrated a robust 15-fold leftward shift of the glutamate concentration response curve (EC50 shifts from 493 nM to 32 nM) with an increase in glutamate max (Fig. 2).
Figure 2.

Compound 16b potentiates mGlu5 activation by glutamate. In the absence of glutamate, 16b does not activate mGlu5. In the presence of a subthreshold quantity of glutamate, 16b potentiates mGlu5 in a concentration-dependent manner. Compound 16b's potentiation of response to glutamate is manifested as increased mGlu5 agonist sensitivity. The glutamate EC50 value is shifted from 493 nM to 32 nM, or a 15-fold shift with 10 μM 16b.
In the regiosiomeric pyrimidine series 17, the 4-Me congener 17c was inactive. The unsubstitiuted phenyl analog 17a was a moderately potent mGlu5 NAM (IC50 = 195±65 nM). Unlike series 16, the 3-Me NAM ‘switch’ performed as expected in series 17, significantly increasing mGlu5 NAM activity (IC50 = 10.8±2.7 nM) for 17b. Moreover, 17b was selective for mGlu5 (>10 μM vs. mGlu1,2,3,4,7,8).
With a potent mGlu5 PAM 16b and a potent mGlu5 NAM 17b, we were poised to determine if the modes of mGlu5 modulation observed in our in vitro cellular assays would be mirrored in standard in vivo behavioral paradigms. To evaluate the PAM 16b, we chose to study the ability of 16b to reverse amphetamine-induced hyperlocomotion in rats, as 6 and 7 displayed robust efficacy in this preclinical model where other known antipsychotic agents show similar positive results.10-13 In the event, 16b was dosed i.p. at 3, 10 or 30 mg/kg thirty minutes prior to s.c. administration of 1 mg/kg amphetamine. As shown in Figure 3, a modest dose response is observed with 16b, with significant reversal noted at the 30 mg/kg dose, and no effect (i.e, sedation) of 16b/vehicle alone. Thus, the mGlu5 PAM activity observed in cell-based in vitro assays is mirrored in vivo with 16b, and comparable to the effects seen with both 6 and 7.10-13 Moreover, the reversal of amphetamine-induced hyperlocomotion with 16b is important, as 16b lacks the intrinsic agonism of the ago-potentiators 6 and 7, suggesting for the first time that positive allosteric modulation alone is sufficient for an antipsychotic profile in this preclinical model.
Figure 3.

Reversal of amphetamine-induced hyperlocomotion with mGlu5 PAM 16b in dose-dependent manner with the non-toxic vehicle, 10% Tween 80.
Previously antagonists for mGlu5 NAMs such as 1 and 2 have demonstrated anxiolytic activity in numerous preclinical models. Therefore compound 17b was tested in a modified Geller-Seifter conflict model wherein an increase in punished responding is consistent with an anxiolytic-like profile.20 As seen in Figure 4, 17b produced a significant dose-dependent increase in punished responding with the 30 mg/kg dose approaching a 300% increase in response rate [F(4,17) = 22.69, p<0.0001] (upper panel) with no significant effect on unpunished responding (lower panel). Post hoc analysis indicated that both the 10 and 30 mg/kg doses in the punished component of the schedule differed significantly from vehicle (p < 0.05, Newman-Keul). Therefore, the NAM activity observed in cell-based in vitro assays was again paralleled in a standard anxiolytic in vivo assay where classical mGlu5 NAMs display similar positive results.3-6,20
Figure 4.

Dose-response curves for the effects of 17b on punished (upper panel) and unpunished (lower panel) responding. The data are the mean number of punished and unpunished responses that animals made when tested on 1, 3, 10, 30 mg/kg 17b and vehicle. Each value represents the mean ± SEM for eighteen animals. For punished responding animals tested on 10 and 30 mg/kg 17b made significantly greater number of responses than animals tested on vehicle (p < 0.05). Unpunished responding did not change significantly at any of the doses tested.
In summary, slight structural changes to an mGlu5 allosteric partial antagonist lead resulted in a shift in activity from partial antagonist to potent full antagonist to potent positive allosteric modulator. Two new molecular switches were elucidated through these changes. A regiosiomeric pyrimidine congener 17b resulted in full NAM activity in vitro and in vivo. The incorporation of an amino methyl group into the 2-position of the pyrimidine core resulted in PAM activity and this new molecular ‘switch’ was able to override previously identified NAM molecular ‘switches’. In this series, 16b represents the most potent mGlu5 PAM reported to date, and the first example of in vivo efficacy of a pure mGlu5 PAM in reversing amphetamine-induced hyperlocomotion. The resulting mGlu5 NAM 17b and PAM 16b showed in vivo efficacy in rodent models of anxiety and schizophrenia, respectively, which mirrored the observed in vitro mode of pharmacology. With such subtle structural modifications capable of fully reversing modes of pharmacology, lead optimization campaigns focused on ligands that bind to the allosteric site occupied by 1 are especially challenging. Further work in this area is in progress and will be reported in due course.
Supplementary Material
Chart I. mGlu5 Allosteric Ligands.

Acknowledgments
The authors thank the NIH and NIDA (RO1DA023947-01) for support of our work.
Footnotes
Abbreviations: mGluR, metabotropic glutamate receptor; NAM, negative allosteric modulator; PAM, positive allosteric modulator; GPCR, G protein-coupled receptor
Supporting Information Available: Experimental procedure and analytical data for compounds 8a-f and 10a, b are provided as well as details of the in vitro and in vivo assays. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Schoepp DD, Jane DE, Monn JA. Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology. 1999;38:1431–1476. doi: 10.1016/s0028-3908(99)00092-1. [DOI] [PubMed] [Google Scholar]
- 2.Conn PJ, Pin JP. Pharmacology and functions of metabotropic glutamate receptors. Annu Rev Pharmacol Toxicol. 1997;37:205–237. doi: 10.1146/annurev.pharmtox.37.1.205. [DOI] [PubMed] [Google Scholar]
- 3.Gasparini F, Lingenhohl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, Varney MA, Johnson EC, Hess SD, Rao SP, Sacaan AI, Santori EM, Velicelebi G, Kuhn R. 2-Methyl-6-(phenylethynyl)-pyrimidine (MPEP), a potent, selective and systemically active mGluR5 receptor antagonist. Neuropharmacology. 1999;38:1493–1503. doi: 10.1016/s0028-3908(99)00082-9. [DOI] [PubMed] [Google Scholar]
- 4.Roppe JR, Wang B, Huang D, Tehrani L, Kamenecka T, Schweiger EJ, Anderson JJ, Brodkin J, Jiang X, Cramer M, Chung J, Reyes-Manalo G, Munoz B, Cosford NDP. 5-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]-2,3′-bipyridine: a highly potent, orally active metabotropic glutamate subtype 5 (mGluR5) receptor antagonist with anxiolytic activity. Bioorg Med Chem Lett. 2004;14:3993–3996. doi: 10.1016/j.bmcl.2004.05.037. [DOI] [PubMed] [Google Scholar]
- 5.Lea PM, IV, Faden AI. Metabotropic glutamate receptor subtype 5 antagonists MPEP and MTEP. CNS Drug Rev. 2006;12:149–166. doi: 10.1111/j.1527-3458.2006.00149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tatarczynska E, Klodzinska A, Chojnacka-Wojcik E, Palucha A, Gasparini F, Kuhn R, Pilc A. Potential anxiolytic- and antidepressant-like effects of MPEP, a potent selective and systemically active mGluR5 receptor antagonist. Br J Pharmacol. 2001;132:1423–1430. doi: 10.1038/sj.bjp.0703923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology. 2005;49:1053–1066. doi: 10.1016/j.neuropharm.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 8.Porter RHP, Jaeschke G, Spooren W, Ballard TM, Buttelmann B, Kolczewski S, Peters JU, Prinseen E, Wichman J, Vieira E, Muhlemann A, Gatti S, Mutel V, Malherbe P. Fenobam: a clinically validated nonbenzodiazepine anxiolytic is a potent, selective, and noncompetitive mGlu5 receptor antagonist with inverse agonist activity. J Pharm Exp Ther. 2005;315:711–721. doi: 10.1124/jpet.105.089839. [DOI] [PubMed] [Google Scholar]
- 9.O'Brien JA, Lemaire W, Chen TB, Chang RSL, Jacobson MA, Ha SN, Lindsley CW, Sur C, Pettibone DJ, Conn PJ, Williams DL. A family of highly selective allosteric modulators of the metabotropic glutamate subtype 5. Mol Pharmacol. 2003;64:731–740. doi: 10.1124/mol.64.3.731. [DOI] [PubMed] [Google Scholar]
- 10.Lindsley CW, Wisnoski DD, Leister WH, O'Brien JA, Lemiare W, Williams DL, Burno M, Sur C, Kinney GG, Pettibone DJ, Tiller PR, Smith S, Duggan ME, Hartman GD, Conn PJ, Huff JR. Discovery of positive allosteric modulators for the metabotropic glutamate receptor subtype 5 from a series of N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamides that potentiate receptor function in vivo. J Med Chem. 2004;47:5825–5828. doi: 10.1021/jm049400d. [DOI] [PubMed] [Google Scholar]
- 11.Kinney GG, O'Brien JA, Lemaire W, Burno M, Bickel DJ, Clements MK, Wisnoski DD, Lindsley CW, Tiller PR, Smith S, Jacobson MA, Sur C, Duggan ME, Pettibone DJ, Williams DW. A novel A novel selective positive allosteric modulator of metabotropic glutamate receptor subtype 5 has in vivo activity and antipsychotic-like effects in rat behavioral models. J Pharmacol Exp Ther. 2005;313:199–206. doi: 10.1124/jpet.104.079244. [DOI] [PubMed] [Google Scholar]
- 12.O'Brien JA, Lemaire W, Wittman M, Jacobson MA, Ha SN, Wisnoski DD, Lindsley CW, Wisnoski DD, Schaffhauser HJ, Sur C, Duggan ME, Pettibone DJ, Conn PJ, Williams DL. A novel selective allosteric modulator potentiates the activity of native metabotropic glutamate receptor subtype 5 in rat forebrain. J Pharmacol Exp Ther. 2004;309:568–577. doi: 10.1124/jpet.103.061747. [DOI] [PubMed] [Google Scholar]
- 13.a Farina M, Gagliardi S, Le Poul E, Mutel V, Palombi G, Poli SM, Rocher JP. Novel oxadiazole derivatives and their use as positive allosteric modulators of metbotropic glutamate receptors and their preparation pharmaceutical compositions and use in the teatment of central peripheral nervous system disorders. 2006 WO123255. [Google Scholar]; b Epping-Jordan M, Le Poul M, Rocher JP. Allosteric modulation: a novel approach to drug discovery. Innov Pharm Technol. 2007;24:24–26. [Google Scholar]
- 14.Engers DW, Rodriguez AL, Oluwatola O, Hammond AS, Venable DF, Williams R, Sulikowski GA, Conn PJ, Lindsley CW. Synthesis SAR and unanticipated pharmacological profiles of analogs of the mGluR5 ago-potentiator ADX-47273. ChemMedChem. 2009;4:505–511. doi: 10.1002/cmdc.200800357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rodriguez AL, Nong Y, Sekaran NK, Alagille D, Tamagnan GD, Conn PJ. A close structural analog of 2-methyl-6-(phenylethynyl)-pyridine acts a neutral allosteric site ligand on metabotropic glutamate receptor subtype 5 and blocks the effects of multiple allosteric modulators. Mol Pharmacol. 2005;68:1793–1802. doi: 10.1124/mol.105.016139. [DOI] [PubMed] [Google Scholar]
- 16.Sharma S, Rodriguez AL, Conn PJ, Lindsley CW. Synthesis and SAR of a mGlu5 allosteric partial antagonist lead: unexpected modulation of pharmacology with slight structural modifications to a 5-(phenylethynyl)pyrimidine scaffold. Bioorg Med Chem Lett. 2008;18:4098–5101. doi: 10.1016/j.bmcl.2008.05.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kennedy JP, Williams L, Bridges TM, Daniels RN, Weaver D, Lindsley CW. Application of combinatorial chemistry science on modern drug discovery'. J Comb Chem. 2008;10:345–354. doi: 10.1021/cc700187t. [DOI] [PubMed] [Google Scholar]
- 18.For full description of the 24-member library, please see Supporting Information Section.
- 19.Leister W, Strauss K, Wisnoski D, Zhao Z, Lindsley C. Development of a custon high-throughput preparative liquid chromaotgraphy/mass spectrometer platform for the preparative purification and anlaytical analysis of comound libraries. J Comb Chem. 2003;5:322–329. doi: 10.1021/cc0201041. [DOI] [PubMed] [Google Scholar]
- 20.Busse CS, Brodkin J, Tattersall D, Anderson JJ, Warren N, Tehrani L, Bristow LJ, Varney MA, Cosford NDP. The behavioral profile of the potent and selective mGluR5 receptor anatgonist 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine (MTEP) in rodent models of anxiety. Neuropsychopharmacology. 2004;29:1971–1979. doi: 10.1038/sj.npp.1300540. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.