

Abstract
Osteoclasts resorb the mineralized matrices formed by chondrocytes or osteoblasts. The cytokine receptor activator of NFκB ligand (RANKL) is essential for osteoclast formation and thought to be supplied by osteoblasts or their precursors. However, RANKL is expressed by a variety of cell types and it is unclear which of them are essential sources for osteoclast formation. Here we have used a conditional mouse RANKL allele and a series of Cre-deleter strains to demonstrate that hypertrophic chondrocytes and osteocytes, both of which are embedded in matrix, are essential sources of the RANKL that controls mineralized cartilage resorption and bone remodeling, respectively. Moreover, osteocyte RANKL is responsible for the bone loss associated with unloading. Contrary to the current paradigm, RANKL produced by osteoblasts or their progenitors does not contribute to bone remodeling. These results suggest that the rate-limiting step of matrix resorption is controlled by cells embedded within the matrix itself.
Resorption of cartilage and bone is essential for development and regeneration of the skeleton. During longitudinal bone growth, calcified cartilage produced by chondrocytes is resorbed and replaced by a bone matrix made by osteoblasts in a process known as endochondral bone formation1. After growth, older bone is periodically resorbed and newer bone is deposited in the resulting cavities by osteoblasts in a process known as remodeling2. Osteoclasts, multinucleated cells derived from the monocyte/macrophage lineage, are responsible for resorption of the mineralized matrices in each of these processes3. Excessive bone resorption causes the most common bone disorders including osteoporosis, Paget’s disease, and osteolysis from cancer2, 4, 5.
The TNF-family cytokine RANKL (encoded by the Tnfsf11 gene) initiates osteoclast differentiation and is essential for the development, function, and survival of osteoclasts6, 7. According to the prevailing paradigm, osteoblasts on the bone surface, or their progenitors in the marrow, supply the RANKL responsible for osteoclast generation8-11. But this idea is based primarily on in vitro experiments demonstrating that osteoblast progenitors support osteoclast formation12. In spite of the lack of in vivo evidence, the concept that osteoblasts or their progenitors control osteoclast generation has gained wide acceptance over the last 30 years and has been used to explain how bone formation is linked to bone resorption during remodeling9, 11, 13. However, several observations suggest that matrix synthesizing osteoblasts are not essential for osteoclast formation, and therefore may not be a major source of RANKL. First, targeted ablation of osteoblasts in transgenic mice does not reduce osteoclast number or RANKL expression14, 15. Second, a variety of genetic modifications in mice alters osteoblast number without changing osteoclast number16-18. And third, administration of glucocorticoids potently reduces osteoblast number on bone, as well as the abundance of their precursors in the bone marrow, but not the number of osteoclasts19, 20.
To identify the cellular sources of RANKL during bone growth and remodeling, we generated mice with a conditional RANKL allele and crossed them with several lines of transgenic mice expressing the Cre recombinase in genetically-defined cell populations representing different stages of osteoblast and chondrocyte differentiation. We report that hypertrophic chondrocytes, which are buried within mineralized cartilage, supply RANKL during bone growth. Moreover, osteocytes - former osteoblasts buried within mineralized bone that sense and respond to changes in mechanical forces - are an essential source of RANKL during bone remodeling; and consistent with this finding, mice lacking RANKL in osteocytes are protected from bone loss due to unloading. Thus, the resorption that occurs during both bone development and remodeling is orchestrated by matrix-embedded cells via production of the rate limiting factor for osteoclast differentiation and function.
RESULTS
Mesenchymal cell RANKL is essential for osteoclastogenesis
To allow deletion of RANKL in various genetically-defined cell populations, we generated mice harboring a Tnfsf11 allele in which exons 3 and 4 were flanked by loxP sites, hereafter referred to as RANKLf/f mice (Supplementary Fig. 1). To determine whether RANKL expression in cells of the mesenchymal lineage is required for osteoclast formation, we crossed RANKLf/f mice with transgenic mice expressing the Cre recombinase under the control of Prrx1 regulatory elements, hereafter referred to as Prx1-Cre mice. Prx1-Cre mice express the Cre recombinase in the mesenchymal condensations that form the developing limbs and parts of the skull, but not in the spine or other organs21. RANKL mRNA was significantly lower in the tibia and calvaria, but not in the vertebra or spleen, of 5-week-old Prx1-Cre;RANKLf/f mice compared to RANKLf/f littermates, confirming deletion in the expected tissues (Fig. 1a).
Figure 1. Deletion of RANKL in Prx1-Cre expressing cells causes osteopetrosis.
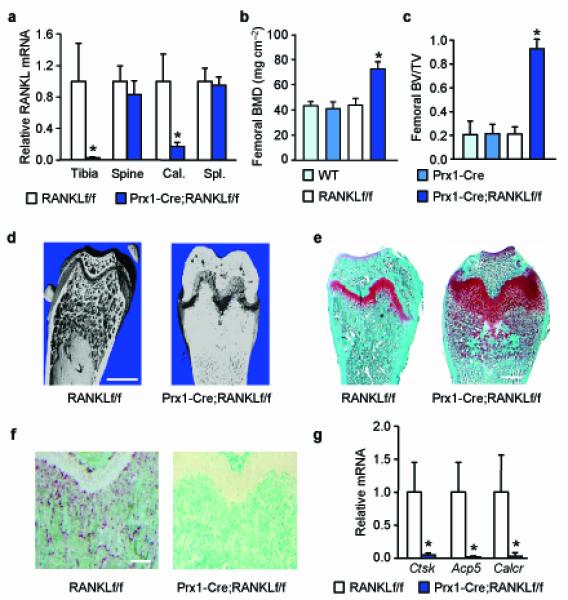
(a) RANKL mRNA levels in bone and spleen of RANKLf/f (n = 9) and Prx1-Cre;RANKLf/f (n = 10) littermates (here and throughout, values are the mean ± s.d.). *P < 0.05 versus RANKLf/f, using Student’s t-test. (b) Femoral BMD of WT (n = 7), Prx1-Cre (n = 7), RANKLf/f (n = 9), and Prx1-Cre;RANKLf/f (n = 10) littermates. *P < 0.05 versus WT, Prx1-Cre, and RANKLf/f, using 2-way ANOVA. (c) Cancellous bone volume in the distal femur of WT (n = 8), Prx1-Cre (n = 8), RANKLf/f (n = 9), and Prx1-Cre;RANKLf/f (n = 10) littermates. *P < 0.05 versus WT, Prx1-Cre, and RANKLf/f, using 2-way ANOVA. (d) Representative μCT images of the distal femur of 5-week-old RANKLf/f and Prx1-Cre;RANKLf/f mice. Scale bar, 1 mm. (e) Histological sections of distal femurs of 5-week-old RANKLf/f and Prx1-Cre;RANKLf/f mice stained with safranin-O (cartilage stains red). Scale bar, 0.5 mm. (f) Histological sections of distal femurs of 5-week-old RANKLf/f and Prx1-Cre;RANKLf/f mice stained for tartrate-resistant acid phosphatase (TRAP) activity (osteoclasts stain red). Scale bar, 200 μm. (g) Cathepsin K (Ctsk), TRAP (Acp5), and calcitonin receptor (Calcr) mRNA levels in tibial RNA of RANKLf/f (n = 9) and Prx1-Cre;RANKLf/f (n = 10) mice. *P < 0.05 versus RANKLf/f, using Student’s t-test. All values were determined in 5-week-old mice, including both sexes.
Consistent with the changes in RANKL expression, bone mineral density (BMD) and cancellous bone volume were increased in the femur (Fig. 1b,c), but not in the spine (Supplementary Fig. 2), of Prx1-Cre;RANKLf/f mice compared to RANKLf/f littermates. Bone mass and volume were not altered in Prx1-Cre or RANKLf/f mice compared to wild-type (WT) littermates (Fig. 1b,c), demonstrating that the Prx1-Cre transgene and the RANKL conditional allele had no impact on the skeleton on their own. Therefore, all subsequent analyses were performed by comparing only RANKLf/f and Prx1-Cre;RANKLf/f littermates. High resolution micro-computed tomography (μCT) analysis of the distal femur revealed severe osteopetrosis in Prx1-Cre;RANKLf/f mice (Fig. 1d). Safranin-O staining, which specifically detects cartilage, revealed large amounts of unresorbed cartilage and widened growth plates in the femurs of Prx1-Cre;RANKLf/f mice (Fig. 1e), findings similar to those observed in mice with germline deletion of either RANKL or its receptor RANK6, 22. Moreover, osteoclasts were completely absent in the femurs of Prx1-Cre;RANKLf/f mice, whereas they were abundant in control littermates (Fig. 1f). Consistent with this, osteoclast-specific gene expression was notably lower in the conditional knock-out mice compared with control littermates (Fig. 1g). These results demonstrate that RANKL expression in mesenchyme-derived cells is essential for all osteoclast formation in growing long bones. Tooth eruption, which also requires osteoclast activity6, was normal in Prx1-Cre;RANKLf/f mice (Supplementary Fig. 2), demonstrating that the Prx1-Cre expression in the skull does not occur in cells that produce the RANKL required for this process.
Loss of chondrocyte RANKL is associated with osteopetrosis
The Prx1-Cre transgene causes deletion in both chondrocytes and osteoblast-lineage cells (Supplementary Fig. 2). To determine the contribution of these cell types, we compared the skeletal phenotype of mice in which RANKL was deleted using transgenes in which the Cre recombinase was under the control of regulatory elements of the following genes: Sp7, hereafter referred to as osterix 1 (Osx1)-Cre mice, Bglap, hereafter referred to as osteocalcin (Ocn)-Cre mice, or Dmp1, hereafter referred to as dentin matrix protein 1 (Dmp1)-Cre mice. The Osx1-Cre transgene becomes active at the earliest stages of osteoblast differentiation as well as in late proliferating chondrocytes23, 24. Ocn-Cre has been reported to be active specifically in mature osteoblasts25, and Dmp1-Cre specifically in osteocytes26. Because these transgenes become active at different stages of osteoblast differentiation, and because only some have activity in chondrocytes, we reasoned that their use with the RANKLf/f allele might identify essential cellular sources of RANKL for osteoclast formation.
RANKL mRNA was lower in bones from Osx1-Cre;RANKLf/f and Ocn-Cre;RANKLf/f mice, but not Dmp1-Cre;RANKLf/f mice, compared to control littermates (Fig. 2a). However, deletion of the RANKL conditional allele was detectable in cortical bone from Dmp1-Cre;RANKLf/f mice (Fig. 2b). RANKL mRNA was unaffected in the spleens of all three models demonstrating the specificity of deletion (Supplementary Fig. 3). Cancellous bone volume, measured by μCT at 5 weeks of age, was much higher in Osx1-Cre;RANKLf/f and Ocn-Cre;RANKLf/f mice, but not in Dmp1-Cre;RANKLf/f mice, compared to control littermates (Fig. 2c). Comparison of the BMD of WT littermates with littermates harboring only the Cre transgenes demonstrated that the high bone mass was not associated with the transgenes alone (Supplementary Fig. 3). Tooth eruption was completely absent in Osx1-Cre;RANKLf/f mice, occurred in some but not all Ocn-Cre;RANKLf/f mice, but was normal in Dmp1-Cre;RANKLf/f mice (Fig. 2d). Large amounts of unresorbed calcified cartilage and widened growth plates were present in Osx1-Cre;RANKLf/f and Ocn-Cre;RANKLf/f femurs, whereas normal cancellous bone and growth plates were present in Dmp1-Cre;RANKLf/f mice (Fig. 2d). Consistent with this, osteoclast abundance (Fig. 2d) and osteoclast-specific gene expression (Supplementary Fig. 3) were lower in Osx1-Cre;RANKLf/f and Ocn-Cre;RANKLf/f mice, but not in Dmp1-Cre;RANKLf/f mice, compared to control littermates.
Figure 2. Deletion of RANKL in Osx1-Cre and Ocn-Cre expressing cells causes osteopetrosis.
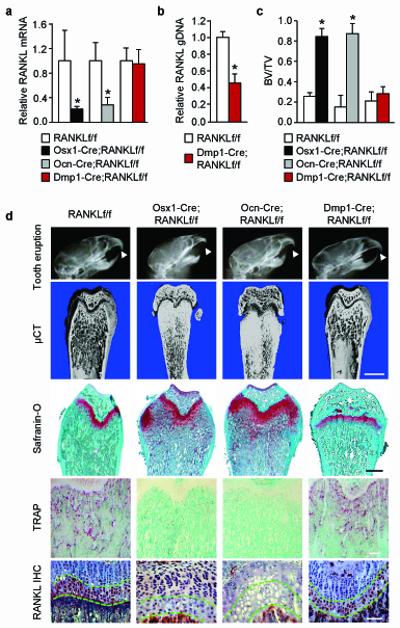
(a) RANKL mRNA levels in whole tibia of Osx1-Cre;RANKLf/f (n = 8), Ocn-Cre;RANKLf/f (n = 6), Dmp1-Cre;RANKLf/f (n = 9), and their respective RANKLf/f littermates (n = 4 to 11). *P < 0.05 versus RANKLf/f littermates, using Student’s t-test. (b) Quantitative PCR of loxP-flanked genomic DNA isolated from collagenase-digested femoral and tibial cortical bone of Dmp1-Cre;RANKLf/f (n = 9) mice and their RANKLf/f (n = 11) littermates. *P < 0.05 using Student’s t-test. (c) Cancellous bone volume of the distal femurs of Osx1-Cre;RANKLf/f (n = 8), Ocn-Cre;RANKLf/f (n = 6), Dmp1-Cre;RANKLf/f (n = 9), and their RANKLf/f littermates (n = 4 to 11). *P < 0.05 versus RANKLf/f littermates, using Student’s t-test. (d) X-ray images, representative μCT images of the distal femur (scale bar, 1 mm), safranin-O-stained histological sections of the distal femur (scale bar, 0.5 mm), anti-RANKL immunohistochemistry (IHC) (scale bar, 100 μm), and TRAP-stained histological sections of the distal femur (scale bar, 200 μm) from Osx1-Cre;RANKLf/f, Ocn-Cre;RANKLf/f, Dmp1-Cre;RANKLf/f, and a representative RANKLf/f littermate. Arrowheads in the X-rays indicate position of erupted incisors. μCT images for each of the RANKLf/f control littermates are presented in Supplementary Fig. 3. The region of the growth plate containing hypertrophic chondrocytes in the IHC images is outlined by green dashed lines and non-immune IgG controls are presented in Supplementary Fig. 3. All values and images are from 5-week-old mice and include both sexes.
Based on the reported cell type-specificity of the Osx1-Cre, Ocn-Cre, and Dmp1-Cre transgenes23, 25, 26, these results suggested that RANKL produced by mature osteoblasts, but not osteocytes or chondrocytes, is required for generating the osteoclasts that resorb the primary spongiosa during bone growth. However, hypertrophic chondrocytes express high levels of RANKL and have been suggested as a possible source of the RANKL required for calcified cartilage resorption27, 28. Therefore, we determined whether the Ocn-Cre transgene also deleted RANKL from hypertrophic chondrocytes and found this to be the case, as determined by immunohistochemistry (Fig. 2d). Analysis of Osx1-Cre and Ocn-Cre mice with R26R reporter mice confirmed that both result in Cre-mediated reporter gene activation in hypertrophic chondrocytes (Supplementary Fig. 4).
These results demonstrate that deletion of RANKL in hypertrophic chondrocytes and osteoblasts (Prx1-Cre, Osx1-Cre, and Ocn-Cre) but not osteocytes (Dmp1-Cre) leads to retention of calcified cartilage. Cartilage retention was also observed when RANKLf/f mice were crossed with transgenic mice expressing the Cre recombinase under the control of Col10a1 regulatory elements, hereafter referred to as collagen X (ColX)-Cre mice, which have been shown to activate a reporter gene efficiently in hypertrophic chondrocytes29 (Supplementary Fig. 5). Consistent with this, RANKL immunostaining was abolished in hypertrophic chondrocytes of these mice (Supplementary Fig. 5). However, analysis of ColX-Cre mice crossed with R26R reporter mice revealed reporter gene activation in scattered osteoblasts and osteocytes in the distal femur (Supplementary Fig. 5), suggesting that either a subset of osteoblast-lineage cells in this region expresses sufficient levels of the ColX-Cre transgene to mediate loxP recombination or that the transgene is active in a subset of osteo-chondro progenitors.
Osteocyte RANKL is essential for bone remodeling
Normal resorption of the primary spongiosa, and thereby production of cancellous bone, in the Dmp1-Cre;RANKLf/f mice allowed us to use this model to determine whether RANKL expression in osteocytes contributes to cancellous bone remodeling. The BMD of Dmp1-Cre;RANKLf/f mice became progressively higher than control littermates up to 6 months of age (Fig. 3a), at which time cancellous bone volume in the femur and spine was higher than control littermates (Fig. 3b,c). The RANKL conditional allele was reduced by more than 70% in genomic DNA isolated from cortical bone enriched in osteocytes (Fig. 3d). However, RANKL mRNA levels measured in whole tibia were not reduced and only about a 40% reduction occurred in whole L5 vertebra (Fig. 3d). This absent or small change in RANKL mRNA in whole bone, despite efficient deletion of the gene in osteocytes, suggests that the contribution of RANKL mRNA produced by osteocytes to the total RANKL mRNA in whole bone is small. Nonetheless, osteoclast number was reduced by more than 70% in femoral cancellous bone of Dmp1-Cre;RANKLf/f mice compared to control littermates (Fig. 3e). Consistent with this, circulating markers of bone resorption and bone formation were significantly lower in the Dmp1-Cre;RANKLf/f mice compared to control mice (Fig. 3f). However, the concentration of circulating soluble RANKL was unchanged, highlighting the importance of locally-produced RANKL (Fig. 3f). These results suggest that RANKL produced by osteocytes is responsible for the majority of the osteoclast differentiation that drives remodeling of cancellous bone.
Figure 3. Deletion of RANKL from Dmp1-Cre expressing cells reduces bone remodeling.
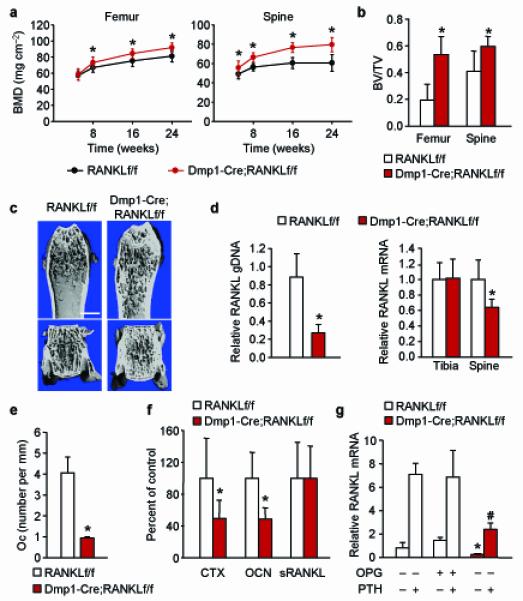
(a) Serial BMD of Dmp1-Cre;RANKLf/f (n = 14) and RANKLf/f (n = 19) littermates. *P < 0.05 using Student’s t-test comparing the two genotypes at a given age. (b) Cancellous bone volume in the distal femur or in L4 vertebra of 6-month-old Dmp1-Cre;RANKLf/f (n = 11) and RANKLf/f (n = 7) littermates. *P < 0.05 using Student’s t-test. (c) Representative μCT images of the distal femur and L4 vertebra of 6-month-old Dmp1-Cre;RANKLf/f and RANKLf/f littermates. Scale bar, 1 mm. (d) Left, quantitative PCR of loxP-flanked RANKL genomic DNA using genomic DNA isolated from collagenase-digested femoral cortical bone of 6-month-old Dmp1-Cre;RANKLf/f (n = 11) and RANKLf/f (n = 7) littermates. Right, quantitative RT-PCR for RANKL mRNA in tibia and L5 vertebra of the same mice as in the left panel. *P < 0.05 using Student’s t-test. (e) Osteoclast number per mm bone surface in cancellous bone of the distal femur of 6-month-old Dmp1-Cre;RANKLf/f (n = 4) and RANKLf/f (n = 4) littermates. *P < 0.05 using Student’s t-test. (f) Carboxy-terminal crosslinked telopeptide of type I collagen (CTX), osteocalcin (Ocn), or soluble RANKL in the blood plasma of 6-month-old Dmp1-Cre;RANKLf/f (n = 9) and RANKLf/f (n = 8) littermates. *P < 0.05 using Student’s t-test. (g) RANKL mRNA levels in tibial cortical bone of 6-month-old Dmp1-Cre;RANKLf/f and RANKLf/f littermates, pretreated with vehicle or OPG and then injected with vehicle or PTH(1-34) (n = 6 to 8 per group). *P < 0.05 versus RANKLf/f mice pretreated with vehicle or OPG and then injected with vehicle using 2-way ANOVA. #P < 0.05 versus RANKLf/f mice pretreated with vehicle or OPG and then injected with PTH using 2-way ANOVA. All values include data from both sexes.
Based on our experience with the Ocn-Cre transgene, we sought to confirm the cell-type specificity of the Dmp1-Cre transgene using R26R mice. We found that, in addition to osteocytes, the transgene was active in cells on the cancellous bone surface (Supplementary Fig. 6). The LacZ-positive cells were present on bone matrix, had morphology consistent with osteoblasts, and were located on top of newly formed bone (Supplementary Fig. 6). These results demonstrate that the Dmp1-Cre transgene is active in both osteoblasts and osteocytes. Since previous studies by us and others have shown that ablation of matrix-synthesizing osteoblasts had no effect on RANKL expression or osteoclast number, the reduction in RANKL and osteoclast number that occurred in Dmp1-Cre;RANKLf/f mice must, therefore, be due to loss of RANKL in osteocytes. This conclusion is supported by evidence that reduction of osteoblasts in mice pretreated with osteoprotegerin (OPG), verified by reduced osteocalcin mRNA levels (Supplementary Fig. 6), did not reduce basal or parathyroid hormone (PTH)-stimulated RANKL expression in cortical bone; whereas deletion of RANKL with the Dmp1-Cre transgene reduced both basal and PTH-stimulated RANKL mRNA levels (Fig. 3g).
Osteoprogenitor RANKL does not contribute to remodeling
Because deletion of RANKL in Dmp1-Cre mice did not completely eliminate osteoclasts on cancellous bone, we sought to determine whether RANKL produced by committed osteoblast progenitors plays a role in osteoclast formation at this site. To do this, we took advantage of the ability to suppress expression of the Osx1-Cre transgene with doxycycline23. We reasoned that suppression of RANKL deletion in hypertrophic chondrocytes until after 4 months of age, a time at which the activity of the growth plate is greatly reduced, would allow the formation of normal cancellous bone architecture and that any reduction in osteoclast formation after this time would be the result of loss of RANKL in the entire osteoblast lineage.
Exposure of Osx1-Cre;RANKLf/f mice to doxycycline in utero and until 4 months of age allowed normal tooth eruption to occur, demonstrating suppression of Osx1-Cre transgene expression (Fig. 4a). At 4 months of age the mice were switched to a doxycycline-free diet and maintained for an additional 2 months to allow deletion of RANKL in Osx1-Cre expressing cells. Histological analysis of femurs at 6 months of age revealed a normal cancellous architecture with no evidence of cartilage retention (Fig. 4a). RANKL deletion occurred in osteocytes (Fig. 4b), but to a lesser extent than with the Dmp1-Cre transgene (Fig. 3d). RANKL mRNA was significantly reduced in whole tibia, but not in L5 vertebra, of Osx1-Cre;RANKLf/f mice compared to RANKLf/f littermates (Fig. 4b). More importantly, both basal and PTH-stimulated RANKL mRNA levels were reduced in osteoblast progenitors from Osx1-Cre;RANKLf/f mice compared to RANKLf/f mice, whereas a control mRNA, IL-6, was unaffected by the deletion (Fig. 4c). Notably, the loss of RANKL from this cell population had no effect on osteoclast number in cancellous bone, osteoclast gene expression measured in whole bones, or bone mass (Fig. 4d-f).
Figure 4. Osx1-Cre mediated RANKL deletion in adult mice does not alter osteoclast number in cancellous bone.
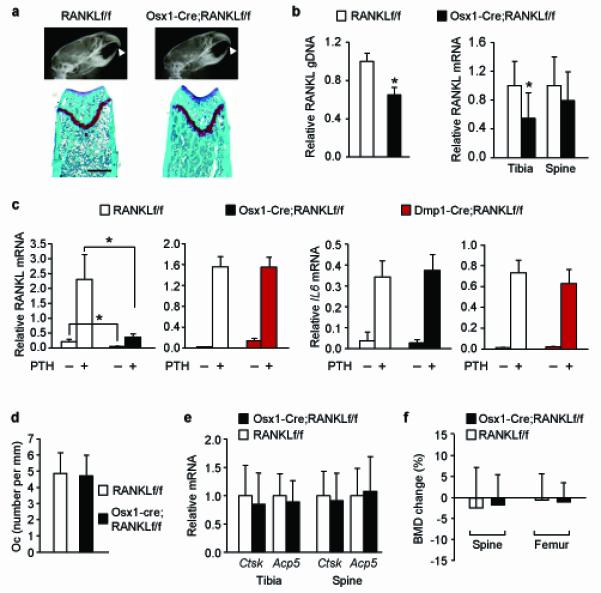
(a) X-ray images (top) and histological sections of femurs stained with safranin-O (bottom) of 6-month-old Osx1-Cre;RANKLf/f and RANKLf/f littermates that were exposed to doxycycline in utero and maintained on a doxycycline-containing diet until 4 months of age. Scale bar, 500 μm. (b) Quantitative PCR of loxP-flanked genomic DNA isolated from collagenase-digested femoral cortical bone (left) and quantitative RT-PCR of RANKL mRNA in tibia and L5 vertebra (right panel) of Osx1-Cre;RANKLf/f (n = 12) mice and their RANKLf/f littermates (n = 10) exposed to doxycycline as described in (a). *P < 0.05 using Student’s t-test. (c) RANKL (left) and interleukin-6 (IL6) (right) mRNA levels in bone marrow cultures from Osx1-Cre;RANKLf/f and RANKLf/f littermates exposed to doxycycline as described in (a) or from 6-month-old Dmp1-Cre;RANKLf/f and RANKLf/f littermates. Each value represents the mean of 3 wells. *P < 0.05 with the comparisons indicated by the brackets using 2-way ANOVA. (d) Osteoclast number per mm bone perimeter in cancellous bone of the distal femur of Osx1-Cre;RANKLf/f (n = 4) and RANKLf/f (n = 4) littermates exposed to doxycycline as described in (a). (e) Cathepsin K (Ctsk) and TRAP (Acp5) mRNA levels in tibia and L5 vertebra of Osx1-Cre;RANKLf/f (n = 12) and RANKLf/f (n = 10) littermates exposed to doxycycline as described in (a). (f) Percent change in BMD between 4 and 6 months of age in Osx1-Cre;RANKLf/f (n = 14) and RANKLf/f (n = 8) littermates exposed to doxycycline as described in (a). All values include data from both sexes.
Osteocyte RANKL controls unloading-induced bone loss
Finally, to determine whether RANKL produced by osteocytes contributes to pathological bone resorption, Dmp1-Cre;RANKLf/f mice and control littermates were tail-suspended for 3 weeks to induce bone loss. Mice lacking RANKL in osteocytes were protected from the loss of bone, as well as the changes in cancellous architecture, caused by tail-suspension (Fig. 5a,b). In this mixed genetic background, tail-suspension did not induce a significant loss of cancellous bone volume in control mice, but did increase trabecular spacing and decrease cortical thickness (Fig. 5b). Consistent with the preservation of bone mass, the increase in RANKL expression that occurred in the cortical bone of tail-suspended control mice was absent in Dmp1-Cre;RANKLf/f mice (Fig. 5c). Finally, even though tail-suspension did not decrease cancellous bone volume in control mice, it did induce a small but significant increase in osteoclast number which did not occur in Dmp1-Cre;RANKLf/f mice (Fig. 5d).
Figure 5. Tail-suspension of mice lacking RANKL in osteocytes.
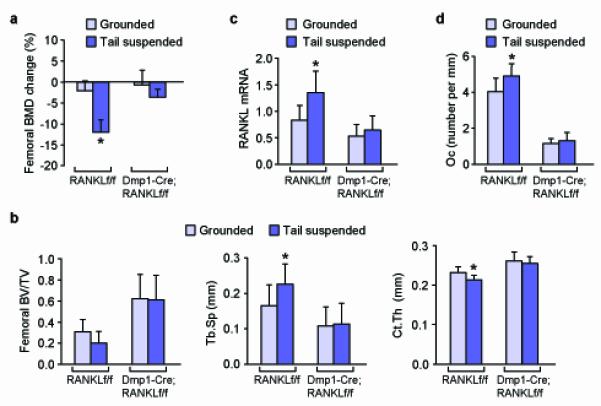
(a) Percent change of femoral BMD after 3 weeks of tail-suspension or normal loading (grounded control) in RANKLf/f and Dmp1-Cre;RANKLf/f littermates. (b) Cancellous bone volume (left), trabecular spacing (Tb.Sp.) (center), and cortical thickness (Ct.Th.) (right), in the femur of tail-suspended or grounded control RANKLf/f and Dmp1-Cre;RANKLf/f littermates. (c) RANKL mRNA levels in RNA prepared from collagenase-digested tibial cortical bone of tail-suspended or grounded control RANKLf/f and Dmp1-Cre;RANKLf/f littermates. (d) Osteoclast number per mm of cancellous bone surface in the distal femur of tail-suspended or grounded control RANKLf/f and Dmp1-Cre;RANKLf/f littermates. All values are from 6-month-old mice, include data from both sexes, and represent the following numbers of animals per group: grounded RANKLf/f (n = 8), suspended RANKLf/f (n = 7), grounded Dmp1-Cre;RANKLf/f (n = 8), and suspended Dmp1-Cre;RANKLf/f (n = 7), with the exception of the panel d, in which n = 5 for each group. *P < 0.05 versus grounded control of the same genotype by 2-way ANOVA.
DISCUSSION
The idea that osteoblasts or their progenitors control osteoclast formation has become a central tenet of skeletal biology and is often used as an explanation for the linkage of bone formation to bone resorption. Nonetheless, there have been suggestions that a variety of other cell types are able to support osteoclastogenesis, including osteocytes30, vascular-associated cells31, and lymphocytes32. Our study represents the first functional identification of osteoclast support cells in vivo. In contrast to the prevailing dogma, our results reveal that osteocytes, but not osteoblasts or their precursors, are an essential cellular source of RANKL for osteoclast formation in remodeling bone. Hence, RANKL expression in osteoblast-lineage cells does not contribute to the coupling of bone formation to bone resorption since osteocytes are not generated until after bone formation is complete. Moreover, the demonstration that unloading-induced bone loss is prevented in mice lacking RANKL in osteocytes suggests that osteocytes are critical players in the pathogenesis of diseases caused by abnormal remodeling.
Our results also strongly suggest that hypertrophic chondrocytes are the source of the RANKL required for resorption of the primary spongiosa. Indeed, even though none of the Cre-deleter strains used in our study has activity exclusively in growth plate chondrocytes, calcified cartilage retention was associated only with those transgenes that deleted RANKL from hypertrophic chondrocytes. Conversely, RANKL deletion by the Dmp1-Cre transgene, which was active in osteoblasts and osteocytes but not in chondrocytes, had no effect on cartilage resorption. Furthermore, RANKL deletion mediated by the ColX-Cre transgene prevented cartilage resorption to a similar extent as the Osx1-Cre and Ocn-Cre transgenes even though it was less effective at deleting RANKL from osteoblast-lineage cells. These findings, coupled with the robust expression of RANKL by hypertrophic chondrocytes in vivo, support a role for RANKL expression by this cell type in the resorption of calcified cartilage.
Cell preparations enriched in osteoblast progenitors express RANKL in vitro and this expression is stimulated by hormones known to stimulate bone resorption33, 34. In spite of this, the results of the present study demonstrate that RANKL expression in these cells does not participate in physiological bone remodeling. It remains possible that RANKL produced by such cells contributes to osteoclast differentiation under different conditions, such as dietary calcium deficiency or lactation. However, it is also possible that RANKL expression in these cultures is an in vitro phenomenon and does not reflect a physiological role.
Deletion of RANKL from osteocytes led to an increase in bone mass that is similar to that observed by pan-inhibition of RANKL in adult mice. Specifically, administration of denosumab, a monoclonal anti-human RANKL antibody, to 10-month-old human RANKL knock-in mice increased femoral cancellous bone volume by almost 2-fold and decreased osteoclast surface by more than 95%35. In our study, deletion of RANKL from osteocytes increased femoral cancellous bone volume by about 2-fold and decreased osteoclast surface by 75%. The greater reduction in osteoclast surface by denosumab suggests either that RANKL deletion in osteocytes in our model was not complete or that RANKL produced by cells other than osteocytes also contributes to osteoclast formation. Nonetheless, even if the latter is the case, our results reveal that RANKL produced by osteocytes is responsible for most of the osteoclastogenesis in cancellous bone.
We expected that RANKL deletion in adult Osx1-Cre mice would reduce osteoclast number in cancellous bone to at least the same extent as RANKL deletion in Dmp1-Cre mice, since both were expected to result in deletion from osteocytes. However, the extent of RANKL deletion in osteocyte-enriched bone was much lower in Osx1-Cre compared to Dmp1-Cre mice. Although the reason for this is unclear, one possibility is that the Osx1-Cre transgene is not active in osteocytes. If this were the case, then the RANKL gene would have been deleted only in osteocytes formed during the 2 months after transgene activation. Since the majority of bone growth has already occurred by 4 months of age in mice, then the proportion of newly formed osteocytes lacking RANKL was likely too small to significantly reduce osteoclast formation.
A recent study used an inducible osteocyte ablation model to show that osteocyte apoptosis led to bone resorption associated with increased RANKL expression36. Moreover this study showed that osteocyte ablation prevented bone loss due to unloading as well as the increase in RANKL expression associated with unloading36. While the latter observation is consistent with our conclusion that osteocytes are an important source of RANKL, the ablation-induced increase in RANKL expression in the grounded controls suggests that other cell types produce RANKL in this situation. An alternative explanation, supported by our findings, is that the remaining live osteocytes respond to the death of their neighboring osteocytes by increasing RANKL expression.
Our studies raise the question of how RANKL expressed by matrix-embedded cells reaches osteoclast progenitors. Both soluble and membrane-bound forms of RANKL are produced37. Osteocytes possess an average of 50 cellular projections, extending throughout the bone matrix, that connect osteocytes to one another and to cells at the bone surface forming the so-called lacunocanalicular network38. Imaging studies have shown that these projections can extend beyond the bone surface so that membrane-bound RANKL expressed by osteocytes may bind osteoclast progenitors39. It is less clear how the membrane-bound form produced by hypertrophic chondrocytes could contact such progenitors. The soluble form of RANKL has a molecular weight of approximately 31 kDa37, which is sufficiently small that it would easily pass throughout the osteocyte lacunocanalicular network40. Moreover, soluble proteins of similar size produced by hypertrophic chondrocytes are able to act in a paracrine manner41. Thus production of either form of RANKL by matrix-embedded cells would be consistent with our contention that they are the predominant sources of the RANKL controlling osteoclast formation.
In closing, demonstration of the essential role of osteocyte- and chondrocyte-derived RANKL for osteoclastogenesis reveals that these cells orchestrate resorption of the matrix in which they are embedded. This evidence provides a mechanistic explanation for the long-held view that at least some remodeling is a targeted, as opposed to a stochastic, process in which cells capable of sensing mechanical strains42 or matrix damage43 act as beacons for excavation and repair of a specific region of bone. Similarly, hypertrophic chondrocytes may sense signals that initiate transformation of the cartilage scaffold into bony trabeculae. In both cases, the cells best able to detect the need for matrix removal directly control the process.
Supplementary Material
ACKNOWLEDGEMENTS
We thank P.E. Cazer, S.B. Berryhill, W. Webb, R. Shelton, A. Deloose, L. K. Climer, K. Vyas, L. Han, A.D. Warren, E.A. Hogan, and J.J. Goellner for technical support and advice; M. Almeida and H. Zhao for helpful discussions; C.J. Tabin (Harvard Medical School), H.M. Kronenberg (Harvard Medical School), K. von der Mark (University of Erlangen-Nuremberg), B. de Crombrugghe (M. D. Anderson Cancer Center), T.L. Clemens (Johns Hopkins University School of Medicine), and J.Q. Feng (Baylor College of Dentistry) for providing Cre-deleter strains; P.D. Pajevic (Harvard Medical School) for the collagenase digestion protocol; D. Chen (University of Rochester School of Medicine) for the X-gal staining protocol; T. Bellido (Indiana University School of Medicine) and I. Aguirre (Univsersity of Florida) for advice on tail-suspension; the staff of the UAMS Department of Laboratory Animal Medicine; and L. Suva and R. Skinner of the UAMS Skeletal Imaging Core. This work was supported by the following grants from the US National Institutes of Health: AR049794 (to C.A.O.) and AG13918 (to S.C.M.). Support was also provided by the Central Arkansas Veteran’s Healthcare System (Merit Reviews to C.A.O., S.C.M., R.L.J., and R.S.W.), by the Arkansas Center for Clinical and Translational Research, and by UAMS tobacco settlement funds.
APPENDIX
METHODS
Mice
We created mice harboring a RANKL conditional allele using homologous recombination in ES cells (see Supplementary Methods). We used a targeting vector in which exons 3 and 4 of the Tnfsf11 (RANKL) gene were flanked by loxP sites (Supplementary Fig. 1). The generation of transgenic mice expressing the Cre recombinase in different cell populations and R26R reporter gene mice have been described previously: Prx1-Cre (ref. 21), Osx1-Cre (ref. 23), Ocn-Cre (ref. 25), Dmp1-Cre (ref. 26), ColX-Cre (ref. 29), and R26R (ref. 44). We housed all mice in the animal facility of the University of Arkansas for Medical Sciences. The Institutional Animal Care and Use Committees of the University of Arkansas for Medical Sciences and the Central Arkansas Veterans Healthcare System approved protocols involving these mice.
Skeletal analysis
We obtained whole body X-rays of anesthetized mice using an AXR 110 Minishot X-ray machine (Associated X-Ray Corporation) and measured BMD of the lumbar spine and femur by dual-energy X-ray absorptiometry using a PIXImus Densitometer (GE-Lunar Corp.) and the manufacturer’s software as previously described45. We measured three dimensional bone volume and architecture of L4 vertebra and femurs using μCT (model μCT40, Scanco Medical). Details are included in the Supplementary Methods.
Histology
We fixed femurs for 24 hours in 10% Millonig’s formalin, decalcified them in 14% EDTA for 1 week, embedded them in paraffin, and then obtained 5 μm longitudinal sections. After removal of paraffin and rehydration, we stained sections for TRAP activity or with safranin-O and counter-stained with fast green. We performed RANKL immunostaining with an anti-RANKL antibody (R & D Systems) using 5 μm frozen sections of fixed and decalcified femurs. We performed quantitative histomorphometry to determine osteoclast number using the TRAP-stained femur sections and a computer and digitizer tablet (OsteoMetrics) interfaced to a Zeiss Axioscope (Carl Zeiss) with an attached drawing tube. We directly measured the number of TRAP-positive cells on the cancellous perimeter (osteoclast number). We used terminology recommended by the Histomorphometry Nomenclature Committee of the American Society for Bone and Mineral Research 46. Details are included in the Supplementary Methods.
Quantitative PCR
We harvested organs and whole bones, from which we removed soft tissues, from animals and stored them immediately in liquid nitrogen. We prepared osteocyte-enriched bone by removing the ends of femurs and tibias and then flushing the bone marrow with PBS. We then scraped the bone surface with a scalpel, after which we cut the bones into small pieces (about 3 mm in length), and digested the pieces with collagenase (1 mg/ml type I:II, ratio 1:3, Worthington Biochemical Corporation) in Hank’s balanced salt solution that also contained 0.1 % bovine serum albumin and 1 mM CaCl2. We performed a total of 6 consecutive 15 minute digestions at 37°C in a water bath shaker to remove the cells on the bone surface. After the final digestion, we washed the bone pieces with PBS and froze them in liquid nitrogen for later RNA isolation, or decalcified them for genomic DNA isolation. We isolated total RNA using Ultraspec reagent (Biotecx Laboratories), according to the manufacturer’s instructions and prepared cDNA as previously described15. We performed quantitative RT-PCR using the following Taqman assays from Applied Biosystems: RANKL (Tnfsf11) (Mm0041908-m1); IL-6 (Mm00446190-m1); Ctsk (Mm01255862-g1); Acp5 (Mm00475698_m1); Calcr (Mm00432271_m1); and ribosomal protein S2 (Mrps2) (for, 5′-CCCAGGATGGCGACGAT-3′, rev, 5′-CCGAATGCTGTAATGGCGTAT-3′, probe, 5′-FAM-TCCAGAGCAGGATCC-NFQ-3′). We calculated relative mRNA amounts using the ΔCt method47. We isolated genomic DNA from decalcified bone fragments after digestion with proteinase K and phenol/chloroform extraction. We obtained two custom Taqman assays from Applied Biosystems for quantifying RANKL gene deletion efficiency: one specific for sequences between the loxP sites and the other specific for sequences downstream from the 3′ loxP site. Details are included in the Supplementary Methods.
Tail suspension
We performed mechanical unloading of hindlimbs by tail suspension for 3 weeks. We suspened mice by attaching a straightened paperclip to the tail with superglue and first-aid tape, and then attaching the paperclip to a rod in the top of a modified shoe-box mouse cage. We adjusted suspension to maintain the mouse at a ~30° head-down tilt. We used fully ambulatory littermates in the same cage, separated by a divider, as controls. Details are included in the Supplementary Methods.
Statistics
We used two-way analysis of variance (ANOVA) or Student’s t-test to detect statistically significant treatment effects, after determining that the data were normally distributed and exhibited equivalent variances. In some cases, we used log transformation to obtain normally-distributed data. All t-tests were two-sided. We used Bonferroni or Holm-Sidak corrections for multiple comparisons. We considered P-values less than 0.05 as significant. Error bars in all figures represent s.d..
Footnotes
COMPETING FINANICAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Kronenberg HM. Developmental regulation of the growth plate [Review] Nature. 2003;423:332–336. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 2.Parfitt AM. Targeted and nontargeted bone remodeling: relationship to basic multicellular unit origination and progression. Bone. 2002;30:5–7. doi: 10.1016/s8756-3282(01)00642-1. [DOI] [PubMed] [Google Scholar]
- 3.Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat. Rev. Genet. 2003;4:638–649. doi: 10.1038/nrg1122. [DOI] [PubMed] [Google Scholar]
- 4.Whyte MP, et al. Osteoprotegerin deficiency and juvenile Paget’s disease. N. Engl. J. Med. 2002;347:175–184. doi: 10.1056/NEJMoa013096. [DOI] [PubMed] [Google Scholar]
- 5.Manolagas SC. Birth and death of bone cells: Basic regulatory mechanisms and implications for the pathogenesis and treatment of osteoporosis [Review] Endocr. Rev. 2000;21:115–137. doi: 10.1210/edrv.21.2.0395. [DOI] [PubMed] [Google Scholar]
- 6.Kong YY, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 7.Sobacchi C, et al. Osteoclast-poor human osteopetrosis due to mutations in the gene encoding RANKL. Nat. Genet. 2007;39:960–962. doi: 10.1038/ng2076. [DOI] [PubMed] [Google Scholar]
- 8.Kearns AE, Khosla S, Kostenuik PJ. Receptor activator of nuclear factor kappaB ligand and osteoprotegerin regulation of bone remodeling in health and disease. Endocr. Rev. 2008;29:155–192. doi: 10.1210/er.2007-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Brien CA. Control of RANKL gene expression. Bone. 2010;46:911–919. doi: 10.1016/j.bone.2009.08.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kobayashi Y, Udagawa N, Takahashi N. Action of RANKL and OPG for osteoclastogenesis. Crit Rev. Eukaryot. Gene Expr. 2009;19:61–72. doi: 10.1615/critreveukargeneexpr.v19.i1.30. [DOI] [PubMed] [Google Scholar]
- 11.Sims NA, Gooi JH. Bone remodeling: Multiple cellular interactions required for coupling of bone formation and resorption. Semin. Cell Dev. Biol. 2008;19:444–451. doi: 10.1016/j.semcdb.2008.07.016. [DOI] [PubMed] [Google Scholar]
- 12.Takahashi N, et al. Osteoblastic cells are involved in osteoclast formation. Endocrinology. 1988;123:2600–2602. doi: 10.1210/endo-123-5-2600. [DOI] [PubMed] [Google Scholar]
- 13.Rodan GA, Martin TJ. Role of osteoblasts in hormonal control of bone resorption--a hypothesis. Calcif. Tissue Int. 1981;33:349–351. doi: 10.1007/BF02409454. [DOI] [PubMed] [Google Scholar]
- 14.Corral DA, et al. Dissociation between bone resorption and bone formation in osteopenic transgenic mice. Proc. Natl. Acad. Sci. U. S. A. 1998;95:13835–13840. doi: 10.1073/pnas.95.23.13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Galli C, et al. Commitment to the osteoblast lineage is not required for RANKL gene expression. J. Biol. Chem. 2009;284:12654–12662. doi: 10.1074/jbc.M806628200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J. Bone Miner. Res. 2008;23:860–869. doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- 17.Akune T, et al. PPARgamma insufficiency enhances osteogenesis through osteoblast formation from bone marrow progenitors. J. Clin. Invest. 2004;113:846–855. doi: 10.1172/JCI19900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ogata N, Kawaguchi H, Chung UI, Roth SI, Segre GV. Continuous activation of G alpha q in osteoblasts results in osteopenia through impaired osteoblast differentiation. J. Biol. Chem. 2007;282:35757–35764. doi: 10.1074/jbc.M611902200. [DOI] [PubMed] [Google Scholar]
- 19.Weinstein RS, Jilka RL, Parfitt AM, Manolagas SC. Inhibition of osteoblastogenesis and promotion of apoptosis of osteoblasts and osteocytes by glucocorticoids. Potential mechanisms of their deleterious effects on bone. J. Clin. Invest. 1998;102:274–282. doi: 10.1172/JCI2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weinstein RS, et al. Promotion of osteoclast survival and antagonism of bisphosphonate-induced osteoclast apoptosis by glucocorticoids. Journal of Clinical. Investigation. 2002;109:1041–1048. doi: 10.1172/JCI14538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Logan M, et al. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002;33:77–80. doi: 10.1002/gene.10092. [DOI] [PubMed] [Google Scholar]
- 22.Dougall WC, et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13:2412–2424. doi: 10.1101/gad.13.18.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rodda SJ, McMahon AP. Distinct roles for Hedgehog and canonical Wnt signaling in specification, differentiation and maintenance of osteoblast progenitors. Development. 2006;133:3231–3244. doi: 10.1242/dev.02480. [DOI] [PubMed] [Google Scholar]
- 24.Kobayashi T, et al. Dicer-dependent pathways regulate chondrocyte proliferation and differentiation. Proc. Natl. Acad. Sci. U. S. A. 2008;105:1949–1954. doi: 10.1073/pnas.0707900105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang M, et al. Osteoblast-specific knockout of the insulin-like growth factor (IGF) receptor gene reveals an essential role of IGF signaling in bone matrix mineralization. J Biol Chem. 2002;277:44005–44012. doi: 10.1074/jbc.M208265200. [DOI] [PubMed] [Google Scholar]
- 26.Lu Y, et al. DMP1-targeted Cre expression in odontoblasts and osteocytes. J. Dent. Res. 2007;86:320–325. doi: 10.1177/154405910708600404. [DOI] [PubMed] [Google Scholar]
- 27.Masuyama R, et al. Vitamin D receptor in chondrocytes promotes osteoclastogenesis and regulates FGF23 production in osteoblasts. J. Clin. Invest. 2006;116:3150–3159. doi: 10.1172/JCI29463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Usui M, et al. Murine and chicken chondrocytes regulate osteoclastogenesis by producing RANKL in response to BMP2. J. Bone Miner. Res. 2008;23:314–325. doi: 10.1359/JBMR.071025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gebhard S, et al. Specific expression of Cre recombinase in hypertrophic cartilage under the control of a BAC-Col10a1 promoter. Matrix Biol. 2008;27:693–699. doi: 10.1016/j.matbio.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao S, et al. MLO-Y4 osteocyte-like cells support osteoclast formation and activation. J. Bone Miner. Res. 2002;17:2068–2079. doi: 10.1359/jbmr.2002.17.11.2068. [DOI] [PubMed] [Google Scholar]
- 31.Collin-Osdoby P, et al. Receptor activator of NF-kappa B and osteoprotegerin expression by human microvascular endothelial cells, regulation by inflammatory cytokines, and role in human osteoclastogenesis. J. Biol. Chem. 2001;276:20659–20672. doi: 10.1074/jbc.M010153200. [DOI] [PubMed] [Google Scholar]
- 32.Kong YY, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304–309. doi: 10.1038/46303. [DOI] [PubMed] [Google Scholar]
- 33.Yasuda H, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. U. S. A. 1998;95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.O’Brien CA, Gubrij I, Lin SC, Saylors RL, Manolagas SC. STAT3 activation in stromal osteoblastic cells is required for induction of the receptor activator of NF- kappa B ligand and stimulation of osteoclastogenesis by gp130-utilizing cytokines or interleukin-1 but not 1,25-dihydroxyvitamin D-3 or parathyroid hormone. J. Biol. Chem. 1999;274:19301–19308. doi: 10.1074/jbc.274.27.19301. [DOI] [PubMed] [Google Scholar]
- 35.Kostenuik PJ, et al. Denosumab, a fully human monoclonal antibody to RANKL, inhibits bone resorption and increases BMD in knock-in mice that express chimeric (murine/human) RANKL. J. Bone Miner. Res. 2009;24:182–195. doi: 10.1359/jbmr.081112. [DOI] [PubMed] [Google Scholar]
- 36.Tatsumi S, et al. Targeted ablation of osteocytes induces osteoporosis with defective mechanotransduction. Cell Metab. 2007;5:464–475. doi: 10.1016/j.cmet.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 37.Lacey DL, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 38.Bonewald LF. The amazing osteocyte. J. Bone Miner. Res. 2011;26:229–238. doi: 10.1002/jbmr.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kamioka H, Honjo T, Takano-Yamamoto T. A three-dimensional distribution of osteocyte processes revealed by the combination of confocal laser scanning microscopy and differential interference contrast microscopy. Bone. 2001;28:145–149. doi: 10.1016/s8756-3282(00)00421-x. [DOI] [PubMed] [Google Scholar]
- 40.Knothe Tate ML. “Whither flows the fluid in bone?” An osteocyte’s perspective. J. Biomech. 2003;36:1409–1424. doi: 10.1016/s0021-9290(03)00123-4. [DOI] [PubMed] [Google Scholar]
- 41.Zelzer E, et al. VEGFA is necessary for chondrocyte survival during bone development. Development. 2004;131:2161–2171. doi: 10.1242/dev.01053. [DOI] [PubMed] [Google Scholar]
- 42.Aguirre JI, et al. Osteocyte apoptosis is induced by weightlessness in mice and precedes osteoclast recruitment and bone loss. J. Bone Miner. Res. 2006;21:605–615. doi: 10.1359/jbmr.060107. [DOI] [PubMed] [Google Scholar]
- 43.Herman BC, Cardoso L, Majeska RJ, Jepsen KJ, Schaffler MB. Activation of bone remodeling after fatigue: differential response to linear microcracks and diffuse damage. Bone. 2010;47:766–772. doi: 10.1016/j.bone.2010.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain [letter] Nat. Gen. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 45.O’Brien CA, et al. IL-6 is not required for parathyroid hormone stimulation of RANKL expression, osteoclast formation, and bone loss in mice. Am. J. Physiol Endocrinol. Metab. 2005;289:E784–E793. doi: 10.1152/ajpendo.00029.2005. [DOI] [PubMed] [Google Scholar]
- 46.Parfitt AM, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. J. Bone Miner. Res. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- 47.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.