

Abstract
Background
Spasticity and rigidity are serious complications associated with spinal traumatic or ischemic injury. Clinical studies show that Tizanidine (Tiz) is an effective anti-spasticity agent, however, the mechanism of this effect is still not clear. Tiz binds not only to α2-adrenoreceptors (AR) but also to imidazoline (I) receptors. Both receptor systems (AR+I) are present in the spinal cord interneurons and α-motoneurons. The aim of the present study was to evaluate the therapeutic potency of systematically or spinally (intrathecally) delivered Tiz on stretch reflex activity (SRA) in animals with ischemic spasticity, and to delineate supraspinal or spinal sites of Tiz action.
Methods
Animals were exposed to 10 min of spinal ischemia to induce an increase in SRA. Increase in SRA was identified by simultaneous increase in recorded EMG activity and ankle resistance measured during computer-controlled ankle dorsiflexion (40°/3 sec) in fully awake animals. Animals with increased SRA were divided into several experimental subgroups and treated as follows: i) Tiz administered systemically at the dose of 1 mg kg-1, or intrathecally (IT) at 10 μg or 50 μg delivered as a single dose; ii) Treatment with systemic Tiz was followed by the systemic injection of vehicle, or by non-selective AR antagonist without affinity for imidazoline receptors; Yohimbine (Yoh), α2A AR antagonist; BRL44408 (BRL), α2B AR antagonist; ARC239 (ARC), non-selective AR and I1 receptor antagonist; Efaroxan (Efa), or non-selective AR and I2 receptor antagonist; Idazoxan (Ida); iii) Treatment with IT Tiz was followed by the IT injection of selective α2A AR antagonist; Atipamezole (Ati). In a separate group of spastic animals the effect of systemic Tiz treatment (1 mg/kg) or isoflurane anesthesia on H-reflex activity was also studied.
Results
Systemic and/or IT treatment with Tiz significantly suppressed SRA. This Tiz-mediated anti-SRA effect was reversed by BRL (5 mg kg-1), Efa (1 mg kg-1) and Ida (1 mg kg-1). No reversal was seen after Yoh (3 mg kg-1) or ARC (5 mg kg-1) treatment. Anti-SRA induced by IT Tiz (50 μg) was reversed by IT injection of Ati (50 μg). Significant suppression of H-reflex was measured after systemic Tiz treatment (1mg/kg) or isoflurane (2%) anesthesia, respectively. Immunofluoresecene staining of spinal cord sections taken from animals with spasticity showed upregulation of α-2A receptor in activated astrocytes.
Conclusions
These data suggest that α2A AR and I receptors, but not α2B AR primarily mediate the Tiz-induced anti-spasticity effect. This effect involves spinal and potentially supraspinal sites and likely targets α2A receptor present on spinal neurons, primary afferents and activated astrocytes Further studies using highly selective antagonists are needed to elucidate the involvement of specific subtypes of the AR and imidazoline receptors in the anti-spasticity effect seen after Tiz treatment.
Introduction
Spinal cord ischemic or traumatic injury can lead to a time-dependent development of muscle spasticity and rigidity (Adams and Hicks, 2005). Spasticity is defined as velocity-dependent increase in muscle tone, i.e., muscle resistance is increasing progressively with increased velocity of muscle stretch while the rigidity is characterized by the presence of velocity-independent (i.e., continuous) increase in muscle tone (Lance, 1980, Maurice et al., 2001). Systematic clinical statistical data show that up to 36% of patients with spinal traumatic injury show progressive appearance of muscle spasticity or rigidity (Kita and Goodkin, 2000, Noreau et al., 2000).
Previous experimental data using several animal models of spinal ischemic injury showed that periods of injurious intervals of spinal ischemia leads to a selective loss of spinal inhibitory interneurons in previously ischemia-exposed spinal segments (Van Harreveld and Spinelli, 1965, Matsushita and Smith, 1970, Kakinohana et al., 2006). Functional deficit resulting from a selective loss of spinal inhibitory neurons is typically expressed as spasticity and rigidity. Qualitatively similar appearance of muscle spasticity and rigidity in human patients suffering from spinal ischemic injury has been described (Tarlov, 1967).
In our previous studies we have developed and characterized an aortic balloon occlusion model of transient spinal cord ischemia in rats. Using this model we have demonstrated i) a selective loss of spinal GABA-ergic interneurons but persisting survival of α-motoneurons after 10 min of transient spinal cord ischemia (Taira and Marsala, 1996), ii) a significant increase in myogenic motor evoked potentials and Hoffmann reflex activity (Marsala et al., 2004, Kakinohana et al., 2006), iii) progressive appearance of spasticity and rigidity as measured by a computer-controlled ankle rotational muscle resistance meter (Marsala et al., 2005), and iv) effective suppression of spasticity, rigidity and H-reflex activity by spinal delivery of GABA-mimetic compounds such as nipecotic acid, GABA B receptor agonist baclofen and AMPA receptor antagonist NGX424 (Kakinohana et al., 2006, Hefferan et al., 2007). In addition, using the same spinal ischemia model, we have characterized in spastic animals the development of baclofen tolerance after chronic intrathecal infusion of baclofen (Hefferan et al., 2006) and have demonstrated effective suppression of spasticity in baclofen-tolerant animals after intrathecal or systemic treatment with AMPA receptor antagonist NGX424 (Oshiro et al., 2010).
In addition to extensive clinical data demonstrating a potent anti-spasticity effect after systemic or spinal delivery of baclofen, a comparable success in the use of systemic tizanidine as an anti-spasticity agent has been reported (Hassan and McLellan, 1980, Sie and Lakke, 1980). The mechanism of tizanidine-mediated anti-spasticity effect has been demonstrated to be the result of potentiation of presynaptic inhibition, suppression of flexor reflexes as well as it direct action on α-motoneurons (Delwaide and Pennisi, 1994). In addition, using animal experimental models of neuropathic and visceral pain, it has been demonstrated that intrathecal treatment with tizanidine has a potent anti-nociceptive effect suggesting its direct spinal mechanism of action (Danzebrink and Gebhart, 1990, McCarthy et al., 1990, Leiphart et al., 1995).
At present there are no systematic experimental data available which would define the effect of intrathecal or systemic tizanidine treatment on spasticity resulting from loss of local spinal segmental inhibition. Accordingly, the aim of present study was to evaluate the therapeutic potency of systematically or spinally (intrathecally; IT) delivered Tiz on stretch reflex activity (SRA) in animals with ischemic spasticity, and to delineate supraspinal or spinal sites of Tiz anti-spasticity action.
Materials and Methods
All procedures were approved by the Animal Care Committee at the University of California, San Diego. Male Sprague Dawley rats (300–350 g) were obtained from Harlan (Indianapolis, IN) and were housed in standard cages with corncob bedding. Animals had access to food and water ad libitum and were housed separately after surgery. A 12 h light/dark cycle (lights on at 7:00 A.M.) was used throughout.
Induction of spinal ischemia
Spinal ischemia was induced using the previously described technique (Taira and Marsala, 1996). Briefly, rats were anesthetized with 4% isoflurane in air and maintained with 1.5–2% isoflurane. Body temperature was maintained at 37°C using a homeothermic blanket system. Distal arterial blood pressure was monitored by a tail PE-50 artery catheter. The left carotid artery was cannulated with a 20 gauge polytetrafluoroethylene catheter for blood withdrawal. To induce spinal ischemia, a 2F Fogarty catheter (Am. V. Mueller, CV 1035; Baxter, Irvine, CA) was passed through the left femoral artery to the descending thoracic aorta so that the tip reached the level of the left subclavian artery (10.8–11.4 cm from site of insertion). The intra-aortic balloon catheter was inflated with 0.05 ml of saline for 10 minutes and occlusion confirmed by an immediate and sustained drop in distal arterial blood pressure measured in the tail artery. Systemic hypotension (40 mmHg) was induced during occlusion by a blood withdrawal from the carotid artery to the glass collecting circuit and kept at 37.5°C. After ischemia, the balloon was deflated, and the blood was reinfused during a 60 sec period. After blood reinfusion, 4 mg of protamine sulfate was administered subcutaneously. Stabilization of arterial blood pressure was then monitored for an additional 10 min, after which the arterial lines were removed and wounds were closed. Rats were then allowed to recover.
Identification of changes in stretch reflex activity (SRA) after ischemia
Seven to ten days after ischemia, animals were tested for changes in SRA. Increase in SRA was identified as an increase in ankle resistance during computer-controlled ankle dorsiflexion, which correlated with increased EMG activity measured in the gastrocnemius muscle during the same time frame (Fig. 1A,B).
Fig. 1. Methodology for qualitative and quantitative assessment of changes in stretch reflex activity in rats with spinal ischemia-induced spastic paraplegia.
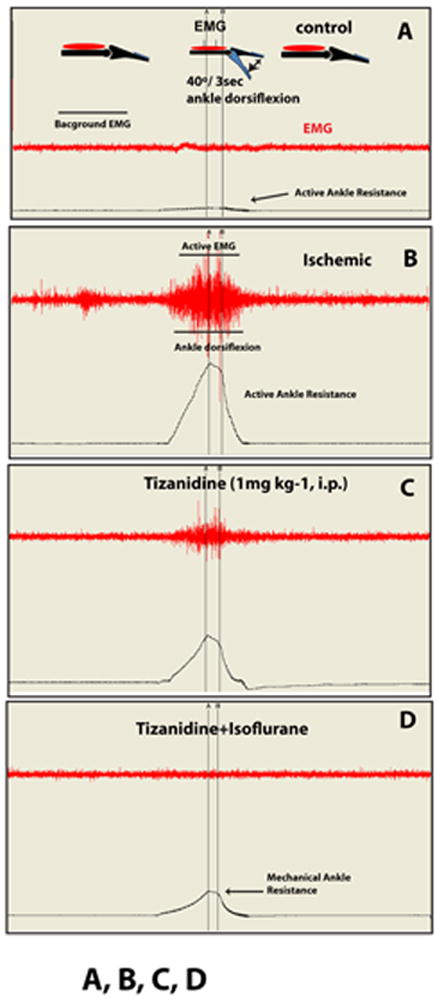
A, B: At 4-10 days after induction of spinal ischemic injury, a progressive increase in stretch reflex activity is seen as evidenced by the appearance of burst EMG activity (Active EMG) and corresponding increase in ankle resistance (Active Ankle Resistance) measured during computer-controlled ankle dorsiflexion (40°/3 sec).
C: Systemic injection of tizanidine (1 mg kg-1) leads to a potent suppression of active EMG and active ankle resistance measured during ankle dorsiflexion.
D: To identify the mechanical component in measured ankle resistance (Mechanical Ankle Resistance) during ankle dorsiflexion, animals are anesthetized with isoflurane at the end of the experiment and the magnitude of active ankle resistance suppression measured.
Direct measurement of ankle resistance during computer-controlled ankle dorsiflexion was performed as described previously (Marsala et al., 2005). Briefly, rats with developed spinal ischemia-induced paraplegia were individually placed in a plastic restrainer, and one hindpaw was securely fastened to the paw attachment metal plate, which is interconnected loosely to the “bridging” force transducer (LCL454G, 0–454 g range; or LCL816G, 0–816 g range; Omega, Stamford, CT). After a 20 min acclimation period, rotational force was applied to the paw attachment unit using a computer-controlled stepping motor (MDrive 34 with onboard electronics; microstep resolution to 256 microsteps/full step; Intelligent Motion Systems, Marlborough, CT), causing the ankle to dorsiflex (Fig. 1A). The resistance of the ankle was measured during 40° of dorsiflexion during 3 sec (13.3° s-1), and data were collected directly to a computer using custom software (Spasticity version 2.01; Ellipse, Kosice, Slovak Republic).
To identify the mechanical component of measured ankle resistance, all animals were anesthetized with 2.5-3% isoflurane at the end of the experiment and the relative contribution of mechanical vs. neurogenic component (isoflurane-sensitive) was calculated. To record EMG activity, a pair of tungsten electrodes were inserted percutaneously into the gastrocnemius muscle 1 cm apart. EMG signals were bandpass filtered (100 Hz to 10 kHz) and recorded before, during, and after ankle dorsiflexion. EMG responses were recorded with an alternating current-coupled differential amplifier (model DB4; World Precision Instruments, Sarasota, FL) and stored on a computer for subsequent analysis. EMG was recorded concurrently with ankle resistance measurement during ankle dorsiflexion, and expressed as the average of three measurements. Digitized EMG signal was full-wave rectified, and values within given time interval (bin) were averaged and used for statistical analysis.
Intrathecal catheterization
At 10–15 days after spinal cord ischemia, intrathecal catheters were implanted in some animals with identified increase in SRA as described previously (Yaksh and Rudy, 1976). Under isoflurane anesthesia, an 8.5 cm PE-5 catheter (Spectranetics, Colorado Springs, CO) connected to a 6 cm long polyethylene-10 tube was placed into lumbar intrathecal space through a cut-opened atlanto-occipital membrane. Polyethylene-10 tube was then externalized on the neck and plugged with 23G stainless steel wire (2 cm in length) and animals were allowed to recover.
Hoffman reflex (H-reflex) recording
In some animals H-reflex was recorded as previously described (Schwarz et al., 1994, Kakinohana et al., 2006). Under ketamine anesthesia (100 mg/kg/hr, i.m.) the right hind limb of the animal was secured and a pair of stimulating needle electrodes was transcutaneously inserted into the surroundings of the tibial nerve. For recording a pair of silver needle electrodes was placed into the interosseous muscles between the fourth and the fifth or the first and the second metatarsal right foot muscles. The tibial nerve was stimulated using square pulses with increasing stimulus intensity (0.1-10 mA in 0.5 mA increments, 0.1 Hz, 0.2 ms; WPI; Isostim A320) and responses were recorded with an A/C-coupled differential amplifier (Model DB4; DPI, Sarasota, FL). H-reflex was recorded in spastic animals before and after systemic Tiz treatment (1 mg/kg) and after isoflurane (2%) anesthesia (n=5).
Immunofluorescence staining
After completion of drug treatment studies, animals (n=9) were deeply anesthetized with pentobarbital and phenytoin and transcardially perfused with 200 ml of heparinized saline followed by 250 ml of 4% paraformaldehyde in PBS. The spinal cords were dissected and postfixed in 4% formaldehyde in PBS overnight at 4°C and then cryoprotected in 30% sucrose PBS until transverse sections (30-μm-thick) were cut on a cryostat and stored in PBS.
Sections were then placed in primary antibody overnight at 4°C with primary antibodies made in PBS with 0.2% triton-X100: rabbit anti-glial fibrillary acidic protein (GFAP, 1:1000, Sigma-Aldrich Corp. St. Louis, MO); biotinylated mouse anti-NeuN (Millipore, Temecula, CA); mouse anti-synaptophysin (SYN; 1:1000; Millipore, Temecula, CA); goat anti-α2A-C (C-terminus; 1:200; Santa Cruz Biotechnology); or rabbit anti- α2A-m (immunogen sequence: KASRWRGRGNREKR; 1:100; Neuromics).
After incubation with primary antibodies, sections were washed 3x in PBS and incubated with fluorescent-conjugated secondary antibodies raised in donkey (Alexa 488, 546; 647; 1:250; Invitrogen Corp., Carlsbad, CA, USA) and DAPI for general nuclear staining. Once staining was complete, sections were mounted on slides, dried at room temperature and covered with Prolong anti-fade kit (Invitrogen Corp., Carlsbad, CA, USA). Images were captured using a Leica SP2 or Olympus Fluoview 100 confocal microscope. Any image post-processing was done with Adobe CS3 (Adobe Systems, Inc., San Jose, CA) with equal changes to any images being compared.
Experimental timeline/drug treatment design
Four principal experimental treatment studies were performed. In all studies animals with identified increase in SRA were used. Before and after drug treatment changes in SRA were measured in 10 min intervals for total of 60 min as described.
Study A (n=6), animals were treated systematically with Tiz (1 mg kg-1; i.p.)
Study B (n=6 for each dose), animals were treated intrathecally with Tiz (10 μg or 50 μg delivered as single bolus injection).
Study C (n=6 for each treatment group), animals were treated systematically with Tiz (1 mg kg-1; i.p.) followed by systemic (i.p.) injection of one of the following: i) vehicle, ii) one of non-selective AR antagonists without affinity for imidazoline receptors: Yohimbine (Yoh; 3 mg kg-1; i.p.), α2A AR antagonist; BRL44408 (BRL; 5 mg kg-1; i.p.), α2B AR antagonist; ARC239 (ARC; 5 mg kg-1; i.p.), non-selective AR and I1 receptor antagonist; Efaroxan (Efa; 1 mg kg-1; i.p.), or non-selective AR and I2 receptor antagonist; Idazoxan (Ida; 1 mg kg-1; i.p.).
Study D (n=6), animals were treated intrathecally with Tiz (50 μg delivered as single bolus injection) followed by the IT injection of selective α2A AR antagonist; Atipamezole (Ati; 50 μg).
Statistical analysis
Comparisons between treatment groups was performed using t-test with Bonferroni correction. All results are shown as mean ± S.D. P < 0.05 was considered statistically significant.
Results
Spinal ischemic injury leads to progressive increase in stretch reflex activity (SRA)
Rats previously exposed to 10 min of spinal ischemia showed a progressive increase in SRA at 4-10 days after ischemic injury. The increase in SRA was identified by i) burst EMG activity, and ii) concomitant increase in ankle resistance measured during computer-controlled ankle dorsiflexion from 0-40°(compare Fig. 1A-control to B-ischemic; Active EMG and Active Ankle Resistance). Systemic treatment with Tizanidine (1 mg kg-1) suppressed SRA measured during ankle dorsiflexion (Fig. 1C). To identify the mechanical component contributing to measured ankle resistance during ankle dorsiflexion, animals were anesthetized with isoflurane at the end of experiment and the magnitude of residual resistance measured (Fig. 1D-Mechanical Ankle Resistance). In control non-ischemic animals, only low-level EMG activity (0.1-0.5 mV) was measured before (Background EMG) and during ankle dorsiflexion and was similar to isoflurane-anesthetized ischemic rats (compare Fig. 1A and D). In control naive animals, ankle resistance measured during ankle dorsiflexion did not exceed 6-10 g (Fig. 1A; Active Ankle Resistance). These data are similar to our previously reported observations (Marsala et al., 2005, Kakinohana et al., 2006).
Systemic or intrathecal treatment with Tizanidine leads to a potent anti-spasticity effect
Intraperitoneal injection of Tizanidine (1 mg kg-1) led to a significant suppression in SRA as identified by changes in EMG activity and ankle resistance measured during ankle dorsiflexion. A significant effect was measured at 10 min after drug injection and was still present at 60 min (Fig.2A, B). A similar significant anti-SRA effect was measured after IT delivery of tizanidine (10 μg or 50 μg), (Fig.2 A, B). In both treatment groups the magnitude of SRA returned back to baseline at 24 hrs after treatment (not shown).
Fig. 2. Potent anti SRA effect after systemic or intrathecal treatment with tizanidine.
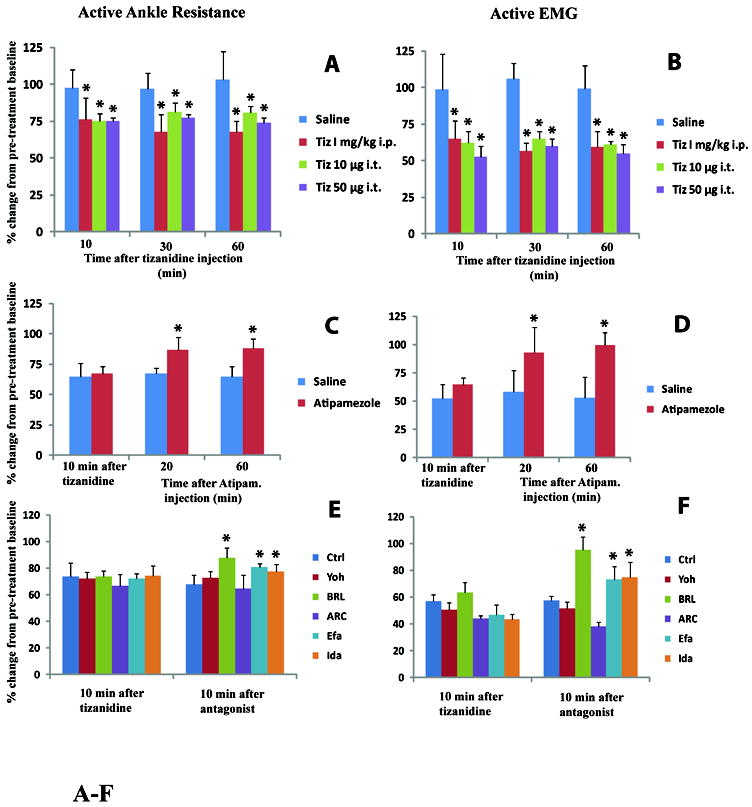
A, B: Systemic (1 mg kg-1; i.p.) or intrathecal injections (10μg or 50 μg) of tizanidine led to a significant anti-SRA effect at 10-30 min after drug injection if compared to saline-injected animals. This effect persisted for a minimum of 60 min (*-p<0.05; t-test with Bonferroni correction).
C, D: Anti-SRA effect measured in animals after intrathecal injections of tizanidine (50 μg) was near completely reversed by IT injections of selective α2A AR antagonist Atipamezole (Ati; 50 μg). A significant reversal effect was seen at 20-60 min after Ati injection if compared to saline-injected animals (*-p<0.05; t-test with Bonferroni correction).
E, F: Similarly, in animals treated systematically with Tiz (1 mg kg-1), the anti-SRA effect was reversed by BRL44408 (BRL; 5 mg kg-1, i.p.), Efaroxan (Efa; 1 mg kg-1; i.p.) and Idazoxan (Ida; 1 mg kg-1; i.p.) administration if compared to saline-injected (Ctrl) animals. No significant reversal effect was seen after Yohimbine (Yoh; 3 mg kg-1; i.p.), or ARC239 (ARC; 5 mg kg-1; i.p.) administration (*-p<0.05; t-test with Bonferroni correction).
Reversal of Tizanidine mediated anti-spasticity affect by α2-adrenoceptors antagonists
Anti-SRA effect induced by intrathecal Tiz (50 μg) was reversed by IT injection of Ati (50 μg), (Fig.2C, D).
The anti-SRA effect seen after systemic treatment with Tiz (1 mg kg-1) was significantly reversed by BRL (5 mg kg-1), Efa (1 mg kg-1) and Ida (1 mg kg-1). No significant reversal effect was seen after Yoh (3 mg kg-1) or ARC (5 mg kg-1) treatment (Fig. 2E, F).
Systemic treatment with Tizanidine or isoflurane anesthesia leads to a suppression of spinal Hoffmann reflex
In animals with spinal ischemic injury, a significant increase in H-reflex activity was measured if compared to control animals (H/M ratio 0.24±0.03 vs. 0.35±0.05 mV; Fig. 3A, B; Table 1). Injection of systemic Tizanidine (1 mg kg-1) led to a significant suppression of otherwise increased H-reflex activity (H/M ratio after Tizanidine injection 0.23±0.03; Fig. 3C; Table 1) at 10-15 min after drug injection. No suppression of H-reflex activity in animals injected with vehicle (saline) only was seen (not shown). In the absence of Tizanidine treatment isoflurane anesthesia (2%) led to a significant suppression of H-reflex in spastic animals (H/M ratio 0.35±0.05 vs. 0.20±0.04 mV; Fig. 3D, E; Table 1).
Fig. 3. Systemic treatment with tizanidine or isoflurane anesthesia suppress Hoffmann reflex activity in animals with spinal ischemic injury.
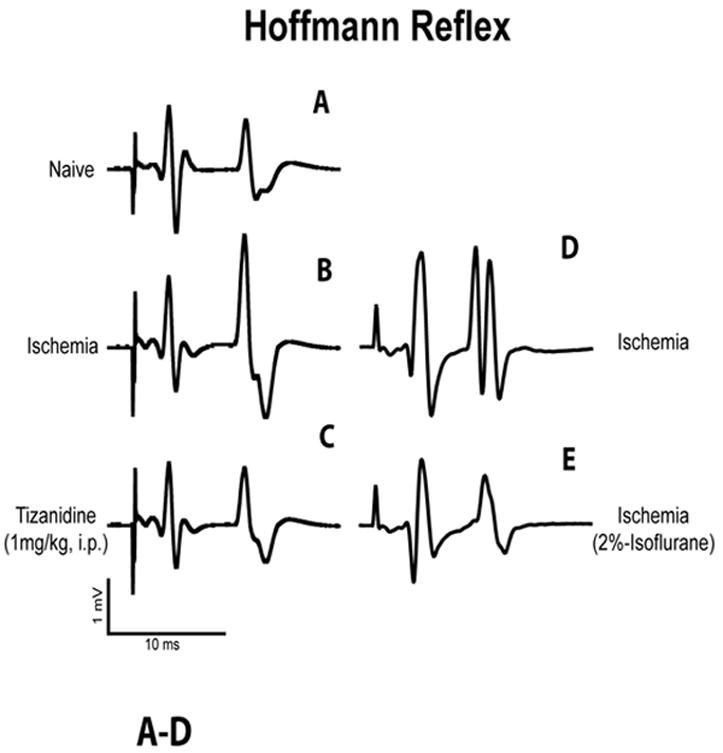
A, B: In comparison to control naive animals (A), rats with spinal ischemic injury, which showed increase in stretch reflex activity, had significantly increased H-wave response after electrical stimulation of the tibial nerve (see Table 1 for details).
C: In animals with spinal ischemic injury, systemic administration of tizanidine (1 mg kg-1; i.p.) effectively suppressed otherwise increased H-wave response (Table 1).
D, E: In animals with spinal ischemic injury but without tizanidine treatment isoflurane anesthesia (2%) lead to a potent reduction in H-reflex amplitude (Table 1).
Table 1.
| Naïve Ctrl | Spastic Pre | Spastic Tiz (#) | Spastic Iso (##) | |
|---|---|---|---|---|
| M max (mV) | 8.61 ± 0.36 | 11.22 ± 1.10 | 9.39 ± 0.85 | 9.68 ± 1.03 |
| H max (mV) | 2.06 ± 0.26 | 3.94 ± 0.22 | 2.15 ± 0.33 | 1.95 ± 0.32 |
| H / M ratio | 0.24 ± 0.03 | 0.35 ± 0.05* | 0.23 ± 0.03** | 0.20 ± 0.04*** |
Tizanidine was administered i.p. (1 mg kg -1)
Animals were anesthetized with 2% Isoflurane
p<0.05 vs. Naïve Control rats
p<0.05 vs. spastic non-treated animals
p<0.05 vs. spastic non-treated animals
(t-test with Bonferroni correction)
Increased expression of α2A receptor in activated astrocytes in animals with identified increase in stretch reflex activity
In control naive animals, staining of transverse lumbar spinal cord sections with α2A-c antibody showed a punctuate-like immunoreactivity in virtually all neurons including small and medium-sized interneurons and α-motoneurons (Fig. 4A). Co-staining of the same sections with GFAP antibody showed only weak expression of α2A-c in astrocytes (Fig. 4B, C). In contrast to control animals, rats with ischemic injury showed a clear increase in α2A-c expression. This increase was primarily seen in activated astrocytes localized at the peri-ischemic regions in the vicinity of persisting α-motoneurons (Fig. 4D-H).
Fig. 4. Increased expression of α2A-c receptor in activated astrocytes in lumbar spinal cord in rats with increased stretch reflex activity.
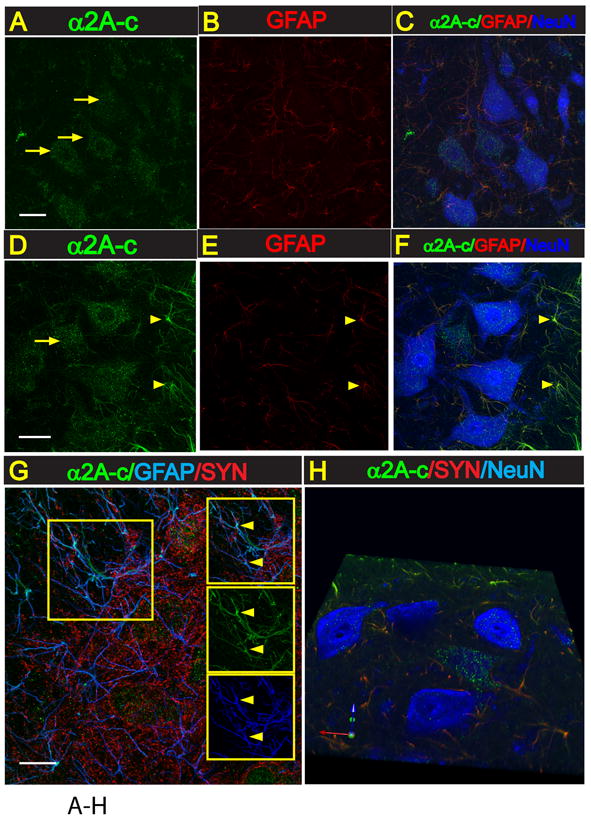
A-F: Transverse lumbar (L3) spinal cord section taken from naive non-ischemic animal (A-C) or from an animal with spinal ischemic spasticity and triple-stained with α2A-c, GFAP and NeuN antibody. In control naive animals a punctuate-like α2A-c immunoreactivity in virtually all neurons including small and medium-sized interneurons and α-motoneurons can be seen (A). Co-staining of the same sections with GFAP antibody showed only weak expression of α2A-c in astrocytes (A, B). In contrast to control animals, in rats with ischemic injury a clear increase in α2A-c expression in activated astrocytes localized at the peri-ischemic regions in the vicinity of persisting α-motoneurons can be identified (D-F; yellow arrows).
G: Confocal images taken from transverse spinal cord lumbar section from an animal with spinal ischemic injury and stained with α2A-c, GFAP and synaptophysin antibody. A clear colocalization of α2A-c and GFAP expression in astrocyte processes can be seen (G-yellow arrows).
H: 3-D image of α2A-c overexpressing astrocytes (green) in the vicinity of lumbar α-motoneurons (blue).
Staining with α2A-m antibody showed the presence of α2A-m immunoreactivity on neuronal membranes of both interneurons and α-motoneurons. No detectable differences in the staining pattern between control and spinal ischemia-injured animals was seen. Confocal analysis of α2A-m and synaptophysin co-stained sections show a spatial colocalization of both proteins (Fig. 5A-F).
Fig. 5. Expression of α2A-m receptor in lumbar spinal cord interneurons and α-motoneurons.
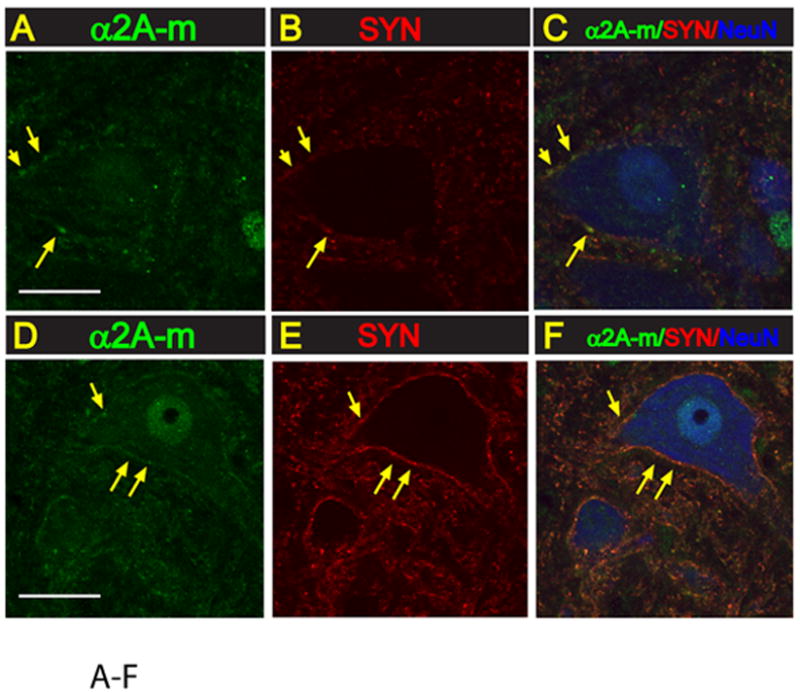
A-F:Transverse lumbar (L3) spinal cord section taken from naive non-ischemic animal (A-C) or from an animal with spinal ischemic spasticity and triple-stained with α2A-m, synaptophysin and NeuN antibody. In both control and spinal ischemic animals a punctate-like α2A-m immunoreactivity can readily be identified on α-motoneuronal membranes (yellow arrows). A spatial colocalization of α2A-m punctata and synaptophysin expression can also be seen (yellow arrows).
Discussion
Systemic or spinal Tizanidine treatment has a potent anti-spasticity effect in animals with spinal ischemic spasticity
In our present study, a potent anti-spasticity effect was seen after both systemic or spinal (intrathecal) treatment with tizanidine and this effect was still present at 60 min after treatment.
Systemically delivered selective and non-selective AR antagonists with or without I 1/2 receptor affinity were then tested to identify receptor specificity of the systemic tizanidine anti-spastic effect. While the lack of reversal after yohimbine treatment was unexpected, a significant reversal was seen after α2A AR (BRL44408) and I 1/2 receptor antagonism (Efaroxan, Idazoxan).
In addition, the anti-spasticity effect measured after intrathecal tizanidine was effectively reversed by IT α2A AR antagonist (Atipamezole). Jointly, these data show that the antispasticity effect seen after tizanidine treatment is mediated in part by the spinal α2A receptor system and potentially by supraspinal and spinal I 1/2 receptors. These data are consistent with previous reports which demonstrate a preferential supraspinal imidazoline receptor and mixed spinal α2 and imidazoline receptor contribution in tizanidine-mediated suppression of spinal polysynaptic reflexes (Honda et al., 2002, Kino et al., 2005).
Consistent with our current data demonstrating tizanidine-mediated antispasticity effect, clinical data in placebo-controlled trials show that systemic treatment with tizanidine reduces muscle tone and the incidence of muscle spasms in patients with multiple sclerosis (Group, 1994). Comparable anti-spasticity effect in patients with chronic spinal trauma, traumatic brain injury or stroke was reported after systemic or spinal treatment with tizanidine or clonidine (Nance et al., 1994, Middleton et al., 1996, Gelber et al., 2001, Meythaler et al., 2001, Kamen et al., 2008).
Suppression of H-reflex activity after systemic Tizanidine treatment
Previous experimental animal studies using systemically or iontophoretically applied tizanidine have demonstrated that tizanidine preferentially depresses peripherally evoked spinal polysynaptic excitation and has modest suppressive effect on spinal monosynaptic reflexes (Davies, 1982, Corboz et al., 1991, Ono et al., 1993). Clinical electrophysiological data in patients with multiple sclerosis show a potent suppression of hyperactive stretch reflexes and ankle clonus after systemic treatment with tizanidine (Lapierre et al., 1987).
In our current study, a significant suppression of H-reflex activity was seen after systemic tizanidine treatment. While the magnitude of this suppressive effect was not as pronounced as we have demonstrated after intrathecal baclofen treatment (Kakinohana et al., 2006), this effect is consistent with a documented tizanidine-mediated potentiation of presynaptic inhibition, suppression of flexor reflexes as well as its direct action on α-motoneurons (Delwaide and Pennisi, 1994).
Effective suppression of spasticity and H-reflex activity after isoflurane anesthesia
Induction of isoflurane anesthesia led to potent suppression of spasticity and significant reduction of H-reflex activity. These data indicate a preferential centrally mediated mechanism in observed ischemia-induced spasticity. In previous studies we have demonstrated a selective degeneration of small inhibitory interneurons in animals with ischemic spasticity and a potent anti-spasticity effect after intrathecal delivery of glycine or baclofen (GABAB receptor agonist) (Marsala et al., 2004, Kakinohana et al., 2006). Jointly, these data show that increased excitability of lumbar α-motoneurons resulting from the loss of local segmental inhibition is primarily responsible for the observed spasticity response measured during the period of muscle stretch. Similarly, clinical data show that isoflurane and sevoflurane anesthesia suppress brisk hyperactive responses during selective dorsal rhizotomy in children with spasticity (Konya et al., 2009) and suppress the H-reflex amplitudes by 65% after 1.2MAC isoflurane anesthesia in normal healthy patients (Zhou et al., 1997, Zhou et al., 1998).
Upregulation of α2A receptor in spinal astrocytes in animals with ischemic spasticity
Previous immunostaining studies in adult rats have shown the presence of the α2A receptors in small and large DRG neurons, both myelinated and unmyelinated axons of the sciatic nerve trunk and on the terminals of capsaicin-sensitive, substance P (SP)-containing primary afferents in the dorsal horn (Stone et al., 1998, Shinder et al., 1999). Similar cellular distribution of α2A mRNA in the adult rat superficial dorsal horn was seen (Zeng and Lynch, 1991). In addition to a prevalent expression of α2A receptors on primary afferents, electrophysiological analysis of extracellularly recorded activity in nociceptive dorsal horn neurons after treatment with medetomidine (a selective α2 receptor agonist) have suggested the presence of α2A on these classes of postsynaptic neurons (Pertovaara et al., 1991). Similarly, in our current study a punctate-like immunoreactivity in spinal cord interneurons but also α-motoneurons was seen with no apparent differences in the expression profile if compared between control animals and animals with induced ischemic spasticity.
In addition to neuronal populations expressing α2A receptors, a comparable expression in astrocytes was reported. Using immunostaining and electron microscopy, α2A receptor was identified in a subpopulation of hippocampal astrocytes (Milner et al., 1998). A similar expression in cultured rat astrocytes as measured by [3H]rauwolscine (alpha 2-receptor antagonist) binding assay, and which was significantly increased after dibutyryl cyclic AMP treatment, was reported (Enkvist et al., 1996). Comparably, in our current study, prominent upregulation of α2A receptor in activated astrocytes localized in the vicinity of lumbar α-motoneurons was seen in spastic animals.
Mechanism of tizanidine anti-spasticity action
Previous studies have demonstrated that adrenergic α 2A, B, C receptors are G protein-coupled receptors and couple to the Giα subunit (a heterotrimeric G protein subunit). Activation of Giα subunit inhibits the production of cAMP through adenylate cyclase inactivation and leads to inhibition of neurotransmitter release (Coward, 1994, Birnbaumer, 2007). Accordingly, several pathological states known to be associated with increased supraspinal or spinal excitatory transmission have been shown to be effectively modulated by treatment with α2 adrenergic agonists through presumed decrease in excitatory amino acid release.
First, noxious afferent stimulation induced by the injection of formalin into the hindpaw leads to a typical biphasic flinching behavior and a rapid and transient increase in glutamate concentration measured in the lumbar intrathecal space (Marsala et al., 1995). Biphasic flinching behavior is effectively suppressed by intrathecal pre-treatment with clonidine (3-12 nmol) (Hao et al., 2001). Consistent with this behavioral effect, other studies have demonstrated that α2 adrenoreceptor agonists can hyperpolarize dorsal horn neurons and act presynaptically on primary afferents to reduce C fiber neurotransmitter release (Davies and Quinlan, 1985, Yaksh et al., 1993) and diminish glutamate release measured in spinal cord synaptosomes in vitro (Kamisaki et al., 1993).
Second, increased primary afferent drive (glutamatergic in nature) has been identified to play a key role in the evolution and maintenance of spasticity state after spinal trauma or ischemia. Accordingly, dorsal rhizotomy provides a potent and long lasting anti-spasticity effect in both human patients and animal models of experimental spasticity. We have recently demonstrated a potent anti-spasticity effect after systemic or intrathecal treatment with AMPA receptor antagonist NXG424 (Hefferan et al., 2007, Oshiro et al., 2010). Thus a potent anti-spasticity effect seen in our current tizanidine study can in part result from suppression of excitatory amino acid (glutamate) release from primary afferents and excitatory interneurons.
Third, previous in vitro studies have demonstrated that activation of cultured astrocytes with NMDA or AMPA leads to a significant increase in intracellular Ca2+, induces glutamate release from activated astrocytes and that such an increased glutamate release is associated with increased frequency of AMPA receptor-mediated miniature EPSPs in neighboring neurons in the hippocampus (Hu et al., 2004, Matsui et al., 2005). Recently, we have demonstrated a comparable AMPA-evoked glutamate release in cultured astrocytes which was effectively blocked by the AMPA receptor antagonist NGX424 (Hefferan et al., 2007). In our current study, a significant upregulation of α2A receptor in activated astrocytes in animals with ischemic spasticity was seen. Using cultured rat cortical astrocytes, is has been demonstrated that the α2 agonist dexmedetomidine inhibits forskolin-induced increases in cyclic AMP, an observation consistent with the demonstrated α2A receptor-mediated adenylate cyclase inactivation. This effect was abolished by the alpha 2-receptor antagonist rauwolscine (Enkvist et al., 1996). Thus, decreased glutamate release from astrocytes after activation of astrocyte-coupled α2A receptors can contribute to the observed anti-spasticity effect (see Fig.6 drawing).
Fig. 6. Schematic diagram of a potential tizanidine-mediated anti-spasticity action.
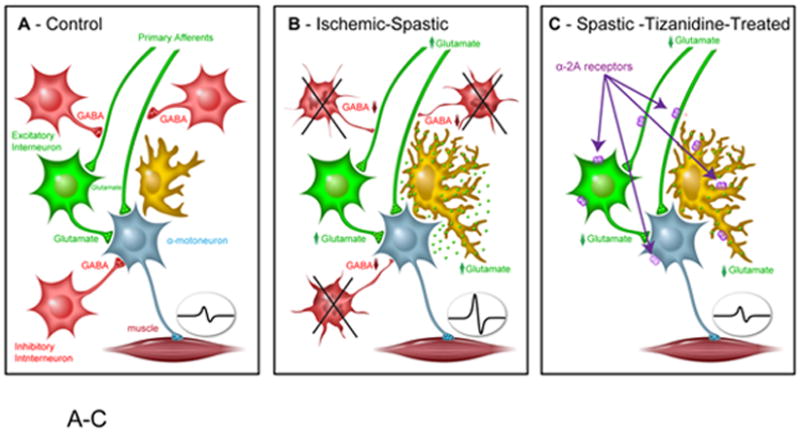
A: Under physiological conditions the magnitude of spinal afferent input and resulting activation of α-motoneuron activity is under control of local inhibitory GABA-ergic interneurons (i.e., pre-, post-synaptic inhibition).
B: In animals with spinal ischemic injury there is a significant loss of local inhibitory GABA-ergic interneurons and resulting loss of pre- and post-synaptic inhibition. In addition, the presence of activated-hypertrophic astrocytes may be associated with increased glutamate release from astrocytes and further potentiate α-motoneuron activity.
C: Activation of α2A receptor may induce its anti-spasticity effect by increasing pre- and post-synaptic inhibition and resulting decrease in glutamate release (Glu) as well as by blocking astrocyte glutamate release.
In summary, our current data using a rat spinal ischemia-induced model of chronic spasticity demonstrate a potent anti-spasticity effect after systemic or spinal treatment with the α2A receptor agonist tizanidine. This effect is primarily mediated by α2A AR and I receptors, but not α2B receptors. Increased expression of α2A receptors in astrocytes also suggests that the tizanidine-mediated anti-spasticity effect can in part be mediated by suppression of secondary glutamate release from activated astrocytes.
Research Highlights.
> Tizanidine (α2-adrenoceptor agonist) is used clinically as a potent anti-spasticity drug. > Tizanidine treatment has comparable anti-spasticity effect in rat model of spasticity. > Tizanidine anti-spasticity effect is mediated by α2A adreno- and imidazoline receptor.
Acknowledgments
This study was supported by grants from the US National Institutes of Health (NS051644 to M.M.) and the Slovak Research and Development Agency (APVV-0314-06 to M.M. and N.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams MM, Hicks AL. Spasticity after spinal cord injury. Spinal Cord. 2005;43:577–586. doi: 10.1038/sj.sc.3101757. [DOI] [PubMed] [Google Scholar]
- Birnbaumer L. Expansion of signal transduction by G proteins. The second 15 years or so: from 3 to 16 alpha subunits plus betagamma dimers. Biochim Biophys Acta. 2007;1768:772–793. doi: 10.1016/j.bbamem.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corboz M, Palmer CI, Palmeri A, Wiesendanger M. Tizanidine-induced depression of polysynaptic cutaneous reflexes in nonanesthetized monkeys is mediated by an alpha 2-adrenergic mechanism. Exp Neurol. 1991;111:210–216. doi: 10.1016/0014-4886(91)90009-2. [DOI] [PubMed] [Google Scholar]
- Coward DM. Tizanidine: neuropharmacology and mechanism of action. Neurology. 1994;44:S6–10. discussion S10-11. [PubMed] [Google Scholar]
- Danzebrink RM, Gebhart GF. Antinociceptive effects of intrathecal adrenoceptor agonists in a rat model of visceral nociception. J Pharmacol Exp Ther. 1990;253:698–705. [PubMed] [Google Scholar]
- Davies J. Selective depression of synaptic transmission of spinal neurones in the cat by a new centrally acting muscle relaxant, 5-chloro-4-(2-imidazolin-2-yl-amino)-2, 1, 3-benzothiodazole (DS103-282) Br J Pharmacol. 1982;76:473–481. doi: 10.1111/j.1476-5381.1982.tb09242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J, Quinlan JE. Selective inhibition of responses of feline dorsal horn neurones to noxious cutaneous stimuli by tizanidine (DS103-282) and noradrenaline: involvement of alpha 2-adrenoceptors. Neuroscience. 1985;16:673–682. doi: 10.1016/0306-4522(85)90200-3. [DOI] [PubMed] [Google Scholar]
- Delwaide PJ, Pennisi G. Tizanidine and electrophysiologic analysis of spinal control mechanisms in humans with spasticity. Neurology. 1994;44:S21–27. discussion S27-28. [PubMed] [Google Scholar]
- Enkvist MO, Hamalainen H, Jansson CC, Kukkonen JP, Hautala R, Courtney MJ, Akerman KE. Coupling of astroglial alpha 2-adrenoreceptors to second messenger pathways. J Neurochem. 1996;66:2394–2401. doi: 10.1046/j.1471-4159.1996.66062394.x. [DOI] [PubMed] [Google Scholar]
- Gelber DA, Good DC, Dromerick A, Sergay S, Richardson M. Open-label dose-titration safety and efficacy study of tizanidine hydrochloride in the treatment of spasticity associated with chronic stroke. Stroke. 2001;32:1841–1846. doi: 10.1161/01.str.32.8.1841. [DOI] [PubMed] [Google Scholar]
- Group UKTT. A double-blind, placebo-controlled trial of tizanidine in the treatment of spasticity caused by multiple sclerosis. United Kingdom Tizanidine Trial Group. Neurology. 1994;44:S70–78. [PubMed] [Google Scholar]
- Hao S, Takahata O, Iwasaki H. Antinociceptive interaction between spinal clonidine and lidocaine in the rat formalin test: an isobolographic analysis. Anesth Analg. 2001;92:733–738. doi: 10.1097/00000539-200103000-00034. [DOI] [PubMed] [Google Scholar]
- Hassan N, McLellan DL. Double-blind comparison of single doses of DS103-282, baclofen and placebo for suppression of spasticity. J Neurol Neurosurg Psychiatry. 1980;43:1132–1136. doi: 10.1136/jnnp.43.12.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefferan MP, Fuchigami T, Marsala M. Development of baclofen tolerance in a rat model of chronic spasticity and rigidity. Neurosci Lett. 2006;403:195–200. doi: 10.1016/j.neulet.2006.04.048. [DOI] [PubMed] [Google Scholar]
- Hefferan MP, Kucharova K, Kinjo K, Kakinohana O, Sekerkova G, Nakamura S, Fuchigami T, Tomori Z, Yaksh TL, Kurtz N, Marsala M. Spinal astrocyte glutamate receptor 1 overexpression after ischemic insult facilitates behavioral signs of spasticity and rigidity. J Neurosci. 2007;27:11179–11191. doi: 10.1523/JNEUROSCI.0989-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honda M, Sekiguchi Y, Sato N, Ono H. Involvement of imidazoline receptors in the centrally acting muscle-relaxant effects of tizanidine. Eur J Pharmacol. 2002;445:187–193. doi: 10.1016/s0014-2999(02)01664-3. [DOI] [PubMed] [Google Scholar]
- Hu B, Sun SG, Tong ET. NMDA and AMPA receptors mediate intracellular calcium increase in rat cortical astrocytes. Acta Pharmacol Sin. 2004;25:714–720. [PubMed] [Google Scholar]
- Kakinohana O, Hefferan MP, Nakamura S, Kakinohana M, Galik J, Tomori Z, Marsala J, Yaksh TL, Marsala M. Development of GABA-sensitive spasticity and rigidity in rats after transient spinal cord ischemia: a qualitative and quantitative electrophysiological and histopathological study. Neuroscience. 2006;141:1569–1583. doi: 10.1016/j.neuroscience.2006.04.083. [DOI] [PubMed] [Google Scholar]
- Kamen L, Henney HR, 3rd, Runyan JD. A practical overview of tizanidine use for spasticity secondary to multiple sclerosis, stroke, and spinal cord injury. Curr Med Res Opin. 2008;24:425–439. doi: 10.1185/030079908x261113. [DOI] [PubMed] [Google Scholar]
- Kamisaki Y, Hamada T, Maeda K, Ishimura M, Itoh T. Presynaptic alpha 2 adrenoceptors inhibit glutamate release from rat spinal cord synaptosomes. J Neurochem. 1993;60:522–526. doi: 10.1111/j.1471-4159.1993.tb03180.x. [DOI] [PubMed] [Google Scholar]
- Kino Y, Tanabe M, Honda M, Ono H. Involvement of supraspinal imidazoline receptors and descending monoaminergic pathways in tizanidine-induced inhibition of rat spinal reflexes. J Pharmacol Sci. 2005;99:52–60. doi: 10.1254/jphs.fp0050520. [DOI] [PubMed] [Google Scholar]
- Kita M, Goodkin DE. Drugs used to treat spasticity. Drugs. 2000;59:487–495. doi: 10.2165/00003495-200059030-00006. [DOI] [PubMed] [Google Scholar]
- Konya D, Gercek A, Dagcinar A, Baykan N, Ozek MM. Prevention of brisk hyperactive response during selective dorsal rhizotomy in children with spasticity: isoflurane versus sevoflurane maintenance anesthesia. J Clin Neurosci. 2009;16:241–245. doi: 10.1016/j.jocn.2008.02.007. [DOI] [PubMed] [Google Scholar]
- Lance JW. Spasticity: Disordered motor control. In: Feldman RG, Young RR, Koella WP, editors. Symposium synopsis. Chicago: Year Book Medical Publishers; 1980. pp. 485–494. [Google Scholar]
- Lapierre Y, Bouchard S, Tansey C, Gendron D, Barkas WJ, Francis GS. Treatment of spasticity with tizanidine in multiple sclerosis. Can J Neurol Sci. 1987;14:513–517. doi: 10.1017/s0317167100038026. [DOI] [PubMed] [Google Scholar]
- Leiphart JW, Dills CV, Zikel OM, Kim DL, Levy RM. A comparison of intrathecally administered narcotic and nonnarcotic analgesics for experimental chronic neuropathic pain. J Neurosurg. 1995;82:595–599. doi: 10.3171/jns.1995.82.4.0595. [DOI] [PubMed] [Google Scholar]
- Marsala M, Hefferan MP, Kakinohana O, Nakamura S, Marsala J, Tomori Z. Measurement of peripheral muscle resistance in rats with chronic ischemia-induced paraplegia or morphine-induced rigidity using a semi-automated computer-controlled muscle resistance meter. J Neurotrauma. 2005;22:1348–1361. doi: 10.1089/neu.2005.22.1348. [DOI] [PubMed] [Google Scholar]
- Marsala M, Kakinohana O, Yaksh TL, Tomori Z, Marsala S, Cizkova D. Spinal implantation of hNT neurons and neuronal precursors: graft survival and functional effects in rats with ischemic spastic paraplegia. Eur J Neurosci. 2004;20:2401–2414. doi: 10.1111/j.1460-9568.2004.03702.x. [DOI] [PubMed] [Google Scholar]
- Marsala M, Malmberg AB, Yaksh TL. The spinal loop dialysis catheter: characterization of use in the unanesthetized rat. Journal of Neuroscience Methods. 1995;62:43–53. doi: 10.1016/0165-0270(95)00053-4. [DOI] [PubMed] [Google Scholar]
- Matsui K, Jahr CE, Rubio ME. High-concentration rapid transients of glutamate mediate neural-glial communication via ectopic release. J Neurosci. 2005;25:7538–7547. doi: 10.1523/JNEUROSCI.1927-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsushita A, Smith CM. Spinal cord function in postischemic rigidity in the rat. Brain Research. 1970;19:395–410. doi: 10.1016/0006-8993(70)90382-3. [DOI] [PubMed] [Google Scholar]
- Maurice V, Allan HR, Raymond DA. Adams & Victor’s Principles of Neurology. 7. McGraw-Hill Companies; 2001. [Google Scholar]
- McCarthy RJ, Kroin JS, Lubenow TR, Penn RD, Ivankovich AD. Effect of intrathecal tizanidine on antinociception and blood pressure in the rat. Pain. 1990;40:333–338. doi: 10.1016/0304-3959(90)91130-B. [DOI] [PubMed] [Google Scholar]
- Meythaler JM, Guin-Renfroe S, Johnson A, Brunner RM. Prospective assessment of tizanidine for spasticity due to acquired brain injury. Arch Phys Med Rehabil. 2001;82:1155–1163. doi: 10.1053/apmr.2001.25141. [DOI] [PubMed] [Google Scholar]
- Middleton JW, Siddall PJ, Walker S, Molloy AR, Rutkowski SB. Intrathecal clonidine and baclofen in the management of spasticity and neuropathic pain following spinal cord injury: a case study. Arch Phys Med Rehabil. 1996;77:824–826. doi: 10.1016/s0003-9993(96)90264-6. [DOI] [PubMed] [Google Scholar]
- Milner TA, Lee A, Aicher SA, Rosin DL. Hippocampal alpha2a-adrenergic receptors are located predominantly presynaptically but are also found postsynaptically and in selective astrocytes. J Comp Neurol. 1998;395:310–327. [PubMed] [Google Scholar]
- Nance PW, Bugaresti J, Shellenberger K, Sheremata W, Martinez-Arizala A. Efficacy and safety of tizanidine in the treatment of spasticity in patients with spinal cord injury. North American Tizanidine Study Group. Neurology. 1994;44:S44–51. discussion S51-42. [PubMed] [Google Scholar]
- Noreau L, Proulx P, Gagnon L, Drolet M, Laramee MT. Secondary impairments after spinal cord injury: a population-based study. Am J Phys Med Rehabil. 2000;79:526–535. doi: 10.1097/00002060-200011000-00009. [DOI] [PubMed] [Google Scholar]
- Ono H, Fukushima C, Fukuda H. Effect of the centrally acting muscle relaxant tizanidine on spinal reflexes: involvement of descending noradrenergic systems. Jpn J Pharmacol. 1993;62:357–362. doi: 10.1254/jjp.62.357. [DOI] [PubMed] [Google Scholar]
- Oshiro M, Hefferan MP, Kakinohana O, Lukacova N, Sugahara K, Yaksh TL, Marsala M. Suppression of stretch reflex activity after spinal or systemic treatment with AMPA receptor antagonist NGX424 in rats with developed baclofen tolerance. British Journal of Pharmacology. 2010;161:976–985. doi: 10.1111/j.1476-5381.2010.00954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertovaara A, Kauppila T, Jyvasjarvi E, Kalso E. Involvement of supraspinal and spinal segmental alpha-2-adrenergic mechanisms in the medetomidine-induced antinociception. Neuroscience. 1991;44:705–714. doi: 10.1016/0306-4522(91)90089-7. [DOI] [PubMed] [Google Scholar]
- Schwarz M, Block F, Pergande G. N-methyl-D-aspartate (NMDA)-mediated muscle relaxant action of flupirtine in rats. Neuroreport. 1994;5:1981–1984. doi: 10.1097/00001756-199410000-00036. [DOI] [PubMed] [Google Scholar]
- Shinder V, Govrin-Lippmann R, Cohen S, Belenky M, Ilin P, Fried K, Wilkinson HA, Devor M. Structural basis of sympathetic-sensory coupling in rat and human dorsal root ganglia following peripheral nerve injury. J Neurocytol. 1999;28:743–761. doi: 10.1023/a:1007090105840. [DOI] [PubMed] [Google Scholar]
- Sie OG, Lakke JP. The spasmolytic properties of 5-chloro-4-(2-imidazolin-2-yl-amino)-2, 1,3- benzothiadiazole hydrochloride (DS 103-282): a pilot study. Clin Neurol Neurosurg. 1980;82:273–279. doi: 10.1016/0303-8467(80)90020-7. [DOI] [PubMed] [Google Scholar]
- Stone LS, Broberger C, Vulchanova L, Wilcox GL, Hokfelt T, Riedl MS, Elde R. Differential distribution of alpha2A and alpha2C adrenergic receptor immunoreactivity in the rat spinal cord. J Neurosci. 1998;18:5928–5937. doi: 10.1523/JNEUROSCI.18-15-05928.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taira Y, Marsala M. Effect of proximal arterial perfusion pressure on function, spinal cord blood flow, and histopathologic changes after increasing intervals of aortic occlusion in the rat. Stroke. 1996;27:1850–1858. doi: 10.1161/01.str.27.10.1850. [DOI] [PubMed] [Google Scholar]
- Tarlov IM. Rigidity in man due to spinal interneuron loss. Arch Neurol. 1967;16:536–543. doi: 10.1001/archneur.1967.00470230088012. [DOI] [PubMed] [Google Scholar]
- Van Harreveld A, Spinelli D. Reflex activity in spinal cats with postasphyxial rigidity. Arch Int Physiol Biochim. 1965;73:209–230. doi: 10.3109/13813456509084248. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Jage J, Takano Y. Pharmacokinetics and pharmacodynamics of medullar agents. III. The spinal actions of alpha 2-adrenergic agonists as analgesic. In: Aitkenhead AR, Benad G, Brown BR, Cousins MJ, Jones JG, Struninn L, Thomson D, Van Aken H, editors. Bailliere’s clinical anesthesiology. 3. Vol. 7. London: Bailliere Tindall; 1993. pp. 597–614. [Google Scholar]
- Yaksh TL, Rudy TA. Analgesia mediated by a direct spinal action of narcotics. Science. 1976;192:1357–1358. doi: 10.1126/science.1273597. [DOI] [PubMed] [Google Scholar]
- Zeng DW, Lynch KR. Distribution of alpha 2-adrenergic receptor mRNAs in the rat CNS. Brain Res Mol Brain Res. 1991;10:219–225. doi: 10.1016/0169-328x(91)90064-5. [DOI] [PubMed] [Google Scholar]
- Zhou HH, Jin TT, Qin B, Turndorf H. Suppression of spinal cord motoneuron excitability correlates with surgical immobility during isoflurane anesthesia. Anesthesiology. 1998;88:955–961. doi: 10.1097/00000542-199804000-00015. [DOI] [PubMed] [Google Scholar]
- Zhou HH, Mehta M, Leis AA. Spinal cord motoneuron excitability during isoflurane and nitrous oxide anesthesia. Anesthesiology. 1997;86:302–307. doi: 10.1097/00000542-199702000-00005. [DOI] [PubMed] [Google Scholar]