

1. Introduction
The serine hydrolase (SH) superfamily consists of >200 enzymes in humans characterized by the presence of an active site serine that is used for the hydrolysis of substrates. The membership of this enzyme class is near-equally split between serine proteases (trypsin/chymotrypsin/subtilisin enzymes) and ‘metabolic’ SHs that cleave ester, amide, or thioester bonds in small molecules, peptides, or proteins.1 The serine proteases have been the subject of several books and reviews.2–5 This article will instead focus on the metabolic SHs (Fig. 1).
Fig. 1.

Dendrogram showing the primary sequence alignment of the human mammalian metabolic SHs, where alignment was generated by anchoring sequences at the site of their catalytic Ser residues.
The nucleophilicity of the active site serine of metabolic SHs arises from its participation in a catalytic dyad (e.g. Ser-Lys or Ser-Asp) or triad (e.g. Ser-His-Asp or Ser-Ser-Lys).6,7 The SH catalytic mechanism proceeds by formation of an acyl-enzyme intermediate at the active site serine, followed by water-induced saponification of the product, and regeneration of the free serine residue for entry into the next reaction cycle (Fig. 2).8,9 Owing to the enhanced reactivity of the active site serine, the functional state of most SHs can be assessed using active-site directed affinity labels such as fluorophosphonates (FPs, Fig. 2).1,10
Fig. 2.

(A) Mechanism of SH catalysis. (B) Mechanism of SH labeling by the active site-directed activity-based probe fluorophosphonate-biotin (FP-biotin). (C) Three dimensional structure of MGLL, a SH with a canonical α/β-hydrolase fold.
The serine nucleophile of metabolic SHs is generally, though not exclusively embedded within a GXSXG motif and a majority these enzymes adopt an α/β hydrolase fold that consists of a central β-sheet surrounded by α-helicies (Fig. 2).11 This superfamily also encompasses other smaller subsets of structurally distinct enzymes such as the phospholipase A2s, the amidase signature enzymes, and the dipeptidylpeptidases.12,13 Metabolic SHs have been shown to participate in virtually all (patho)physiological processes in mammals, including neurotransmission,14 metabolism,15 pain sensation,16 inflammation,17 oxidative stress,18 cancer,19 and bacterial infection.20
Many excellent reviews have described the structure and function of individual SHs.15,19,21–23 Here, we attempt to provide a comprehensive summary that captures our state of knowledge about mammalian metabolic SHs in their entirety, including those enzymes that remain mostly or completely uncharacterized. Particular emphasis will be placed on relating the biochemistry and enzymology of individual SHs to the physiological substrates and products that they regulate in living systems, and how SHs, through the regulation of specific metabolic pathways impact health and disease. If selective and efficacious inhibitors are available for a particular SH, we will also include a discussion of their use. The majority of this review will be organized by substrate class. Later, we will discuss SHs for which putative endogenous substrates have not been identified, as well as emerging chemoproteomic and metabolomic methods aimed at assigning functions to these enzymes. For the sake of consistency, we have elected to refer to SHs by their proper gene names throughout this review (rather than their common name or abbreviation), but have also attempted to include other aliases if possible.
2. Small-molecule hydrolases
The largest category of substrates for metabolic SHs is small molecules, which include neutral fatty acyl esters, acyl thioesters (e.g., acyl CoAs), phospholipids, lipid amides, and other ester metabolites (e.g., acetylcholine). As will be described in this section, the small molecules themselves may be structural components of cells and tissues, as is the case for some phospholipids, or important stores of energy, as is the case for triglycerides, or signaling molecules, as is the case for acetylcholine.
2.1. Intracellular neutral lipases
Intracellular triglyceride and cholesteryl ester stores in organs such as adipose tissue and brain are hydrolyzed by multiple SHs, including LIPE, PNPLA2, MGLL, and DAGLα and β (Fig. 3). The resultant free fatty acid products are an important fuel in mammals and can be converted by the β-oxidation pathway to acetyl-CoA, which can enter the citric acid cycle for oxidative phosphorylation to generate ATP. These hydrolytic reactions also generate signaling molecules, such as the neuromodulatory lipid 2-arachidonoylglycerol (2-AG), which activates cannabinoid receptors.
Fig. 3.

The enzymatic catabolism of triglycerides into fatty acids and glycerol by PNPLA2, HSL, DAGLα/β, and MGLL.
2.1.1. LIPE (Hormone-sensitive lipase)
In humans, LIPE, also called hormone-sensitive lipase (HSL), is an 84 kDa intracellular enzyme predominantly expressed in adipocytes and adrenal glands, with lower expression in cardiac and skeletal muscle and macrophages.24,25 In vitro, LIPE hydrolyzes triglycerides (TGs), diglycerides (DGs), monoglycerides (MGs), cholesteryl esters, and retinyl esters, with ~5–10-fold higher activity for DGs over TGs and MGs, but has no phospholipase activity.26,27 An unusual feature of LIPE is the modulation of its activity by phosphorylation by protein kinase A (PKA).27 In adipocytes, LIPE phosphorylation is stimulated by catecholamines or suppressed by insulin, causing translocation of HSL from the cytosol to the surface of lipid droplets, where its hydrolytic activity is substantially increased.28,29
For many years, LIPE was thought to be the principal TG hydrolase in adipocytes that participates in the first and rate-limiting step of TG lipolysis. This hypothesis was convincingly disproven by the generation of multiple LIPE(−/−) mouse lines.30–32 While the neutral cholesterol ester hydrolase (NCEH) activities in white and brown adipocyte tissues (WAT and BAT) and testis of LIPE(−/−) mice are completely ablated, WAT retains a substantial amount (~40%) of the TG lipase activity.32 LIPE(−/−) mice are neither obese nor cold sensitive, but show adipocyte hypertrophy which does not change WAT mass, but causes a ~1.7-fold increase in BAT mass. Fat pads treated with the β1- and β2-adrenergic agonist isoproterenol from LIPE(−/−) mice show a decrease in released free fatty acids, an absence of released glycerol, and an accumulation of intracellular DG. Male LIPE(−/−) mice are infertile owing to the lack of spermatozoa. DGs are elevated in multiple tissues from LIPE(−/−) mice, confirming that DGs are a major endogenous substrates for LIPE.30 Taken together, these data suggest that LIPE participates in the second step of TG lipolysis in adipose tissue by hydrolyzing DG in vivo.
Several chemical classes of synthetic LIPE inhibitors have been described, including carbamates,33 heterocyclic ureas,34,35 and pyrrolopyrazinediones,36 although their selectivity and pharmacological effects in mammals have not been extensively reported.
2.1.2. PNPLA2 (Adipose triglyceride lipase)
The observation that LIPE is not the principal TG hydrolase in adipose tissue prompted a search for enzymes that could perform this chemistry and resulted in the identification of patatin-like phospholipase domain containing 2 (PNPLA2), also called adipose triglyceride lipase (ATGL),37 desnutrin,38 TTS-2.2,39 iPLA2ζ, or PLA2G6E.40 In mice, PNPLA2 is a ~54 kDa protein principally expressed in white and brown adipose tissues, with lower levels in skeletal and cardiac muscle and testis. When overexpressed in COS-7 cells, PNPLA2 is detected in both the soluble and pellet fractions and has activity for triolein but not cholesterol oleate, retinyl palmitate, or PC.41 Unlike LIPE, PNPLA2 activity is unaffected by PKA phosphorylation or β-adrenergic stimulation, but is co-activated by another protein ABHD5, also known as CGI-58, which too possesses an α/β-hydrolase fold, but lacks a serine nucleophile required for hydrolytic catalysis.41,42
The generation of PNPLA2(−/−) mice confirmed that PNPLA2 is the rate-limiting step for TG lipolysis in adipose tissue.43 TG hydrolysis activity of WAT, skeletal muscle, and liver from PNPLA2(−/−) mice is decreased by >80%, 44%, and 73%, respectively, and fat pads from these animals show reduced release of free fatty acids following stimulation by isoproterenol. Anatomically, PNPLA2(−/−) mice have two-fold increases in body fat mass, enlarged lipid droplets in both WAT and BAT, adipocyte hypertrophy, and mild obesity. These animals do not survive prolonged cold exposure or fasting, presumably because their impairment in lipolysis precludes the production of fatty acids for energy generation. PNPLA2(−/−) mice suffer premature death from a mechanical contraction defect in the heart which arises from the >20-fold increase in cardiac TG. These data suggest that, in the absence of PNPLA2, TG lipolysis is dramatically impaired, leading to the accumulation of TG in multiple tissues, a decrease in available free fatty acids for energy production, and premature death from an inappropriate cardiac accumulation of TG.
Selective inhibitors of ATGL have not, to our knowledge, been described.
2.1.3. MGLL (Monoacylglycerol lipase)
MGLL, also called monoglyceride lipase (MGL) and sometimes abbreviated MAGL, is a ~33 kDa enzyme in mice ubiquitously expressed in all tissues, with highest expression in brain, white adipose tissue, and liver.44,45 MGLL is a soluble enzyme that peripherally associates with membranes by means of a hydrophobic lid domain.46,47 In vitro, MGLL is capable of hydrolyzing a variety of monoglycerides into free fatty acids and glycerol, and has strong preference for MG over DG or TG.48 One such monoglyceride is the endocannabinoid 2-AG,49–51 an endogenous ligand for the cannabinoid receptors CB1 and CB2, two G-protein coupled receptors that also respond to the psychoactive component of marijuana Δ9-tetrahydrocannabinol.52,53
A principal role of MGLL in the deactivation of 2-AG signaling at cannabinoid receptors has been demonstrated using the selective MGLL inhibitor JZL18454,55 and MGLL(−/−) mice (Fig. 4).56,57 JZL184-treated or MGLL(−/−) mice have >80% reductions in 2-AG hydrolysis activity in most tissues including brain and concomitant elevations in 2-AG and other MGs. In brain, these mice also show an approximately 50% decrease in free arachidonic acid. Acute MGLL disruption with JZL184 produces CB1-dependent anti-nociception and hypomotility,45,58 two cannabimimetic behaviors classically associated with activation of CB1 receptors.59 Interestingly, similar effects are not observed in mice treated chronically with JZL184 or in MGLL(−/−) mice because CB1 receptors become desensitized in these animals.56 MGLL is also highly expressed in aggressive human cancer cells and primary tumors, where it regulates fatty acids and downstream fatty acid-derived oncogenic signaling metabolites.60 Overexpression of MGLL in less aggressive cancer cells elevates fatty acids and promotes cell proliferation and in vivo tumor growth. Conversely, JZL184-treatment or short hairpin RNA-mediated silencing of MGLL in aggressive cancer cells decreases MG hydrolysis activity, reduces intracellular fatty acids, and reduces their pathogenicity. Together, these data demonstrate that MGLL serves specialized metabolic functions in different cell types and tissues. In the nervous system, MGLL terminates 2-AG signaling at CB1 receptors and possibly also arachidonic acid pathways. In peripheral tissues, such as WAT, MGLL mainly controls the levels of non-cannabinoid MGs as part of its role in the TG lipolysis pathway. In certain pathological systems, such as in cancer cells, MGLL regulates, not only MGs, but also free fatty acids, which in turn feed into downstream pathways that produce pro-tumorigenic signaling lipids.
Fig. 4.

(A) MGLL hydrolyzes the endocannabinoid 2-arachidonoylglycerol (2-AG) to generate arachidonic acid and glycerol. (B) Structure of the MGLL-selective inhibitor JZL184.
2.1.4. DAGLA and DAGLB (Diacylglycerol lipase α and β)
In humans, DAGLA and DAGLB, also called diacylglycerol lipase α and β (DAGLα and DAGLβ), are ~120 kDa and ~70 kDa, four-pass transmembrane enzymes that share ~30% identity.61 DAGLA is highly expressed in brain and pancreas, whereas DAGLB is strongly expressed in bone marrow and liver.62 In vitro, the DAGLs have DG hydrolase activity with a several-fold preference for the sn-1 position over the sn-2 position, are stimulated by calcium, and have negligible MG or TG hydrolase, phospholipase, or lipid amidase activity.61
The generation of both DAGLA and DAGLB(−/−) have established a role for the DAGLs in the biosynthesis of the endocannabinoid 2-AG.62,63 Consistent with their relative expression levels in organs, DAGLA(−/−) mice show 80% reductions in brain 2-AG and DAGLB(−/−) mice show 90% reductions in liver 2-AG. Concurrent decreases in arachidonic acid are also observed in these tissues. In the hippocampus, a phenomenon called depolarization-induced suppression of inhibition (DSI) occurs when retrograde endocannabinoid signaling at the CB1 receptor attenuates GABA release from presynaptic termini. DSI is absent in the DAGLA(−/−) mice, demonstrating that 2-AG is the endocannabinoid ligand responsible for this form of synaptic plasticity. Lastly, both DAGLA and DAGLB(−/−) show decreases in adult neurogenesis in the brain.62 These data support the hypothesis that, by hydrolyzing sn-2 arachidonoyl-containing DGs, the DAGLs directly regulate basal levels of 2-AG in multiple tissues including brain, and, indirectly, also modulate free arachidonic acid by limiting the availability of one of its major metabolic precursors 2-AG.
Both DAGLA and DAGLB are inhibited by the non-selective lipase inhibitors tetrahydrolipstatin (THL) and RHC80267 (Fig. 5).64 Pharmacological studies with these inhibitors support the results from DAGL-deficient mice that DAGL enzymes regulate DSI and other forms of endocannabinoid-mediated retrograde signaling in the nervous system.65
Fig. 5.
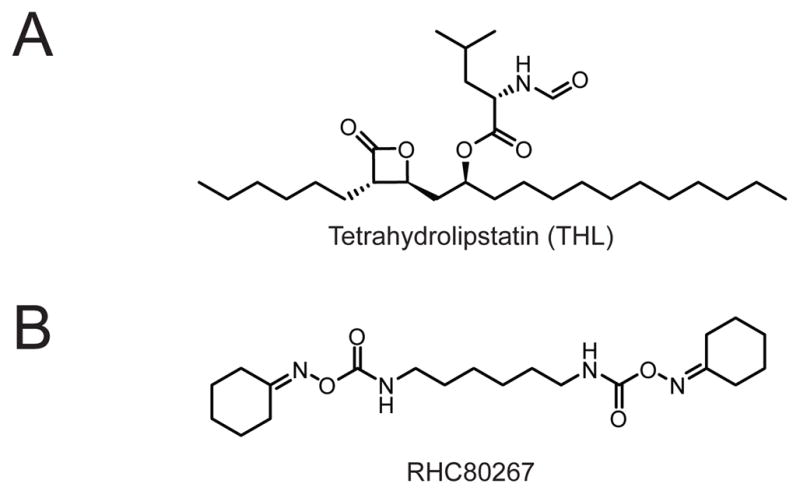
(A) Structure of the non-selective lipase inhibitor tetrahydrolipstatin (THL, also called Orlistat, Xenical, or Alli). (B) Structure of the non-selective DAGL inhibitor RHC80267.
2.1.5. CES3 (Carboxylesterase 3)
CES3, also known as triglyceride hydrolase or triacylglycerol hydrolase (TGH), whose ortholog in humans is CES1, is a 60 kDa microsomal enzyme with in vitro activity for a variety of short- and long-chain TGs, but no activity for fatty acyl-CoAs or phospholipids.66–69 In mice, CES3 is highly expressed in the liver, with moderate amounts of expression in the lung, heart, kidney, and adipose tissues, and no expression in brain, spleen, or muscle.70
The cells of the intestine deliver dietary TGs to peripheral tissues by secreting TG-rich chylomicron particles into the plasma. In contrast, the liver delivers endogenously synthesized TGs by packaging and secreting TGs in apoB-containing very low density lipoprotein (VLDL) particles, a process that does not occur by direct transport of hepatic TGs but rather requires the sequential lipolysis of intracellular hepatic TG stores from lipid droplets followed by re-esterification of the acylglycerols onto VLDL particles by endoplasmic reticulum-localized acyltransferases.66 CES3(−/−) mice has provided evidence that CES3 contributes to the production of very low density lipoprotein (VLDL) particles.71 Livers from CES3(−/−) mice show ~30% decrease in total carboxylesterase activity. VLDL-sized particles and apoB100 are reduced by ~50% in plasma from CES3(−/−) versus wild-type littermates, which correlates with a decreased secretion of TG from cultured CES3(−/−) hepatocytes of comparable magnitude. Despite decreased TG secretion, CES3(−/−) mice show no evidence of hepatic TG accumulation, an observation that is, in part, explained by a combination of reduced TG lipolysis in white adipose tissue, which limits TG accumulation in the liver, and elevations in hepatic β-oxidation enzymes which redirects hepatic TG to catabolic processes. CES3(−/−) mice show a variety of improved metabolic parameters, including enhanced insulin and glucose sensitivity and pancreatic islet function. The in vitro TG hydrolase activity of CES3 and the reduced hepatic VLDL production in CES3(−/−) mice, taken together, support the hypothesis that CES3 hydrolyzes hepatic TGs, thereby supplying acylglycerol substrates for the re-esterification and formation of VLDL particles. These data also suggest that CES3 may also play an important role in TG lipolysis in white adipose tissue, an issue that may be resolved by the generation of tissue-specific CES3(−/−) mice.
A few CES3 inhibitors have been reported, but their selectivity, especially among other CES enzymes, has not been extensively examined.33
2.1.6. AADACL1 (Arylacetamide deacetylase-like 1)
AADACL1, also called neutral cholesterol ester hydrolase 1 (NCEH1)72 or KIAA1363,73 is a lumenally-oriented, integral membrane ~50 kDa glycoprotein highly expressed in brain, macrophages, heart, and the adrenal gland.74 In vitro, AADACL1 can hydrolyze an unusual class of neutral lipids, 2-acetyl-monoalkylglycerol ethers (2-Ac-MAGEs), to generate MAGEs (Fig. 6).75,76 AADACL1 has also been reported to hydrolyze cholesteryl esters in vitro,74 though another group has been unable to reproduce this activity.76
Fig. 6.

(A) KIAA1363 hydrolyzes 2-AcMAGE to generate MAGE, which can be further converted to alkyl-LPC (left) or alkyl-LPA (right). (B, C) Structures of the lead KIAA1363 inhibitor AS115 (B) and the more advanced KIAA1363 inhibitor JW480 (C), which shows improved selectivity and in vivo activity.
The endogenous biochemical function of AADACL1 in normal tissues remains enigmatic, but the enzyme has been shown to serve an important metabolic role in cancer. AADACL1 is highly expressed in aggressive cancer cells73 and primary tumors,77 where, at least in certain instances, its gene is highly amplified.78 Blockade of AADACL1 in aggressive cancer cells by shRNA knockdown or the carbamate inhibitors, AS115 or JW480, causes reductions in MAGEs and downstream metabolites such as alkyl-LPAs (a known chemoattractant) and alkyl-LPCs (Fig. 6).75,79 shRNA knockdown of AADACL1 also impairs cell migration and tumor growth in vivo.75 These findings are particularly intriguing when evaluated in the context of a number of other reports, dating back to the 1960s, that cancer cells possess fundamental laterations in neutral ether lipid metabolism. These studies have shown strong correlations between neutral ether lipid levels and enhanced tumor-forming potential in cancer cells and have postulated that MAGEs, in particular, may originate from 2-Ac-MAGE hydrolysis.80,81 It appears that AADACL1 represents the principal 2-Ac-MAGE hydrolase in many cancer cells.
Curiously, despite having nearly ablated 2-Ac-MAGE hydrolysis activity in multiple tissues, including the brain, heart, and lung, AADACL1(−/−) mice do not show reduced MAGE levels82, suggesting that the enzyme may serve a distinct metabolic function in normal tissues. One possibility is that AADACL1 plays a role in cholesteryl ester (CE) metabolism in macrophages. In support of this hypothesis, AADACL1(−/−) mice show significant (but not complete) reductions in CE hydrolysis activity and aggravated atherosclerosis when crossed into the APOE(−/−) line and fed a high fat diet.72 These findings, however, have been challenged by another group, which found that LIPE (hormone sensitive lipase) is the principal cholesteryl ester hydrolase in mouse macrophages and that AADACL1(−/−) macrophages do not show any deficiency in this activity.76 These discrepancies and their interpretation are further complicated by differences in the SH composition of mouse and human macrophages, the latter of which has recently been reported to express high levels of AADACL1, but not LIPE.83 Finally, beyond its roles in neutral ether lipid and CE metabolism, AADACL1 has been found to represent the principal reactive SH in the brain for the organophosphorous insecticide metabolite chlorpyrifos oxon (CPO), and AADACL1(−/−) are more sensitive than wild-type littermates to poisoning by organophosphorous reagents.82,84
2.1.7. ABHD6 (Alpha/beta hydrolase domain containing 6)
ABHD6 is a ~30 kDa hydrolase with a single transmembrane domain near the N-terminus and a cytosolically-oriented catalytic domain.85 ABHD6 is highly expressed in multiple human tissues by RT-PCR, including brain, lung, liver, muscle, kidney, pancreas, and testis.86 In vitro, ABHD6 can hydrolyze the endocannabinoid 2-arachidonoylglycerol (2-AG),85 and blockade of ABHD6 in prefrontal cortical slices from adult mice using the selective carbamate inhibitor WWL70 (Fig. 7) and concurrent sub-threshold stimulation produces significant cannabinoid receptor-dependent long-term depression (LTD).87 These data support the hypothesis that ABHD6 regulates the duration and magnitude of 2-AG signaling in some brain areas, despite its small contribution to the total brain 2-AG hydrolysis activity.87 To date, ABHD6(−/−) mice have not been generated, and no reports have described if WWL70 or other ABHD6 inhibitors33 such as WWL123 (Fig. 7) produces CB1-dependent behaviors in rodents similar to inhibitors of other endocannabinoid hydrolases including MAGL and FAAH.88 Whether ABHD6 can also cleave other lipids in addition to 2-AG, and whether it regulates the levels of other lipids in vivo remain to be determined.
Fig. 7.

(A, B) Structures of the ABHD6 inhibitors WWL70 (A) and WWL123 (B).
2.1.8. ABHD12 (Alpha/beta hydrolase domain containing 12)
ABHD12 is a ~45 kDa transmembrane glycoprotein with high expression by mRNA in multiple murine tissues and cells, including brain, microglia, macrophages, and white adipose tissue.89 In vitro, ABHD12 has 2-AG hydrolase activity.85 Recently, the neurodegenerative disease polyneuropathy, hearing loss, ataxia, retinitis pigmentosa, and cataract (PHARC) has been mapped to mutations and deletions in the Abhd12 gene in humans.89 PHARC patients also present with hearing loss, demyelination, and cerebellar ataxia. Neither ABHD12(−/−) mice nor selective inhibitors have yet been described. The precise mechanism by which loss of ABHD12 function causes PHARC remains to be determined, and possibilities include ABHD12 regulation of 2-AG tone in particular cell types such as macrophages and microglia (where ABHD12 is highly expressed and MGLL is absent), or ABHD12 regulation of other signaling or structural lipids. The full spectrum of lipids substrates accepted by ABHD12 has not been extensively investigated.
Selective inhibitors of ABHD12 have not, to our knowledge, been described.
2.2. Extracellular neutral lipases
The absorption of dietary triglycerides (TGs) and the subsequent distribution of intestinal and hepatic TGs to peripheral tissues occur by the action of multiple SHs. The enzymes involved in dietary TG absorption (LIPF, CEL, PNLIP, and PNLIPRP2) hydrolyze TGs into free fatty acids that can be absorbed by the intestines.90–93 Once absorbed, the fatty acids are re-esterified into TGs in enterocytes, and these “exogenous” TGs are then packaged and secreted to the plasma as lipoprotein particles called chylomicrons for distribution to peripheral tissues. In parallel, TGs from the liver (endogenous TGs) are similarly transported in the form of very low density lipoprotein (VLDL) particles.94 The transport of TGs in the form of lipoprotein particles reduces diffusion of hydrophobic molecules in the plasma and also allows targeting of TG delivery to specific tissues through interactions with the protein components of lipoprotein particles.
The distribution of TGs and recycling of lipoprotein remnants occurs by the action of LPL, LIPC, LCAT, and LIPA. Lumenally oriented lipoprotein lipase and hepatic lipase hydrolyze TGs and cholesteryl esters from chylomicron and VLDL particles to generate free fatty acids that can be absorbed and used by tissues. The remaining lipoprotein particles, called remnant chylomicrons and LDL particles, respectively, are loaded with cholesterol from peripheral tissues by a process dependent on the lecithin-cholesterol acyltransferase, and are targeted to and recycled in the liver by lysosomal acid lipase.94–97
2.2.1. PNLIP (Pancreatic lipase)
PNLIP, also known as pancreatic triglyceride lipase,98 or colipase-dependent pancreatic lipase,99 and sometimes abbreviated PL, is a 51 kDa glycoprotein in humans.100,101 PNLIP is only expressed in the pancreas and is secreted from there into the intestinal lumen.100 In vitro, PNLIP has no activity on phospholipids or cholesteryl esters, but can hydrolyze TGs to monoglycerides and free fatty acids.102,103 This activity can be inhibited by bile salts, which raises an interesting question of how PNLIP can function in the bile salt-enriched environment of the intestinal lumen.103 This issue was resolved by the discovery of a ~10 kDa protein cofactor secreted by the pancreas called colipase, which binds the C-terminal domain of PNLIP and preserves its activity in the presence of bile salts.104–106 In humans, it appears that colipase secretion is the limiting factor in PNLIP activity, since addition of exogenous colipase to pancreatic juices in vitro can further stimulate PNLIP activity.107
Multiple studies of deficiency in PNLIP have been reported, though none have conclusively demonstrated the extent of the contribution of PNLIP to dietary TG absorption. First, individuals with deficiencies in total lipolytic activity from duodenal juices present with steatorrhea (oily stools), but, in these cases, the Pnlip gene was not sequenced and it is unclear whether the subjects had deficiencies in PNLIP and/or other lipases of the gastrointestinal tract.108,109 Second, administration of tetrahydrolipstatin (THL), an inhibitor of PNLIP,110 induces modest weight loss and steatorrhea in humans and has been used as a drug to treat obesity for many years.111 THL, however, has also been shown to inhibit other lipases in the digestive tract including carboxyl ester lipase (CEL) and gastric lipase (LIPG),112 and therefore the extent to which the inhibition of PNLIP is responsible for the weight loss effects of THL remains unresolved. Third, pancreatic extracts from PNLIP(−/−) mice98 have >80% decreased TG hydrolysis activity in the absence of taurocholate, a stimulator of another lipase in the intestinal lumen, carboxyl ester lipase (CEL), but, when taurocholate was added to the assay, TG hydrolysis activity was reduced by ~50% in the PNLIP(−/−) mice. These mice show delayed and reduced TG and cholesterol absorption when given a fatty meal by oral gavage, but do not show steatorrhea on chow or high fat diet and are of normal weight during post-natal development. These data, taken together, support the hypothesis that PNLIP is a principal, but not sole enzyme responsible for the hydrolysis and absorption of dietary TG.
2.2.2. PNLIPRP1 and PNLIPR2 (Pancreatic lipase-related proteins 1 and 2)
PNLIPRP2 was originally cloned from cytotoxic T lymphocytes as an interleukin-4-inducible factor with homology to PNLIP.113 Later, human pancreatic lipase-related proteins 1 and 2 (PNLIPRP1 and PNLIPRP2) were cloned from a pancreatic cDNA library using oligonucleotide probes to conserved regions of canine and porcine PNLIP.114 Of the two related proteins, catalytic activity has only been found for PNLIPRP2, an enzyme that can hydrolyze TG, phospholipids, and monogalactosyldiglycerides in vitro.93,115,116 The effect of bile salt concentrations and the presence of colipase in modulating PNLIPRP2 activity are unclear but appear to vary across species.114–116
PNLIPRP2 is highly expressed in the pancreas and has essentially no expression in other tissues.113 In the rat, PNLIPRP2 expression begins at E17, peaks at P0.5, and is maintained at a lower level in adult,117 a profile complementary to rat PNLIP, which is expressed only after P21. These data suggested that PNLIPRP2 might be involved in neonatal but not adult dietary TG digestion, a hypothesis confirmed by PNLIPRP2(−/−) mice.118 Slightly fewer than expected PNLIPRP2(−/−) mice are born from heterozygous matings. At P10, PNLIPRP2(−/−) mice have a 2.5-fold increase in stool weight and a 5-fold increase in stool fat content. The steatorrhea of P10 knockouts is absent by P23, consistent with the increased expression of PNLIP during postnatal development. The fat in feces from wild-type animals at P10 is principally nonesterified fatty acids, whereas PNLIPRP2(−/−) mice accumulate DGs and TGs, demonstrating incomplete lipolysis of dietary TGs. Lastly, these mice gain weight slower than their wild-type littermates. Thus PNLIPRP2 is a major contributor to the pancreatic lipase activity in early postnatal development and its role is diminished in adults owing to the upregulated expression and secretion of PNLIP after P21.
THL is a non-selective inhibitor of PNLIPRP2.93
2.2.3. CEL (Carboxyl ester lipase)
CEL is also known by the names carboxyl ester lipase, cholesterol esterase,119 bile salt-stimulated lipase,120 bile salt-dependent lipase,121 and bile salt-activated lipase.122 In humans, CEL is an ~80 kDa protein123 that is primarily synthesized in pancreatic acinar cells and lactating mammary glands, with much lower levels of expression in liver, macrophages, endothelial cells, and eosinophils.91 Mammary-derived CEL is highly abundant in the milk of most species except rats and cows, constituting ~1% of the protein component.120 In the intestinal lumen, CEL is attached to the brush border of enterocytes via a heparin binding domain,124 and in pancreatic juice CEL is present as ~5% of the total protein.91 In vitro, CEL has broad substrate scope and is capable of hydrolyzing cholesteryl esters, TG, diglycerides (DGs), monoglycerides (MGs), phospholipids, lysophospholipids, and ceramide.125–127 The name bile salt-stimulated lipase (BSSL) originated from the finding that CEL has an absolute requirement for bile salts with 3α, 7α hydroxyl groups in hydrolyzing TGs and other long chain fatty acyl substrates.128 The Kd for bile salts and CEL is approximately 20 3M,91 and therefore CEL is thought to be active principally in the intestinal lumen where it is exposed to high bile salt concentrations.128 Consistent with this premise, CEL has been shown to be stable under acidic conditions and therefore mammary-derived CEL is thought to be stable as it passes through the stomach to the intestines where it actively cleaves TGs in milk.128
Multiple lines of evidence have established a role for both mammary-derived maternal CEL and pancreas-derived pup CEL in dietary TG hydrolysis and absorption in neonates. Kittens fed formula gain weight at only half the rate of kittens fed breast milk or kittens fed formula supplemented with recombinant human CEL.129 CEL activity from milk produced by CEL(−/−) mothers or from the intestinal lumen of P5 pups nursed by CEL(−/−) dams is dramatically reduced, and this coincides with an accumulation of lipid droplets along the endothelial lining of the distal small intestine and damage to the villus architecture in the pups.130 Using all combinations of CEL(−/−) pups and dams, it was demonstrated that fecal fat levels were only elevated when CEL(−/−) dams nursed CEL(−/−) pups but not under any other pair-wise combination, demonstrating the importance of both maternal CEL and pancreas-derived CEL.131 Despite steatorrhea in knockout pups nursed by knockout mothers, weight gain is normal during neonatal development.131 The role of CEL in adult dietary TG absorption is less clear though studies with CEL(−/−) mice have provided some evidence that CEL plays a role in the absorption of esterified cholesterol and the determination of chylomicron particle size.119,132
CEL is inhibited by THL112 as well as multiple carbamates;33 however, these compounds are non-selective towards other lipases.
2.2.4. LIPF (Gastric lipase)
LIPF, also abbreviated GL, in humans is a glycoprotein with a de-glycosylated molecular weight of 43 kDa.133 It is secreted by the chief cells of the gastric mucosa134 and has hydrolytic activity for triglycerides of varying acyl chain lengths with no dependence on bile salts.90 Interestingly, LIPF TG hydrolytic activity has a pH optimum of ~3–5 which contrasts with the pH optimum of ~7–9 displayed by most SHs. Crystal structures reveal that LIPF adopts a standard α/β-hydrolase fold and did not provide any insight into its unusual activity at acidic pH.135 This issue was resolved when it was shown that LIPF binds to TG emulsions optimally at low pH, suggesting that its optimal activity was derived in large part from productive substrate interactions.136 To date, LIPF(−/−) mice have not been generated, but in human studies, estimations based on total amounts of LIPF and PNLIP suggest that LIPF contributes to the hydrolysis of ~10–20% of TG in stomach and intestinal lumen.137
THL is a non-selective LIPF inhibitor.
2.2.5. LPL (Lipoprotein lipase)
LPL is a ~55 kDa glycoprotein primarily expressed in adipocytes, cardiac and skeletal muscles, and the lactating mammary gland, with lower expression in macrophages, adrenal glands, ovaries, some neuronal cells, spleen, testes, and lung.15,138–140 LPL is bound to the lumen of capillary endothelial cells by heparin sulfate-proteoglycans (HSPGs) and consequently it can be displaced and collected following an intravenous injection of heparin (i.e., post-heparin plasma). LPL mRNA has not been detected directly in endothelial cells and it is therefore thought that LPL is synthesized by parenchymal cells and then translocated to the lumen.141
Exogenous TG from the intestines and endogenous TG from the liver, after acquiring the apolipoproteins apoE and apoC-II from high density lipoproteins (HDL), become mature chylomicrons and mature VLDL particles, respectively, and circulate through the blood to peripheral tissues. LPL in the capillary endothelial lumen hydrolyzes TG from these lipoprotein particles into MG and non-esterified fatty acid so that these catabolic products can be taken up by organs. The active form of LPL is a homodimer that requires the cofactor apoC-II for activity,142 and the initial binding of chylomicrons or VLDL to LPL is also mediated by the LPL-apoC-II interaction. After LPL processing, chylomicrons and VLDL particles become chylomicron remnants and intermediate density lipoprotein (IDL) particles, respectively, and are targeted to the liver by ApoE.
Deficiencies in LPL have been reported in both humans and rodents. Familial LPL deficiency occurs in approximately one out one million individuals and results from a collection of heterogeneous loss-of-function mutations in Lpl.143–145 These patients have dramatically reduced LPL activity in postheparin plasma with concomitant elevations in plasma chylomicrons, TG, and VLDL. Similar phenotypes are also found in patients with deficiency in the obligate LPL cofactor apoC-II, and together these disorders are classified as familial chylomicronemia syndromes or type I hyperlipoproteinemia.146 Mice deficient for LPL have dramatically reduced LPL activity, three-fold greater TG, seven-fold greater cholesterol in VLDL-C, accumulation of chylomicrons in capillaries, and reduced HDL compared to controls.147 Upon suckling, LPL(−/−) pups become cyanotic (blue discoloration of the skin caused by lack of blood oxygenation) and die at approximately 18 h, at which point plasma TG is elevated eighty-fold and VLDL-C is elevated forty-fold. Gross morphological analysis revealed severe reductions in adipocyte mass in LPL(−/−) pups. Transgenic expression of LPL only in skeletal and cardiac muscle entirely rescues the LPL(−/−) phenotypes demonstrating that the presence of LPL in muscle is sufficient to normalize whole body VLDL metabolism. Though LPL(−/−) pups do not entirely phenocopy LPL-deficient humans, together these data provide strong evidence for LPL participation in the catabolism of chylomicron and VLDL particles in vivo.
A series of covalent urea-based inhibitors of LPL have been described though these compounds are also cross-reactive with endothelial lipase (LIPG) in vitro (Fig. 8).148
Fig. 8.
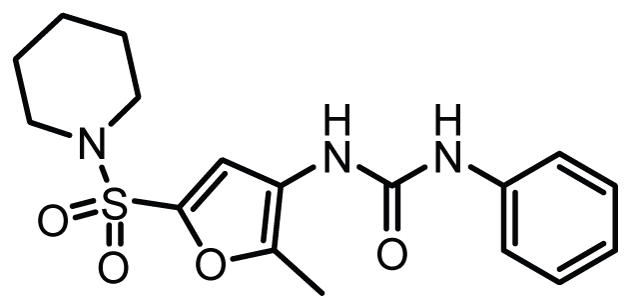
Structure of a urea-based dual LPL/LIPG inhibitor.
2.2.6. LIPC (Hepatic lipase)
LIPC, also called hepatic lipase or hepatic triglyceride lipase,149 is a ~60 kDa glycoprotein secreted by the liver and localized to the lumenal surface of hepatic endothelial cells by HSPGs.150 In vitro, LIPC can hydrolyze TG and phospholipids in all lipoproteins.151,152 Like LPL, LIPC is eluted in post-heparin plasma and its activity can be differentiated from that of LPL by including high salt concentrations in the in vitro assay buffer, which stimulates LIPC but inhibits LPL activity.152
LIPC deficiency or inhibition in humans and other organisms has revealed complex role for LIPC in lipoprotein metabolism. Several cases of familial LIPC deficiency in humans have been reported.153–155 In two brothers, β-VLDL is markedly elevated and both LDL and HDL are enriched in TG and PC,154 phenotypes that are nearly identical to those described in another investigation.155 In cynomolgus monkeys, the administration of LIPC antisera dramatically inhibits LIPC activity but not LPL, which causes elevations in VLDL and intermediate-density lipoprotein (IDL) mass and a decrease in LDL mass. In this same study, VLDL TG, cholesterol, and phospholipids, as well as HDL2 phospholipids were also elevated.149 In rats, administration of anti-rLIPC that does not inhibit rat LPL caused a 4-fold elevation in plasma TGs due to an increase in VLDL and LDL.156 In LIPC(−/−) mice, total basal plasma cholesterol and phospholipids are elevated but plasma TG is unchanged, and HDL1 was elevated. Taken together these studies suggest that, by hydrolyzing lipoprotein TG and phospholipids in vivo, LIPC plays a dual role in converting VLDL to LDL particles in a manner complementary to that of LPL and in the remodeling of HDL TG.95
Selective inhibitors of LIPC have not, to our knowledge, been described.
2.2.7. LIPA (Lysosomal acid lipase)
LIPA, also abbreviated LAL, is a ~40 kDa glycoprotein in humans that is highly expressed in brain, lung, kidney, and mammary gland, with lower levels in liver and heart.157–159 In vitro, LIPA hydrolyzes cholesterol esters, TGs, DGs, and MGs with maximal activity at pH ~4.160,161 LIPA co-localizes to the membrane fractions of early and late endosomes, consistent with its in vitro acidic pH optimum.162
The available data from both humans and rodents support a role for LIPA in the hydrolysis of cholesteryl esters and TG from internalized LDL particles. By doing so, LIPA supplies cells with the cellular cholesterol required to stimulate cellular cholesteryl ester formation and suppression of hydroxymethylglutaryl-CoA reductase, the rate limiting step for cholesterol biosynthesis.97 In humans, LIPA deficiencies manifest themselves as either Wolman disease or cholesteryl ester storage disease (CESD).163–165 Newborns with Wolman disease have complete loss of LIPA activity and present with intra-lysosomal accumulation of TG and cholesteryl esters in most tissues which is accompanied by vomiting, diarrhea, and hepatosplenomegaly. Typically these individuals do not live past their first year. Patients with CESD typically have a residual 10–30% remaining LIPA activity and present with hypercholesterolemia and elevated LDL-cholesterol levels, but can typically survive past middle age. LIPA(−/−) mice are normal at birth, fertile, and survive into adulthood.166,167 However, adult LIPA(−/−) mice have massive accumulations of TG and cholesteryl esters in the liver, adrenal glands, and small intestines, with a reduction in the mass of white and brown adipose tissues.
THL and a series of thiadiazole carbamate inhibitors have been shown to covalently inactivate LIPA, though the selectivity and in vivo efficacy of these carbamates remains unknown.168
2.2.8. LCAT (Lecithin-cholesterol acyltransferase)
LCAT, also called lecithin-cholesterol acyltransferase, is a ~60 kDa glycoprotein in humans that is predominantly synthesized in the liver and found in the plasma reversibly bound to lipoproteins.96,169,170 LCAT is activated by ApoA-I,171 a major protein component of HDL, and transfers acyl chains from phosphatidylcholines (PCs, or lecithins) to cholesterol to form cholesteryl esters on the surface of HDL particles.172 In vitro, other sterols such as 25-hydroxycholesterol, pregnenolone, and dehydroepiandrosterone can also serve as acyl acceptors.173 It appears that there are two distinct binding pockets for PCs and sterols, since it has been shown that both can simultaneously be bound to LCAT.174
In vivo, by generating cholesteryl esters, LCAT promotes the efflux of cholesterol from peripheral tissues such as white fat onto HDL particles, a process called reverse cholesterol transport. In a subsequent step, cholesterol ester transfer protein (CETP) moves the cholesteryl esters from HDL to LDL particles and chylomicron remnants for transport back to the liver.96 Human patients with familial LCAT deficiency (FLD) show near complete loss of LCAT activity, reduced plasma cholesteryl esters, HDL deficiency, and lipid changes in the VLDL and LDL fractions.175–177 Clinically, these individuals present with corneal opacification, anemia, proteinuria, and renal disease. Patients with partial LCAT deficiency, also known as fish-eyed disease (FED), show HDL deficiency but present with few clinical signs except for age-dependent corneal opacification. In LCAT(−/−) mice, plasma LCAT activity is ablated and > 90% reductions in plasma HDL and HDL cholesteryl ester content are observed. HDL particles are also of a heterogeneous size and morphology, and the adrenal glands showed reduced lipid content.178 No reports have described whether LCAT(−/−) mice present with any of the physiological defects observed in humans with FLD or FED. Together, these studies provide strong evidence that in the absence of LCAT, cholesterol efflux from peripheral tissues to HDL is impaired, resulting in the reduction of HDL and cholesteryl esters in plasma.
Selective inhibitors of LCAT have, to our knowledge, not been described.
2.3. Phospholipase A2 enzymes
The phospholipase A2 (PLA2) enzymes are a diverse collection of hydrolases that are all characterized by the ability to cleave the sn-2 acyl chain from phospholipids to produce a fatty acid and a lysophospholipid.12,40 The sn-2 chain falls into one of three categories: 1) short (i.e., acetyl), for example in the bioactive signaling lipid platelet activating factor (PAF); 2) intermediate (i.e., butyryl or azeloyl), for example in oxidatively truncated phospholipids generated from reactive oxygen species and polyunsaturated phospholipids; or 3) long, such as the saturated (e.g., stearoyl), monounsaturated (e.g., oleoyl), or polyunsaturated (e.g. arachidonoyl), acyl chains commonly found in phospholipids that form structural components of cell membranes.12 Thus, the PLA2 reaction can inactivate signaling lipids such as PAF or produce lipids such as arachidonate that serve as metabolic precursors for bioactive lipid transmitters like prostanoids. PLA2s are classified into groups and subgroups by homology. The PLA2s use either an active site histidine or serine for catalysis, and therefore those in the SH superfamily only include the active site serine-containing PLA2s belonging to groups 4, 6, 7, 8, and 15. In the remainder of this section, G will be used to abbreviate the group.
PLA2G4 consists of the cytosolic PLA2s (cPLA2s), of which there are six members, A–F or α–ζ. G4B–F have ~30–37% identity with G4A and all members of G4 contain a Ser/Asp catalytic dyad. The G4 members, with the exception of 4C, are two domain proteins with a C-terminal catalytic domain and an N-terminal C2 domain. C2 domains generally serve as calcium-binding domains in proteins, and, for the PLA2G4 enzymes, are thought to assist these enzymes in anchoring to membranes. PLA2G4A is the only well-characterized member of this group (see below). PLA2G6 consists of the six calcium-independent PLA2s (iPLA2s), many of which also belong to the patatin-like phospholipase domain containing (PNPLA) family: G6A (iPLA2), G6B (iPLA2γ or PNPLA8), G6C (iPLA2δ or PNPLA6 or NTE), G6D (iPLA2ε or PNPLA3 or adiponutrin), G6E (PNPLA2 or ATGL), and G6F (iPLA2η or PNPLA4 or GS2). The patatin-like phospholipase domain is characterized by an α/β/α sandwich architecture distinct from the α/β-hydrolase fold.37 While all PLA2G6 members exhibit calcium-independent phospholipase activities, some have been shown to also hydrolyze other classes of lipids in vitro. PLA2G7 and PLA2G8 contain two members each and are characterized by the ability to hydrolyze the short sn-2 acetyl from platelet activating factor (PAF), a lipid molecule that binds a G-protein coupled PAF receptor. PLA2G15 only has one member with an acidic pH optimum and lysosomal localization.
For consistency, the official gene symbol will be used to name the corresponding serine hydrolase, but common names and PLA2G nomenclature will also be included where possible.
2.3.1. PLA2G4A (Cytosolic PLA2)
PLA2G4A, also abbreviated cPLA2 or cPLA2α,12 is an 85 kDa protein that is ubiquitously expressed throughout mammalian tissues, with highest levels in immune cells such as macrophages and mast cells.179–181 PLA2G4A hydrolyzes PCs and PEs in a calcium-dependent manner and shows a 20-fold selectivity for arachidonate in the sn-2 position.182 In addition, PLA2G4A also possesses lysophospholipase A1 and transacylase activities.183 Upon stimulation by calcium, cPLA2 translocates from the cytoplasm to the membrane fraction of cells.179
Arachidonic acid is the precursor for the cyclooxygenase (COX)- and lipoxygenase-mediated generation of a variety of bioactive eicosanoids that can initiate downstream signaling by binding to and activating cognate eicosanoid receptors.184 The use of COX inhibitors in humans for the treatment of inflammation and pain underscores the importance of this pathway in immunobiology and inflammation.184 The contribution of arachidonic acid originating from cPLA2-mediated hydrolysis of phospholipids to the eicosanoid biosynthetic pathway has been elucidated by the generation of PLA2G4A(−/−) mice.17,185–187 Peritoneal macrophages from PLA2G4A(−/−) mice show basally lower concentrations of prostaglandin E2 (PGE2) and leukotriene B4 and C4 (LTB4 and LTC4) and a blunted production of PGE2 following a lipopolysaccharide (LPS) challenge.17 Similarly, bone-marrow derived mast cells from PLA2G4A(−/−) mice fail to generate leukotrienes upon stimulation by IgE or stem cell factor (SCF).187 When tested in an ischemia-reperfusion model of stroke by occlusion of the middle cerebral artery (MCA), PLA2G4A(−/−) mice demonstrate reduced neurological deficit and reduced infarct size compared to wild-type mice.17 PLA2G4A(−/−) mice also show reduced anaphylactic responses induced by ovalbumin and reduced bronchial reactivity to methacholine.185 Taken together, these studies demonstrate that, in macrophages and other monocytes, PLA2G4A produces the arachidonic acid that feeds into downstream biosynthetic pathways for pro-inflammatory eicosanoids (Fig. 9). To what extent PLA2G4A also provides arachidonic acid for eicosanoid pathways in other cells and tissues remains mostly unknown. That brain tissue from PLA2G4A(−/−) mice show unaltered levels of arachidonic acid and prostaglandins suggests the involvement of additional hydrolases in eicosanoid biosynthetic pathways in the nervous system, and possibly other tissues.188
Fig. 9.

PLA2G4A liberates arachidonic acid from sn-2 arachidonoyl-containing phospholipids in macrophages. The arachidonate generated by this reaction can be further oxidized by cyclooxygenases, lipoxygenases, and prostaglandin synthases into eicosanoid such as PGE2 (shown) and other oxidative derivatives.
Because blockade of PLA2G4A provides a therapeutically viable alternative to inhibition of COX enzymes, PLA2G4A inhibitors have become the target of intense pharmaceutical interest.189–195 To date, the most potent and selective compounds have arisen from substituted indoles such as ecopladib,192 efipladib,194 and WAY-196025 (Fig. 10),194 which at least in vitro show good selectivity over related PLA2s. These compounds also show efficacy in rodent models of inflammation such as in carrageenan-induced paw edema,192,194 but a correlation of target occupancy, eicosanoid reduction, and efficacy in rodents remains to be demonstrated. Whether such compounds can reduce inflammatory responses in humans is unknown.
Fig. 10.

Structures of indole-derived PLA2G4A inhibitors.
2.3.2. PLA2G4B–F (Cytosolic PLA2β, γ, δ, ε, and ζ)
The remaining members of the PLA2G4 family, PLA2G4B–F, are relatively uncharacterized in comparison to PLA2G4A. PLA2G4B, also called cPLA2β, is a ~100–110 kDa membrane-bound protein ubiquitously expressed in human tissues, with highest levels in pancreas, brain, heart, and liver.196,197 PLA2G4B shows calcium-dependent phospholipase A1 and A2 activity with phosphatidylcholine substrates, including the PLA2G4A-preferred substrate palmitoyl-arachidonoyl-PC. In the human lung epithelial line BEAS-2B, PLA2G4B is found as at least three splice isoforms, cPLA2β1–3, which contain internal deletions in the catalytic domain.198
PLA2G4C, also called cPLA2γ, is a 61 kDa farnesylated protein localized to the endoplasmic reticulum and mitochondria. PLA2G4C mRNA is predominantly expressed in skeletal and cardiac muscle of human tissues, with lower expression levels in brain.197,199,200 In vitro, PLA2G4C can hydrolyze both the sn-1 and sn-2 chains of PC in a calcium-independent manner by sequential PLA2 and lysophospholipase activity, but does not have PLA1 activity on intact phospholipids.201 An immortalized mouse lung fibroblast line from PLA2G4A(−/−) mice stably expressing PLA2G4C and loaded with radiolabeled fatty acids released equal amounts of C16, C18:1, C18:2, and 20:4 fatty acids following serum stimulation, whereas stable expression of PLA2G4A showed preference for C20:4 fatty acid, demonstrating that PLA2G4C has broader PC acyl chain substrate specificity compared to PLA2G4A.201 Unlike other members of the PLA2G4 family, PLA2G4C also has transesterification activity, generating PCs from fatty acyl-CoAs and LPC, and a related dismutase activity, generating glycerophosphatidylcholine and PC from two equivalents of LPC.199 Some preliminary evidence from HEK293 cells overexpressing PLA2G4C suggests that it may play a role in oxidative stress responses.200
PLA2G4D (cPLA2δ), PLA2G4E (cPLA2ε), and PLA2G4F (cPLA2ζ) form a gene cluster on mouse chromosome 2E5 along with PLA2G4B.202,203 PLA2G4D is a 90 kDa protein localized to the cytoplasm and is only expressed in human placenta, fetal skin, and cervix. PLA2G4E is a ~100 kDa protein that colocalizes with lysosomes but not the endoplasmic reticulum or Golgi, and is expressed in the thyroid, heart, and skeletal muscle of mice.203 PLA2G4F is a ~96 kDa cytosolic protein highly expressed in the mouse thyroid, with lower levels of expression in the stomach and essentially no expression in other tissues.203 Following ionomycin treatment, both PLA2G4D and PLA2G4F translocate from the cytoplasm to perinuclear regions, similar to PLA2G4A.203,204 PLA2G4D, E, and F all show calcium-dependent PLA2 activity with PCs and PEs, without any strong acyl chain preferences at the sn-2 position.203
The pyrazole derivative Lf-80 inhibits PLA2G4B selectively over other PLA2G4 members in vitro, but its activity in vivo is unknown.205 With the exception of this compound, neither knockouts nor selective inhibitors have been, to our knowledge, described for any of the PLA2G4B–F family members. While assignment of their physiological substrates and functions must await further investigations with selective genetic or pharmacological tools, the localized tissue distributions of PLA2G4B–F enzymes compared to PLA2G4A suggest that they may perform tissue-specific roles in controlling phospholipid and/or arachidonate metabolism.
2.3.3. PLA2G6 (Calcium-independent PLA2)
PLA2G6, also called PLA2G6A, calcium-independent PLA2 (iPLA2), or iPLA2β, is an 85–88 kDa protein with ubiquitous expression in mice with highest levels in testis.206,207 Multiple splice isoforms have been identified for human iPLA2,208 though it appears that rats and mice only have a single transcript by Northern blot analysis.207,209 PLA2G6 displays a broad substrate scope and can hydrolyze a variety of PCs, including PAF, dihexanoyl-PC, dipalmitoyl-PC, and sn-2 arachidonoyl-containing PCs.207,210 PLA2G6 also shows acyltransferase and lysophospholipase activity in vitro.210 The activity of PLA2G6 is stabilized by ATP and glycerol, but not dependent on calcium.210
The generation of PLA2G6(−/−) mice has helped to elucidate physiological functions for this enzyme.211,212 Palmitoyl-linoleoyl-PC hydrolase activity is reduced ~50% in muscle, kidney, liver, and epididymis, and ~80% in brain tissue of PLA2G6(−/−) mice, but is unchanged in other tissues.211 Various PC species are unaltered in testis, brain, and pancreatic islets between wild type and PLA2G6(−/−) mice. Male PLA2G6(−/−) mice have reduced fertility owing to the impaired motility of spermatozoa, but had testis of normal weight and anatomy. Excepting male infertility, adult PLA2G6(−/−) mice are generally indistinguishable from their wild-type littermates, but, at ~2 years of age, PLA2G6(−/−) mice gradually lose weight and prematurely die.212 This death is accompanied by severe neurological deficits, including gait problems and clasping behaviors when suspended by the tail, and, at an anatomical level, peripheral demyelination and the accumulation of spheroids and vacuoles localized to axons and synapses. These neurological phenotypes in rodents agree well with data from humans, where a heterogeneous collection of insertion, deletion, or missense mutations in the Pla2g6 gene cause infantile neuroaxonal dystrophy (INAD), a fatal autosomal recessive neurodegenerative disease that presents clinically within the first two years of life as cerebellar ataxia, hypotonia of the trunk, spastic quadriplegia, and hyperreflexia and is associated with axonal swelling and an accumulation of spheroid bodies in the CNS.213,214 When several of these human mutations were examined biochemically, they were found to abolish catalytic activity in recombinant PLA2G6 protein.215 In mice, a G1117A missense mutation in PLA2G6 generated by ENU mutagenesis does not change protein expression but abolishes catalytic activity and also causes INAD-like symptoms.216 The endogenous substrates of PLA2G6, and how loss of PLA2G6 activity promotes the pathogenesis of INAD-like sympotoms, remains to be determined.
Selective inhibitors of PLA2G6 have not, to our knowledge, been described.
2.3.4. PNPLA8 (Calcium-independent PLA2γ)
PNPLA8, also called PLA2G6B or calcium-independent PLA2γ (iPLA2γ), is a 63 kDa, mitochondrial-localized enzyme highly expressed in heart and skeletal muscle.37,217 PNPLA8 displays calcium-independent PLA1 and PLA2 activity with multiple PCs containing saturated or monounsaturated sn-1 and sn-2 acyl chains,218,219 but minimal activity toward PCs bearing polyunsaturated fatty acids (PUFAs) at the sn-2 position. Consequently, an accumulation of lysophosphatidylcholines (LPCs) with polyunsaturated sn-2 acyl chains is observed when PNPLA8 is incubated with the appropriate PC substrate, or when PNPLA8 is overexpressed in the myocardium of mice by transgenic methods.219,220
The physiological consequences of PNPLA8 deficiency in mice have been reported.221–223 Myocardial mitochondria from PNPLA8(−/−) mice show ~60% lower hydrolysis of palmitoyl-arachidonoyl-PC. In addition to growth retardation and a kyphotic posture by age two months, PNPLA8(−/−) mice show a variety of bioenergetic defects, including cold intolerance, reduced exercise capacity, reduced mitochondrial oxygen consumption, muscle weakness, and myofilament atrophy in skeletal muscles.221,223 Biochemically, these phenotypes are accompanied by a reduction in myocardial cardiolipin content, alterations in cardiolipin and glycerophospholipid species distribution, and an increase in oxidized lipids.221–223 Together, these data provide evidence that PNPLA8 participates in the remodeling of mitochondrial lipids and that its absence causes mitochondrial dysfunction from perturbations in glycerophospholipids and increased oxidative stress.
Selective inhibitors of PNPLA8 have not, to our knowledge, been described.
2.3.5. PNPLA6 (Neuropathy target esterase)
PNPLA6, also called PLA2G6C, iPLA2δ, or neuropathy target esterase (NTE), is the mammalian homolog of the Drosophila gene swisscheese (sws) and is a ~150 kDa integral membrane enzyme anchored to the endoplasmic reticulum by an N-terminal transmembrane domain. PNPLA6 is highly expressed throughout the brain and in some cells of the testis.224,225 A truncated form of PNPLA6 containing the esterase domain has been shown to hydrolyze phospholipids, lysophospholipids, and MGs, in vitro, but not DGs, TGs, or lipid amides. Historically, the activity of PNPLA6 has been detected in tissues with the unnatural substrate phenyl valerate.226,227
In humans, PNPLA6 is a target for organophosphorous nerve agents that cause axonal degradation and onset of ataxia and lower limb paralysis typically 1–3 weeks after initial exposure.228 PNPLA6(−/−) mice are lethal at mid-gestation because of failed placental development and consequently massive apoptosis in the developing embryo.229,230 Brain-specific deletion of PNPLA6 in mice causes near complete loss of PNPLA6 activity in brain and vacuolation in the hippocampus, neuronal loss in the thalamus, partial loss of Purkinje cells, and reduced performance on rotorod.224 These mammalian CNS phenotypes are nearly identical to those shown by Drosophila mutants for swisscheese (sws).231 In S. cerevisiae, Drosophila, and mammalian cell lines, overexpression or genetic deletion of PNPLA6 or PNPLA6 homologs modulates PC levels.231,232 However, the endogenous biochemical role for PNPLA6 in the mammalian nervous system and how the regulation of PNPLA6 substrates or products protects CNS integrity remain unanswered.
Selective inhibitors of PNPLA6 have not, to our knowledge, been described.
2.3.6. PNPLA3 (Adiponutrin)
PNPLA3, also called PLA2G6D, iPLA2ε, or adiponutrin, is a ~45 kDa transmembrane protein with highest expression in white and brown adipose tissue in mice.39,233 PNPLA3 displays TG lipase activity, acylglycerol transferase activity (the generation of DG and glycerol from two MG molecules, or TG and glycerol from MG and DG), and very modest calcium-independent PLA2 activity in vitro.39,234 PNPLA3 expression is dramatically elevated upon the differentiation of the preadipocyte line 3T3-L1.233 In white adipose tissue from mice, PNPLA3 expression is decreased upon fasting and restored by refeeding and is highly elevated in white fat from obese Zucker rats (fa/fa) relative to lean controls.233
The in vivo biochemical and physiological processes regulated by PNPLA3 remain elusive. In humans, hepatic steatosis, or the excessive accumulation of lipids in the liver, is associated with multiple adverse metabolic events including insulin resistance and dyslipidemia. Several genome-wide association studies have provided evidence that some PNPLA3 polymorphisms are strongly correlated with increased hepatic fat, and others are strongly correlated with lower hepatic fat content.235–237 However, PNPLA3(−/−) mice are viable, fertile, and show no differences compared to wild-type littermates in multiple metabolic parameters including liver enzyme levels and liver fat content on either normal chow or fatty liver-inducing diets.238 Further characterization of the lipidomic changes and metabolic phenotypes of PNPLA3(−/−) mice should help to elucidate the physiological functions of this enzyme.
Selective inhibitors of PNPLA3 have not, to our knowledge, been described.
2.3.7. PLA2G7 (Plasma platelet-activating factor acetylhydrolase)
PLA2G7, also called lipoprotein-associated phospholipase A2 (Lp-PLA2), plasma platelet-activating factor acetylhydrolase (pPAFAH), or PLA2G7A, is a ~45 kDa glycoprotein that is principally produced and secreted by monocytes and found in the plasma associated with both LDL and HDL particles.239–241 PLA2G7 is also highly expressed in rodent brain.85 In vitro, PLA2G7 cleaves the sn-2 acetyl group on PAF, a bioactive signaling lipid that can bind the G-protein coupled PAF receptor and initiate downstream signaling as part of immune and inflammatory responses.240 PLA2G7 can also hydrolyze PCs that are oxidatively truncated at the sn-2 position such as sn-2 azeloyl (az-PAF) species, which are thought to originate from oxidative damage to sn-2 unsaturated phospholipids (Fig. 11). PLA2G7 has no activity on medium (i.e., C9:0) or long chain (i.e., C20:4) sn-2 PCs without oxidative modification.242,243 In addition to PCs, PLA2G7 has also been shown to cleave short chain DG and TG and displays transacetylation activity from PAF to lysophospholipids.244,245
Fig. 11.

Structures of oxidatively-truncated phosphatidylcholine species.
PLA2G7 has been implicated in multiple pathological conditions including allergy, inflammation, and atherosclerosis, but the data regarding the endogenous biochemical and physiological functions of PLA2G7 remain inconclusive.246,247 Four percent of the Japanese population is homozygous for a loss-of-function missense V279F mutation in the Pla2g7 gene, a mutation that completely ablates plasma PAF hydrolase activity. While this polymorphism is correlated with respiratory problems,248 studies using recombinant PLA2G7 in humans have failed to demonstrate efficacy at alleviating clinical symptoms.249 Several studies have shown that recombinant PLA2G7 or overexpression of PLA2G7 reduces inflammation and oxidative stress associated with atherosclerosis in rodents.240,250,251 However, in humans, it appears that the opposite is true: Pla2g7 null mutations are protective for coronary heart disease, and the PLA2G7 inhibitor darapladib is currently in clinical trials for lowering the risk of cardiovascular events in patients with coronary heart disease, where the drug has shown promising phase II results with minimal adverse effects (Fig. 12).252–254
Fig. 12.
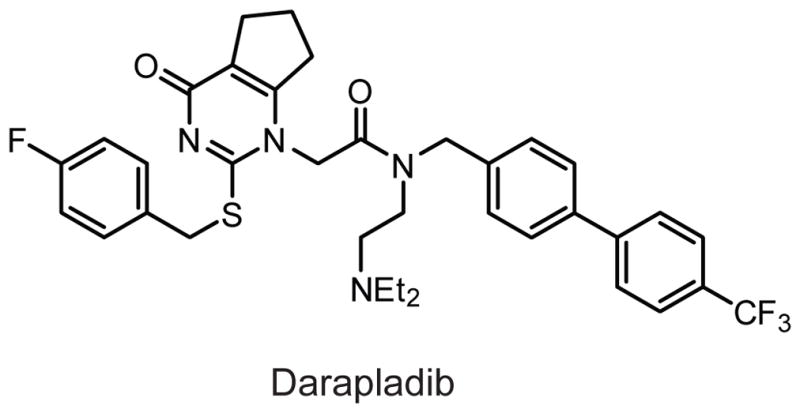
Structure of the PLA2G7 inhibitor darapladib.
While the in vitro biochemical substrate specificity for PLA2G7 is well defined, it remains unclear whether PLA2G7 modulates plasma levels of PAF or other oxidatively truncated PCs in vivo, and, if so, how this process might relate to the cardiovascular effects of PLA2G7 inhibition in humans. One report has shown that darapladib reduces arterial plaque size in hypercholesterolemic swine, but causes little change in arterial PC or oxidatively truncated PC composition, while modestly reducing in total arterial LPC content.255 The generation of PLA2G7(−/−) mice may answer these questions.
2.3.8. PAFAH2 (Platelet-activating factor acetylhydrolase 2)
PAFAH2, also called PLA2G7B, is a ~40 kDa intracellular enzyme that, in mice, is highly expressed in mast cells, liver, kidney, large intestines, and testis, with lower expression in most other tissues.18,181,256,257 PAFAH2 shows similar substrate selectivity to PLA2G7 in that it can also hydrolyze PAF, oxidatively truncated PCs, and transacylate the 2-acetyl group from PAF to lipid acceptors such as lysophospholipids or sphingosine.256,258 When overexpressed in cells, PAFAH2 is found in both the soluble and particulate fractions, and the enzyme translocates from the cytoplasm to the membrane following the application of oxidative stressors.257,259 Overexpression of PAFAH2 but not a catalytically dead PAFAH2 is protective to peroxide-induced apoptosis in CHO-K1 cells.259
These experiments point to a role for PAFAH2 in the response to oxidative stress, a hypothesis supported by studies with PAFAH2(−/−) mice.18 PAFAH2(−/−) mice show > 90% reductions in PAF hydrolysis in kidney and liver, but not brain or plasma, two places where PLA2G7 is highly expressed. Hydrolysis of azelaoyl-PAF(1-O-hexadecyl-2-O-(9-carboxyoctanoyl)-sn-glyceryl-3-phosphocholine), an oxidatively truncated PC, is also modestly reduced in liver and kidney of PAFAH2(−/−) mice. Mouse embryonic fibroblasts derived from PAFAH2(−/−) mice are more sensitive to peroxide-induced cell death than those derived from wild-type littermates. Upon challenge by carbon tetrachloride, PAFAH2(−/−) mice show greater plasma alanine transferase (ALT) levels indicative of hepatic damage and impaired recovery from hepatic tissue injury. Together, these data support the hypothesis that PAFAH2 participates in the clearance of oxidatively damaged phospholipids that may perturb membrane structure and function. Direct measurements of PC species, especially oxidatively cleaved PCs, in PAFAH2(+/+) and (−/−) mice would provide additional evidence to support this conclusion.
2.3.9. PAFAH1b2 and PAFAH1b3 (Type Ib platelet-activating factor acetylhydrolases 2 and 3)
The PAFAH1b complex has been purified from bovine brain cytosol as a ~100 kDa complex that resolves on SDS-PAGE as three bands of size 45, 30, and 29 kDa, which were originally named PAFAHIb α, β, and γ,260 respectively, and later renamed β, α1, and α2, respectively, when it was discovered that the overall structure of the enzyme was similar to that of a trimeric G-protein.261 The catalytic subunit α1 is also called PAFAH1b2 or PLA2G8A and similarly, the other catalytic subunit α2 is called PAFAH1b3 or PLA2G8B. The non-catalytic, non-serine hydrolase β regulatory subunit, also called LIS1, is encoded by the Pafah1b1 gene. PAFAH1b2 and PAFAH1b3 are ~60% identical by amino acid sequence.262,263 In the remainder of this section the PAFAH1bx nomenclature will be used.
The PAFAH1b complex displays calcium-independent hydrolytic activity for PAF and oxidized PCs, but not PC, PE, or LPC.260 Interestingly, when recombinantly expressed in E. coli, both PAFAH1b2 and PAFAH1b3 show PAF acetylhydrolase activity and label with the active site electrophile diisopropyl fluorophosphonate (DIFP), even though incubation of DIFP with the PAFAH1b complex only produced labeling of the PAFAH1b3 subunit.260,262,263 Biochemical analyses of the PAFAH1b2/1b2, PAFAH1b2/1b3, and PAFAH1b3/1b3 dimers show differential substrate preferences and modulation by the PAFAH1b1 subunit. PAFAH1b2/1b2 and PAFAH1b2/1b3 complexes hydrolyze PAF similarly and are not affected by the presence of PAFAH1b1. In contrast, PAFAH1b3/1b3 dimers hydrolyze PAF with approximately five-fold better activity than the other two dimers and is activated by over four-fold in the presence of PAFAH1b1.264
In adult mice, PAFAH1b1 and PAFAH1b3 are ubiquitously expressed with highest levels in brain, lung, and testis, whereas the expression of PAFAH1b2 is principally restricted to brain and testis.265 The PAFAH1b3/1b3 dimer is the major catalytic unit in the adult brain, whereas the PAFAH1b2/1b3 dimer is abundantly expressed in the embryonic brain, and, therefore, subunit switching during nervous system development may represent a regulatory mechanism for PAFAH1b activity.266
Despite the generation of gene targeted mice for all three PAFAH1b subunits, the physiological functions and endogenous substrates for PAFAH1b remain largely unknown.265,267 PAFAH1b1(−/−) mice are embryonic lethal at ~E9.5 and develop type I lissencephaly, a cephalic disorder characterized by lack of brain fold and groove development. PAFAH1b1(+/−) littermates show delayed neuronal migration that causes disorganization of neuronal structures.267 In contrast, PAFAH1b2(−/−), PAFAH1b3(−/−), and PAFAH1b2(−/−)/PAFAH1b3(−/−) double knockout mice are overtly normal and born in the expected Mendelian ratio.265 PAFAH1b2(−/−)/PAFAH1b3(−/−) mice have a ~35% reduction in PAF hydrolytic activity in brain and testes, with the remaining activity likely due to PLA2G7, PAFAH2, or other unidentified PAFAH subtypes. PAFAH1b3(−/−) and PAFAH1b2(−/−)/PAFAH1b3(−/−) knockouts show >50% reduction in testis weight and impaired spermatogenesis causing infertility in the males, but females reproductivity is normal in these animals. PAFAH1b catalytic subunit knockouts show no obvious abnormalities in the lamination of neurons or other nervous system defects.265
2.3.10. PLA2G15 (Lysosomal PLA2)
PLA2G15, also called lysophospholipase 3 (LYPLA3), lysosomal PLA2 (LPLA2), 1-O-acylceramide synthase (ACS), or LCAT-like lysophospholipase (LLPL), is a ~40 kDa glycoprotein localized to the lysosome and ubiquitously expressed in human tissues, with highest levels in heart and skeletal muscle.268,269 PLA2G15 was originally purified from calf brain and characterized to have calcium- and magnesium-independent transacylation activity involving transfer of the sn-2 fatty acyl chain from PC or PE to the primary hydroxyl of N-acetylsphingosine with a pH optimum ~4.5.270 Other primary alcohols, such as those in MGs or 2-acetyl-MGs, can also be acylated,271 and, in the absence of an acyl acceptor, PLA2G15 performs PLA2 hydrolytic chemistry on PC or PE.270
PLA2G15(−/−) mice have shown how loss of PLA2G15 may contribute to phospholipidosis, a condition characterized by the intralysosomal accumulation of phospholipids and concentric lamellar bodies within cells.272 Alveolar macrophages from PLA2G15(−/−) mice show complete loss of acidic PLA2 hydrolysis activity and N-acetylsphingosine acyltransferase activity.273 Lipidomic analysis of alveolar macrophage lipids from these animals showed an approximately two-fold elevation in PC, LPC, and PE species, while phosphatidylserine, phosphatidylinositol, and sphingomyelin were unchanged, consistent with the established in vitro substrate specificity of PLA2G15. One year, but not four month old PLA2G15(−/−) mice have splenomegaly, elevations in spleen PE and PC content, and an accumulation of foam cells in multiple tissues. Alveolar macrophages from PLA2G15(−/−) mice are enlarged and contain lamellar inclusion bodies. These data demonstrate that PLA2G15 is involved in the lysosomal catabolism of phospholipids, and that deficiency of PLA2G15 causes phospholipid accumulation and promotes phospholipidosis.
2.4. Other phospholipases
2.4.1. LIPG (Endothelial lipase)
LIPG, also abbreviated EDL or EL, is a ~60 kDa protein with ~30–40% homology to other members of the extracellular TG lipases, including LPL (lipoprotein lipase), LIPC (hepatic lipase), and PNLIP (pancreatic lipase).274,275 LIPG exhibits modest TG hydrolase activity in vitro, but considerably more phospholipase activity relative to homologous extracellular lipases.276 LIPG has biochemical characteristics of both LIPC and LPL; like LIPC, neither the TG nor phospholipase activity of LIPG is activated by the requisite LPL cofactor apoC-II, but like LPL, LIPG activity is inhibited by 1 M NaCl. LIPG exhibits a preference for substrates on HDL particles.276 In contrast to LPL or LIPC, which are synthesized by parenchymal cells and then translocated to the endothelium, LIPG is so named because it is directly synthesized by endothelial cells. LIPG is expressed in liver, lung, kidney, and placenta but not in skeletal muscle.274
LIPG(−/−) and LIPG-overexpressing mice have been instrumental in elucidating the role of LIPG in HDL metabolism.274,277,278 Heparin-releasable plasma phospholipase activity is reduced by > 50% in LIPG(−/−) mice. These animals have unchanged plasma TGs, but show two-fold elevations in plasma cholesterol, HDL cholesterol, and plasma phospholipids. ApoAI and apoE are also elevated in LIPG(−/−) mice. While one study found that the number, not the composition, of HDL particles is increased in LIPG(−/−) versus control mice,277 another study found that the size of HDL particles is increased.278 The clearance rate of radiolabeled cholesterol ester from HDL particles is reduced in LIPG(−/−) mice. Conversely, transgenic or adenoviral overexpression of LIPG dramatically reduces plasma HDL cholesterol. Taken together, these studies suggest that LIPG makes a physiological contribution to the regulation of HDL cholesterol levels in rodents, but future studies will be required to understand the underlying mechanism by which this occurs. Whether the pharmacological blockade of LIPG in humans is a viable strategy for elevating HDL cholesterol and protecting against atherosclerosis remains an important unanswered question.
Lead covalent sulfonylfuran urea-based inhibitors of LIPG have been described (Fig. 8), but these compounds cross-react with LPL and their efficacy for inhibiting LIPG in vivo remains unknown.148
2.4.2. PLA1A (Phosphatidylserine-specific PLA1)
PLA1A, also called NMD279 or phosphatidylserine-specific phospholipase A1 (PS-PLA1),280 is a 55 kDa secreted protein with ~30% homology to the TG lipases LPL, LIPC, and PNLIP.280 PLA1A exhibits phospholipase A1 activity specifically for glycerolipids containing a phosphoserine head group such as phosphatidylserine (PS) or 1-acyl-2-lysophosphatidylserine (lysoPS), but cannot hydrolyze PE, PC, PA, PI, or TG.280,281 A splice variant of PLA1A that lacks ~70 AA from the C-terminus is expressed in multiple human tissues and maintains equivalent activity as full length PLA1A for lysoPS, but has dramatically reduced PS hydrolysis activity.282 In humans and mice, PLA1A is highly expressed in kidney, heart, and liver, with significantly lower expression in spleen, lung, small intestines, skeletal muscle, and brain.283
PS and lysoPS have been implicated in multiple physiological processes. For example, PS, which usually resides on the inner leaflet of plasma membranes, becomes cell-surface exposed upon initiation of apoptosis or following stimulation by cytokines and serves as a signal for macrophage engulfment.284,285 LysoPS has been shown to activate mast cells and inhibit mitogen-induced T cell activation.286,287 PLA1A might therefore be a direct regulator of PS or lysoPS signaling in certain inflammatory responses. Some evidence for this function has been obtained in the THP-1 human macrophage cell line, where PLA1A expression is stimulated by the toll-like receptor (TLR) ligand LPS and inhibited by corticosteroids,288 and in rat peritoneal mast cells, where addition of recombinant PLA1A elevates lysoPS levels and stimulates histamine release,289 the biochemical and physiological functions of this enzyme in vivo remain unknown.
Neither knockouts nor selective inhibitors have been, to our knowledge, described for PLA1A.
2.4.3. LIPH and LIPI (Phosphatidic acid-specific PLA1α and β)
LIPH is also called PLA1B, phosphatidic acid-specific PLA1α (PA-PLA1α), lpd lipase related lipase (LPDLR), or lacrimal lipase, and LIPI is also called phosphatidic acid-specific PLA1β (PA-PLA1β) or lpd lipase (LPDL).290–292 LIPH and LIPI share > 40% sequence identity and are ~55 kDa plasma membrane-localized enzymes that are also homologous to PLA1A.293 LIPH is ubiquitously expressed with highest expression in skeletal muscle and heart and, in humans, whereas LIPI is expressed exclusively in testis.294 LIPH and LIPI both show phospholipase A1 activity in vitro with preference for the substrate PA to generate LPA, but cannot hydrolyze PC, PS, or PE, or TG.293
LPA is an important signaling lipid that activates cognate G-protein coupled receptors to modulate various (patho)physiological processes.295 When LIPH or LIPI are overexpressed in Sf9 cells, LPA is elevated and PA is reduced, but these changes also require the presence of a phospholipase D, a phosphodiesterase that generates PA from phospholipids.293 The extent to which LIPH or LIPI contributes to the biosynthesis of LPA in vivo, however, remains unknown. Some insights into the physiological function of LIPI have been made using a mouse transgenic line harboring a recessive mutation called lpd (lipid defect).296 The lpd locus is on the distal part of chromosome 16 and shows duplication of host genomic sequences at the site of integration. Homozygous lpd mice have stunted growth and die at P10–15, presenting with highly vacuolated livers and elevated TGs in liver and plasma. One of the genes in the lpd locus is LIPI, hence the origin of its alternative name LPDL, and LIPH’s alternative name LPDLR.297 However, the lpd locus could contain other genes that contribute to the lpd phenotype.
Neither knockouts nor selective inhibitors have been, to our knowledge, described for LIPH or LIPI.
2.4.4. PLB1 (Phospholipase B)
PLB1, also called phospholipase B or phospholipase B/lipase (PLB/LIP), is a ~180 kDa membrane-associated protein with an unusual domain structure, consisting of an N-terminal signal peptide, four tandem repeats of ~350 AA each, followed by a C-terminal hydrophobic domain, with the second of the four repeats harboring the catalytic serine.298 In rat tissues, PLB1 is selectively expressed in the upper and lower ileum of the intestine, but has not been detected in any other tissues.298 Purified PLB1 displays calcium-independent phospholipase activity on a variety of phospholipids (PC, PE, PG), lysophospholipids, and neutral lipids including TGs, DGs, and MGs.299 The endogenous biochemical and physiological functions of PLB1 remain unknown.
Neither knockouts nor selective inhibitors have been, to our knowledge, described for PLB1.
2.4.5. DDHD1 and DDHD2 (DDHD domain containing 1 and 2)
DDHD1, also called phosphatidic acid-selective phospholipase A1 (PA-PLA1), is a ~100 kDa cytosolic hydrolase expressed in multiple human tissues, with highest levels in testis, and lower levels in brain, lung, spleen, and thymus.300 Despite its name, DDHD1 only shows modest preference for hydrolyzing PA in vitro, and exhibits significant activity on other phospholipids including PI, PE, and PC.301,302 Upon cellular stimulation by ionomycin, DDHD1 undergoes translocation from the cytosol to the membrane,302 and DDHD1 expression is expressed in mature, but not newborn calf testis,300 but the relevance of these two observations to the physiological functions of DDHD1 remain unknown. DDHD2, also called KIAA0725p, is an ~80 kDa Golgi-localized hydrolase highly expressed in multiple human tissues including brain, lung, spleen, and testis, but not heart, liver, or pancreas.303 Like DDHD1, DDHD2 displays PLA1 activity with a variety of phospholipid substrates in vitro, including PA, PS, PC, and PE, and its overexpression in Vero cells causes disorganization of the ER and Golgi. Whether alterations in phospholipid content are responsible for the morphological changes induced by DDHD2 overexpression remains unknown.
Neither knockouts nor selective inhibitors have been, to our knowledge, described for DDHD1 or DDHD2.
2.4.6. ABHD4 (Alpha/beta hydrolase domain containing 4)
ABHD4 is a 40 kDa cytosolic hydrolase highly expressed in the spinal cord and testes of mice, with lower expression in the brain, kidney, and liver, and no expression in heart.304 ABHD4 can sequentially deacylate both O-acyl chains from an unusual class of phospholipids, the N-acylphosphatidylethanolamines (NAPEs).304 Thus ABHD4 displays both NAPE phospholipase and lysoNAPE lysophospholipase activities, and was not found to have activity with other lysophospholipids including lysoPE, lysoPC, and lysoPS (Fig. 13). The NAPEs, by the action of a phospholipase D called NAPE-PLD, are hypothesized to be the biosynthetic precursors to N-acylethanolamines (NAEs) including the endocannabinoid anandamide (N-arachidonoylethanolamine). However, NAPE-PLD(−/−) mice only show reductions in a subset of brain NAEs and do not show any differences in brain anandamide,305 leading to the hypothesis that ABHD4 may participate in an alternative NAPE catabolic process for the biosynthesis of anandamide.306
Fig. 13.

ABHD4 sequentially hydrolyzes O-acyl chains from NAPEs to generate lysoNAPEs and glycerophospho-NAEs (GP-NAEs).
Neither knockouts nor selective inhibitors have been, to our knowledge, described for ABHD4.
2.5. Small-molecule amidases
2.5.1. FAAH and FAAH2 (Fatty acid amide hydrolase and FAAH2)
Fatty acid amide hydrolase (FAAH) is a ~60 kDa integral membrane enzyme highly expressed in the brain, liver, kidney, and testis, but not in the heart or skeletal muscle.9 Unlike most other mammalian SHs, which use a Ser-His-Asp triad for catalysis, FAAH possesses an unusual Ser-Ser-Lys triad characteristic of enzymes from the amidase signature (AS) class and adopts a fold consisting of a twisted β-sheet surrounded by α-helicies.307 FAAH cleaves and inactivates a broad range of lipid amides in vitro and can tolerate variations in the amide head group and lipid acyl chain. Bioactive lipid amide substrates for FAAH include the endocannabinoid anandamide (N-arachidonoylethanolamine)308 and related N-acylethanolamine (NAE) congeners, the sleep-inducing factor oleamide (cis-9,10-octadecanoamide),309 and the transient receptor potential (TRP) ion channel agonists the N-acyl taurines (NATs) (Fig. 14).310,311
Fig. 14.

(A-C) Structure of the FAAH substrates anandamide (A), N-arachidonoyltaurine (B), and oleamide (C).
The endogenous biochemical and physiological functions of FAAH have been established by FAAH inhibitors312,313 and FAAH(−/−) mice.16 Blockade of FAAH ablates anandamide hydrolysis activity and causes >10-fold elevations in brain anandamide (similar elevations are observed for other NAEs and NATs in multiple tissues).310 An untargeted lipidomic profiling platform did not uncover other lipid changes, suggesting a specific function for FAAH in regulating lipid amides.314 In rodents, the heightened anandamide tone generated by FAAH disruption results in cannabinoid receptor-dependent anxiolytic,312 anti-depressive,315 anti-nociceptive phenotypes.16 FAAH disruption also causes cannabinoid-receptor independent effects such as anti-inflammation in the carrageenan assay.316 Taken together, these data demonstrate that FAAH regulates the endogenous levels and signaling activity of multiple classes of lipid amides and further suggest that FAAH inhibitors could offer a new way to treat pain and neuropsychiatric disorders. Toward this end, several classes of FAAH inhibitors that display in vivo activity have been generated, including OL-135, a prototypical α-ketoheterocyclic reversible inhibitor,317 covalent carbamate inhibitors including URB597,312 and highly selective covalent urea inhibitors such as PF-3845 or PF-04457845 (Fig. 15).313,318,319
Fig. 15.
(A-D) Structure of the FAAH inhibitors OL-135 (A), URB597 (B), PF-3845 (C), and PF-04457845 (D).
A second fatty acid amide hydrolase (FAAH2) with ~20% sequence identity to FAAH has also been identified.320 FAAH2 is a ~60 kDa membrane-associated enzyme with an AS sequence containing the Ser-Ser-Lys catalytic triad, and is expressed in primates and humans, but not in lower placental mammals including mouse and rat. FAAH2 hydrolyzes primary fatty acid amides (i.e., oleamide) at a comparable rate to FAAH, but shows greatly reduced activity with other lipid amide substrates such as anandamide and C18:1 NAT. Mechanism-based inhibitors of FAAH, such as URB597 or OL-135, also inhibit FAAH2 at roughly similar potency, while urea inhibitors, such as PF-3845, are not active against FAAH2. Owing to the restricted expression of FAAH2 in higher mammals, very little is known about its endogenous substrates or physiological functions, but its existence suggests that the metabolism of lipid amides may be more complex in higher mammals than what would be predicted from rodent models.
2.5.2. AFMID (Arylformamidase)
Arylformamidase (AFMID), also called kynurenine formamidase (KF), is a ~33 kDa enzyme with high expression in liver and kidney.321,322 Recombinant AFMID hydrolyzes N-formyl-L-kynurenine, the immediate downstream metabolite of tryptophan catabolism, to generate kynurenine, a precursor for the bioactive metabolites kynurenic and quinolinic acid (Fig. 16). 321,323 AFMID uses a Ser-His-Asp catalytic triad and is predicted to have an α/β hydrolase fold, structural features that make AFMID more similar to other serine esterases than to amidase signature enzymes like FAAH.324
Fig. 16.

AFMID hydrolyzes N-formyl-L-kynurenine to generate kynurenine.
The functions of AFMID in vivo remain unclear. The Afmid and thymidine kinase (Tk) genes share a bidirectional promoter325 and therefore TK(−/−) mice are also deficient in AFMID.326 Livers and kidneys from AFMID(−/−)/TK(−/−) double knockout mice show little residual formylkynurenine amidase activity. In the plasma, both upstream (i.e., formylkynurenine) and downstream (i.e., kynurenine and kynurenic acid) metabolites of AFMID are elevated several fold compared to wild-type littermates, and plasma tryptophan remains unchanged, suggesting that other metabolic pathways or enzymes besides AFMID may participate in the catabolism of tryptophan. AFMID(−/−)/TK(−/−) mice show various phenotypic abnormalities, including shortened life span, development of sclerosis of kidney glomeruli, and reduced cloning efficiencies of spleen lymphocytes. The generation of AFMID(−/−) by gene targeting of the AFMID catalytic exon, and studies on the in vitro substrate selectivity of AFMID, should enhance our understanding of this enzyme’s biochemical and physiological functions.
Selective inhibitors of AFMID have not, to our knowledge, been described.
2.6. Acyl-CoA hydrolases
2.6.1. FASN (Fatty acid synthase)
In eukaryotic systems, FASN (type I) is a large homodimer of two 270 kDa polypeptides each containing seven distinct domains (listed from N- to C-terminus): β-ketoacyl synthase (KS), malonyl-CoA-/acetyl-CoA-ACP-transacylase (MAT), dehydratase (DH), β-enoyl reductase (ER), β-ketoacyl reductase (KR), acyl carrier protein (ACP), and thioesterase (TE).327–330 The ACP domain has a requisite prosthetic 4′-phosphopantetheine attached to Ser2156 (human amino acid numbering) that supplies the sulfydryl residue required for ACP-mediated catalysis. In comparison, prokaryotes use type II FASN consisting of structurally independent proteins, the composition of which is species-dependent. FASN is responsible for the de novo biogenesis of palmitic acid by sequentially coupling seven equivalents of malonyl-CoA to acetyl-CoA by decarboxylative Claisen condensations and NADPH-mediated reductions: the overall relevant reaction is acetyl-CoA + 7 malonyl-CoA + 14 NADPH → palmitic acid. When written with all reactants and products, the reaction becomes acetyl-CoA + 7 malonyl-CoA + 14 NADPH + 14 H+ → palmitic acid + 8 CoA + 14 NADP+ + 6 H2O + 7 CO2 (Fig. 17).
Fig. 17.

The FASN catalytic cycle. The seven domains are abbreviated β-ketoacyl synthase (KS), malonyl-CoA-/acetyl-CoA-ACP-transacylase (MAT), dehydratase (DH), β-enoyl reductase (ER), β-ketoacyl reductase (KR), acyl carrier protein (ACP), and thioesterase (TE). The residues are abbreviated –OH (serine), –SH (cysteine), and
 SH (4′-phosphopantetheine). MAT* and TE* indicate that these two are SH domains. Following attachment of the initial acetate, the cycle is repeated seven times to generate one equivalent of palmitic acid.
SH (4′-phosphopantetheine). MAT* and TE* indicate that these two are SH domains. Following attachment of the initial acetate, the cycle is repeated seven times to generate one equivalent of palmitic acid.
During the catalytic cycle, substrates are covalently attached to FASN at one of three sites: Ser581 of the MAT domain, the sulfhydryl of the 4′-phosphopantetheine group on ACP, or Cys161 of the KS domain (human amino acid numbering). Briefly, the catalytic cycle begins by the loading of an acetyl unit, a process that involves sequential transfers of the acetyl from acetyl-CoA to Ser581 on MAT, then to the ACP-phosphopantetheine sulfhydryl, and finally to Cys161 on the KS domain. Malonate from malonyl-CoA is then similarly loaded onto ACP-phosphopantetheine via Ser581 (MAT). Energetically favorable malonyl decarboxylation drives the condensation of the malonyl-derived C2:0 enolate and the KS-acetyl, generating a β-ketothioester on ACP-phosphopantetheine. The β-ketothioester reduced to the saturated C4:0 thioester using two equivalents of NADPH by the sequential action of KR, DH and ER. This chain is transferred from ACP-phosphopantetheine onto the KS domain at Cys161, and the cycle is repeated with additional equivalents of malonyl-CoA and NADPH. In the final step, the formation of C16:0 thioester on ACP-phosphopantetheine causes the TE domain to liberate palmitate and the catalytic cycle begins again.
Of the seven domains, only the MAT331–333 and TE334,335 domains use nucleophilic serines embedded in GXSXG motifs for catalysis. The MAT domain adopts an unusual variant of the α/β hydrolase fold using a Ser-His dyad.332,333 The TE domain has maximal activity with C16:0 phosphopantetheine thioesters, with activity dropping precipitously for shorter or longer acyl chain lengths.336 Interestingly, FASN variants lacking the TE domain generate predominantly C20:0 or C22:0 fatty acids, demonstrating that this domain is responsible for the palmitic acid product specificity of FASN.337 The TE domain consists of two subdomains, the larger of which adopts an overall α/β hydrolase fold, contains a standard Ser-His-Asp triad, and also has a hydrophobic groove with a geometry consistent with specificity towards C16:0 fatty acid products.335
The importance of FASN in cell and organismal physiology has been elucidated by FASN inhibitors and FASN(−/−) mice. A hallmark of human cancers is an increase in de novo fatty acid synthesis compared to normal tissues19 to create a “lipogenic phenotype,” which is correlated with upregulated FASN338 in cancers from multiple tissues of origin, including carcinomas of the prostate, breast, ovary, and colon.339 Concurrently, multiple independent studies have demonstrated that FASN expression in carcinomas is correlated with disease reoccurrence and death in humans.340–342 Though the causes and consequences of FASN expression in cancer cells are still under active investigation, pharmacological blockade of FASN has been shown to cause cancer cell death in vitro and retards the growth of tumors in xenograft mouse studies, underscoring the utility of FASN inhibitors for the treatment of certain types of carcinomas.343 C75 and cerulenin are two such inhibitors that bind the KS domain of FASN but their utility is limited by their inhibition of other targets.344,345 The TE domain of FASN is sensitive to inhibition by the general lipase inhibitor THL, possibly accounting for the anti-cancer activity displayed by this inhibitor in cell and animal models.334
The function of FASN in normal tissues has been elucidated by genetic models in mice. FASN(−/−) mice are embryonic lethal at a pre-implanation stage and fewer than expected FASN(+/–) are born, suggesting a partial haploinsufficiency.346 Liver-specific deletion of FASN (FASNKOL) show nearly ablated hepatic FASN activity and several fold elevations in hepatic malonyl-CoA, demonstrating that FASN is a principal mediator of malonyl-CoA catabolism in this organ.347 On a zero fat diet or following prolonged fasting, FASNKOL mice develop hypoglycemia and fatty liver, phenotypes that correlate with decreased expression of PPARα target genes. Administration of PPARα agonist reverses these phenotypes, consistent with the hypothesis that hepatic FASN generates fatty acid products that can act as endogenous ligands for PPARα to regulate glucose and fat metabolism. Deletion of FASN from the brain and pancreatic β-cells by Cre-mediated recombination under the control of rat insulin II promoter (RIP-Cre) abolishes FAS activity in the pancreas and elevates malonyl-CoA by approximately two-fold in isolated islet cells and the hypothalamus.348 While the pancreatic function in these mice is normal, there is an overall decrease in food intake, fat weight, and total weight, and pair feeding studies show that reduced adiposity is due to the reduced caloric intake. Similarly, either systemic or intracerebroventricular administration of the FASN inhibitor C75 reduced food intake and causes weight loss by a leptin-independent, neuropeptide Y-dependent process.349 The effects of C75 are attenuated by co-treatment with TOFA, an inhibitor of acetyl-CoA carboxylase, the enzyme that creates malonyl-CoA units for FASN consumption, suggesting that changes in upstream malonyl-CoA, not downstream fatty acids, mediates the weight loss caused by FASN blockade. Taken together, these data provide evidence for a central role of FASN in the catabolism of malonyl-CoA and the de novo biogenesis of fatty acids in regulating a variety of cell and organismal phenotypes under normal and pathological conditions.
2.6.2. ACOT1–6 (Acyl-CoA thioesterases 1–6)
The acyl-CoA thioesterases (ACOTs) are enzymes of varying tissue and subcellular distribution that hydrolyze acyl-CoA molecules to generate fatty acids and free CoA.350,351 In mice, there are thirteen distinct ACOT proteins classified as either type I (mACOT1–6) or type II (mACOT7–13), but only the type I mACOTs use nucleophilic serines for catalysis and are members of the serine hydrolase superfamily. The type I ACOTs, which in pair-wise comparisons share > 65% identity with each other, are all inducible in a PPARα-dependent manner by peroxisome proliferation reagents (i.e., clofibrate) and other PPARα ligands (i.e, WY-14,643), suggesting that the ACOTs are transcriptional targets for the PPARα nuclear receptor.352,353 Humans only express three type I ACOTs, ACOT1, 2, and 4. Whereas human ACOT1 and ACOT2 are homologous to mouse ACOT1 and ACOT2, respectively, human ACOT4, while being most homologous to mouse ACOT4, appears to have acquired the functions of mouse ACOT3–5 by convergent evolution.
ACOT1, also called acyl-CoA hydrolase 2 (ACH2), long-chain acyl-CoA hydrolase 2 (LACH2), or cytosolic thioesterase 1 (CTE-I) is a 43 kDa cytosolic enzyme highly expressed in rat heart, liver, and kidney, with lower expression in rat testis.354 ACOT2, also called mitochondrial thioesterase 1 (MTE-I) or arachidonic acid-related thioesterase involved in steroidogenesis (ARTISt) is a 45 kDa mitochondrial phosphoprotein with >90% identity to ACOT1 that is expressed in the kidney, muscle, heart, and brown adipose tissue.353,355 In vitro, both ACOT1 and ACOT2 have overlapping substrate scope and can hydrolyze a variety of saturated and unsaturated long chain fatty acyl-CoAs from C12–C20.355,356 The expression of hepatic ACOT1, but not ACOT2, is highly induced by fasting.352
Whereas ACOT1 and ACOT2 are localized to the cytosol and mitochondria, respectively, ACOT3–6 are localized to the peroxisomes. ACOT3 is also called peroxisome thioesterase 1a (PTE-Ia) and is highly expressed in the kidney, with lower expression in most other tissues.357 ACOT3 hydrolyzes C12–C20 fatty acyl-CoAs.357 ACOT4, also called peroxisome thioesterase 1b (PTE-Ib), is highly expressed in the kidney and upregulated in the same tissue by fasting.358 Mouse ACOT4 displays unusual substrate specificity and hydrolyzes succinyl- and glutaryl-CoA, but not any other long chain fatty-acyl CoAs, and, conversely, the other type I ACOTs in mice cannot hydrolyze succinyl- or glutaryl-CoA. ACOT5, which is also called peroxisome thioesterase 1c (PTE-Ic), is highly expressed in spleen, brain, testis, and intestines. ACOT5 has an overlapping substrate selectivity with ACOT3 and can hydrolyze C10–C16 acyl-CoA substrates.357 In humans, hACOT4 displays not only succinyl-/glutaryl-CoA hydrolysis activity, but also activity towards other long chain fatty acyl-CoA substrates, thus accounting for its ability to functionally replace mouse ACOT3–5.359
ACOT6 in mice is 48 kDa peroxisomal protein highly expressed in white adipose tissue with lower levels in liver, kidney, brown adipose tissue, and brain.360 Mouse ACOT6 can hydrolyze branched fatty acyl-CoAs including pristanoyl- and phytanoyl-CoA, two metabolites derived from dietary fatty acids. In contrast, the Acot6 gene in humans, while containing all three exons similar to the other family members, is predicted based on expressed sequence tag sequences to encode a truncated protein of ~20 kDa that contains the catalytic domain, but it remains unknown whether such a large truncation affects the catalytic activity. The tissue distribution and hydrolysis activity of human ACOT6 have not yet been described.359
Taken together, these studies elucidate the biochemical properties of the type I ACOTs. Whether disruption of ACOTs will cause accumulations of acyl-CoA substrates, and whether such acyl-CoA elevations will disrupt cell or physiological processes, remain open questions. Neither knockouts nor selective inhibitors have been, to our knowledge, described for any of the ACOT members.
2.7. Cholinesterases
In mammals, two related serine hydrolases catalyze the hydrolysis of choline ester conjugates. Acetylcholinesterase (ACHE) has strict specificity for acetylcholine (ACh) and has essentially no activity on choline esters with longer chains. Butyrylcholinesterase (BCHE), also called pseudocholinesterase, nonspecific cholinesterase, or serum cholinesterase, can hydrolyze ACh in addition to other choline and non-choline conjugates (Fig. 18).361 ACHE and BCHE have overlapping yet distinct roles in mammalian physiology and behavior, which have only been recently elucidated through methods that selectively disrupt either enzyme.
Fig. 18.

(A-C) Structure of the ACHE substrate acetylcholine (A) and of the BCHE substrates butyrylcholine (B) and succinylcholine (C).
ACHE is a ~75 kDa glycoprotein that deactivates ACh signaling at nicotinic and muscarinic acetylcholine receptors (AChRs), which are located at synapses in the central nervous system and at neuromuscular junctions throughout the body. Unusually, ACHE uses a Ser-His-Glu triad for catalysis, and engages ACh in the active site by cation-π interactions instead of electrostatics.362,363 The human ACHE enzyme is produced from a single gene in three distinct alternatively spliced isoforms that differ in their carboxy terminal regions.364,365 All three ACHE isoforms have similar enzymatic properties, but show distinct subcellular and tissue distributions. The tailed form, ACHE-T, also known as the synaptic form of ACHE, has carboxy terminal cysteines that allow for dimerization. ACHE-T dimers can further associate into tetramers or even higher order 8- or 12-mers through association with collagen Q (ColQ) or proline-rich membrane anchor (PRiMA) proteins, resulting in the tethering of extracellular ACHE-T oligomers to the plasma membrane. ACHE-T is the principal form found in the synaptic cleft and neuromuscular junctions.364 The hydrophobic form, ACHE-H, also called the erythrocytic form, is tethered to the plasma membrane by a glycophosphatidylinositol (GPI) anchor and is mostly found in hematopoietic cells. Lastly, the readthrough form, ACHE-R, results from the lack of splicing at the carboxy terminus and therefore remains soluble and monomeric. ACHE-R is expressed in embryonic tissues in mouse, but not human erythrocytes.
BCHE in humans is a ~85 kDa glycoprotein with ~50% identity to ACHE.361 BCHE hydrolyzes a variety of choline conjugates, including acetylcholine, butyrylcholine, and succinylcholine, non-choline conjugates, including cocaine, heroin, and aspirin, and arylamides such as the synthetic substrate o-nitroacetanilide.361,366–369 Unlike the three isoforms of ACHE, BCHE is only generated as the tailed form (BCHE-T) that can associate into dimers or tetramers that act either as catalytic soluble oligomers or are attached to the plasma membrane by ColQ or PRiMA proteins.365 BCHE activity in mice is highest in liver, intestines, and serum, and is lower in heart, lung, muscle, and brain.370
The complementary roles of ACHE and BCHE have been dissected through selective pharmacological inhibition and gene targeting experiments. ACHE(−/−) mice are viable, but do not grow at the same rate as wild-type littermates and die by P21 due to an inability to obtain sufficient nutrition.371 In the intestines, serum, heart, lung, muscle, and brain from ACHE(−/−) mice, ACHE activity is completely ablated with no compensatory changes in BCHE activity. ACHE(−/−) mice have dramatically elevated interstitial hippocampal acetylcholine levels and decreased choline levels, consistent with the function of ACHE in converting acetylcholine into acetate and choline.372,373 Brains from ACHE(−/−) mice also have dramatically reduced levels of muscarinic G-protein coupled ACh receptors (AChRs), but not the nicotinic ion channel AChRs or dopamine receptors, demonstrating that specific components of cholinergic neurotransmission show compensatory changes in response to constitutively heightened ACh signaling.374 Force feeding of a liquid diet to ACHE(−/−) mice allows them to mature into adulthood, where they demonstrate multiple abnormal behaviors characteristic of heightened cholinergic overstimulation, including body tremors, muscle weakness, abnormal gait and posture, decreased pain response, urination and defecation in the nest, sexual dysfunction, and early death at ~100 days from seizures or bowel obstruction.375 The muscle phenotypes in ACHE(−/−) mice qualitatively resemble the consequences of acute pharmacological inhibition of ACHE with fasciculin, a non-competitive ~7 kDa polypeptide inhibitor with > 106-fold selectivity for ACHE over BCHE,376,377 which produces prolonged generalized muscle fasciculations with complete recovery.378 Exogenous ACh causes mortality within one minute in ACHE(−/−) mice but has only mild effects in wild-type littermates.379 Similarly, the BCHE inhibitor bambuterol, which has some selectivity for BCHE over ACHE,380 at 0.3 mg/kg causes mortality in ACHE(−/−) mice but has essentially no effect in wild-type littermates, demonstrating that, in the absence of ACHE, essential esterase activities are performed by BCHE.371
Historically, it had been generally assumed that ACHE is the principal regulator of ACh neurotransmission in vivo and interest in BCHE originated not from cholinergic signaling but rather from the study of succinylcholine, a muscle relaxant, in patients with reduced BCHE activity.381 Patients with low BCHE activity could not sufficiently hydrolyze succinylcholine and would experience life-threatening apnea due to paralysis.382 Recently, the role of BCHE in cholinergic signaling has been reexamined following the characterization of viable ACHE(−/−) mice and the discovery that BCHE is expressed in glia and neurons of limbic system.383,384 Administration of PEC, a carbamate-based small molecule inhibitor with >3,000-fold selectivity for BCHE over ACHE, inhibits brain BCHE by 50%, does not inhibit ACHE, and elevates brain interstitial ACh levels.342 Moreover bambuterol administration to ACHE(−/−) mice can further augment the already elevated interstitial brain ACh levels, demonstrating that BCHE participates in ACHE-independent regulation of ACh.372 BCHE inhibition in rodents by PEC does not cause overt cholinergic toxicity, consistent with the normal phenotypic profile of humans with reduced BCHE activity. Similarly, BCHE(−/−) mice are viable, fertile, and overtly normal, and have dramatically decreased BCHE activity in plasma and tissues with no changes in plasma ACHE.379,385 BCHE(−/−) mice die from respiratory failure following exogenous treatment with succinylcholine, whereas the same dose in wild-type littermates produces essentially no effect, demonstrating that the effects of succinylcholine in humans with reduced BCHE can be replicated in BCHE(−/−) mice.379 Fasciculin produces cholinergic toxicity in BCHE(−/−) mice within 30 min, whereas wild-type mice also show some cholinergic toxicity of later onset at ~18 h, and ACHE(−/−) mice show no additional phenotypes upon treatment with this inhibitor.379 These pharmacological and genetic studies of ACHE and BCHE demonstrate that neither is essential for rodent viability and that both participate in the regulation of cholinergic neurotransmission.
Organophosphate (OP) reagents, which are used as both insecticides and chemical warfare agents, have further elucidated the role of cholinesterases in regulating the duration and magnitude of cholinergic neurotransmission in the brain and at neuromuscular junctions. When administered at high doses to rodents, OP insecticides (for example, paraoxon or chlorpyrifos) and nerve agents (for example, sarin or VX) elevate brain acetylcholine levels and cause death due to paralysis and respiratory failure (Fig. 19).386 In humans, acute high-level exposure causes symptoms characteristic of overactive parasympathetic and/or sympathetic cholinergic signaling, including salivation, lacrimation, urination, defecation, gastrointestinal distress, emesis, miosis, nausea, dizziness, muscle twitching, weakness, and tremor, and in some cases mortality by respiratory paralysis.387 In the literature, it is generally stated that the mechanism of OP toxicity is due to inhibition of ACHE,388 but most OP inhibitors including sarin and chlorpyrifos are not selective for ACHE and also inhibit BCHE.377,389,390 These data, taken together with the data from ACHE(−/−) and BCHE(−/−) mice, as well as the selective inhibitors fasciculin and PEC, suggest that the toxicity of OP reagents in humans and rodents derives from inhibition of both ACHE and BCHE. Regardless of the precise mechanism, cholinergic overstimulation is clearly central to the toxicity of OP reagents since treatment for OP poisoning in humans typically involves administration of: 1) oxime compounds (e.g., pralidoxime) that can turn over OP-inactivated ACHE/BCHE to generate free cholinesterases and 2) muscarinic ACh receptor antagonists (e.g., atropine).391
Fig. 19.

(A-D) Structures of the organophosphorous insecticides paraoxon (A) and chlorpyrifos (B), and the nerve agents sarin (C) and VX (D).
While overstimulation of the cholinergic system from pharmacological inhibition or genetic perturbation produces multiple undesirable effects, several studies have correlated decreases in cholinergic neurotransmission pathways in patients with AD, a disease characterized by gradual cognitive decline, reduced function in daily activities, and behavioral disturbances.392 This finding has engendered the cholinergic hypothesis, which states that loss of cholinergic signaling contributes to the cognitive decline in AD.392 Consequently, inhibitors such as rivastigmine, which block both ACHE and BCHE, have emerged as a frontline therapy for the treatment of dementia in Alzheimer’s disease (AD), presumably producing their beneficial effects by rectifying the deficiency in ACh neurotransmission in AD patients (Fig. 20).393 While approximately one quarter of rivastigmine-treated patients experience adverse events consistent with cholinergic overstimulation (nausea, vomiting),394 overt cholinergic toxicity such as those observed with OP reagents is not observed in these subjects, which likely indicates that efficacious dosing regimes can be achieved by partial inhibition of ACHE/BCHE. Preliminary studies have demonstrated that administration of the BCHE-selective inhibitor PEC to transgenic mice overexpressing human Aβ reduces the amounts of Aβ associated with plaques395 and suggest that selective pharmacological inhibition of BCHE but not ACHE may be efficacious in humans while avoiding some of the adverse events associated with dual ACHE/BCHE inhibitory agents currently used in the clinic.
Fig. 20.
Structure of the dual ACHE/BCHE inhibitor rivastigmine.
Taken together, these studies demonstrate a critical role for ACHE, and possibly also BCHE, in modulating ACh levels and cholinergic transmission in both the central and peripheral nervous system. In addition to its enzymatic activities, ACHE has been proposed to function as a scaffolding protein on the plasma membrane, as discussed in other reviews.364,396
3. Peptidases
In contrast to other peptidases that use aspartate, cysteine, threonine, or metals for catalysis, the serine peptidases use a nucleophilic serine to hydrolyze a variety of peptide bonds and represent over one third of all known proteolytic enzymes.397,398 The serine peptidases are divided into 13 clans, each of which is further divided into groups called subclans or families. Only a subset of peptidases, those from clan SC families S9, S10, S15, S28, and S33, belong to the mammalian metabolic SH superfamily.397 These clan SC peptidases, like other members of the SH superfamily and distinct from serine proteases that belong to clans such as trypsin (PA) or subtilisin (SB), are characterized by a GXSXG motif surrounding the active site serine, a Ser-Asp-His catalytic triad, and adopt or are predicted to adopt an α/β hydrolase fold. Substrates for SH peptidases include polypeptides such as circulating hormones (e.g., glucagon-like peptide-1)23 or neuropeptides (e.g., melanocyte-stimulating hormone),399 as well as intact proteins (e.g., collagen),400 and therefore the action of these peptidases can regulate endocrine signaling or modulate extracellular scaffolding proteins. Clan SC subfamily S9A contains two members, PREP and PREPL. Subfamily S9B contains four members, DPP4, FAP, DPP8, and DPP9, and is also known as the DPP4 family. S9C and S10 each contain one member, APEH and CTSA, respectively. Lastly, S28 contains two members, PRCP and DPP7.
3.1. PREP (Prolyl endopeptidase)
PREP, also called prolyl oligopeptidase (POP) or proline endopeptidase (PE) and often abbreviated PEP, is a ~75 kDa cytosolic peptidase that is ubiquitously expressed with highest levels in brain, heart, muscle, and liver, and lower expression in kidney and ovaries.401,402 PREP cleaves C-terminal to proline residues (i.e., Pro-Xaa, where Xaa is any amino acid except proline) in peptides shorter than 30 AA and can accept nearly any proline-containing peptide (Fig. 21). Hormones and neuropeptides including oxytocin, neurotensin, thyrotropin-releasing hormone (TRH), arginine-vasopressin (AVP), angiotensin II, and substance P (SP) are all PREP substrates in vitro.403 The crystal structure of PREP reveals a two-domain structure consisting of a non-catalytic N-terminal seven-bladed β-propeller domain and catalytic C-terminal α/β hydrolase domain. The active site, located at the interface of the two domains, is a large cavity occluded by the β-propeller domain. The position of the β-propeller suggests that only unstructured polypeptides can gain access to the active side through the central tunnel of the β-propeller, consistent with the observation that PREP cannot cleave larger structured peptides or proteins.402
Fig. 21.

Cleavage positions of some representative PREP substrates, with the scissile bond between the amino acids shown in red. SP, substance P; AVP, arginine-vasopressin; α-MSH, α-melanocyte stimulating hormone.
PREP inhibitors and PREP-deficient mice have demonstrated a broad role for PREP in the regulation of multiple bioactive neuropeptides (Fig. 22). JTP-4819, a substrate mimetic based on a Pro-Pro dipeptide, is a slowly reversible PREP inhibitor with > 105-fold selectivity over other peptidases assayed including DPP4 (dipeptidyl peptidase 4).404 When administered to rats, JTP-4819 modestly decreases brain hydrolysis activity for the synthetic substrate 7-(succinyl-Gly-Pro)-4-methylcoumarinamide and elevates a variety of bioactive neuropeptides including SP, AVP, and TRH.404,405 Similarly, S 17092 is a perhydroindol-based PREP inhibitor with selectivity for PREP over other serine hydrolases406,407 and, following administration to mice, elevates SP and other peptides including α-melanocyte stimulating hormone (α-MSH).407,408 In mice with a gene-trap cassette inserted into the PREP locus, PREP protein is abolished and α-MSH is also elevated.399 Consistent with the observation that exogenous administration of neuropeptides such as SP improves learning and memory in rodents,409 inhibition of PREP in aged rodents410 or monkeys treated with the dopaminergic neurotoxin MPTP411 causes cognitive enhancements.
Fig. 22.

(A, B) Structures of the PREP inhibitors JTP-4819 (A) and S 17092 (B).
Despite these observations, a rigorous demonstration of the relationship between elevated neuropeptides regulated by PREP such as α-MSH or SP and the cognitive enhancements observed in rodents and monkeys is still lacking. Some studies have also suggested a role for PREP in inositol triphosphate (IP3) signaling, but the mechanisms of this regulation remain unclear.412,413 Whether PREP inhibitors can ultimately be used as cognition enhancers in human patients with disorders such as Alzheimer’s or Parkinson’s disease is an exciting prospect, but remains to be determined.414
3.2. PREPL (Prolyl endopeptidase-like)
PREPL is an ~80 kDa cytoplasmic enzyme with ~30% identity to PREP.415 PREPL is expressed as multiple splice isoforms with ubiquitous tissue distribution in humans, with the highest levels in brain, heart, kidney, and skeletal muscle, and lower levels in pancreas, liver, lung, and placenta.415 PREPL is labeled by the active-site probe diisopropyl fluorophosphonate (DIFP), suggesting that this enzyme has catalytic activity, but attempts to identify peptide or non-peptidic substrates have so far been unsuccessful.416
A recessive congenital disorder called hypotonia-cystinuria syndrome (HCS) in human patients has been mapped to microdeletions along chromosome 2p21 in the Slc3a1 and Prepl genes.417,418 Patients present with hypotonia (low muscle tone) at birth, cystinuria type I (formation of cystine stones in the bladder and kidneys), growth hormone (GH) deficiency, facial dysmorphism, and a failure to thrive. Since inactivation of the amino acid transporter SLC3A1 has been shown to cause cystinuria type I, the other deficiencies in these patients have been attributed to PREPL.417 Elucidation of the biochemical functions of this peptidase is an important future area of research for understanding its relationship to HCS.
Neither knockouts nor selective inhibitors have been, to our knowledge, described for PREPL.
3.3. DPP4 (Dipeptidyl peptidase 4)
DPP4, also called CD26, is a ~120 kDa glycoprotein and the archetypical member of the DPP4 peptidase family (clan SC, subfamily S9B).419,420 In rat, DPP4 is highly expressed in kidney and at lower levels in lung, adrenal gland, jejunum, liver, spleen, and testis, and negligible expression in skin, heart, pancreas, and brain.419 At the cellular level, DPP4 is a lumenally-oriented enzyme anchored to the plasma membrane of epithelial cells at sites of physiological barriers, such as the brush border and microvillus fractions of the kidney,421 around the bile duct epithelia and bile caniculi in the liver,422 or in circumventricular organs in the CNS,423 and can also be found as a soluble enzyme in body fluids such as plasma or cerebrospinal fluid.424 DPP4 is composed of two domains, an N-terminal eight-bladed β-propeller and C-terminal α/β-hydrolase domain.425–427 The active site is located between the two domains, similar to what is observed in the crystal structure of PREP. DPP4 can hydrolyze Xaa-Pro or Xaa-Ala (where Xaa is any amino acid) dipeptides from the N-terminus of oligopeptides ranging from 3 to 80 AA (Fig. 23).428
Fig. 23.

Structure of GLP-1, a representative substrate of DPP4, which is cleaved to liberate an N-terminal dipeptide (cleavage site shown in red). Other DPP4 substrates are also cleaved at the same penultimate position.
DPP4 has been the focus of considerable pharmaceutical interest because it regulates the endogenous levels of glucagon-like peptide-1 (GLP-1), a gastrointestinal hormone that is highly elevated in plasma following food intake.23 Circulating GLP-1 is found in two equipotent forms, GLP-1(7–37) or GLP-1(7–36) amide, and is synthesized in enteroendocrine L cells of the distal ileum and colon. GLP-1 acts at the G-protein coupled receptor GLP-1R to promote insulin secretion, insulin sensitivity, and β-cell proliferation,429–431 but its duration of action is short owing to rapid degradation and inactivation in a process mediated by DPP4. Plasma from DPP4(−/−) mice show dramatically reduced hydrolytic activity with the synthetic substrate Gly-Pro-p-nitroanilide or with GLP-1, and a concomitant two-fold increase in plasma insulin and GLP-1 levels, biochemical changes that result in enhanced oral glucose tolerance.432 DPP4 inhibitors,433 such as the reversible and fast binding inhibitor sitagliptin434 or the cyano-containing covalent reversible inhibitors vildagliptin435 or saxagliptin,436 all have good selectivity for DPP4 over closely related peptidases and, following oral dosing to human patients, increase circulating GLP-1 levels, reduce fasting glucose, and improve β-cell function (Fig. 24).437–440 These data, coupled with the minimal adverse events associated with DPP4 inhibitors in humans, has resulted in FDA approval of sitagliptin and saxagliptin for the treatment of type 2 diabetes mellitus.
Fig. 24.

The DPP4 inhibitors sitagliptin (A, also called Januvia), vildagliptin (B), and saxagliptin (C).
Besides the well characterized DPP4/GLP-1 pathway, some evidence points to non-catalytic roles for the cell surface form of DPP4 on T-cells, including the participation in intercellular binding events and immune responses.441 These studies, as well as the determination of other endogenous DPP4 substrates besides GLP-1,442,443 should continue to illuminate new aspects of DPP4-mediated physiology.
3.4. FAP (Fibroblast activation protein)
FAP, also called FAPα, seprase, or antiplasmin-cleaving enzyme (APCE),444 is a ~90 kDa plasma membrane-localized glycoprotein and the second of four members of the S9B subfamily.400,445 FAP has a very limited expression in normal tissues and during development where it is restricted to somites, myotubes, and perichondrial mesenchyme from cartilage primordial.446 FAP is, however, a core component of the “reactive stromal response” and is highly expressed in stromal cells neighboring wound healing events or epithelial tumors.447 A soluble form of FAP has also been found in bovine serum.448 The crystal structure reveals that, like DPP4, FAP adopts a two-domain fold with an N-terminal eight-bladed β-propeller and C-terminal α/β-hydrolase fold.449 FAP has dipeptidyl peptidase activity on synthetic substrates such as Ala-Pro-7-amino-4-trifluoro-methylcoumarin (AFC) and can also hydrolyze gelatin, α2-antiplasmin, and type I collagen,450,451 but whether or not FAP participates in the endogenous degradation of these protein substrates remains unknown. The mechanisms and consequences of FAP upregulation in stromal cells around wounds and tumors remain poorly understood, but, in rodent models, pharmacological blockade of FAP shows antitumor effects.452–454 As a result, α-FAP antibodies (i.e., sibrotuzumab,455 a humanized monoclonal antibody) and FAP inhibitors (i.e., talabostat,452 a nonselective DPP4-family boronic acid inhibitor) are under investigation as treatments for various cancers (Fig. 25). FAP(−/−) mice are fertile, histologically normal, and do not spontaneously develop more tumors than wild-type littermates, but have not been intensively investigated in other cancer models for tumor formation or growth.456
Fig. 25.
Structure of the non-selective DPP-family inhibitor talabostat.
3.5. DPP8 and DPP9 (Dipeptidyl peptidase 8 and 9)
DPP8 and DPP9 are the last two members of the S9B subfamily and share >50% identity with each other and ~25% identity with DPP4.457,458 In human tissues, DPP8 and DPP9 are both ubiquitously expressed, with highest levels of DPP8 in testis and DPP9 in skeletal muscle, liver, and heart. Unlike DPP4 and FAP, DPP8 and DPP9 lack transmembrane domains and, when recombinantly expressed using a baculovirus expression system, are found in the cytosol as ~100 kDa proteins.459 In vitro, DPP8 and DPP9 can cleave dipeptides from the N-terminus of bioactive peptides such as neuropeptide Y (NPY), glucagon-like peptide-1 (GLP-1), GLP-2, and peptide YY (PYY), but with rates ranging from ~1–50% compared to DPP4 depending on the substrate.459 Because gene targeted mice for either DPP8 or DPP9 have not been generated, and small molecule inhibitors such as valine pyrrolidide (Val-Pyr) inhibit DPP8, DPP9, and DPP4 with similar potencies,459 the in vivo functions and endogenous substrates of DPP8 and DPP9 remain largely unknown.
3.6. APEH (Acylpeptide hydrolase)
APEH (APH), also called acylamino acid-releasing enzyme (AARE), is an ~80 kDa enzyme that is ubiquitously expressed in rat tissues with highest expression in immune cells, lung and kidney and lowest expression in muscle and brain.181,460 The crystal structure of APEH from Aeropyrum pernix K1, which shows ~30% sequence identity to human APEH, reveals a two domain enzyme with an N-terminal seven-bladed β-propeller and a C-terminal α/β-hydrolase fold, similar to other serine peptidases.461 When APEH is overexpressed in transfected cells, it is primarily found in the cell lysate but a small amount is also detectable in the conditioned media.462
The N-terminal amino acid of nascent polypeptide chains is often co-translationally acetylated (“blocked”) in eukaryotes and formylated in prokaryotes on the α-amine. The function of this modification remains enigmatic but is hypothesized to affect protein stability, processing, or sorting.460 APEH removes the acetylated amino acid from peptide substrates to generate a peptide that is one amino acid shorter and an N-acetyl amino acid.463–465 APEH cleaves a variety of N-acetyl amino acids from peptide substrates including Ac-Ala-, Ac-Met-, Ac-Ser-, or Ac-Gly-, with the penultimate amino acid having some influence on the rate of cleavage (Fig. 26).466 Besides this activity, APEH has also been reported to possess non-N-terminal cleavage activity on other substrates such as oxidatively damaged proteins467 or the amyloid-β peptide.462
Fig. 26.

APEH cleaves N-acetylated peptides to generate an N-acetyl amino acid and an unblocked peptide.
Neither knockouts nor selective inhibitors have been, to our knowledge, described for APEH.
3.7. CTSA (Cathepsin A)
CTSA, also called lysosomal carboxypeptidase A, lysosomal protective protein, or protective protein/cathepsin A (PPCA), is a 54 kDa lysosomal glycoprotein ubiquitously expressed in all mouse tissues, but with highest levels in kidney, brain, and placenta.468–470 Besides its lysosomal localization, CTSA can also be secreted from platelets.471 Unlike other serine peptidases, CTSA does not have an N-terminal β-propeller domain but instead has a “cap” domain and an α/β-hydrolase domain.472 CTSA is synthesized as a 54 kDa precursor protein that, upon reaching the lysosome, is activated by proteolytic processing into two 32 and 20 kDa polypeptides joined by disulfide linkages.473
The C-terminus of many bioactive peptides is amidated, a modification that affects multiple aspects of peptide function including stability and activity on cognate receptors. CTSA displays both peptidase and deamidase activity with peptides such as angiotensin I, substance P, endothelin I, oxytocin, and bradykinin, and removes the ultimate or penultimate C-terminal amino acid and/or the amidation modification, depending on the substrate.471,474 CTSA also shows esterase activity on synthetic substrates such as benzoyl-tyrosine ethyl ester. Whereas the peptidase activity of CTSA has a pH optimum of 5, the deamidase and esterase activities have pH optima of 7.471
CTSA has been well characterized in human patients presenting with galactosialidosis (also called combined β-galactosidase and neuraminidase deficiency), a lysosomal storage disease that presents in infants or juveniles and causes edema, hepatosplenomegaly, abnormal bone development, an eye abnormality called a cherry-red spot, and distinctive facial features.475,476 In infants, galactosialidosis causes death, whereas the juvenile form results in normal life expectancy but progressive ataxia, seizures, and cognitive disabilities. Galactosialidosis is caused by a heterogeneous collection of mutations in CTSA that impair the association of CTSA with a ~600 kDa complex containing β-galactosidase and neuraminidase, resulting in the rapid degradation of β-galactosidase and a concomitant accumulation of sialylated oligosaccharides.477,478 β-galactosidase and neuraminidase activities can be restored in fibroblasts from galactosialidosis patients by the addition of culture medium from COS-1 cells transfected with either wildtype or a catalytically inactive (S150A) mutant of CTSA, demonstrating that the “protective” effects of this enzyme are independent of its catalytic activity.479
Mice deficient in CTSA have near complete loss of CTSA and neuraminidase activity and partial loss of β-galactosidase activity, are viable and fertile, but weigh 25–40% less than wildtype littermates and die at ~12 months of age.480 CTSA(−/−) mice recapitulate the phenotype of human patients and show hepatosplenomegaly, excessive sialyated oligosaccharides in urine, and vacuolated storage cells in multiple tissues including brain and kidney, which ultimately leads to organ failure and death. Bone marrow transplantation from wildtype littermates or transgenic mice overexpressing CTSA in erythroid precursors partially or completely reversed the disease phenotype, respectively, demonstrating the potential of somatic gene therapy for the treatment of this disorder in human patients.
To dissociate the lysosomal scaffolding functions of CTSA from its catalytic activity, CTSA(S190A) mice harboring a catalytically inactive CTSA knocked-in to the locus of the wild-type Ctsa gene were generated.481 CTSA(S190A) mice have near complete loss of CTSA activity in liver, kidney, and lungs, but no change in galactosidase or neuraminidase activity and normal CTSA complex formation. CTSA(S190A) mice do not show vacuolation or overt tissue abnormalities, consistent with the lysosomal disorders in galactosialidosis arising from secondary deficiencies in β-galactosidase and neuraminidase activities rather than loss of CTSA itself. Instead, elastin-rich tissues such as skin, arteries, and lungs from CTSA(S190A) mice show morphological changes, and skin fibroblasts show decreased deposition of insoluble elastin compared to wildtype littermates. In addition to the presentations of lysosomal dysfunction in patients with galacosialidosis, another clinical feature that could neither be explained by impaired lysosomal catabolism nor sialylated glycoconjugates is arterial hypertension. CTSA(S190A) mice have elevated diastolic and systolic blood pressure and impaired endothelin I degradation rates in plasma following an exogenous injection of endothelin I, a bioactive peptide previously characterized to induce vasoconstriction, and suggest that the catalytic activity of CTSA is important for regulating blood pressure in rodents and humans.482
Taken together, these data provide strong evidence that rodent and human CTSA participates in a scaffolding role to stabilize β-galactosidase and neuramidase activities. The CTSA(S190A) mice suggest that the hypertension in galacosialidosis patients might be caused by enhanced endothelin I signaling due to an absence of CTSA-mediated endothelin I catabolism. Whether the catalytic activity of CTSA is involved in other aspects of galacosialidosis in vivo remains unclear. Elucidation of changes in bioactive peptides of CTSA(−/−) mice should enhance our understanding of the biochemical reactions that this enzyme performs in vivo.
Selective inhibitors for CTSA have not, to our knowledge, been described.
3.8. PRCP (Prolylcarboxypeptidase)
PRCP, also called angiotensinase C, is a ~58 kDa glycoprotein that is highly expressed in human placenta, lung, and liver with lower expression in brain, heart, and pancreas.483 PRCP is localized to the lysosome484 but can also be found extracellularly in urine.485 As its name suggests, PRCP cleaves a variety of bioactive peptides at C-terminal amino acids linked to proline, such as angiotensin II and III and bradykinin, and has an acidic pH optimum but retains significant activity at neutral pH.486 The crystal structure of PRCP reveals that it adopts an α/β-hydrolase fold with a novel helical structural domain that caps the active site.487
The role of PRCP in food intake and metabolism has been demonstrated by both PRCP-deficient mice and PRCP inhibitors. PRCP gene-trapped [PRCP(gt/gt)] mice have complete loss of PRCP mRNA in the hypothalamus and kidney and show reductions in white adipose tissue (WAT) mass on chow or high-fat diet due to a reduction in food intake. PRCP(gt/gt) mice also have two-fold elevation in hypothalamic α-melanocyte-stimulating hormone (α-MSH) concentrations compared to wildtype littermates.488 Consistent with a direct role for PRCP in regulating α-MSH in vivo, purified recombinant PRCP cleaves between Pro-Val and liberates the C-terminal valine from α-MSH(1–13) to generate α-MSH(1–12), and exogenous α-MSH(1–13), but not α-MSH(1–12), reduces food intake when injected intracerebroventricularly in mice. In wildtype mice, administration of a non-selective inhibitor of PRCP and other peptidases, N-benzyloxycarbonyl-prolyl-prolinal (ZPP), reduces food intake, whereas coadministration of a melanocortin receptor antagonist, the agouti mimetic SHU9119, blocks this effect, indirectly demonstrating a role for melanocortin receptors in PRCP-dependent effects on feeding. Recently, several heterocycle-substituted pyrrolidines have also been developed that selectively and reversibly inhibit PRCP but not other related peptidases or ion channels/receptors.489 These compounds, despite having poor penetration into the central nervous system (CNS), nevertheless reduce food intake and body fat mass in rodents following a chronic dosing regime with no overt signs of toxicity, and suggest that inhibition of PRCP in peripheral tissues may also be therapeutically beneficial for weight loss. Whether PRCP also regulates other bioactive peptides490 and whether PRCP inhibitors could be efficacious for causing weight loss in humans remain open questions.
3.9. DPP7 (Dipeptidyl peptidase 7)
DPP7, also called dipeptidyl peptidase 2 (DPP2) or quiescent cell proline dipeptidase (QPP), is a ~58 kDa glycoprotein found in intracellular vesicles and, in rats, is highly expressed in kidney, heart, and testis, with lower expression in brain and lung.491–493 Despite having essentially no sequence homology with DPP4, DPP7 also cleaves dipeptides from the N-terminus of peptides and shows a preference for proline or alanine in the P1 position.494
DPP7 has been implicated in a variety of physiological processes including apoptosis and glucose homeostasis. Inhibition of DPP7 using nonselective inhibitors of post-proline-cleaving aminodipeptidases such as L-valinyl-L-boroproline (VbP) causes apoptosis-like phenotypes in peripheral blood monocuclear cells (PBMCs).495 Conditional knockdown of DPP7 in vivo under the neurogenin 3 promoter (NGN3-DPP7 kd mice) produces viable animals with alterations in glucose homeostatis.496 The viral construct in NGN3-DPP7 kd mice is expressed in enteroendocrine cells of the small intestines, pancreatic islets, and several regions of the brain including the ventromedial nucleus and the arcuate nucleus, but not the liver or muscle. These mice have an ~80% knockdown of DPP7 mRNA in the pancreas but no changes in liver or skeletal muscle DPP7. NGN3-DPP7 kd have normal plasma GLP-1 levels, normal body weight on chow or high-fat diet (HFD), but present with fasting hyperglycemia, glucose intolerance, hyperinsulinemia, and insulin resistance. On a HFD, NGN3-DPP7 kd have exacerbated liver steatosis and show increased liver lipid content compared to wild-type controls.
Despite the phenotypes caused by modulating DPP7 activity, the endogenous substrates regulated by DPP7 remain unknown. Recently, some piperidine- and azabicyclo[3.3.0]octane- derivatives have been developed as selective DPP7 inhibitors.497–499 The latter category of compounds shows good selectivity over other peptidases including DPP4, FAP, DPP8, DPP9, and PREP.499 Since global knockout of DPP7 results in embryonic lethality in mice,496 these inhibitors should help to elucidate the endogenous biochemical function and physiological role of DPP7 in vivo.
4. Protein- and glycan-modifying hydrolases
Besides small organic molecules and peptides, SHs can also act on larger (i.e., > 10 kDa) classes of macromolecules including esterified or thioesterified proteins or glycans. Like many small molecule and peptide hydrolases, these “macromolecular” hydrolases still adopt α/β-hydrolase domains, 500–502 demonstrating the versatility of this general fold for enabling catalysis on a variety of substrates of widely differing structures and physicochemical properties. SHs of this category, by directly acting on proteins or glycans, can modulate their activity, localization, and/or signaling.
4.1. Protein hydrolases
4.1.1. PPME1 (Protein phosphate methylesterase 1)
PPME1, also called protein methylesterase-1 (PME-1), is a 45 kDa enzyme localized to the nucleus.503,504 PPME1 shows ubiquitous tissue expression in mice, with highest mRNA levels found in the brain, kidney, ovary, and testis, and lower levels found in the heart, skeletal muscle, and lung.505 The best studied substrate of PPME1 is protein phosphatase 2A (PP2A), a major serine/threnonine phosphatase in cells.506,507 PP2A is a trimer composed of a catalytic subunit (C subunit), a constant regulatory subunit (A subunit), and one of several variable regulatory subunits (B subunits). It is thought that the B subunits affect PP2A function by targeting PP2A to different phosphoprotein substrates or distinct cellular localizations.507,508 In addition to changes in subunit composition, the PP2A catalytic subunit (PP2AC) is subject to an unusual post-translational modification, the methylesterification of its C-terminal leucine residue, which occurs by the transfer of a methyl group from S-adenosyl methionine (SAM) to PP2A by the action of cytosolic leucine carboxyl methyltransferase (LCMT1).505 PPME1 hydrolyzes this carboxymethylation to create a free C-terminal acid on PP2AC (Fig. 27).505
Fig. 27.

PPME hydrolyzes the carboxymethylated C-terminal leucine residue of the PP2A catalytic subunit (PP2AC). Shown in purple, PP2AC.
Overexpression of PPME1 in COS-7 cells increases demethylated PP2AC504 whereas knockdown of PPME1 in multiple cancer cell lines increased methylated PP2AC,509 demonstrating that PPME1 regulates the methylation status of PP2AC. In cells, carboxymethylation of PP2AC promotes the assembly of the PP2A holoenzyme,508,510 but there remains considerable debate about the functional consequences of this modification on PP2A activity. The crystal structure of PPME1 in complex with PP2AC shows that PPME1 directly binds the active site of PP2AC and, by doing so, dislodges the manganese ions required for PP2A activity,500 and mutations in the active site histidines of PP2AC result in the formation of inactive PP2A complexes with PPME1 and other cellular proteins.505,511 These lines of evidence suggest that the demethylation of PP2Ac by PPME1 is an inactivating modification. However, PPME1(−/−) mice show an absence of demethylated PP2AC and, surprisingly, a decrease in PP2A activity on a phosphopeptide substrate,503 demonstrating that PPME1 demethylation may also activate PP2A in some contexts. Regardless of the precise mechanism, PPME1 modification of PP2A carboxymethylation causes multiple changes in the phosphorylation status of PP2A substrates,503 which in turn affects cellular processes including cancer cell survival509 and differentiation.512
PPME1(−/−) mice are overtly normal throughout development but die soon after birth, precluding a functional assessment of PPME1 deficiency in adult mice.503 The recent disclosure of aza-β-lactams as potent and highly selective covalent inhibitors of PPME1 that can be used to disrupt PPME1 function in cells and mice513 should aid in the elucidation of how this enzyme affects PP2A function in vivo (Fig. 28).
Fig. 28.
(A, B) Structure of the aza-β-lactam-based PPME inhibitors ABL127 (A) and ABL103 (B).
4.1.2. LYPLA1 and LYPLA2 (Lysophospholipase 1 and 2)
LYPLA1, more commonly called acyl-protein thioesterase 1 (APT1), is a 24 kDa cytosolic protein that is highly expressed in the brain, kidney, and testis of human tissues, with lower expression in heart and liver.514,515 LYPLA1 is so named because it hydrolyzes a variety of lysophospholipids, including lysoPC, lysoPE, lysoPI, lysoPS, and PAF-like substrates, but does not have activity on phosphatidylcholines.516 LYPLA1 also shows thioesterase/esterase activity with a variety of peptide and protein substrates that are acylated on cysteine or serine residues, two post-translational modifications that can be used to regulate the signaling of peptide hormones or to localize proteins to specific membrane compartments of the cell.515 One such substrate in vitro is the peptide hormone ghrelin, which contains an n-octanoyl esterification on Ser3. Removal of this acyl chain (des-acyl ghrelin) ablates the activity of this hormone at the ghrelin receptor.517 Additional protein substrates of LYPLA1 include G-protein α subunit Giα1, members of the oncogenic Ras protein subfamily, and the enzyme endothelial nitric-oxide synthase (eNOS), which all contain cysteines thioesterified by palmitate groups.517–519 There is some selectivity in the depalmitoylation activity of LYPLA1 since caveolin, another membrane-bound palmitoylated protein, is not an in vitro substrate.519
To date LYPLA1(−/−) mice have not been described. The β-lactone derivative Palmostatin B is a LYPLA1 inhibitor that shows some selectivity for PLA1, PLA2, PLD, PLCbeta activities but has not been comprehensively evaluated for selectivity amongst SHs. Cells treated with Palmostatin B show elevated palmitoylated NRas levels and shifts in the localization of NRas from predominantly plasma membrane/Golgi to all cell membranes.520 Palmostatin B also caused a partial phenotypic reversion of HRasG12V-transformed MDCK-F3 cells, suggesting that targeting the palmitoylation status of oncogenes such as members of the Ras subfamily may be a viable strategy for the treatment of certain cancers. A microRNA (miRNA) screen in primary neuronal cultures revealed that LYPLA1 is a target for miR-138, and knockdown of LYPLA1 in these cells alters dendritic spine morphology, but the precise mechanism of this process remains poorly understood.521
LYPLA2, also called acyl-protein thioesterase 2 (APT2) or lysophospholipase 2, is a 25 kDa cytosolic protein with 60% identity to APT1. In mice, LYPLA2 is ubiquitously expressed with slightly higher levels in brain and liver by RT-PCR.522 Like APT1, LYPLA2 has both lysophospholipase and protein thioesterase activities, and can hydrolyze LPC, LPE, LPS, LPI, and PAF-like molecules, but not phosphatidylcholine.522 In CHO-K1 or HeLa cells, overexpression of LYPLA2, but not overexpression of LYPLA1, increases the deacylation rate of growth-associated protein 43 (GAP43).523 LYPLA2 overexpression also increases the deacylation rate of HRasG12V, demonstrating that LYPLA1 and 2 may have overlapping but distinct palmitoylated protein substrate profiles.523 Neither knockouts nor selective inhibitors have been, to our knowledge, been described for LYPLA2.
Taken together, these data suggest that LYPLA1 and LYPLA2 can regulate the palmitoylation status of certain proteins in cell culture models, but whether this activity extends more broadly to the hundreds of palmitoylated proteins found in mammalian cells and tissues remains unknown. It also remains to be determined whether the lysophospholipase activities of LYPLA1 or 2 contribute to lysophospholipid metabolism in physiological contexts.
4.1.3. PPT1 and PPT2 (Protein palmitoyl thioesterase 1 and 2)
PPT1 is a 37 kDa lumenally-oriented lysosomal glycoprotein that is highly expressed in rat testis, brain, lung, spleen, and at lower levels in white adipose tissue, kidney, liver, heart.524–527 PPT1 can depalmitoylate HRas in vitro and shows an atypical insensitivity to the general serine-reactive inhibitors phenylmethylsulfonyl fluoride (PMSF) or diisopropyl fluorophosphonate (DIFP).524,528 This latter observation is explained by the crystal structure of PPT1, which shows that it adopts an α/β-hydrolase fold with an unusually narrow hydrophobic channel that precludes the large aryl or isopropyl groups of PMSF and DIFP, respectively, from accessing the PPT1 active site.502,528 PPT1 also shows activity for the palmitoylated α subunit of the G-protein Go.524 Besides its protein thioesterase activity, PPT1 can cleave fatty acyl-CoAs, with a preference for myristoyl to stearoyl (C14 to C18) acyl chains and markedly lower activity on both shorter (C12) or longer (C20) fatty acyl CoAs.525
Mutations in PPT1 causes infantile neuronal ceroid lipofuscinosis (INCL), a lysosomal storage disease that occurs in 1:12,500 people and is characterized by loss of vision, mental retardation, a flat electroencephalogram at 3 years, and death by 8–11 years. Upon autopsy, the typical finding is the accumulation of granular autofluorescent material throughout the brain as well as other tissues.529,530 PPT1 activity using palmitate-labelled HRas is absent in brain tissues from INCL patients.530 Similarly, PPT1(−/−) mice that have near complete loss of PPT1 activity in brain lysates on the synthetic substrate 4-methylumbelliferyl-6-thiopalmitoyl-β-D-glucoside, and also accumulate autofluorescent depots consisting of granular spheroid structures throughout the brain as early as 4 weeks of age.531 PPT1(−/−) mice exhibit abnormal clasping behavior in the tail suspension test, seizures and other convulsions, neuronal degeneration, and die earlier than wild-type littermates (75% dead by 10 months). Curiously, the cellular and morphological abnormalities of PPT1(−/−) mice are largely restricted to the brain, despite the expression of PPT1 in multiple peripheral tissues.531
PPT2 is a 30 kDa lysosomal glycoprotein with highest expression in human skeletal muscle and lower expression in heart, brain, placenta, and kidney. PPT2 has ~20% identity with PPT1 and the two enzymes show comparable activity for palmitoyl-CoA as a substrate, but PPT2 does not act on palmitoylated protein substrates of PPT1 such as HRas.532 PPT2(−/−) mice develop an unusual variant of neuronal ceroid lipofuscinosis that, in addition to causing neurological dysfunction, also strongly affects peripheral tissues.533 PPT2(−/−) mice show a clasping phenotype in the tail suspension test with a slower onset than what is observed in PPT1(−/−) mice. Similarly, the lifespan of PPT2(−/−) mice is two years, longer than that of PPT1(−/−) mice but still significantly shorter than wild-type mice, and, at death, PPT2(−/−) brains are ~10% lighter than wild-type controls and show cerebral cortical atrophy and scattered apoptotic bodies. An autofluorescent material also accumulates in multiple tissues of PPT2(−/−) mice, including the brain, pancreas, kidney, lungs, heart, and skeletal muscle. Gross morphological and histological examination of PPT2(−/−) tissues show massive splenomegaly and extramedullary hematopoiesis in the spleen and liver, which is accompanied by macrophage infiltration into the bone marrow. Despite these findings, PPT2(−/−) mice have normal peripheral blood counts, bone radiographs, and pancreatic function. There is no evidence, to date, of human diseases that map to the Ppt2 locus on chromosome 6p21.3.
Taken together, these studies suggest that PPT1 and PPT2 have important roles in the regulation of lysosomal homeostasis in the nervous system and peripheral tissues, respectively, but the precise mechanisms that link the biochemical and enzymatic properties of PPT1 and PPT2 to these lysosomal storage phenotypes remain unclear. For instance, it remains unknown whether protein palmitoylation is increased in the central nervous system of INCL patients or PPT1(−/−) mice, or precisely how the protein thioesterase activity of PPT1 contributes to the accumulation of autofluorescent depots in the brain. It is also unknown whether PPT2 has protein depalmitoylation activity in cells or in mice, since its nomenclature is based solely on homology to PPT1 and not from any biochemical evidence that the enzyme displays protein thioesterase activity. The chemical composition of the autofluorescent material in PPT-deficient humans and rodents, and whether its accumulation is due to an absence of degradation of specific biomolecules by PPT1/2, remain open questions that are under active experimental investigation.
Selective inhibitors of PPT1 or PPT2 have not, to our knowledge, been described. The advent of such inhibitors, combined with further study of PPT1(−/−) and PPT2(−/−) mice, should enhance our understanding of the connection between PPT biochemical activities and diseases like INCL.
4.2. Glycan hydrolases
4.2.1. AOAH (Acyloxyacyl hydrolase)
Gram-negative bacterial lipopolysaccharides (LPS) are large molecules consisting of an O antigen, a core oligosaccharide, and a lipid-containing portion called lipid A.534 LPS produce strong immune responses in animals leading to toxic effects such as fever, hypotension, and death by activating toll-like receptors (TLRs) expressed on B cells.535,536 Lipid A is the bioactive center of LPS and consists of a phosphoglucosamine disaccharide lipidated by four molecules of 3-hydroxymyristate (3-OH-C14:0). Two of these hydroxymyristoyl acyl chains are further modified by saturated fatty acids (laurate, myristate, or palmitate) to form acyloxyacyl groups. By selectively removing the acyloxyacyl groups from lipid A, AOAH from the host converts bioactive, hexaacyl LPS into tetraacyl LPS (“deacylated” LPS or dLPS), a molecule that is several orders of magnitude less potent at stimulating host responses in a variety of assays (Fig. 29).537
Fig. 29.

AOAH hydrolyzes the two acyloxyacyl groups from the lipid A portion of LPS to generate tetraacyl-LPS. The core oligosaccharide and O antigen of LPS are represented by the green hexagon and yellow rectangle, respectively.
AOAH is expressed by neutrophils, dendritic cells, renal cortical epithelial cells, and monocyte-macrophages and its activity can be stimulated or inhibited by LPS or cytokines, respectively.538,539 Purified AOAH from the human promyelocytic line HL-60 is a ~52–60 kDa glycoprotein composed of two disulfide-linked subunits of 50 kDa and 14–20 kDa.540 Recombinant expression of the large subunit is sufficient to recapitulate the lipolytic activity of the purified AOAH complex.541 In addition to its acyloxyacyl hydrolase activity on lipid A, AOAH also has in vitro phospholipase, lysophospholipase, and DG lipase activity, and can catalyze acyl-transfer from LPS to MAGE acceptors.542
The in vivo function of AOAH in modulating host responses to LPS has been established by the generation of both transgenic and gene-targeted mice.539,543 AOAH transgenesis into dendritic cells and macrophages under the human CD68 promoter increases LPS deacylation activity in peritoneal macrophages, shortens the recovery time required following an LPS challenge, and lowers susceptibility to LPS-induced hepatosplenomegaly and death.539 In AOAH(−/−) mice, LPS deacylation activity is completely abolished in peritoneal macrophages and liver, which are unable to convert exogenous LPS to dLPS.543–545 When challenged with the Gram-negative bacteria, AOAH(−/−) mice produce higher titers of immunoglobulin M (IgM) and IgG3 antibodies.545 Lastly, an inflammatory response is typically followed a period of tolerance or reprogramming characterized by infection-induced immunosuppression to LPS or other microbial molecules. The recovery from the tolerant state is delayed in AOAH(−/−) mice and the magnitude and duration of tolerance is correlated to the presence of acylated LPS in peritoneal macrophages, suggesting that a recovery from the tolerant state requires AOAH-mediated hydrolysis of LPS to dLPS.20
Selective inhibitors of AOAH have not, to our knowledge, been described.
4.2.2. PGAP1 (Post-GPI attachment protein 1)
PGAP1, also called GPI inositol deacylase (GPID), is a ~105 kDa, endoplasmic reticulum (ER)-localized multi-pass transmembrane protein with a cytoplasmic N-terminus and a lumenally-oriented lipase motif.546 PGAP1 is expressed and active in the brain85 and also at high levels in B cells.181 PGAP1 function has best been characterized in the context of glycophosphatidylinositol (GPI), a glycolipid attached to the C-termini of proteins to anchors them in cellular membranes. The GPI anchor consists of a phosphatidylinositol (PI) connected to an oligosaccharide, which is in turn covalently bound to the protein via an ethanolamine phosphate linker. At an early stage of GPI biosynthesis in the ER, the inositol ring is palmitoylated, but this modification is rapidly removed following attachment to the C-terminus of the target protein. The removal of palmitate from the inositol of triacyl-GPI to generate mature, diacyl-GPI is performed by PGAP1 (Fig. 30). In a PGAP1-deficient subline of mutagenized CHO-K1 cells expressing CD59 and DAF, an accumulation is observed in triacyl-GPI-modified CD59.546 Analysis of total DAF protein in these cells shows an equal amount of mature, glycosylated DAF, but an accumulation of pro-DAF in the PGAP1-deficient line, and pulse-chase experiments show a delay in the maturation of DAF, supporting the hypothesis that PGAP1-mediated deacylation of palmitoyl GPI expedites the ER-to-Golgi transport of GPI-modified proteins.
Fig. 30.

PGAP1 hydrolyzes palmitate from the inositol of triacyl GPI to generate mature, diacyl-GPI. Shown in green, a protein modified by GPI; shown as a blue hexagon, a mature diacyl-GPI modification.
PGAP1(−/−) mice also support a role for PGAP1 in the deacylation of GPI-anchored proteins in vivo.547 CD48 from wild-type, but not PGAP1(−/−) mice, is readily cleaved by PI-phospholipase C, an enzyme that liberates diglycerides from GPI-modified proteins but not from triacyl-GPI-modified proteins. These mice have severe developmental defects including otocephaly (absence of lower jaw), which causes the majority of knockout pups to die shortly after birth. The few PGAP1(−/−) pups that survive into adulthood have slower growth rates than wild-type littermates and males PGAP1(−/−) mice are infertile. The precise mechanism by which PGAP1 causes these dramatic phenotypes remain to be elucidated, but may relate to differences between “normal” diacyl-GPI-anchored proteins and triacyl-GPI-proteins in the Wnt signaling pathway, which has previously been implicated in multiple developmental abnormalities including otocephaly or agnathia.
Selective inhibitors of PGAP1 have not, to our knowledge, been described.
4.2.3. SIAE (Sialic acid acetylesterase)
SIAE, also called sialate O-acetylesterase, is an enzyme with two splice isoforms differing at the N-terminus.548,549 The lysosomal lumenally-oriented glycosylated isoform, abbreviated LSE, is a 62 kDa protein composed of two disulfide-bonded subunits of 36 kDa and 30 kDa, and can also be found secreted or bound to the cell surface of transfected cells.548–550 LSE is generated by proteolytic processing of a ~65 kDa precursor polypeptide and has widespread tissue expression in mouse tissues.549,551 The cytosolic isoform, abbreviated CSE, lacks the 5′ signal-peptide encoding region found on the mRNA of LSE, and, in mouse tissues, is highly expressed in the brain and ovary, with lower expression levels in the liver and thymus, and essentially no expression in kidney, spleen, testis, and skeletal muscle.549 SIAE cleaves acetate from 9-O-acetylated sialic acids including the parent compound 9-O-acetyl-N-acetylneuraminic acid (Neu5,9Ac2) (Fig. 31). SIAE also shows activity with di- or tri-acetylated sialic acid owing to the spontaneous intramolecular migration of other acetyl groups to the 9-position.548
Fig. 31.

SIAE removes acetate from 9-O-acetyl-N-acetylneuraminic acid (shown), as well as other 9-O-acetylated sialic acids. This activity can occur on both free acetylated sialic acids or on acetylated sialic acid found in glycoproteins.
The function of SIAE as an enzymatic regulator of peripheral B cell tolerance has been elucidated recently by SIAE-deficient mice552 and mutations in this enzyme in humans.553 Siaylated glycoconjugates can be recognized by cell surface receptors called Siglecs, short for sialic acid binding Ig-like lectins. One such receptor is CD22, also called Siglec-2, which is expressed on the surface of B cells and recognizes N-glycans containing 9-OH-sialic acids, but not 9-acetylated sialic acids.554 CD22 is a functional repressor of B cell receptor (BCR) signaling and therefore activation of CD22 reduces the proliferation and differentiation of self-reactive B cells.554 B cells from SIAE(−/−) mice show an accumulation of N-glycans containing 9-O-acetylated sialic acids and concomitant reduction in CD22 activation. Since CD22 negatively regulates BCR signaling, the net effect of SIAE deficiency is hyperactive BCR signaling. Consistent with previously reported phenotypes associated with an absence of negative regulation of BCR signaling, marginal zone (MZ) B cells, MZ B cell precursors, and bone marrow perisinusoidal B cells are significantly reduced in SIAE(−/−) mice. Rag-1(−/−) mice reconstituted with lymphocytes from SIAE(−/−) mice also demonstrated defects in MZ B cell development, demonstrating that the effects on B cells in SIAE(−/−) mice are lymphocyte-intrinsic defects. SIAE(−/−) mice have increases in certain serum immunoglobulins including IgE and IgG2b, increases in serum autoantibodies, and develop glomerulonephritis, an inflammatory disease of the kidneys, demonstrating a break in B cell tolerance. Supporting these rodent studies is the discovery in human patients of multiple polymorphisms in the Siae gene associated with the development of autoimmune disorders.553 Mutant SIAE proteins arising from these alleles are typically defective in either or both esterase activity and secretion, and can function in a dominant negative manner when transfected in cells because both isoforms of SIAE can associate into dimers or higher order oligmers under these conditions.553
Taken together, these data suggest that SIAE directly modulates immunological tolerance in rodents and humans by endogenously regulating the acetylation status of sialic acid-containing glycoconjugates. The function of the cytosolic isoform of SIAE, which does not have access to cell surface glycans, remains unclear, though some speculate that it may modulate the acetylation status of free cytosolic sialic acids.554 Selective inhibitors of SIAE have not, to our knowledge, been described.
5. Partially-characterized hydrolases and emerging methods for their investigation
The profile of the mammalian SHs described above underscores how members of this superfamily regulate a diverse array of biochemical pathways that affect virtually all aspects of physiology and behavior. This basic knowledge of SH function has fostered translational advances in the form of selective inhibitors of SHs such as DPP4, ACHE/BCHE, pancreatic lipases, PLA2G7, and FAAH, which are currently being used or are in clinical development for the treatment of diabetes, neurodegenerative disorders, obesity, cardiovascular disease, and pain, respectively.255,319,394,555 Additional SHs, such as PRCP, MGLL, CES3, and LIPG, just to name a few, represent enticing future drug targets based on preclinical studies.148,489 Considering the importance of the SH superfamily in mammalian physiology and the established potential for inhibitors of these enzymes to positively impact human health and the treatment of disease, it is noteworthy that our aforementioned summary only addresses approximately half of the mammalian metabolic SHs. The other half of mammalian SHs fall into the general category of being too poorly characterized to even predict candidate biochemical substrates or physiological functions. Assignment of in vitro and eventually endogenous substrates for these uncharacterized SHs is critical for elucidating their functions at the cellular and organismal level. Achieving this goal will require the development of selective pharmacological and/or genetic tools to disrupt the function of individual uncharacterized SHs, as well as the implementation of global methods to evaluate the effect of these disruption events on the biochemical composition of cells and tissues.
In the first part of this section, we will discuss a few representative examples of poorly characterized SHs that lack evidence regarding the identity of potential endogenous substrates, and in the second half, we will review recent proteomic and metabolomic technology platforms that may be used to address the challenge of annotating biochemical and physiological functions for these enzymes. For clarity, we have refrained from discussing the following uncharacterized SHs: ABHD8, ABHD10, ABHD12B, ABHD13, ABHD14A/B, FAM108A/B/C, FAM135A/B, SERHL, LYPLAL1, LIPL3, QRSL1, SERAC1, BAT5, THEDC1, FABD, c13orf27, c20orf135, c2orf43, and multiple carboxylesterases.
5.1. Representative examples of poorly characterized hydrolases
5.1.1. AADAC and AADACL2 and 4 (Aryl acetamide deacetylase and aryl acetamide deacetylase-like 2 and 4)
AADAC is a 45 kDa membrane-associated hydrolase highly expressed in the human liver that has been implicated in the hydrolysis of arylacetamide xenobiotics including flutamide and phenacetin.556–560 AADAC is highly homologous to several related lipases, AADACL1, 2, and 4, the latter two of which remain completely unannotated. Overexpression of AADAC in a rat hepatoma cell line produces changes in cellular TG levels, apoplipoprotein B secretion, and fatty acid oxidation, but the mechanism for these effects remains unclear.561 Candidate endogenous substrates, selective inhibitors, or knockout mice have not, to our knowledge, been described for AADAC or the related enzymes AADACL2 and AADACL4.
5.1.2. ABHD1, ABHD2, and ABHD3 (Alpha/beta hydrolase domain containing 1, 2, and 3)
ABHD1, 2, and 3, also called lung α/β-hydrolase 1, 2, and 3 (LABH), are three 46–48 kDa hydrolases that were originally cloned from lung cDNA. ABHD1 and ABHD2 share ~20% identity, whereas ABHD1 and ABHD3 share ~45% identity.562 In mice, the three hydrolases are ubiquitously expressed, with highest expression of ABHD1 in liver, ABHD2 in liver, lung, and intestines and ABHD3 in brain, kidney, and liver.562 Of these three hydrolases, ABHD2 is best characterized. Vascular smooth muscle cells from gene-trapped ABHD2 mice [ABHD2(gt/gt)] mice show an increase in both in vitro and in vivo migration,563 and ABHD2(gt/gt) mice also spontaneously develop emphysema due to macrophage infiltration, increased cytokine production, and increased apoptosis in the lung.564 Interestingly, the bronchoalveolar lavage fluids, but not the lung homogenate, of ABHD2(gt/gt) mice showed a modest decrease in PCs compared to wild-type littermates, but the mechanism for this change in PCs is unknown.564 In kidneys from mice lacking the D5 dopamine receptor, ABHD1 expression is elevated >10-fold, and overexpression of ABHD1 in an immortalized proximal tubule cell line decreases O2− production by NADPH oxidase.565 There are no reports of ABHD3 function in the literature. To date no knockouts have been reported for ABHD1 or ABHD3 and no inhibitors have been reported for any of these three enzymes. Candidate endogenous substrates or selective inhibitors have not, to our knowledge, been described for ABHD1–3.
5.1.3. ABHD11 (Alpha/beta hydrolase domain containing 11)
ABHD11 is an ~30 kDa protein ubiquitously expressed in mouse tissues, with highest expression levels in heart, skeletal muscle, brown and white fat, lung, and brain.33,181 Proteomic studies indicate that ABHD11 is a mitochondrial protein.566 ABHD11 is one of multiple genes deleted in Williams-Beuren syndrome, a developmental disorder characterized by congenital heart and vascular disease, dental abnormalities, cognitive defects, and premature aging of the skin, but the contribution of ABHD11 to this disorder remains unknown.567 Recent competitive ABPP studies have identified the 2-ethyl piperidine carbamate WWL222 as a potent, selective, and in vivo-active inhibitor of ABHD11.33 Candidate endogenous substrates or knockout mice have not, to our knowledge, been described for ABHD11.
5.1.4. ABHD14B (Alpha/beta hydrolase domain containing 14B)
ABHD14B, also called CCB1/TAFII250-interacting factor B (CIB), is a ~25 kDa hydrolase expressed roughly equally in most human tissues, with lowest expression in brain, testis, and placenta by Northern blot.568 ABHD14B adopts an α/β-hydrolase fold with an unusual SPSLS motif surrounding the nucleophilic serine and has diffuse cytosolic and nuclear localization when transfected into COS cells.568 ABHD14B was originally identified in a yeast two-hybrid screen as a binder to the TFIID subunit CCG1/TAFII250 and therefore is postulated to have some role in transcriptional initiation,568 though direct evidence supporting this claim is lacking. In vitro, ABHD14B can turn over the general hydrolase substrate p-nitrophenyl butyrate but has not been tested for activity on any potentially natural substrates. ABHD14B inhibitors or knockout mice have not, to our knowledge, been described.
5.1.5. BPHL (Biphenyl hydrolase-like protein)
BPHL, also called valacyclovirase, is a ~30 kDa protein highly expressed in human liver and kidney.569–571 BPHL has been found to convert the prodrug ester forms of multiple nucleoside-containing compounds into their active products, including antiviral produgs valacyclovir and valganciclovir.572 However, candidate endogenous substrates, selective inhibitors, or knockout mice have not, to our knowledge, been described for BPHL.
5.1.6. SEC23IP (Sec23p-interacting protein)
SEC23IP, also called p125, is a 111 kDa ER- and Golgi-localized protein closely related in sequence to DDHD1 and DDHD2, two intracellular phospholipase A1 enzymes.573–577 SEC23IP has not been found to show phospholipase A1 activity itself. SEC23IP is ubiquitously expressed in human tissues with highest levels found in heart, placenta, skeletal muscle, and pancreas.574 SEC23IP is so named because its N-terminal proline-rich domain interacts with Sec23p, a protein involved in the transport of vesicles from the ER membrane.574 RNA-mediated silencing of SEC23IP in cells produces morphological changes and dysfunction of the ER and Golgi,575,576 but it is unclear to what extent its catalytic activity contributes to this process. Candidate endogenous substrates, selective inhibitors, or knockout mice have not, to our knowledge, been described for SEC23IP.
5.1.7. OVCA2 (Ovarian tumor suppressor candidate 2)
OVCA2 is a ~25 kDa protein originally identified as a gene on human chromosome 17p13.3,578 a region deleted in 80% of ovarian cancers.579 However, besides this correlational observation, there is little evidence to support a role for OVCA2 in tumor suppression. Candidate endogenous substrates, selective inhibitors, or knockout mice have not, to our knowledge, been described for OVCA2.
5.1.8. RBBP9 (Retinablastoma-binding protein 9)
RBBP9, also called Bog, is a ~20 kDa protein with ubiquitous expression in rat tissues and highest levels found in spleen, testis, kidney, and brain.580,581 Despite having its three-dimensional structure solved as part of the Structural Genomics initiative and being subjected to multiple broad enzymatic assays for phosphatase, phosphodiesterase, dehydrogenase, oxidase, protease, carboxylesterase, or thioesterase activities, in vitro substrates have not yet been identified for RBBP9.580 RBBP9 is so named because it directly binds the tumor suppressor gene retinoblastoma (Rb), and elevated levels of RBBP9 are associated with the transformation process and carcinogenesis in vivo for epithelial and pancreatic cells, respectively.581,582 This process occurs in part by RBBP9-induced resistance to TGF-β-mediated antiproliferative signaling, but the endogenous biochemical role for RBBP9 in these processes remains unclear.581 Competitive ABPP studies have identified the natural product emetine as a reversible inhibitor of RBBP9, as well as a family of oxime esters that covalently inhibit RBPP9 with good selectivity over other SHs in cell and tissue proteomes.583,584 Whether these inhibitors can be used to selectively disrupt RBBP9 function in living systems remains unknown. Candidate endogenous substrates or knockout mice have not, to our knowledge, been described for RBBP9.
5.1.9. PNLIPRP1 (Pancreatic lipase-related protein 1)
PNLIPRP1, also abbreviated PLRP1, is a ~50 kDa secreted protein with >65% identity to PNLIP and high expression in the pancreas.585 Unlike PNLIP or PNLIPRP2, PNLIPRP1 does not appear to show any hydrolytic activity with a wide variety of lipid substrates.585 PNLIPRP1(−/−) mice have greater fat mass and less lean mass, impaired glucose and insulin tolerance, and a higher levels of serum insulin compared to littermate controls.586 Interestingly, PNLIPRP1(−/−) mice show a higher lipase activity in pancreatic juice but no difference in PNLIP or PNLIPRP2 by RT-PCR. Because previous studies have demonstrated that PNLIPRP1 can also bind the PNLIP obligate cofactor colipase,585 these data support the hypothesis that PNLIPRP1 basally sequesters a fraction of colipase and thus attenuates the activity of PNLIP. In the absence of PNLIPRP1, PNLIP would then have greater access to colipase to produce higher activity for absorption of dietary fats, resulting in the obesity phenotype observed in PNLIPRP1(−/−) mice. The potential contribution of PNLIPRP1 activity itself, versus colipase binding, to this obesity phenotype remains unknown. Candidate endogenous substrates or selective inhibitors have not, to our knowledge, been described for PNLIPRP1.
5.1.10. LACTB (Serine β-lactamase-like protein)
LACTB is a ~60 kDa protein with an active site motif similar to bacterial C-class β-lactamases, and, in mice, is highly expressed in heart, liver, skeletal muscle, kidney, and testes, with lower expression levels in brain, spleen, and lung.587 It is unknown whether LACTB has lactamase activity. LACTB is localized to the intracristal areas of the mitochondrial intermembrance space, where it polymerizes into straight or slightly curved ordered filaments.588 The function of this unusual mitochondrial structure remains unknown. Candidate endogenous substrates, selective inhibitors, or knockout mice have not, to our knowledge, been described for LACTB.
5.1.11. PNPLA5 (Patatin-like phospholipase domain containing 5)
PNPLA5, also called GS2-like, is a ~315 AA protein that, upon overexpression in HEK293 cells, reduces intracellular TGs.234,589 It is not known whether recombinant PNPLA5 also has TG lipase activity in vitro, and its tissue distribution in rodents is also unknown due to low levels of PNPLA5 mRNA.234 Candidate endogenous substrates, selective inhibitors, or knockout mice have not, to our knowledge, been described for PNPLA5.
5.1.12. SCPEP1 and CPVL (Serine carboxypeptidase 1 and carboxypeptidase, vitellogenic-like)
SCPEP1, also called retinoid-induced serine carboxypeptidase (RISC), is a ~51 kDa lysosomal and secreted glycoprotein that is proteolytically processed into a ~35 kDa mature form.590–592 SCPEP1 expression is found in most murine tissues except heart, with highest levels in kidney, liver, spleen, and stomach, and its expression can be induced by all-trans-retinoid acid in cultured smooth muscle cells (SMCs) or in mouse carotid artery following ligation injury.590,591,593 Overexpression of SCPEP1 but not a catalytically inactive mutant enhances SMC growth and migration; conversely, knockdown of endogenous SCPEP1 impairs SMC growth, and SCPEP1(−/−) mice show reductions in medial and intimal cell proliferation following arterial injury. These data demonstrate a role for SCPEP1 activity in modulating smooth muscle proliferation, migration, and vascular remodeling. SCPEP1 has ~20% identity to the lysosomal carboxypeptidase CTSA, but cannot hydrolyze any of the known CTSA or other N-terminally blocked peptide substrates.592
CPVL is a 57 kDa ER-localized hydrolase with ~40% identity to SCPEP1 and high expression in multiple human tissues including heart, spleen, and kidney, as well as human macrophages but not lymphocytes or neutrophils.594,595 It is not known whether CPVL has carboxypeptidase activity. Candidate endogenous substrates or selective inhibitors have not, to our knowledge, been described for SCPEP1 or CPVL.
5.2. Chemoproteomic and metabolomic platforms for functional assignment of poorly characterized hydrolases
As the functional roles of SHs continue to be elucidated, of particular importance will be to develop an understanding of the endogenous substrates and products for these enzymes and how they relate to physiology and disease. Elucidating these pathways, however, remains challenging for multiple reasons. First, the substrates or substrate class for individual SHs cannot, in general, be deduced a priori by primary amino acid sequence or even three-dimensional structure. In the PNLIP, PNLIPRP1 and PNLIPRP2 subfamily, where the three hydrolases have >50% pair-wise identity to each other, only PNLIP and PNLIPRP2 have TG lipase activity,114,596 whereas PNLIPRP has yet to show in vitro hydrolytic activity with any tested small molecule substrate, including TGs.585 Even broad substrate classes cannot necessarily be predicted by primary sequence identity: PPME1 clusters with MGLL, ABHD4, ABHD6, and several uncharacterized SHs, yet PPME1 substrates are carboxymethylated proteins505 whereas MGLL and ABHD6 substrates are neutral small molecules85 and ABHD4 substrates are phospholipids.304 Second, many SHs accept multiple substrate classes in vitro, and it is nearly impossible to predict which of these substrates are relevant to the enzymes’ functions in vivo. PPT1, in addition to displaying protein palmitoyl thioesterase activity, also hydrolyzes fatty acyl-CoAs,525 and, to date, it remains unknown whether a deficiency in one or both of these activities (or another as of yet unidentified activity) causes the lysosomal storage disorder observed in PPT1(−/−) mice531,533 and patients with infantile neuronal ceroid lipofuscinosis.529,530 More generally, in vitro substrate specificity is often not predictive of the relevant in vivo substrates for an enzyme due to other competitive metabolic pathways, substrate accessibility within the cell, or lack of coordinated substrate/enzyme expression. For instance, FAAH, in addition to its N-acylethanolamine amidase activity, also can hydrolyze lipid esters such as MGs, but these lipids are unaltered in FAAH-disrupted animals likely because other co-expressed enzymes, such as MGLL, are much more active as MG hydrolases.314
These issues highlight the limitations of traditional biochemical techniques for the assignment of endogenous substrates to SHs and underscore the need for technologies that embrace the complexity of endogenous biochemical networks by, for example, globally profiling metabolic pathways in the relevant native environments. Concurrently, there is also a need for methods to efficiently discover and optimize inhibitors to perturb SH function in cells and animals. Such chemical probes are important complements to genetic methods for perturbing SHs because targeted genetic disruption of some SHs in mice has already been shown to cause embryonic lethality (e.g., PPME1503) or adapative changes that mask phenotypes induced by acute pharmacological blockade (e.g., MGLL56).
We do not mean to imply that emerging technology platforms for discovering pharmacological inhibitors and profiling metabolic changes ought to be used in place of more conventional biochemical and genetic methodologies, but rather suggest that the approaches are highly complementary and can, when used together, provide powerful platforms for forging functional connections between SHs, their physiological substrates, and higher-level physiological processes.
5.2.1. Chemoproteomic platforms for discovery and optimization of SH inhibitors
Even though members of the SH superfamily accept a wide range of substrates, the vast majority of these enzymes react with the fluorophosphonate (FP) class of affinity labels, which covalently label the conserved catalytic serine residue in SH active sites.10,33 When conjugated to reporter tags, such as fluorophores or biotin, FPs take the form of activity-based protein profiling (ABPP) probes that can be used directly in native biological samples to detect, quantify, and/or enrich and identify SHs (Fig. 2). FP probes have been used to globally profile the activity of SHs in a wide range of cell and animal models, as well as primary human tumors.77,597–599 The successful FP labeling of uncharacterized SHs like RBBP9 and PNLIPRP1 has provided the first biochemical evidence that these enzymes are catalytically active, even though substrates have not yet been identified.33
ABPP with FP probes can also be performed in a competitive manner for the discovery and optimization of small molecule inhibitors of SHs (Fig. 32). Competitive ABPP for inhibitor discovery typically involves preincubation of cell or tissue lysates with a library of small-molecules followed by addition of a fluorescent FP probe.600,601 Following separation of probe-labeled proteomes by SDS-PAGE, inhibition of one or multiple SHs is detected by a decrease in their fluorescent signals. Competitive ABPP can concurrently optimize inhibitor potency and selectivity directly in native proteomes and, when performed under kinetically controlled conditions, can identify reversible or irreversible SH inhibitors.600
Fig. 32.

(A) Competitive ABPP. The potency and selectivity of a single inhibitor (green square) can be evaluated against SHs in a native tissue proteome by chasing the inhibitor with a SH-directed activity probe (red circle) and visualization by in-gel fluorescence following SDS-PAGE separation. Inhibitor targets are detected as bands with reduced fluorescence. (B) Fluopol-ABPP. A single SH can be evaluated against many compounds by chasing inhibitors (yellow hexagon or green triangle) with a SH-directed activity probe (red circle). Active (green triangle) or inactive (yellow hexagon) inhibitors will produce low or high fluorescence polarization signals, respectively.
Gel-based competitive ABPP is limited, however, by resolving power and compound throughout. A typical gel-based ABPP experiment can only resolve ~20 SHs per proteome, leaving many low-abundance and co-migrating enzymes undetected. Substantial increases in proteome-coverage can be achieved by liquid chromatography-mass spectrometry (LC-MS) methods for enzyme detection. In this shotgun LC-MS approach, often referred to as ABPP-MudPIT, control- and inhibitor-treated proteomes are incubated with a biotinylated FP probe and then probe-labeled enzymes are enriched by avidin chromatography, digested with trypsin, and the corresponding tryptic peptides analyzed by LC-MS.77 The activity levels of SHs can be quantified in this method by label-free (e.g., spectral counting) or isotopic (e.g. SILAC) techniques.
To address the limited throughput of competitive gel-based ABPP, which can assay at most several hundred compounds per day, a complementary platform termed fluorescence polarization ABPP (fluopol-ABPP) has been developed.583 This technology relies on the emission from fluorescent FP probes of depolarized light when the probe is free in solution and highly polarized light when it is bound to large macromolecules such as proteins. Fluopol-ABPP is a homogeneous assay compatible with high-throughput screening (HTS), where inhibitors are scored by causing reductions in fluorescence polarization.
The aforementioned suite of competitive ABPP technologies has provided an emerging workflow for SH inhibitor development, wherein, fluopol-ABPP is first used to screen a large compound library like the NIH small-molecule collection, and then hits from this screen are evaluated for selectivity against several SHs in native proteomes by gel-based ABPP. Inhibitors that show promising potency and selectivity can then have their target specificities confirmed against a large set of SHs by ABPP-MudPIT. This workflow has greatly expanded the number of SHs for which potent, selective, and in vivo-active inhibitors are now available, including FAAH,313,317 MGLL,54 AADACL1,75 ABHD6,601 ABHD11,33 and PPME1.513
5.2.2. Specialized ABPP probes that mimic SH substrate classes
Targeted ABPP probes can be synthesized to label particular subsets of SHs and inform on their potential substrates. For instance, FP electrophiles can be placed at the sn-1 or sn-2 position of phosphatidylcholine lipid scaffolds with latent alkyne handles for click-chemistry mediated enrichment (Fig. 33).602 These probes label some proteins in tissue homogenates preferentially over a general FP-alkyne probe, demonstrating that such a targeted chemical strategy can distinguish between SHs that preferentially react with phospholipid-like substrates versus those that are generally reactive with undecorated FP electrophiles. Moreover, DDHD1, an sn-1-selective phospholipase, preferentially reacts with the sn-1 over the sn-2 FP phospholipid probe. Ultimately the full potential of such a platform for SH functional characterization will require the chemical synthesis of a diverse suite of substrate-like probe scaffolds and the identification of SHs that are labeled by these probes.
Fig. 33.

(A) Structure of the general SH-directed probe FP-alkyne. (B, C) Structure of the sn-1-selective (B) or sn-2-selective (C) phospholipase-directed probes.
5.2.3. Untargeted metabolomics and peptidomics for SH substrate discovery
In a direct effort to comprehensively profile the lipidomic changes in tissues following pharmacological or genetic perturbation of a single enzyme, Saghatelian and colleagues developed a standard-free untargeted liquid chromatography-mass spectrometry (LC-MS) method termed discovery metabolite profiling (DMP).314 In this method, many lipids are analyzed by broad mass-scanning mode and their abundances are relatively quantified in parallel by integration of mass ion intensities, thereby obviating the need for isotope-labeled standards. Metabolites whose signals are increased in the enzyme-disrupted systems are candidate substrates, whereas those whose signals are decreased are candidate products (Fig. 34). DMP allows for the identification of metabolomic changes by two parameters, mass over charge (m/z) and retention time, without prior knowledge of the metabolite’s chemical structure. This broad unbiased analysis, however, requires follow-up structure elucidation methods, such as tandem MS, nuclear magnetic resonance (NMR), or chemical synthesis for structural assignment of the changing metabolites.
Fig. 34.

DMP can identify metabolite peaks in LC-MS analyses with relative levels that are either increasing (A) or decreasing (B) between control and enzyme-disrupted samples. The increasing or decreasing peaks correspond to candidate substrates and products, respectively. Standard-free, relative quantitation is achieved by comparison to the many other peaks that are not changing (shown in grey).
In the original application, DMP was used to study tissues from FAAH(−/−) and FAAH(+/+) littermates.310,314 Prior to the DMP analysis, the in vivo biochemical role of FAAH was thought to be limited to the catabolism of the endocannabinoid anandamide (N-arachidonoylethanolamine) and its N-acylethanolamine (NAE) cogeners. DMP, in conjunction with chemical synthesis and tandem MS studies, identified a structurally novel class of metabolites, the fatty acyl-conjugated taurines, which were dramatically elevated in multiple tissues from FAAH(−/−) mice.311 In vitro, FAAH hydrolyzes N-acyltaurines, but in some cases with rates multiple orders of magnitude slower than for the NAE substrates, underscoring how poor in vitro substrates might still be regulated by an enzyme if alternative catabolic routes are absent in vivo.
For KIAA1363, a cancer-related SH highly elevated in multiple aggressive human cancer cell lines and primary tumors,73,77 DMP identified a family of monoalkylglycerol ethers (MAGEs) that were decreased following treatment with a selective inhibitor AS115.75 Consistent with the DMP results, KIAA1363 hydrolyzed acetate from 2-acetyl-MAGEs, and additional experiments demonstrated how KIAA1363, by producing MAGEs in these cells, could contribute to downstream metabolites, including the chemotaxic metabolite alkyl-lysophosphatidic acid and promote cancer pathogenesis.
Though in most cases the metabolic changes from enzyme disruption are proximal to the enzymatic reaction (i.e., elevation of substrates or decreases in products), DMP can also be used to identify secondary metabolic changes. In attempts to understand why MGLL is highly expressed in aggressive versus less aggressive human cancer cells and why its pharmacological or genetic perturbation decreases cancer cell proliferation, invasion, and tumor growth, DMP was applied to cancer cells where MGGL had been disrupted by RNA-interference or pharmacological methods.60 These studies revealed that, in addition to the expected changes in the proximal MGLL reaction of elevated MGs and decreased fatty acids, many other fatty acid-derived metabolites, including lysophosphatidic acid and eicosanoids, were also indirectly decreased following blockade of MGLL. DMP thus helped to build a model for MGLL action where this enzyme supports pathogenicity, at least in part, by regulating a fatty acid network enriched in pro-tumorigenic lipids.
A DMP-like platform has also been developed for the unbiased analysis of peptide substrates of SHs (“global peptide profiling”).442,443 This platform adapts chromatography and tandem MS conditions traditionally used in the proteomic identification of trypsin-digested proteins for the analysis of the “peptidome” fraction of biological samples. Global peptide profiling has helped to elucidate new renal peptide substrates for DPP4, a peptidase that had been highly studied in the context of GLP-1 regulation and glucose homeostasis but virtually uncharacterized in the kidney where its expression is very high.442 These experiments revealed multiple endogenous substrate or product peptides that were elevated in DPP4(−/−) and DPP4(+/+) kidneys, respectively, and established that DPP4 participates in the renal catabolism of peptides generated from protein catabolism. Global peptide profiling has also been applied to brain tissue from vehicle-treated or PREP inhibitor-treated mice to identify both known and previously unrecognized PREP-regulated peptides, as well as novel cleavage specificity in peptides containing multiple prolines.407
Although DMP has successfully been used to annotate the endogenous biochemical functions for multiple enzymes, the technology has several important limitations. First, DMP is far from comprehensive in its coverage of the metabolome, but instead is limited to the particular extractable portion of the metabolome under analysis (e.g. hydrophilic, lipophilic, peptides) and therefore SHs that act on protein or glycan substrates may not be amenable to these types of untargeted MS analyses. Second, as with any other mass spectrometry technology, DMP trades sensitivity for increased breadth and changes in low abundance metabolites may not be detectable. Despite these constraints, DMP nevertheless remains an important technology platform for the direct analysis of biochemical networks in vivo.
6. Summary and key outstanding questions
Here, we have attempted to comprehensively review all members of the mammalian metabolic SH superfamily, with special emphasis on understanding the biochemical and physiological functions of SHs in living systems and comparing these functions to the biochemical and enzymological properties displayed by SHs in vitro. We have also highlighted emerging technology platforms that, in conjunction with traditional biochemical and genetic techniques, are being used to illuminate the activities of poorly-characterized SHs. Moving forward, we suggest that there are three broad, yet related areas for future emphasis in SH research.
The first area is the continued development of selective chemical inhibitors to complement gene targeting studies for interrogating SH functions in vivo. While both substrate-dependent and substrate-independent platforms (e.g. competitive ABPP, clickable-ABPP probes) have greatly expanded the availability of lead small molecule inhibitors and advanced chemical probes,603 some SHs remain recalcitrant to these technologies, such as those that label poorly with existing FP probes (e.g., PPT1) or enzymes, often membrane-bound, that cannot yet be expressed and purified for fluopol-ABPP screening. Future modifications to the current ABPP platforms, such as modifications in probe design to improve activity and selectivity for individual SHs, might overcome some of these current limitations. Nevertheless, the available competitive and clickable ABPP technologies for broadly evaluating inhibitor selectivity across the proteome in a particular cell line or tissue will continue to become more integral to future inhibitor development programs.33,601 We also note that, for irreversible inhibitors of SHs, these competitive ABPP assays can also be readily applied to assess selectivity in vivo by, for instance, ex vivo profiling of SH activities in tissues taken from inhibitor-treated animals. One could even envision such a platform being adapted for confirming target engagement by inhibitors in human clinical trial studies by performing ex vivo competitive ABPP studies on patients’ blood samples.
The second future area of emphasis is on increasing the characterization of physiological substrates and products of SHs in addition to their in vitro biochemical substrates, an area particularly relevant for SHs where in vitro assays have not yet succeeded in identifying candidate natural substrates. Analyzing the metabolomic changes following selective pharmacological and/or genetic blockade of a given SH directly in cells or tissues may provide insight into unanticipated endogenous biochemical pathways. Such information can subsequently be used to create testable hypotheses for the physiological functions of SHs. For example, metabolomic profiling following disruption of ABHD2 or RBBP9, two SHs that have been shown to regulate smooth muscle migration and cancer cell growth, respectively, might reveal changes, such as an increase in chemotaxic molecules, which in turn might implicate particular biochemical pathways and connect these SH activities to higher order cellular properties.
The third area of future research emphasis relates to technology development. As illustrated by the DMP examples highlighted in this review, small molecules and peptides are particularly amenable to MS-based identification and analysis, and thus the biochemical functions of most SHs with small molecule or peptide substrates are, in principle, tractable with current technologies. However, exemplary SHs such as SIAE, AOAH, PPT1, and PPME1 already indicate that the substrate scope for this enzyme class extends well beyond low-molecular weight molecules to include macromolecular proteins and carbohydrates. Some progress has been made for profiling lipid and sugar modifications on proteins using metabolic labeling with clickable probes or chemoenzymatic tagging of these modifications with reporter groups.604–607 Yet, platforms for the broad and unbiased analysis of other important post-translational modifications (PTMs) on proteins and glycans remain lacking. For instance, how would one globally assess changes in protein methylation following disruption of an enzyme like PPME1? In the absence of such profiling tools, it will not only be difficult to outline the full scope of substrates for SHs like PPME1 or PPT1, but also to assign functions to additional members of the protein/glycan-acting subset of SHs. Creative strategies for the global analysis of these “macromolecular” substrates will provide a systematic tractability to an area where biochemical characterization has only been performed in a highly targeted way.
That only a handful of SHs have been “fully” characterized with regard to their in vitro biochemistry, endogenous substrate profile, and physiological functions underscores how far researchers are from understanding the full breadth of the metabolic and physiological activities for SHs in living systems. Nevertheless, the data available to date already suggests that SHs can profoundly influence human health and disease and designate these enzymes as being integral parts of most, if not all major physiological processes in mammals. In certain cases, the roles played by SHs in disease have translated into valuable therapeutics, such as inhibitors of DPP4 and cholinesterases for the treatment of type II diabetes and cognitive deficiencies in Alzheimer’s disease, respectively. The continued development of pharmacological, genetic, and proteomic/metabolomic methods for perturbing and annotating SH activities in biological systems should help to assign functions to members of this large and diverse enzyme superfamily and hopefully uncover new targets for treating human disorders with selective inhibitors.
Acknowledgments
We thank D. A. Bachovchin for helpful discussions and gratefully acknowledge the support of the NIH (Grants CA132630, DA017259, DA009789), the Helen L. Dorris Child and Adolescent Neuropsychiatric Disorder Institute, and the Skaggs Institute for Chemical Biology.
Biographies

Jonathan Long obtained his B.A. in biochemistry from Columbia University in 2007. He is currently pursuing his Ph.D. with B. F. Cravatt at The Scripps Research Institute. His research interests include inhibitor development and annotation of enzymes in the mammalian nervous system.

Benjamin Cravatt obtained his undergraduate education at Stanford University, receiving a B.S. in the Biological Sciences and a B.A. in History. He then trained with Drs. Dale Boger and Richard Lerner and received a Ph.D. in Macromolecular and Cellular Structure and Chemistry from The Scripps Research Institute (TSRI) in 1996. Professor Cravatt joined the faculty at TSRI in 1997 as a member of the Skaggs Institute for Chemical Biology and the Department of Chemical Physiology. His research group is interested in developing and applying new technologies to elucidate the roles that enzymes play in physiological and pathological processes, especially as pertains to the nervous system and cancer.
References
- 1.Simon GM, Cravatt BF. J Biol Chem. 2010;285:11051. doi: 10.1074/jbc.R109.097600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Powers JC, Asgian JL, Ekici OD, James KE. Chem Rev. 2002;102:4639. doi: 10.1021/cr010182v. [DOI] [PubMed] [Google Scholar]
- 3.Perona JJ, Craik CS. Protein Sci. 1995;4:337. doi: 10.1002/pro.5560040301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Siezen RJ, Leunissen JA. Protein Sci. 1997;6:501. doi: 10.1002/pro.5560060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barrett AJ, Rawlings ND, Woessner JF, editors. Handbook of Proteolytic Enzymes. Academic Press; Bath: 1998. [Google Scholar]
- 6.Dodson G, Wlodawer A. Trends Biochem Sci. 1998;23:347. doi: 10.1016/s0968-0004(98)01254-7. [DOI] [PubMed] [Google Scholar]
- 7.Patricelli MP, Lovato MA, Cravatt BF. Biochemistry. 1999;38:9804. doi: 10.1021/bi990637z. [DOI] [PubMed] [Google Scholar]
- 8.Abdel-Aal YA, Hammock BD. Science. 1986;233:1073. doi: 10.1126/science.3738525. [DOI] [PubMed] [Google Scholar]
- 9.Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB. Nature. 1996;384:83. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- 10.Liu Y, Patricelli MP, Cravatt BF. Proc Natl Acad Sci U S A. 1999;96:14694. doi: 10.1073/pnas.96.26.14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ollis DL, Cheah E, Cygler M, Dijkstra B, Frolow F, Franken SM, Harel M, Remington SJ, Silman I, Schrag J, Sussman JL, Verschueren KHG, Goldman A. Protein Eng. 1992;5:197. doi: 10.1093/protein/5.3.197. [DOI] [PubMed] [Google Scholar]
- 12.Six DA, Dennis EA. Biochim Biophys Acta. 2000;1488:1. doi: 10.1016/s1388-1981(00)00105-0. [DOI] [PubMed] [Google Scholar]
- 13.Yu DM, Yao TW, Chowdhury S, Nadvi NA, Osborne B, Church WB, McCaughan GW, Gorrell MD. FEBS J. 2010;277:1126. doi: 10.1111/j.1742-4658.2009.07526.x. [DOI] [PubMed] [Google Scholar]
- 14.Lane RM, Potkin SG, Enz A. Int J Neuropsychopharmacol. 2006;9:101. doi: 10.1017/S1461145705005833. [DOI] [PubMed] [Google Scholar]
- 15.Merkel M, Eckel RH, Goldberg IJ. J Lipid Res. 2002;43:1997. doi: 10.1194/jlr.r200015-jlr200. [DOI] [PubMed] [Google Scholar]
- 16.Cravatt BF, Demarest K, Patricelli MP, Bracey MH, Giang DK, Martin BR, Lichtman AH. Proc Natl Acad Sci U S A. 2001;98:9371. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonventre JV, Huang Z, Taheri MR, O’Leary E, Li E, Moskowitz MA, Sapirstein A. Nature. 1997;390:622. doi: 10.1038/37635. [DOI] [PubMed] [Google Scholar]
- 18.Kono N, Inoue T, Yoshida Y, Sato H, Matsusue T, Itabe H, Niki E, Aoki J, Arai H. J Biol Chem. 2008;283:1628. doi: 10.1074/jbc.M708622200. [DOI] [PubMed] [Google Scholar]
- 19.Menendez JA, Lupu R. Nat Rev Cancer. 2007;7:763. doi: 10.1038/nrc2222. [DOI] [PubMed] [Google Scholar]
- 20.Lu M, Varley AW, Ohta S, Hardwick J, Munford RS. Cell Host Microbe. 2008;4:293. doi: 10.1016/j.chom.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ahn K, McKinney MK, Cravatt BF. Chem Rev. 2008;108:1687. doi: 10.1021/cr0782067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dennis EA. J Biol Chem. 1994;269:13057. [PubMed] [Google Scholar]
- 23.Drucker DJ, Nauck MA. Lancet. 2006;368:1696. doi: 10.1016/S0140-6736(06)69705-5. [DOI] [PubMed] [Google Scholar]
- 24.Holm C, Kirchgessner TG, Svenson KL, Fredrikson G, Nilsson S, Miller CG, Shively JE, Heinzmann C, Sparkes RS, Mohandas T, Lusis AJ, Belfrage P, Schotz MC. Science. 1988;241:1503. doi: 10.1126/science.3420405. [DOI] [PubMed] [Google Scholar]
- 25.Reue K, Cohen RD, Schotz MC. Arterioscler Thromb Vasc Biol. 1997;17:3428. doi: 10.1161/01.atv.17.12.3428. [DOI] [PubMed] [Google Scholar]
- 26.Fredrikson G, Stralfors P, Nilsson NO, Belfrage P. J Biol Chem. 1981;256:6311. [PubMed] [Google Scholar]
- 27.Kraemer FB, Shen WJ. J Lipid Res. 2002;43:1585. doi: 10.1194/jlr.r200009-jlr200. [DOI] [PubMed] [Google Scholar]
- 28.Egan JJ, Greenberg AS, Chang MK, Wek SA, Moos MC, Jr, Londos C. Proc Natl Acad Sci U S A. 1992;89:8537. doi: 10.1073/pnas.89.18.8537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stralfors P, Bjorgell P, Belfrage P. Proc Natl Acad Sci U S A. 1984;81:3317. doi: 10.1073/pnas.81.11.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Haemmerle G, Zimmermann R, Hayn M, Theussl C, Waeg G, Wagner E, Sattler W, Magin TM, Wagner EF, Zechner R. J Biol Chem. 2002;277:4806. doi: 10.1074/jbc.M110355200. [DOI] [PubMed] [Google Scholar]
- 31.Wang SP, Laurin N, Himms-Hagen J, Rudnicki MA, Levy E, Robert MF, Pan L, Oligny L, Mitchell GA. Obes Res. 2001;9:119. doi: 10.1038/oby.2001.15. [DOI] [PubMed] [Google Scholar]
- 32.Osuga J, Ishibashi S, Oka T, Yagyu H, Tozawa R, Fujimoto A, Shionoiri F, Yahagi N, Kraemer FB, Tsutsumi O, Yamada N. Proc Natl Acad Sci U S A. 2000;97:787. doi: 10.1073/pnas.97.2.787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bachovchin DA, Ji T, Li W, Simon GM, Blankman JL, Adibekian A, Hoover H, Niessen S, Cravatt BF. Proc Natl Acad Sci U S A. 2010;107:20941. doi: 10.1073/pnas.1011663107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lowe DB, Magnuson S, Qi N, Campbell AM, Cook J, Hong Z, Wang M, Rodriguez M, Achebe F, Kluender H, Wong WC, Bullock WH, Salhanick AI, Witman-Jones T, Bowling ME, Keiper C, Clairmont KB. Bioorg Med Chem Lett. 2004;14:3155. doi: 10.1016/j.bmcl.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 35.Ebdrup S, Sorensen LG, Olsen OH, Jacobsen P. J Med Chem. 2004;47:400. doi: 10.1021/jm031004s. [DOI] [PubMed] [Google Scholar]
- 36.Slee DH, Bhat AS, Nguyen TN, Kish M, Lundeen K, Newman MJ, McConnell SJ. J Med Chem. 2003;46:1120. doi: 10.1021/jm020460y. [DOI] [PubMed] [Google Scholar]
- 37.Kienesberger PC, Oberer M, Lass A, Zechner R. J Lipid Res. 2009;50(Suppl):S63. doi: 10.1194/jlr.R800082-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Villena JA, Roy S, Sarkadi-Nagy E, Kim KH, Sul HS. J Biol Chem. 2004;279:47066. doi: 10.1074/jbc.M403855200. [DOI] [PubMed] [Google Scholar]
- 39.Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW. J Biol Chem. 2004;279:48968. doi: 10.1074/jbc.M407841200. [DOI] [PubMed] [Google Scholar]
- 40.Schaloske RH, Dennis EA. Biochim Biophys Acta. 2006;1761:1246. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 41.Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, Neuberger G, Eisenhaber F, Hermetter A, Zechner R. Science. 2004;306:1383. doi: 10.1126/science.1100747. [DOI] [PubMed] [Google Scholar]
- 42.Lass A, Zimmermann R, Haemmerle G, Riederer M, Schoiswohl G, Schweiger M, Kienesberger P, Strauss JG, Gorkiewicz G, Zechner R. Cell Metab. 2006;3:309. doi: 10.1016/j.cmet.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 43.Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, Heldmaier G, Maier R, Theussl C, Eder S, Kratky D, Wagner EF, Klingenspor M, Hoefler G, Zechner R. Science. 2006;312:734. doi: 10.1126/science.1123965. [DOI] [PubMed] [Google Scholar]
- 44.Karlsson M, Contreras JA, Hellman U, Tornqvist H, Holm C. J Biol Chem. 1997;272:27218. doi: 10.1074/jbc.272.43.27218. [DOI] [PubMed] [Google Scholar]
- 45.Long JZ, Nomura DK, Cravatt BF. Chem Biol. 2009;16:744. doi: 10.1016/j.chembiol.2009.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Labar G, Bauvois C, Borel F, Ferrer JL, Wouters J, Lambert DM. Chembiochem. 2010;11:218. doi: 10.1002/cbic.200900621. [DOI] [PubMed] [Google Scholar]
- 47.Bertrand T, Auge F, Houtmann J, Rak A, Vallee F, Mikol V, Berne PF, Michot N, Cheuret D, Hoornaert C, Mathieu M. J Mol Biol. 2010;396:663. doi: 10.1016/j.jmb.2009.11.060. [DOI] [PubMed] [Google Scholar]
- 48.Vaughan M, Berger JE, Steinberg D. J Biol Chem. 1964;239:401. [PubMed] [Google Scholar]
- 49.Stella N, Schweitzer P, Piomelli D. Nature. 1997;388:773. doi: 10.1038/42015. [DOI] [PubMed] [Google Scholar]
- 50.Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, Pertwee RG, Griffin G, Bayewitch M, Barg J, Vogel Z. Biochem Pharmacol. 1995;50:83. doi: 10.1016/0006-2952(95)00109-d. [DOI] [PubMed] [Google Scholar]
- 51.Sugiura T, Kondo S, Sukagawa A, Nakane S, Shinoda A, Itoh K, Yamashita A, Waku K. Biochem Biophys Res Commun. 1995;215:89. doi: 10.1006/bbrc.1995.2437. [DOI] [PubMed] [Google Scholar]
- 52.Matsuda LA, Lolait SJ, Brownstein MJ, Young AC, Bonner TI. Nature. 1990;346:561. doi: 10.1038/346561a0. [DOI] [PubMed] [Google Scholar]
- 53.Munro S, Thomas KL, Abu-Shaar M. Nature. 1993;365:61. doi: 10.1038/365061a0. [DOI] [PubMed] [Google Scholar]
- 54.Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavon FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF. Nat Chem Biol. 2009;5:37. doi: 10.1038/nchembio.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Long JZ, Jin X, Adibekian A, Li W, Cravatt BF. J Med Chem. 2010;53:1830. doi: 10.1021/jm9016976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, Nguyen PT, Ramesh D, Booker L, Burston JJ, Thomas EA, Selley DE, Sim-Selley LJ, Liu QS, Lichtman AH, Cravatt BF. Nat Neurosci. 2010;13:1113. doi: 10.1038/nn.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pan B, Wang W, Long JZ, Sun D, Hillard CJ, Cravatt BF, Liu QS. J Pharmacol Exp Ther. 2009;331:591. doi: 10.1124/jpet.109.158162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kinsey SG, Long JZ, O’Neal ST, Abdullah RA, Poklis JL, Boger DL, Cravatt BF, Lichtman AH. J Pharmacol Exp Ther. 2009;330:902. doi: 10.1124/jpet.109.155465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Little PJ, Compton DR, Johnson MR, Melvin LS, Martin BR. J Pharmacol Exp Ther. 1988;247:1046. [PubMed] [Google Scholar]
- 60.Nomura DK, Long JZ, Niessen S, Hoover HS, Ng SW, Cravatt BF. Cell. 2010;140:49. doi: 10.1016/j.cell.2009.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bisogno T, Howell F, Williams G, Minassi A, Cascio MG, Ligresti A, Matias I, Schiano-Moriello A, Paul P, Williams EJ, Gangadharan U, Hobbs C, Di Marzo V, Doherty P. J Cell Biol. 2003;163:463. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gao Y, Vasilyev DV, Goncalves MB, Howell FV, Hobbs C, Reisenberg M, Shen R, Zhang MY, Strassle BW, Lu P, Mark L, Piesla MJ, Deng K, Kouranova EV, Ring RH, Whiteside GT, Bates B, Walsh FS, Williams G, Pangalos MN, Samad TA, Doherty P. J Neurosci. 2010;30:2017. doi: 10.1523/JNEUROSCI.5693-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tanimura A, Yamazaki M, Hashimotodani Y, Uchigashima M, Kawata S, Abe M, Kita Y, Hashimoto K, Shimizu T, Watanabe M, Sakimura K, Kano M. Neuron. 2010;65:320. doi: 10.1016/j.neuron.2010.01.021. [DOI] [PubMed] [Google Scholar]
- 64.Hoover HS, Blankman JL, Niessen S, Cravatt BF. Bioorg Med Chem Lett. 2008;18:5838. doi: 10.1016/j.bmcl.2008.06.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hashimotodani Y, Ohno-Shosaku T, Kano M. J Neurosci. 2007;27:1211. doi: 10.1523/JNEUROSCI.4159-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dolinsky VW, Gilham D, Alam M, Vance DE, Lehner R. Cell Mol Life Sci. 2004;61:1633. doi: 10.1007/s00018-004-3426-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lehner R, Verger R. Biochemistry. 1997;36:1861. doi: 10.1021/bi962186d. [DOI] [PubMed] [Google Scholar]
- 68.Lehner R, Vance DE. Biochem J. 1999;343(Pt 1):1. [PMC free article] [PubMed] [Google Scholar]
- 69.Lehner R, Cui Z, Vance DE. Biochem J. 1999;338(Pt 3):761. [PMC free article] [PubMed] [Google Scholar]
- 70.Dolinsky VW, Sipione S, Lehner R, Vance DE. Biochim Biophys Acta. 2001;1532:162. doi: 10.1016/s1388-1981(01)00133-0. [DOI] [PubMed] [Google Scholar]
- 71.Wei E, Ben Ali Y, Lyon J, Wang H, Nelson R, Dolinsky VW, Dyck JR, Mitchell G, Korbutt GS, Lehner R. Cell Metab. 2010;11:183. doi: 10.1016/j.cmet.2010.02.005. [DOI] [PubMed] [Google Scholar]
- 72.Sekiya M, Osuga J, Nagashima S, Ohshiro T, Igarashi M, Okazaki H, Takahashi M, Tazoe F, Wada T, Ohta K, Takanashi M, Kumagai M, Nishi M, Takase S, Yahagi N, Yagyu H, Ohashi K, Nagai R, Kadowaki T, Furukawa Y, Ishibashi S. Cell Metab. 2009;10:219. doi: 10.1016/j.cmet.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 73.Jessani N, Liu Y, Humphrey M, Cravatt BF. Proc Natl Acad Sci U S A. 2002;99:10335. doi: 10.1073/pnas.162187599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Okazaki H, Igarashi M, Nishi M, Sekiya M, Tajima M, Takase S, Takanashi M, Ohta K, Tamura Y, Okazaki S, Yahagi N, Ohashi K, Amemiya-Kudo M, Nakagawa Y, Nagai R, Kadowaki T, Osuga J, Ishibashi S. J Biol Chem. 2008;283:33357. doi: 10.1074/jbc.M802686200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chiang KP, Niessen S, Saghatelian A, Cravatt BF. Chem Biol. 2006;13:1041. doi: 10.1016/j.chembiol.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 76.Buchebner M, Pfeifer T, Rathke N, Chandak PG, Lass A, Schreiber R, Kratzer A, Zimmermann R, Sattler W, Koefeler H, Frohlich E, Kostner GM, Birner-Gruenberger R, Chiang KP, Haemmerle G, Zechner R, Levak-Frank S, Cravatt B, Kratky D. J Lipid Res. 2010;51:2896. doi: 10.1194/jlr.M004259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jessani N, Niessen S, Wei BQ, Nicolau M, Humphrey M, Ji Y, Han W, Noh DY, Yates JR, 3rd, Jeffrey SS, Cravatt BF. Nat Methods. 2005;2:691. doi: 10.1038/nmeth778. [DOI] [PubMed] [Google Scholar]
- 78.Haverty PM, Hon LS, Kaminker JS, Chant J, Zhang Z. BMC Med Genomics. 2009;2:21. doi: 10.1186/1755-8794-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chang JW, Nomura DK, Cravatt BF. Chem Biol. 2011;18:476. doi: 10.1016/j.chembiol.2011.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Blank ML, Smith ZL, Cress EA, Snyder F. Biochim Biophys Acta. 1990;1042:153. doi: 10.1016/0005-2760(90)90001-e. [DOI] [PubMed] [Google Scholar]
- 81.Roos DS, Choppin PW. Proc Natl Acad Sci U S A. 1984;81:7622. doi: 10.1073/pnas.81.23.7622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nomura DK, Fujioka K, Issa RS, Ward AM, Cravatt BF, Casida JE. Toxicol Appl Pharmacol. 2008;228:42. doi: 10.1016/j.taap.2007.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Igarashi M, Osuga J, Uozaki H, Sekiya M, Nagashima S, Takahashi M, Takase S, Takanashi M, Li Y, Ohta K, Kumagai M, Nishi M, Hosokawa M, Fledelius C, Jacobsen P, Yagyu H, Fukayama M, Nagai R, Kadowaki T, Ohashi K, Ishibashi S. Circ Res. 2010;107:1387. doi: 10.1161/CIRCRESAHA.110.226613. [DOI] [PubMed] [Google Scholar]
- 84.Nomura DK, Leung D, Chiang KP, Quistad GB, Cravatt BF, Casida JE. Proc Natl Acad Sci U S A. 2005;102:6195. doi: 10.1073/pnas.0501915102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Blankman JL, Simon GM, Cravatt BF. Chem Biol. 2007;14:1347. doi: 10.1016/j.chembiol.2007.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li F, Fei X, Xu J, Ji C. Mol Biol Rep. 2009;36:691. doi: 10.1007/s11033-008-9230-7. [DOI] [PubMed] [Google Scholar]
- 87.Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, Bodor AL, Muccioli GG, Hu SS, Woodruff G, Fung S, Lafourcade M, Alexander JP, Long JZ, Li W, Xu C, Moller T, Mackie K, Manzoni OJ, Cravatt BF, Stella N. Nat Neurosci. 2010;13:951. doi: 10.1038/nn.2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Long JZ, Nomura DK, Vann RE, Walentiny DM, Booker L, Jin X, Burston JJ, Sim-Selley LJ, Lichtman AH, Wiley JL, Cravatt BF. Proc Natl Acad Sci U S A. 2009;106:20270. doi: 10.1073/pnas.0909411106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Fiskerstrand T, H’Mida-Ben Brahim D, Johansson S, M’Zahem A, Haukanes BI, Drouot N, Zimmermann J, Cole AJ, Vedeler C, Bredrup C, Assoum M, Tazir M, Klockgether T, Hamri A, Steen VM, Boman H, Bindoff LA, Koenig M, Knappskog PM. Am J Hum Genet. 2010;87:410. doi: 10.1016/j.ajhg.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Aloulou A, Carriere F. Cell Mol Life Sci. 2008;65:851. doi: 10.1007/s00018-008-7546-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hui DY, Howles PN. J Lipid Res. 2002;43:2017. doi: 10.1194/jlr.r200013-jlr200. [DOI] [PubMed] [Google Scholar]
- 92.Borgstrom B, Erlanson-Albertsson C. J Clin Invest. 1982;70:30. doi: 10.1172/JCI110599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Roussel A, Yang Y, Ferrato F, Verger R, Cambillau C, Lowe M. J Biol Chem. 1998;273:32121. doi: 10.1074/jbc.273.48.32121. [DOI] [PubMed] [Google Scholar]
- 94.Mead JR, Irvine SA, Ramji DP. J Mol Med. 2002;80:753. doi: 10.1007/s00109-002-0384-9. [DOI] [PubMed] [Google Scholar]
- 95.Connelly PW. Clin Chim Acta. 1999;286:243. doi: 10.1016/s0009-8981(99)00105-9. [DOI] [PubMed] [Google Scholar]
- 96.Jonas A. Biochim Biophys Acta. 2000;1529:245. doi: 10.1016/s1388-1981(00)00153-0. [DOI] [PubMed] [Google Scholar]
- 97.Goldstein JL, Dana SE, Faust JR, Beaudet AL, Brown MS. J Biol Chem. 1975;250:8487. [PubMed] [Google Scholar]
- 98.Huggins KW, Camarota LM, Howles PN, Hui DY. J Biol Chem. 2003;278:42899. doi: 10.1074/jbc.M303422200. [DOI] [PubMed] [Google Scholar]
- 99.Bernback S, Blackberg L, Hernell O. J Clin Invest. 1990;85:1221. doi: 10.1172/JCI114556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Lowe ME, Rosenblum JL, Strauss AW. J Biol Chem. 1989;264:20042. [PubMed] [Google Scholar]
- 101.Lowe ME. Annu Rev Nutr. 1997;17:141. doi: 10.1146/annurev.nutr.17.1.141. [DOI] [PubMed] [Google Scholar]
- 102.Yang LY, Kuksis A, Myher JJ. J Lipid Res. 1990;31:137. [PubMed] [Google Scholar]
- 103.Whitcomb DC, Lowe ME. Dig Dis Sci. 2007;52:1. doi: 10.1007/s10620-006-9589-z. [DOI] [PubMed] [Google Scholar]
- 104.Borgstrom B, Erlanson C. Biochim Biophys Acta. 1971;242:509. [PubMed] [Google Scholar]
- 105.Sternby B, Engstrom A, Hellman U, Vihert AM, Sternby NH, Borgstrom B. Biochim Biophys Acta. 1984;784:75. doi: 10.1016/0167-4838(84)90175-4. [DOI] [PubMed] [Google Scholar]
- 106.van Tilbeurgh H, Egloff MP, Martinez C, Rugani N, Verger R, Cambillau C. Nature. 1993;362:814. doi: 10.1038/362814a0. [DOI] [PubMed] [Google Scholar]
- 107.Gaskin KJ, Durie PR, Hill RE, Lee LM, Forstner GG. J Clin Invest. 1982;69:427. doi: 10.1172/JCI110466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Figarella C, De Caro A, Leupold D, Poley JR. J Pediatr. 1980;96:412. doi: 10.1016/s0022-3476(80)80683-4. [DOI] [PubMed] [Google Scholar]
- 109.Sheldon W. Arch Dis Child. 1964;39:268. doi: 10.1136/adc.39.205.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hadvary P, Lengsfeld H, Wolfer H. Biochem J. 1988;256:357. doi: 10.1042/bj2560357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sjostrom L, Rissanen A, Andersen T, Boldrin M, Golay A, Koppeschaar HP, Krempf M. Lancet. 1998;352:167. doi: 10.1016/s0140-6736(97)11509-4. [DOI] [PubMed] [Google Scholar]
- 112.Borgstrom B. Biochim Biophys Acta. 1988;962:308. doi: 10.1016/0005-2760(88)90260-3. [DOI] [PubMed] [Google Scholar]
- 113.Grusby MJ, Nabavi N, Wong H, Dick RF, Bluestone JA, Schotz MC, Glimcher LH. Cell. 1990;60:451. doi: 10.1016/0092-8674(90)90596-7. [DOI] [PubMed] [Google Scholar]
- 114.Giller T, Buchwald P, Blum-Kaelin D, Hunziker W. J Biol Chem. 1992;267:16509. [PubMed] [Google Scholar]
- 115.Thirstrup K, Verger R, Carriere F. Biochemistry. 1994;33:2748. doi: 10.1021/bi00176a002. [DOI] [PubMed] [Google Scholar]
- 116.Jennens ML, Lowe ME. J Lipid Res. 1995;36:2374. [PubMed] [Google Scholar]
- 117.Payne RM, Sims HF, Jennens ML, Lowe ME. Am J Physiol. 1994;266:G914. doi: 10.1152/ajpgi.1994.266.5.G914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lowe ME, Kaplan MH, Jackson-Grusby L, D’Agostino D, Grusby MJ. J Biol Chem. 1998;273:31215. doi: 10.1074/jbc.273.47.31215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Howles PN, Carter CP, Hui DY. J Biol Chem. 1996;271:7196. doi: 10.1074/jbc.271.12.7196. [DOI] [PubMed] [Google Scholar]
- 120.Blackberg L, Hernell O. Eur J Biochem. 1981;116:221. doi: 10.1111/j.1432-1033.1981.tb05322.x. [DOI] [PubMed] [Google Scholar]
- 121.Lombardo D. Biochim Biophys Acta. 2001;1533:1. doi: 10.1016/s1388-1981(01)00130-5. [DOI] [PubMed] [Google Scholar]
- 122.Wang CS. Biochem J. 1991;279(Pt 1):297. doi: 10.1042/bj2790297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nilsson J, Blackberg L, Carlsson P, Enerback S, Hernell O, Bjursell G. Eur J Biochem. 1990;192:543. doi: 10.1111/j.1432-1033.1990.tb19259.x. [DOI] [PubMed] [Google Scholar]
- 124.Rigtrup KM, Ong DE. Biochemistry. 1992;31:2920. doi: 10.1021/bi00126a011. [DOI] [PubMed] [Google Scholar]
- 125.Hui DY, Hayakawa K, Oizumi J. Biochem J. 1993;291(Pt 1):65. doi: 10.1042/bj2910065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang CS, Hartsuck JA, Downs D. Biochemistry. 1988;27:4834. doi: 10.1021/bi00413a038. [DOI] [PubMed] [Google Scholar]
- 127.Mackay K, Starr JR, Lawn RM, Ellsworth JL. J Biol Chem. 1997;272:13380. doi: 10.1074/jbc.272.20.13380. [DOI] [PubMed] [Google Scholar]
- 128.Hernell O. Eur J Clin Invest. 1975;5:267. doi: 10.1111/j.1365-2362.1975.tb02294.x. [DOI] [PubMed] [Google Scholar]
- 129.Wang CS, Martindale ME, King MM, Tang J. Am J Clin Nutr. 1989;49:457. doi: 10.1093/ajcn/49.3.457. [DOI] [PubMed] [Google Scholar]
- 130.Howles PN, Stemmerman GN, Fenoglio-Preiser CM, Hui DY. Am J Physiol. 1999;277:G653. doi: 10.1152/ajpgi.1999.277.3.G653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Miller R, Lowe ME. J Nutr. 2008;138:927. doi: 10.1093/jn/138.5.927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kirby RJ, Zheng S, Tso P, Howles PN, Hui DY. J Biol Chem. 2002;277:4104. doi: 10.1074/jbc.M107549200. [DOI] [PubMed] [Google Scholar]
- 133.Bodmer MW, Angal S, Yarranton GT, Harris TJ, Lyons A, King DJ, Pieroni G, Riviere C, Verger R, Lowe PA. Biochim Biophys Acta. 1987;909:237. doi: 10.1016/0167-4781(87)90083-2. [DOI] [PubMed] [Google Scholar]
- 134.Moreau H, Bernadac A, Gargouri Y, Benkouka F, Laugier R, Verger R. Histochemistry. 1989;91:419. doi: 10.1007/BF00493829. [DOI] [PubMed] [Google Scholar]
- 135.Roussel A, Canaan S, Egloff MP, Riviere M, Dupuis L, Verger R, Cambillau C. J Biol Chem. 1999;274:16995. doi: 10.1074/jbc.274.24.16995. [DOI] [PubMed] [Google Scholar]
- 136.Chahinian H, Snabe T, Attias C, Fojan P, Petersen SB, Carriere F. Biochemistry. 2006;45:993. doi: 10.1021/bi0518803. [DOI] [PubMed] [Google Scholar]
- 137.Carriere F, Barrowman JA, Verger R, Laugier R. Gastroenterology. 1993;105:876. doi: 10.1016/0016-5085(93)90908-u. [DOI] [PubMed] [Google Scholar]
- 138.Wion KL, Kirchgessner TG, Lusis AJ, Schotz MC, Lawn RM. Science. 1987;235:1638. doi: 10.1126/science.3823907. [DOI] [PubMed] [Google Scholar]
- 139.Semenkovich CF, Chen SH, Wims M, Luo CC, Li WH, Chan L. J Lipid Res. 1989;30:423. [PubMed] [Google Scholar]
- 140.Paradis E, Clavel S, Julien P, Murthy MR, de Bilbao F, Arsenijevic D, Giannakopoulos P, Vallet P, Richard D. Neurobiol Dis. 2004;15:312. doi: 10.1016/j.nbd.2003.09.017. [DOI] [PubMed] [Google Scholar]
- 141.Braun JE, Severson DL. Biochem J. 1992;287(Pt 2):337. doi: 10.1042/bj2870337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Scanu A. Science. 1966;153:640. doi: 10.1126/science.153.3736.640. [DOI] [PubMed] [Google Scholar]
- 143.Ameis D, Kobayashi J, Davis RC, Ben-Zeev O, Malloy MJ, Kane JP, Lee G, Wong H, Havel RJ, Schotz MC. J Clin Invest. 1991;87:1165. doi: 10.1172/JCI115114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Cryer A. Int J Biochem. 1981;13:525. doi: 10.1016/0020-711x(81)90177-4. [DOI] [PubMed] [Google Scholar]
- 145.Mailly F, Palmen J, Muller DP, Gibbs T, Lloyd J, Brunzell J, Durrington P, Mitropoulos K, Betteridge J, Watts G, Lithell H, Angelico F, Humphries SE, Talmud PJ. Hum Mutat. 1997;10:465. doi: 10.1002/(SICI)1098-1004(1997)10:6<465::AID-HUMU8>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 146.Baggio G, Manzato E, Gabelli C, Fellin R, Martini S, Enzi GB, Verlato F, Baiocchi MR, Sprecher DL, Kashyap ML. J Clin Invest. 1986;77:520. doi: 10.1172/JCI112332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Weinstock PH, Bisgaier CL, Aalto-Setala K, Radner H, Ramakrishnan R, Levak-Frank S, Essenburg AD, Zechner R, Breslow JL. J Clin Invest. 1995;96:2555. doi: 10.1172/JCI118319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Goodman KB, Bury MJ, Cheung M, Cichy-Knight MA, Dowdell SE, Dunn AK, Lee D, Lieby JA, Moore ML, Scherzer DA, Sha D, Suarez DP, Murphy DJ, Harpel MR, Manas ES, McNulty DE, Annan RS, Matico RE, Schwartz BK, Trill JJ, Sweitzer TD, Wang DY, Keller PM, Krawiec JA, Jaye MC. Bioorg Med Chem Lett. 2009;19:27. doi: 10.1016/j.bmcl.2008.11.033. [DOI] [PubMed] [Google Scholar]
- 149.Goldberg IJ, Le NA, Paterniti JR, Jr, Ginsberg HN, Lindgren FT, Brown WV. J Clin Invest. 1982;70:1184. doi: 10.1172/JCI110717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kuusi T, Kinnunen PK, Nikkila EA. FEBS Lett. 1979;104:384. doi: 10.1016/0014-5793(79)80858-3. [DOI] [PubMed] [Google Scholar]
- 151.LaRosa JC, Levy RI, Windmueller HG, Fredrickson DS. J Lipid Res. 1972;13:356. [PubMed] [Google Scholar]
- 152.Ehnholm C, Shaw W, Greten H, Brown WV. J Biol Chem. 1975;250:6756. [PubMed] [Google Scholar]
- 153.Connelly PW, Maguire GF, Lee M, Little JA. Arteriosclerosis. 1990;10:40. doi: 10.1161/01.atv.10.1.40. [DOI] [PubMed] [Google Scholar]
- 154.Breckenridge WC, Little JA, Alaupovic P, Wang CS, Kuksis A, Kakis G, Lindgren F, Gardiner G. Atherosclerosis. 1982;45:161. doi: 10.1016/0021-9150(82)90136-8. [DOI] [PubMed] [Google Scholar]
- 155.Carlson LA, Holmquist L, Nilsson-Ehle P. Acta Med Scand. 1986;219:435. doi: 10.1111/j.0954-6820.1986.tb03337.x. [DOI] [PubMed] [Google Scholar]
- 156.Grosser J, Schrecker O, Greten H. J Lipid Res. 1981;22:437. [PubMed] [Google Scholar]
- 157.Ameis D, Merkel M, Eckerskorn C, Greten H. Eur J Biochem. 1994;219:905. doi: 10.1111/j.1432-1033.1994.tb18572.x. [DOI] [PubMed] [Google Scholar]
- 158.Du H, Witte DP, Grabowski GA. J Lipid Res. 1996;37:937. [PubMed] [Google Scholar]
- 159.Anderson RA, Sando GN. J Biol Chem. 1991;266:22479. [PubMed] [Google Scholar]
- 160.Sheriff S, Du H, Grabowski GA. J Biol Chem. 1995;270:27766. doi: 10.1074/jbc.270.46.27766. [DOI] [PubMed] [Google Scholar]
- 161.Zschenker O, Illies T, Ameis D. J Biochem. 2006;140:23. doi: 10.1093/jb/mvj137. [DOI] [PubMed] [Google Scholar]
- 162.Sugii S, Reid PC, Ohgami N, Du H, Chang TY. J Biol Chem. 2003;278:27180. doi: 10.1074/jbc.M300542200. [DOI] [PubMed] [Google Scholar]
- 163.Aslanidis C, Ries S, Fehringer P, Buchler C, Klima H, Schmitz G. Genomics. 1996;33:85. doi: 10.1006/geno.1996.0162. [DOI] [PubMed] [Google Scholar]
- 164.Anderson RA, Byrum RS, Coates PM, Sando GN. Proc Natl Acad Sci U S A. 1994;91:2718. doi: 10.1073/pnas.91.7.2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Hoeg JM, Demosky SJ, Jr, Pescovitz OH, Brewer HB., Jr Am J Hum Genet. 1984;36:1190. [PMC free article] [PubMed] [Google Scholar]
- 166.Du H, Heur M, Duanmu M, Grabowski GA, Hui DY, Witte DP, Mishra J. J Lipid Res. 2001;42:489. [PubMed] [Google Scholar]
- 167.Du H, Duanmu M, Witte D, Grabowski GA. Hum Mol Genet. 1998;7:1347. doi: 10.1093/hmg/7.9.1347. [DOI] [PubMed] [Google Scholar]
- 168.Rosenbaum AI, Cosner CC, Mariani CJ, Maxfield FR, Wiest O, Helquist P. J Med Chem. 2010;53:5281. doi: 10.1021/jm100499s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Glomset JA. Biochim Biophys Acta. 1962;65:128. doi: 10.1016/0006-3002(62)90156-7. [DOI] [PubMed] [Google Scholar]
- 170.McLean J, Fielding C, Drayna D, Dieplinger H, Baer B, Kohr W, Henzel W, Lawn R. Proc Natl Acad Sci U S A. 1986;83:2335. doi: 10.1073/pnas.83.8.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Fielding CJ, Shore VG, Fielding PE. Biochem Biophys Res Commun. 1972;46:1493. doi: 10.1016/0006-291x(72)90776-0. [DOI] [PubMed] [Google Scholar]
- 172.Subbaiah PV, Albers JJ, Chen CH, Bagdade JD. J Biol Chem. 1980;255:9275. [PubMed] [Google Scholar]
- 173.Lavallee B, Provost PR, Belanger A. Biochim Biophys Acta. 1996;1299:306. doi: 10.1016/0005-2760(95)00222-7. [DOI] [PubMed] [Google Scholar]
- 174.Szedlacsek SE, Wasowicz E, Hulea SA, Nishida HI, Kummerow FA, Nishida T. J Biol Chem. 1995;270:11812. doi: 10.1074/jbc.270.20.11812. [DOI] [PubMed] [Google Scholar]
- 175.Kuivenhoven JA, Pritchard H, Hill J, Frohlich J, Assmann G, Kastelein J. J Lipid Res. 1997;38:191. [PubMed] [Google Scholar]
- 176.Glomset JA, Norum KR, King W. J Clin Invest. 1970;49:1827. doi: 10.1172/JCI106400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Norum KR, Gjone E. Biochim Biophys Acta. 1967;144:698. doi: 10.1016/0005-2760(67)90064-1. [DOI] [PubMed] [Google Scholar]
- 178.Ng DS, Francone OL, Forte TM, Zhang J, Haghpassand M, Rubin EM. J Biol Chem. 1997;272:15777. doi: 10.1074/jbc.272.25.15777. [DOI] [PubMed] [Google Scholar]
- 179.Clark JD, Lin LL, Kriz RW, Ramesha CS, Sultzman LA, Lin AY, Milona N, Knopf JL. Cell. 1991;65:1043. doi: 10.1016/0092-8674(91)90556-e. [DOI] [PubMed] [Google Scholar]
- 180.Sharp JD, White DL, Chiou XG, Goodson T, Gamboa GC, McClure D, Burgett S, Hoskins J, Skatrud PL, Sportsman JR, Becker GW, Kang LH, Roberts EF, Kramer RM. J Biol Chem. 1991;266:14850. [PubMed] [Google Scholar]
- 181.Su AI, Cooke MP, Ching KA, Hakak Y, Walker JR, Wiltshire T, Orth AP, Vega RG, Sapinoso LM, Moqrich A, Patapoutian A, Hampton GM, Schultz PG, Hogenesch JB. Proc Natl Acad Sci U S A. 2002;99:4465. doi: 10.1073/pnas.012025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Diez E, Louis-Flamberg P, Hall RH, Mayer RJ. J Biol Chem. 1992;267:18342. [PubMed] [Google Scholar]
- 183.Reynolds LJ, Hughes LL, Louis AI, Kramer RM, Dennis EA. Biochim Biophys Acta. 1993;1167:272. doi: 10.1016/0005-2760(93)90229-3. [DOI] [PubMed] [Google Scholar]
- 184.Vane JR, Bakhle YS, Botting RM. Annu Rev Pharmacool Toxicol. 1998;38:97. doi: 10.1146/annurev.pharmtox.38.1.97. [DOI] [PubMed] [Google Scholar]
- 185.Uozumi N, Kume K, Nagase T, Nakatani N, Ishii S, Tashiro F, Komagata Y, Maki K, Ikuta K, Ouchi Y, Miyazaki J, Shimizu T. Nature. 1997;390:618. doi: 10.1038/37622. [DOI] [PubMed] [Google Scholar]
- 186.Sapirstein A, Bonventre JV. Biochim Biophys Acta. 2000;1488:139. doi: 10.1016/s1388-1981(00)00116-5. [DOI] [PubMed] [Google Scholar]
- 187.Fujishima H, Sanchez Mejia RO, Bingham CO, 3rd, Lam BK, Sapirstein A, Bonventre JV, Austen KF, Arm JP. Proc Natl Acad Sci U S A. 1999;96:4803. doi: 10.1073/pnas.96.9.4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Rosenberger TA, Villacreses NE, Contreras MA, Bonventre JV, Rapoport SI. J Lipid Res. 2003;44:109. doi: 10.1194/jlr.m200298-jlr200. [DOI] [PubMed] [Google Scholar]
- 189.Gopalsamy A, Yang H, Ellingboe JW, McKew JC, Tam S, Joseph-McCarthy D, Zhang W, Shen M, Clark JD. Bioorg Med Chem Lett. 2006;16:2978. doi: 10.1016/j.bmcl.2006.02.067. [DOI] [PubMed] [Google Scholar]
- 190.Kirincich SJ, Xiang J, Green N, Tam S, Yang HY, Shim J, Shen MW, Clark JD, McKew JC. Bioorg Med Chem. 2009;17:4383. doi: 10.1016/j.bmc.2009.05.027. [DOI] [PubMed] [Google Scholar]
- 191.Lee KL, Behnke ML, Foley MA, Chen L, Wang W, Vargas R, Nunez J, Tam S, Mollova N, Xu X, Shen MW, Ramarao MK, Goodwin DG, Nickerson-Nutter CL, Abraham WM, Williams C, Clark JD, McKew JC. Bioorg Med Chem. 2008;16:1345. doi: 10.1016/j.bmc.2007.10.060. [DOI] [PubMed] [Google Scholar]
- 192.Lee KL, Foley MA, Chen L, Behnke ML, Lovering FE, Kirincich SJ, Wang W, Shim J, Tam S, Shen MW, Khor S, Xu X, Goodwin DG, Ramarao MK, Nickerson-Nutter C, Donahue F, Ku MS, Clark JD, McKew JC. J Med Chem. 2007;50:1380. doi: 10.1021/jm061131z. [DOI] [PubMed] [Google Scholar]
- 193.McKew JC, Foley MA, Thakker P, Behnke ML, Lovering FE, Sum FW, Tam S, Wu K, Shen MW, Zhang W, Gonzalez M, Liu S, Mahadevan A, Sard H, Khor SP, Clark JD. J Med Chem. 2006;49:135. doi: 10.1021/jm0507882. [DOI] [PubMed] [Google Scholar]
- 194.McKew JC, Lee KL, Shen MW, Thakker P, Foley MA, Behnke ML, Hu B, Sum FW, Tam S, Hu Y, Chen L, Kirincich SJ, Michalak R, Thomason J, Ipek M, Wu K, Wooder L, Ramarao MK, Murphy EA, Goodwin DG, Albert L, Xu X, Donahue F, Ku MS, Keith J, Nickerson-Nutter CL, Abraham WM, Williams C, Hegen M, Clark JD. J Med Chem. 2008;51:3388. doi: 10.1021/jm701467e. [DOI] [PubMed] [Google Scholar]
- 195.McKew JC, Lovering F, Clark JD, Bemis J, Xiang Y, Shen M, Zhang W, Alvarez JC, Joseph-McCarthy D. Bioorg Med Chem Lett. 2003;13:4501. doi: 10.1016/j.bmcl.2003.08.070. [DOI] [PubMed] [Google Scholar]
- 196.Song C, Chang XJ, Bean KM, Proia MS, Knopf JL, Kriz RW. J Biol Chem. 1999;274:17063. doi: 10.1074/jbc.274.24.17063. [DOI] [PubMed] [Google Scholar]
- 197.Pickard RT, Strifler BA, Kramer RM, Sharp JD. J Biol Chem. 1999;274:8823. doi: 10.1074/jbc.274.13.8823. [DOI] [PubMed] [Google Scholar]
- 198.Ghosh M, Loper R, Gelb MH, Leslie CC. J Biol Chem. 2006;281:16615. doi: 10.1074/jbc.M601770200. [DOI] [PubMed] [Google Scholar]
- 199.Yamashita A, Tanaka K, Kamata R, Kumazawa T, Suzuki N, Koga H, Waku K, Sugiura T. Biochim Biophys Acta. 2009;1791:1011. doi: 10.1016/j.bbalip.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 200.Asai K, Hirabayashi T, Houjou T, Uozumi N, Taguchi R, Shimizu T. J Biol Chem. 2003;278:8809. doi: 10.1074/jbc.M212117200. [DOI] [PubMed] [Google Scholar]
- 201.Stewart A, Ghosh M, Spencer DM, Leslie CC. J Biol Chem. 2002;277:29526. doi: 10.1074/jbc.M204856200. [DOI] [PubMed] [Google Scholar]
- 202.Chiba H, Michibata H, Wakimoto K, Seishima M, Kawasaki S, Okubo K, Mitsui H, Torii H, Imai Y. J Biol Chem. 2004;279:12890. doi: 10.1074/jbc.M305801200. [DOI] [PubMed] [Google Scholar]
- 203.Ohto T, Uozumi N, Hirabayashi T, Shimizu T. J Biol Chem. 2005;280:24576. doi: 10.1074/jbc.M413711200. [DOI] [PubMed] [Google Scholar]
- 204.Ghosh M, Loper R, Ghomashchi F, Tucker DE, Bonventre JV, Gelb MH, Leslie CC. J Biol Chem. 2007;282:11676. doi: 10.1074/jbc.M608458200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ghomashchi F, Naika GS, Bollinger JG, Aloulou A, Lehr M, Leslie CC, Gelb MH. J Biol Chem. 2010;285:36100. doi: 10.1074/jbc.M110.165647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Ackermann EJ, Kempner ES, Dennis EA. J Biol Chem. 1994;269:9227. [PubMed] [Google Scholar]
- 207.Tang J, Kriz RW, Wolfman N, Shaffer M, Seehra J, Jones SS. J Biol Chem. 1997;272:8567. doi: 10.1074/jbc.272.13.8567. [DOI] [PubMed] [Google Scholar]
- 208.Ma Z, Wang X, Nowatzke W, Ramanadham S, Turk J. J Biol Chem. 1999;274:9607. doi: 10.1074/jbc.274.14.9607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Winstead MV, Balsinde J, Dennis EA. Biochim Biophys Acta. 2000;1488:28. doi: 10.1016/s1388-1981(00)00107-4. [DOI] [PubMed] [Google Scholar]
- 210.Lio YC, Dennis EA. Biochim Biophys Acta. 1998;1392:320. doi: 10.1016/s0005-2760(98)00049-6. [DOI] [PubMed] [Google Scholar]
- 211.Bao S, Miller DJ, Ma Z, Wohltmann M, Eng G, Ramanadham S, Moley K, Turk J. J Biol Chem. 2004;279:38194. doi: 10.1074/jbc.M406489200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Shinzawa K, Sumi H, Ikawa M, Matsuoka Y, Okabe M, Sakoda S, Tsujimoto Y. J Neurosci. 2008;28:2212. doi: 10.1523/JNEUROSCI.4354-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Morgan NV, Westaway SK, Morton JE, Gregory A, Gissen P, Sonek S, Cangul H, Coryell J, Canham N, Nardocci N, Zorzi G, Pasha S, Rodriguez D, Desguerre I, Mubaidin A, Bertini E, Trembath RC, Simonati A, Schanen C, Johnson CA, Levinson B, Woods CG, Wilmot B, Kramer P, Gitschier J, Maher ER, Hayflick SJ. Nat Genet. 2006;38:752. doi: 10.1038/ng1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Khateeb S, Flusser H, Ofir R, Shelef I, Narkis G, Vardi G, Shorer Z, Levy R, Galil A, Elbedour K, Birk OS. Am J Hum Genet. 2006;79:942. doi: 10.1086/508572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Engel LA, Jing Z, O’Brien DE, Sun M, Kotzbauer PT. PLoS One. 2010;5:e12897. doi: 10.1371/journal.pone.0012897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Wada H, Yasuda T, Miura I, Watabe K, Sawa C, Kamijuku H, Kojo S, Taniguchi M, Nishino I, Wakana S, Yoshida H, Seino K. Am J Pathol. 2009;175:2257. doi: 10.2353/ajpath.2009.090343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Underwood KW, Song C, Kriz RW, Chang XJ, Knopf JL, Lin LL. J Biol Chem. 1998;273:21926. doi: 10.1074/jbc.273.34.21926. [DOI] [PubMed] [Google Scholar]
- 218.Mancuso DJ, Jenkins CM, Gross RW. J Biol Chem. 2000;275:9937. doi: 10.1074/jbc.275.14.9937. [DOI] [PubMed] [Google Scholar]
- 219.Yan W, Jenkins CM, Han X, Mancuso DJ, Sims HF, Yang K, Gross RW. J Biol Chem. 2005;280:26669. doi: 10.1074/jbc.M502358200. [DOI] [PubMed] [Google Scholar]
- 220.Mancuso DJ, Han X, Jenkins CM, Lehman JJ, Sambandam N, Sims HF, Yang J, Yan W, Yang K, Green K, Abendschein DR, Saffitz JE, Gross RW. J Biol Chem. 2007;282:9216. doi: 10.1074/jbc.M607307200. [DOI] [PubMed] [Google Scholar]
- 221.Mancuso DJ, Sims HF, Han X, Jenkins CM, Guan SP, Yang K, Moon SH, Pietka T, Abumrad NA, Schlesinger PH, Gross RW. J Biol Chem. 2007;282:34611. doi: 10.1074/jbc.M707795200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Mancuso DJ, Kotzbauer P, Wozniak DF, Sims HF, Jenkins CM, Guan S, Han X, Yang K, Sun G, Malik I, Conyers S, Green KG, Schmidt RE, Gross RW. J Biol Chem. 2009;284:35632. doi: 10.1074/jbc.M109.055194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Yoda E, Hachisu K, Taketomi Y, Yoshida K, Nakamura M, Ikeda K, Taguchi R, Nakatani Y, Kuwata H, Murakami M, Kudo I, Hara S. J Lipid Res. 2010;51:3003. doi: 10.1194/jlr.M008060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Akassoglou K, Malester B, Xu J, Tessarollo L, Rosenbluth J, Chao MV. Proc Natl Acad Sci U S A. 2004;101:5075. doi: 10.1073/pnas.0401030101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Moser M, Stempfl T, Li Y, Glynn P, Buttner R, Kretzschmar D. Mech Dev. 2000;90:279. doi: 10.1016/s0925-4773(99)00239-7. [DOI] [PubMed] [Google Scholar]
- 226.van Tienhoven M, Atkins J, Li Y, Glynn P. J Biol Chem. 2002;277:20942. doi: 10.1074/jbc.M200330200. [DOI] [PubMed] [Google Scholar]
- 227.Johnson MK. Arch Toxicol. 1977;37:113. doi: 10.1007/BF00293860. [DOI] [PubMed] [Google Scholar]
- 228.Glynn P. Biochim Biophys Acta. 2005;1736:87. doi: 10.1016/j.bbalip.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 229.Moser M, Li Y, Vaupel K, Kretzschmar D, Kluge R, Glynn P, Buettner R. Mol Cell Biol. 2004;24:1667. doi: 10.1128/MCB.24.4.1667-1679.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Winrow CJ, Hemming ML, Allen DM, Quistad GB, Casida JE, Barlow C. Nat Genet. 2003;33:477. doi: 10.1038/ng1131. [DOI] [PubMed] [Google Scholar]
- 231.Muhlig-Versen M, da Cruz AB, Tschape JA, Moser M, Buttner R, Athenstaedt K, Glynn P, Kretzschmar D. J Neurosci. 2005;25:2865. doi: 10.1523/JNEUROSCI.5097-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Zaccheo O, Dinsdale D, Meacock PA, Glynn P. J Biol Chem. 2004;279:24024. doi: 10.1074/jbc.M400830200. [DOI] [PubMed] [Google Scholar]
- 233.Baulande S, Lasnier F, Lucas M, Pairault J. J Biol Chem. 2001;276:33336. doi: 10.1074/jbc.M105193200. [DOI] [PubMed] [Google Scholar]
- 234.Lake AC, Sun Y, Li JL, Kim JE, Johnson JW, Li D, Revett T, Shih HH, Liu W, Paulsen JE, Gimeno RE. J Lipid Res. 2005;46:2477. doi: 10.1194/jlr.M500290-JLR200. [DOI] [PubMed] [Google Scholar]
- 235.Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, Boerwinkle E, Cohen JC, Hobbs HH. Nat Genet. 2008;40:1461. doi: 10.1038/ng.257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Kotronen A, Johansson LE, Johansson LM, Roos C, Westerbacka J, Hamsten A, Bergholm R, Arkkila P, Arola J, Kiviluoto T, Fisher RM, Ehrenborg E, Orho-Melander M, Ridderstrale M, Groop L, Yki-Jarvinen H. Diabetologia. 2009;52:1056. doi: 10.1007/s00125-009-1285-z. [DOI] [PubMed] [Google Scholar]
- 237.Sookoian S, Castano GO, Burgueno AL, Fernandez Gianotti T, Rosselli MS, Pirola CJ. J Lipid Res. 2009 doi: 10.1194/jlr.P900013-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Chen W, Chang B, Li L, Chan L. Hepatology. 2010;52:1134. doi: 10.1002/hep.23812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Stafforini DM, McIntyre TM, Carter ME, Prescott SM. J Biol Chem. 1987;262:4215. [PubMed] [Google Scholar]
- 240.Tjoelker LW, Wilder C, Eberhardt C, Stafforini DM, Dietsch G, Schimpf B, Hooper S, Le Trong H, Cousens LS, Zimmerman GA, Yamadat Y, McIntyre TM, Prescott SM, Gray PW. Nature. 1995;374:549. doi: 10.1038/374549a0. [DOI] [PubMed] [Google Scholar]
- 241.Stafforini DM, Elstad MR, McIntyre TM, Zimmerman GA, Prescott SM. J Biol Chem. 1990;265:9682. [PubMed] [Google Scholar]
- 242.Stremler KE, Stafforini DM, Prescott SM, Zimmerman GA, McIntyre TM. J Biol Chem. 1989;264:5331. [PubMed] [Google Scholar]
- 243.Stremler KE, Stafforini DM, Prescott SM, McIntyre TM. J Biol Chem. 1991;266:11095. [PubMed] [Google Scholar]
- 244.Min JH, Wilder C, Aoki J, Arai H, Inoue K, Paul L, Gelb MH. Biochemistry. 2001;40:4539. doi: 10.1021/bi002600g. [DOI] [PubMed] [Google Scholar]
- 245.Tsoukatos DC, Liapikos TA, Tselepis AD, Chapman MJ, Ninio E. Biochem J. 2001;357:457. doi: 10.1042/0264-6021:3570457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Karasawa K, Harada A, Satoh N, Inoue K, Setaka M. Prog Lipid Res. 2003;42:93. doi: 10.1016/s0163-7827(02)00049-8. [DOI] [PubMed] [Google Scholar]
- 247.Karabina SA, Ninio E. Biochim Biophys Acta. 2006;1761:1351. doi: 10.1016/j.bbalip.2006.05.008. [DOI] [PubMed] [Google Scholar]
- 248.Stafforini DM, Numao T, Tsodikov A, Vaitkus D, Fukuda T, Watanabe N, Fueki N, McIntyre TM, Zimmerman GA, Makino S, Prescott SM. J Clin Invest. 1999;103:989. doi: 10.1172/JCI5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Henig NR, Aitken ML, Liu MC, Yu AS, Henderson WR., Jr Am J Respir Crit Care Med. 2000;162:523. doi: 10.1164/ajrccm.162.2.9911084. [DOI] [PubMed] [Google Scholar]
- 250.Lee ES, Jiang J, Sund GC, Simonson WT, Graham J, Dietsch G, Schimpf B, Bieg S, Peterman G, Lernmark A. Diabetes. 1999;48:43. doi: 10.2337/diabetes.48.1.43. [DOI] [PubMed] [Google Scholar]
- 251.Noto H, Hara M, Karasawa K, Iso ON, Satoh H, Togo M, Hashimoto Y, Yamada Y, Kosaka T, Kawamura M, Kimura S, Tsukamoto K. Arterioscler Thromb Vasc Biol. 2003;23:829. doi: 10.1161/01.ATV.0000067701.09398.18. [DOI] [PubMed] [Google Scholar]
- 252.Mohler ER, 3rd, Ballantyne CM, Davidson MH, Hanefeld M, Ruilope LM, Johnson JL, Zalewski A, Darapladib I. J Am Coll Cardiol. 2008;51:1632. doi: 10.1016/j.jacc.2007.11.079. [DOI] [PubMed] [Google Scholar]
- 253.Anderson JL. Am J Cardiol. 2008;101:23F. doi: 10.1016/j.amjcard.2008.04.015. [DOI] [PubMed] [Google Scholar]
- 254.Serruys PW, Garcia-Garcia HM, Buszman P, Erne P, Verheye S, Aschermann M, Duckers H, Bleie O, Dudek D, Botker HE, von Birgelen C, D’Amico D, Hutchinson T, Zambanini A, Mastik F, van Es GA, van der Steen AF, Vince DG, Ganz P, Hamm CW, Wijns W, Zalewski A. Circulation. 2008;118:1172. doi: 10.1161/CIRCULATIONAHA.108.771899. [DOI] [PubMed] [Google Scholar]
- 255.Wilensky RL, Shi Y, Mohler ER, 3rd, Hamamdzic D, Burgert ME, Li J, Postle A, Fenning RS, Bollinger JG, Hoffman BE, Pelchovitz DJ, Yang J, Mirabile RC, Webb CL, Zhang L, Zhang P, Gelb MH, Walker MC, Zalewski A, Macphee CH. Nat Med. 2008;14:1059. doi: 10.1038/nm.1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Hattori K, Hattori M, Adachi H, Tsujimoto M, Arai H, Inoue K. J Biol Chem. 1995;270:22308. doi: 10.1074/jbc.270.38.22308. [DOI] [PubMed] [Google Scholar]
- 257.Hattori K, Adachi H, Matsuzawa A, Yamamoto K, Tsujimoto M, Aoki J, Hattori M, Arai H, Inoue K. J Biol Chem. 1996;271:33032. doi: 10.1074/jbc.271.51.33032. [DOI] [PubMed] [Google Scholar]
- 258.Bae K, Longobardi L, Karasawa K, Malone B, Inoue T, Aoki J, Arai H, Inoue K, Lee T. J Biol Chem. 2000;275:26704. doi: 10.1074/jbc.M003951200. [DOI] [PubMed] [Google Scholar]
- 259.Matsuzawa A, Hattori K, Aoki J, Arai H, Inoue K. J Biol Chem. 1997;272:32315. doi: 10.1074/jbc.272.51.32315. [DOI] [PubMed] [Google Scholar]
- 260.Hattori M, Arai H, Inoue K. J Biol Chem. 1993;268:18748. [PubMed] [Google Scholar]
- 261.Ho YS, Swenson L, Derewenda U, Serre L, Wei Y, Dauter Z, Hattori M, Adachi T, Aoki J, Arai H, Inoue K, Derewenda ZS. Nature. 1997;385:89. doi: 10.1038/385089a0. [DOI] [PubMed] [Google Scholar]
- 262.Hattori M, Adachi H, Tsujimoto M, Arai H, Inoue K. J Biol Chem. 1994;269:23150. [PubMed] [Google Scholar]
- 263.Hattori M, Adachi H, Aoki J, Tsujimoto M, Arai H, Inoue K. J Biol Chem. 1995;270:31345. doi: 10.1074/jbc.270.52.31345. [DOI] [PubMed] [Google Scholar]
- 264.Manya H, Aoki J, Kato H, Ishii J, Hino S, Arai H, Inoue K. J Biol Chem. 1999;274:31827. doi: 10.1074/jbc.274.45.31827. [DOI] [PubMed] [Google Scholar]
- 265.Koizumi H, Yamaguchi N, Hattori M, Ishikawa TO, Aoki J, Taketo MM, Inoue K, Arai H. J Biol Chem. 2003;278:12489. doi: 10.1074/jbc.M211836200. [DOI] [PubMed] [Google Scholar]
- 266.Adachi H, Tsujimoto M, Hattori M, Arai H, Inoue K. Biochem Biophys Res Commun. 1997;233:10. doi: 10.1006/bbrc.1997.6383. [DOI] [PubMed] [Google Scholar]
- 267.Hirotsune S, Fleck MW, Gambello MJ, Bix GJ, Chen A, Clark GD, Ledbetter DH, McBain CJ, Wynshaw-Boris A. Nat Genet. 1998;19:333. doi: 10.1038/1221. [DOI] [PubMed] [Google Scholar]
- 268.Abe A, Hiraoka M, Wild S, Wilcoxen SE, Paine R, 3rd, Shayman JA. J Biol Chem. 2004;279:42605. doi: 10.1074/jbc.M407834200. [DOI] [PubMed] [Google Scholar]
- 269.Taniyama Y, Shibata S, Kita S, Horikoshi K, Fuse H, Shirafuji H, Sumino Y, Fujino M. Biochem Biophys Res Commun. 1999;257:50. doi: 10.1006/bbrc.1999.0411. [DOI] [PubMed] [Google Scholar]
- 270.Abe A, Shayman JA. J Biol Chem. 1998;273:8467. doi: 10.1074/jbc.273.14.8467. [DOI] [PubMed] [Google Scholar]
- 271.Abe A, Hiraoka M, Shayman JA. J Lipid Res. 2007;48:2255. doi: 10.1194/jlr.M700277-JLR200. [DOI] [PubMed] [Google Scholar]
- 272.Halliwell WH. Toxicol Pathol. 1997;25:53. doi: 10.1177/019262339702500111. [DOI] [PubMed] [Google Scholar]
- 273.Hiraoka M, Abe A, Lu Y, Yang K, Han X, Gross RW, Shayman JA. Mol Cell Biol. 2006;26:6139. doi: 10.1128/MCB.00627-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Jaye M, Lynch KJ, Krawiec J, Marchadier D, Maugeais C, Doan K, South V, Amin D, Perrone M, Rader DJ. Nat Genet. 1999;21:424. doi: 10.1038/7766. [DOI] [PubMed] [Google Scholar]
- 275.Hirata K, Dichek HL, Cioffi JA, Choi SY, Leeper NJ, Quintana L, Kronmal GS, Cooper AD, Quertermous T. J Biol Chem. 1999;274:14170. doi: 10.1074/jbc.274.20.14170. [DOI] [PubMed] [Google Scholar]
- 276.McCoy MG, Sun GS, Marchadier D, Maugeais C, Glick JM, Rader DJ. J Lipid Res. 2002;43:921. [PubMed] [Google Scholar]
- 277.Ishida T, Choi S, Kundu RK, Hirata K, Rubin EM, Cooper AD, Quertermous T. J Clin Invest. 2003;111:347. doi: 10.1172/JCI16306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Ma K, Cilingiroglu M, Otvos JD, Ballantyne CM, Marian AJ, Chan L. Proc Natl Acad Sci U S A. 2003;100:2748. doi: 10.1073/pnas.0438039100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.van Groningen JJ, Egmond MR, Bloemers HP, Swart GW. FEBS Lett. 1997;404:82. doi: 10.1016/s0014-5793(97)00098-7. [DOI] [PubMed] [Google Scholar]
- 280.Sato T, Aoki J, Nagai Y, Dohmae N, Takio K, Doi T, Arai H, Inoue K. J Biol Chem. 1997;272:2192. doi: 10.1074/jbc.272.4.2192. [DOI] [PubMed] [Google Scholar]
- 281.Yokoyama K, Kudo I, Inoue K. J Biochem. 1995;117:1280. doi: 10.1093/oxfordjournals.jbchem.a124856. [DOI] [PubMed] [Google Scholar]
- 282.Nagai Y, Aoki J, Sato T, Amano K, Matsuda Y, Arai H, Inoue K. J Biol Chem. 1999;274:11053. doi: 10.1074/jbc.274.16.11053. [DOI] [PubMed] [Google Scholar]
- 283.Aoki J, Nagai Y, Hosono H, Inoue K, Arai H. Biochim Biophys Acta. 2002;1582:26. doi: 10.1016/s1388-1981(02)00134-8. [DOI] [PubMed] [Google Scholar]
- 284.Blankenberg FG, Katsikis PD, Tait JF, Davis RE, Naumovski L, Ohtsuki K, Kopiwoda S, Abrams MJ, Darkes M, Robbins RC, Maecker HT, Strauss HW. Proc Natl Acad Sci U S A. 1998;95:6349. doi: 10.1073/pnas.95.11.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, Green DR. J Exp Med. 1995;182:1545. doi: 10.1084/jem.182.5.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Bellini F, Viola G, Menegus AM, Toffano G, Bruni A. Biochim Biophys Acta. 1990;1052:216. doi: 10.1016/0167-4889(90)90079-s. [DOI] [PubMed] [Google Scholar]
- 287.Bellini F, Bruni A. FEBS Lett. 1993;316:1. doi: 10.1016/0014-5793(93)81724-e. [DOI] [PubMed] [Google Scholar]
- 288.Hosono H, Homma M, Ogasawara Y, Makide K, Aoki J, Niwata H, Watanabe M, Inoue K, Ohkohchi N, Kohda Y. Cell Transplant. 2010;19:759. doi: 10.3727/096368910X508861. [DOI] [PubMed] [Google Scholar]
- 289.Hosono H, Aoki J, Nagai Y, Bandoh K, Ishida M, Taguchi R, Arai H, Inoue K. J Biol Chem. 2001;276:29664. doi: 10.1074/jbc.M104597200. [DOI] [PubMed] [Google Scholar]
- 290.Jin W, Broedl UC, Monajemi H, Glick JM, Rader DJ. Genomics. 2002;80:268. doi: 10.1006/geno.2002.6837. [DOI] [PubMed] [Google Scholar]
- 291.Aoki J, Inoue A, Makide K, Saiki N, Arai H. Biochimie. 2007;89:197. doi: 10.1016/j.biochi.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 292.Remington SG, Nelson JD. Invest Ophthalmol Vis Sci. 2002;43:3617. [PubMed] [Google Scholar]
- 293.Hiramatsu T, Sonoda H, Takanezawa Y, Morikawa R, Ishida M, Kasahara K, Sanai Y, Taguchi R, Aoki J, Arai H. J Biol Chem. 2003;278:49438. doi: 10.1074/jbc.M213018200. [DOI] [PubMed] [Google Scholar]
- 294.Sonoda H, Aoki J, Hiramatsu T, Ishida M, Bandoh K, Nagai Y, Taguchi R, Inoue K, Arai H. J Biol Chem. 2002;277:34254. doi: 10.1074/jbc.M201659200. [DOI] [PubMed] [Google Scholar]
- 295.Choi JW, Herr DR, Noguchi K, Yung YC, Lee CW, Mutoh T, Lin ME, Teo ST, Park KE, Mosley AN, Chun J. Annu Rev Pharmacool Toxicol. 2010;50:157. doi: 10.1146/annurev.pharmtox.010909.105753. [DOI] [PubMed] [Google Scholar]
- 296.Wen XY, Bryce DM, Breitman ML. Hum Mol Genet. 1998;7:743. doi: 10.1093/hmg/7.4.743. [DOI] [PubMed] [Google Scholar]
- 297.Wen XY, Hegele RA, Wang J, Wang DY, Cheung J, Wilson M, Yahyapour M, Bai Y, Zhuang L, Skaug J, Young TK, Connelly PW, Koop BF, Tsui LC, Stewart AK. Hum Mol Genet. 2003;12:1131. doi: 10.1093/hmg/ddg124. [DOI] [PubMed] [Google Scholar]
- 298.Takemori H, Zolotaryov FN, Ting L, Urbain T, Komatsubara T, Hatano O, Okamoto M, Tojo H. J Biol Chem. 1998;273:2222. doi: 10.1074/jbc.273.4.2222. [DOI] [PubMed] [Google Scholar]
- 299.Tojo H, Ichida T, Okamoto M. J Biol Chem. 1998;273:2214. doi: 10.1074/jbc.273.4.2214. [DOI] [PubMed] [Google Scholar]
- 300.Higgs HN, Han MH, Johnson GE, Glomset JA. J Biol Chem. 1998;273:5468. doi: 10.1074/jbc.273.10.5468. [DOI] [PubMed] [Google Scholar]
- 301.Higgs HN, Glomset JA. J Biol Chem. 1996;271:10874. doi: 10.1074/jbc.271.18.10874. [DOI] [PubMed] [Google Scholar]
- 302.Yamashita A, Kumazawa T, Koga H, Suzuki N, Oka S, Sugiura T. Biochim Biophys Acta. 2010;1801:711. doi: 10.1016/j.bbalip.2010.03.012. [DOI] [PubMed] [Google Scholar]
- 303.Sato S, Inoue H, Kogure T, Tagaya M, Tani K. FEBS Lett. 2010;584:4389. doi: 10.1016/j.febslet.2010.09.047. [DOI] [PubMed] [Google Scholar]
- 304.Simon GM, Cravatt BF. J Biol Chem. 2006;281:26465. doi: 10.1074/jbc.M604660200. [DOI] [PubMed] [Google Scholar]
- 305.Leung D, Saghatelian A, Simon GM, Cravatt BF. Biochemistry. 2006;45:4720. doi: 10.1021/bi060163l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Simon GM, Cravatt BF. J Biol Chem. 2008;283:9341. doi: 10.1074/jbc.M707807200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Bracey MH, Hanson MA, Masuda KR, Stevens RC, Cravatt BF. Science. 2002;298:1793. doi: 10.1126/science.1076535. [DOI] [PubMed] [Google Scholar]
- 308.Devane WA, Hanus L, Breuer A, Pertwee RG, Stevenson LA, Griffin G, Gibson D, Mandelbaum A, Etinger A, Mechoulam R. Science. 1992;258:1946. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- 309.Cravatt BF, Prospero-Garcia O, Siuzdak G, Gilula NB, Henriksen SJ, Boger DL, Lerner RA. Science. 1995;268:1506. doi: 10.1126/science.7770779. [DOI] [PubMed] [Google Scholar]
- 310.Saghatelian A, McKinney MK, Bandell M, Patapoutian A, Cravatt BF. Biochemistry. 2006;45:9007. doi: 10.1021/bi0608008. [DOI] [PubMed] [Google Scholar]
- 311.Long JZ, LaCava M, Jin X, Cravatt BF. J Lipid Res. 2011;52:337. doi: 10.1194/jlr.M012153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Kathuria S, Gaetani S, Fegley D, Valino F, Duranti A, Tontini A, Mor M, Tarzia G, La Rana G, Calignano A, Giustino A, Tattoli M, Palmery M, Cuomo V, Piomelli D. Nat Med. 2003;9:76. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- 313.Ahn K, Johnson DS, Mileni M, Beidler D, Long JZ, McKinney MK, Weerapana E, Sadagopan N, Liimatta M, Smith SE, Lazerwith S, Stiff C, Kamtekar S, Bhattacharya K, Zhang Y, Swaney S, Van Becelaere K, Stevens RC, Cravatt BF. Chem Biol. 2009;16:411. doi: 10.1016/j.chembiol.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Saghatelian A, Trauger SA, Want EJ, Hawkins EG, Siuzdak G, Cravatt BF. Biochemistry. 2004;43:14332. doi: 10.1021/bi0480335. [DOI] [PubMed] [Google Scholar]
- 315.Naidu PS, Varvel SA, Ahn K, Cravatt BF, Martin BR, Lichtman AH. Psychopharmacology. 2007;192:61. doi: 10.1007/s00213-006-0689-4. [DOI] [PubMed] [Google Scholar]
- 316.Cravatt BF, Saghatelian A, Hawkins EG, Clement AB, Bracey MH, Lichtman AH. Proc Natl Acad Sci U S A. 2004;101:10821. doi: 10.1073/pnas.0401292101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Lichtman AH, Leung D, Shelton CC, Saghatelian A, Hardouin C, Boger DL, Cravatt BF. J Pharmacol Exp Ther. 2004;311:441. doi: 10.1124/jpet.104.069401. [DOI] [PubMed] [Google Scholar]
- 318.Ahn K, Johnson DS, Fitzgerald LR, Liimatta M, Arendse A, Stevenson T, Lund ET, Nugent RA, Nomanbhoy TK, Alexander JP, Cravatt BF. Biochemistry. 2007;46:13019. doi: 10.1021/bi701378g. [DOI] [PubMed] [Google Scholar]
- 319.Johnson DS, Stiff C, Lazerwith SE, Kesten SR, Fay LK, Morris M, Beidler D, Liimatta M, Smith SE, Dudley DT, Sadagopan N, Bhattachar SN, Kesten SJ, Nomanbhoy TK, Cravatt BF, Ahn K. ACS Medicinal Chemistry Letters. 2011;2:91. doi: 10.1021/ml100190t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Wei BQ, Mikkelsen TS, McKinney MK, Lander ES, Cravatt BF. J Biol Chem. 2006;281:36569. doi: 10.1074/jbc.M606646200. [DOI] [PubMed] [Google Scholar]
- 321.Pabarcus MK, Casida JE. Protein Expr Purif. 2005;44:39. doi: 10.1016/j.pep.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 322.Dobrovolsky VN, Bowyer JF, Pabarcus MK, Heflich RH, Williams LD, Doerge DR, Arvidsson B, Bergquist J, Casida JE. Biochim Biophys Acta. 2005;1724:163. doi: 10.1016/j.bbagen.2005.03.010. [DOI] [PubMed] [Google Scholar]
- 323.Bailey CG, Wagner C. J Biol Chem. 1974;249:4439. [PubMed] [Google Scholar]
- 324.Pabarcus MK, Casida JE. Biochim Biophys Acta. 2002;1596:201. doi: 10.1016/s0167-4838(02)00232-7. [DOI] [PubMed] [Google Scholar]
- 325.Schuettengruber B, Doetzlhofer A, Kroboth K, Wintersberger E, Seiser C. J Biol Chem. 2003;278:1784. doi: 10.1074/jbc.M204843200. [DOI] [PubMed] [Google Scholar]
- 326.Dobrovolsky VN, Bucci T, Heflich RH, Desjardins J, Richardson FC. Mol Genet Metab. 2003;78:1. doi: 10.1016/s1096-7192(02)00224-x. [DOI] [PubMed] [Google Scholar]
- 327.Maier T, Jenni S, Ban N. Science. 2006;311:1258. doi: 10.1126/science.1123248. [DOI] [PubMed] [Google Scholar]
- 328.Smith S. FASEB J. 1994;8:1248. [PubMed] [Google Scholar]
- 329.Chang SI, Hammes GG. Acc Chem Res. 1990;23:363. [Google Scholar]
- 330.Maier T, Leibundgut M, Ban N. Science. 2008;321:1315. doi: 10.1126/science.1161269. [DOI] [PubMed] [Google Scholar]
- 331.Serre L, Verbree EC, Dauter Z, Stuitje AR, Derewenda ZS. J Biol Chem. 1995;270:12961. doi: 10.1074/jbc.270.22.12961. [DOI] [PubMed] [Google Scholar]
- 332.Bunkoczi G, Misquitta S, Wu X, Lee WH, Rojkova A, Kochan G, Kavanagh KL, Oppermann U, Smith S. Chem Biol. 2009;16:667. doi: 10.1016/j.chembiol.2009.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Pappenberger G, Benz J, Gsell B, Hennig M, Ruf A, Stihle M, Thoma R, Rudolph MG. J Mol Biol. 2010;397:508. doi: 10.1016/j.jmb.2010.01.066. [DOI] [PubMed] [Google Scholar]
- 334.Pemble CWt, Johnson LC, Kridel SJ, Lowther WT. Nat Struct Mol Biol. 2007;14:704. doi: 10.1038/nsmb1265. [DOI] [PubMed] [Google Scholar]
- 335.Chakravarty B, Gu Z, Chirala SS, Wakil SJ, Quiocho FA. Proc Natl Acad Sci U S A. 2004;101:15567. doi: 10.1073/pnas.0406901101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Lin CY, Smith S. J Biol Chem. 1978;253:1954. [PubMed] [Google Scholar]
- 337.Singh N, Wakil SJ, Stoops JK. J Biol Chem. 1984;259:3605. [PubMed] [Google Scholar]
- 338.Kuhajda FP, Jenner K, Wood FD, Hennigar RA, Jacobs LB, Dick JD, Pasternack GR. Proc Natl Acad Sci U S A. 1994;91:6379. doi: 10.1073/pnas.91.14.6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Kuhajda FP. Cancer Res. 2006;66:5977. doi: 10.1158/0008-5472.CAN-05-4673. [DOI] [PubMed] [Google Scholar]
- 340.Jensen V, Ladekarl M, Holm-Nielsen P, Melsen F, Soerensen FB. J Pathol. 1995;176:343. doi: 10.1002/path.1711760405. [DOI] [PubMed] [Google Scholar]
- 341.Shurbaji MS, Kuhajda FP, Pasternack GR, Thurmond TS. Am J Clin Pathol. 1992;97:686. doi: 10.1093/ajcp/97.5.686. [DOI] [PubMed] [Google Scholar]
- 342.Gansler TS, Hardman W, 3rd, Hunt DA, Schaffel S, Hennigar RA. Hum Pathol. 1997;28:686. doi: 10.1016/s0046-8177(97)90177-5. [DOI] [PubMed] [Google Scholar]
- 343.Lupu R, Menendez JA. Curr Pharm Biotechnol. 2006;7:483. doi: 10.2174/138920106779116928. [DOI] [PubMed] [Google Scholar]
- 344.Kuhajda FP, Pizer ES, Li JN, Mani NS, Frehywot GL, Townsend CA. Proc Natl Acad Sci U S A. 2000;97:3450. doi: 10.1073/pnas.050582897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Pizer ES, Jackisch C, Wood FD, Pasternack GR, Davidson NE, Kuhajda FP. Cancer Res. 1996;56:2745. [PubMed] [Google Scholar]
- 346.Chirala SS, Chang H, Matzuk M, Abu-Elheiga L, Mao J, Mahon K, Finegold M, Wakil SJ. Proc Natl Acad Sci U S A. 2003;100:6358. doi: 10.1073/pnas.0931394100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Chakravarthy MV, Pan Z, Zhu Y, Tordjman K, Schneider JG, Coleman T, Turk J, Semenkovich CF. Cell Metab. 2005;1:309. doi: 10.1016/j.cmet.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 348.Chakravarthy MV, Zhu Y, Lopez M, Yin L, Wozniak DF, Coleman T, Hu Z, Wolfgang M, Vidal-Puig A, Lane MD, Semenkovich CF. J Clin Invest. 2007;117:2539. doi: 10.1172/JCI31183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Loftus TM, Jaworsky DE, Frehywot GL, Townsend CA, Ronnett GV, Lane MD, Kuhajda FP. Science. 2000;288:2379. doi: 10.1126/science.288.5475.2379. [DOI] [PubMed] [Google Scholar]
- 350.Kirkby B, Roman N, Kobe B, Kellie S, Forwood JK. Prog Lipid Res. 2010;49:366. doi: 10.1016/j.plipres.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 351.Hunt MC, Alexson SE. Prog Lipid Res. 2002;41:99. doi: 10.1016/s0163-7827(01)00017-0. [DOI] [PubMed] [Google Scholar]
- 352.Hunt MC, Lindquist PJ, Peters JM, Gonzalez FJ, Diczfalusy U, Alexson SE. J Lipid Res. 2000;41:814. [PubMed] [Google Scholar]
- 353.Svensson LT, Alexson SE, Hiltunen JK. J Biol Chem. 1995;270:12177. doi: 10.1074/jbc.270.20.12177. [DOI] [PubMed] [Google Scholar]
- 354.Yamada J, Matsumoto I, Furihata T, Sakuma M, Suga T. Arch Biochem Biophys. 1994;308:118. doi: 10.1006/abbi.1994.1017. [DOI] [PubMed] [Google Scholar]
- 355.Svensson LT, Engberg ST, Aoyama T, Usuda N, Alexson SE, Hashimoto T. Biochem J. 1998;329(Pt 3):601. doi: 10.1042/bj3290601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Huhtinen K, O’Byrne J, Lindquist PJ, Contreras JA, Alexson SE. J Biol Chem. 2002;277:3424. doi: 10.1074/jbc.M109040200. [DOI] [PubMed] [Google Scholar]
- 357.Westin MA, Alexson SE, Hunt MC. J Biol Chem. 2004;279:21841. doi: 10.1074/jbc.M313863200. [DOI] [PubMed] [Google Scholar]
- 358.Westin MA, Hunt MC, Alexson SE. J Biol Chem. 2005;280:38125. doi: 10.1074/jbc.M508479200. [DOI] [PubMed] [Google Scholar]
- 359.Hunt MC, Rautanen A, Westin MA, Svensson LT, Alexson SE. FASEB J. 2006;20:1855. doi: 10.1096/fj.06-6042com. [DOI] [PubMed] [Google Scholar]
- 360.Westin MA, Hunt MC, Alexson SE. J Biol Chem. 2007;282:26707. doi: 10.1074/jbc.M703718200. [DOI] [PubMed] [Google Scholar]
- 361.Darvesh S, Hopkins DA, Geula C. Nat Rev Neurosci. 2003;4:131. doi: 10.1038/nrn1035. [DOI] [PubMed] [Google Scholar]
- 362.Bourne Y, Taylor P, Marchot P. Cell. 1995;83:503. doi: 10.1016/0092-8674(95)90128-0. [DOI] [PubMed] [Google Scholar]
- 363.Sussman JL, Harel M, Frolow F, Oefner C, Goldman A, Toker L, Silman I. Science. 1991;253:872. doi: 10.1126/science.1678899. [DOI] [PubMed] [Google Scholar]
- 364.Soreq H, Seidman S. Nat Rev Neurosci. 2001;2:294. doi: 10.1038/35067589. [DOI] [PubMed] [Google Scholar]
- 365.Massoulie J. Neurosignals. 2002;11:130. doi: 10.1159/000065054. [DOI] [PubMed] [Google Scholar]
- 366.Lockridge O, Mottershaw-Jackson N, Eckerson HW, La Du BN. J Pharmacol Exp Ther. 1980;215:1. [PubMed] [Google Scholar]
- 367.Gatley SJ. Biochem Pharmacol. 1991;41:1249. doi: 10.1016/0006-2952(91)90665-r. [DOI] [PubMed] [Google Scholar]
- 368.Masson P, Froment MT, Fortier PL, Visicchio JE, Bartels CF, Lockridge O. Biochim Biophys Acta. 1998;1387:41. doi: 10.1016/s0167-4838(98)00104-6. [DOI] [PubMed] [Google Scholar]
- 369.George ST, Balasubramanian AS. Eur J Biochem. 1981;121:177. doi: 10.1111/j.1432-1033.1981.tb06447.x. [DOI] [PubMed] [Google Scholar]
- 370.Li B, Stribley JA, Ticu A, Xie W, Schopfer LM, Hammond P, Brimijoin S, Hinrichs SH, Lockridge O. J Neurochem. 2000;75:1320. doi: 10.1046/j.1471-4159.2000.751320.x. [DOI] [PubMed] [Google Scholar]
- 371.Xie W, Stribley JA, Chatonnet A, Wilder PJ, Rizzino A, McComb RD, Taylor P, Hinrichs SH, Lockridge O. J Pharmacol Exp Ther. 2000;293:896. [PubMed] [Google Scholar]
- 372.Hartmann J, Kiewert C, Duysen EG, Lockridge O, Greig NH, Klein J. J Neurochem. 2007;100:1421. doi: 10.1111/j.1471-4159.2006.04347.x. [DOI] [PubMed] [Google Scholar]
- 373.Hartmann J, Kiewert C, Duysen EG, Lockridge O, Klein J. Neurochem Int. 2008;52:972. doi: 10.1016/j.neuint.2007.10.008. [DOI] [PubMed] [Google Scholar]
- 374.Volpicelli-Daley LA, Hrabovska A, Duysen EG, Ferguson SM, Blakely RD, Lockridge O, Levey AI. Mol Pharmacol. 2003;64:1309. doi: 10.1124/mol.64.6.1309. [DOI] [PubMed] [Google Scholar]
- 375.Duysen EG, Stribley JA, Fry DL, Hinrichs SH, Lockridge O. Brain Res Dev Brain Res. 2002;137:43. doi: 10.1016/s0165-3806(02)00367-x. [DOI] [PubMed] [Google Scholar]
- 376.Radic Z, Duran R, Vellom DC, Li Y, Cervenansky C, Taylor P. J Biol Chem. 1994;269:11233. [PubMed] [Google Scholar]
- 377.Minic J, Chatonnet A, Krejci E, Molgo J. Br J Pharmacol. 2003;138:177. doi: 10.1038/sj.bjp.0705010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Rodriguez-Ithurralde D, Silveira R, Barbeito L, Dajas F. Neurochem Int. 1983;5:267. doi: 10.1016/0197-0186(83)90028-1. [DOI] [PubMed] [Google Scholar]
- 379.Li B, Duysen EG, Carlson M, Lockridge O. J Pharmacol Exp Ther. 2008;324:1146. doi: 10.1124/jpet.107.133330. [DOI] [PubMed] [Google Scholar]
- 380.Bosak A, Gazic I, Vinkovic V, Kovarik Z. Chem Biol Interact. 2008;175:192. doi: 10.1016/j.cbi.2008.04.050. [DOI] [PubMed] [Google Scholar]
- 381.Evans FT, Gray PW, Lehmann H, Silk E. Lancet. 1952;1:1229. doi: 10.1016/s0140-6736(52)92059-x. [DOI] [PubMed] [Google Scholar]
- 382.Lehmann H, Ryan E. Lancet. 1956;271:124. doi: 10.1016/s0140-6736(56)90869-8. [DOI] [PubMed] [Google Scholar]
- 383.Darvesh S, Grantham DL, Hopkins DA. J Comp Neurol. 1998;393:374. [PubMed] [Google Scholar]
- 384.Mesulam MM, Guillozet A, Shaw P, Levey A, Duysen EG, Lockridge O. Neuroscience. 2002;110:627. doi: 10.1016/s0306-4522(01)00613-3. [DOI] [PubMed] [Google Scholar]
- 385.Li B, Duysen EG, Saunders TL, Lockridge O. J Mol Neurosci. 2006;30:193. doi: 10.1385/JMN:30:1:193. [DOI] [PubMed] [Google Scholar]
- 386.Shih TM. Psychopharmacology. 1982;78:170. doi: 10.1007/BF00432257. [DOI] [PubMed] [Google Scholar]
- 387.Brown MA, Brix KA. J Appl Toxicol. 1998;18:393. doi: 10.1002/(sici)1099-1263(199811/12)18:6<393::aid-jat528>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 388.Marrs TC. Pharmacol Ther. 1993;58:51. doi: 10.1016/0163-7258(93)90066-m. [DOI] [PubMed] [Google Scholar]
- 389.Khan WA, Dechkovskaia AM, Herrick EA, Jones KH, Abou-Donia MB. Toxicol Sci. 2000;57:112. doi: 10.1093/toxsci/57.1.112. [DOI] [PubMed] [Google Scholar]
- 390.Amitai G, Moorad D, Adani R, Doctor BP. Biochem Pharmacol. 1998;56:293. doi: 10.1016/s0006-2952(98)00035-5. [DOI] [PubMed] [Google Scholar]
- 391.Bowls BJ, Freeman JM, Jr, Luna JA, Meggs WJ. Acad Emerg Med. 2003;10:286. doi: 10.1111/j.1553-2712.2003.tb02006.x. [DOI] [PubMed] [Google Scholar]
- 392.Terry AV, Jr, Buccafusco JJ. J Pharmacol Exp Ther. 2003;306:821. doi: 10.1124/jpet.102.041616. [DOI] [PubMed] [Google Scholar]
- 393.Rosler M, Anand R, Cicin-Sain A, Gauthier S, Agid Y, Dal-Bianco P, Stahelin HB, Hartman R, Gharabawi M. Br Med J. 1999;318:633. doi: 10.1136/bmj.318.7184.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Emre M, Aarsland D, Albanese A, Byrne EJ, Deuschl G, De Deyn PP, Durif F, Kulisevsky J, van Laar T, Lees A, Poewe W, Robillard A, Rosa MM, Wolters E, Quarg P, Tekin S, Lane R. N Engl J Med. 2004;351:2509. doi: 10.1056/NEJMoa041470. [DOI] [PubMed] [Google Scholar]
- 395.Greig NH, Utsuki T, Ingram DK, Wang Y, Pepeu G, Scali C, Yu QS, Mamczarz J, Holloway HW, Giordano T, Chen D, Furukawa K, Sambamurti K, Brossi A, Lahiri DK. Proc Natl Acad Sci U S A. 2005;102:17213. doi: 10.1073/pnas.0508575102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Silman I, Sussman JL. Curr Opin Pharmacol. 2005;5:293. doi: 10.1016/j.coph.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 397.Page MJ, Di Cera E. Cell Mol Life Sci. 2008;65:1220. doi: 10.1007/s00018-008-7565-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 398.Rawlings ND, Barrett AJ, Bateman A. Nucleic Acids Res. 2010;38:D227. doi: 10.1093/nar/gkp971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Perroud B, Alvarado RJ, Espinal GM, Morado AR, Phinney BS, Warden CH. Mol Brain. 2009;2:14. doi: 10.1186/1756-6606-2-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 400.Park JE, Lenter MC, Zimmermann RN, Garin-Chesa P, Old LJ, Rettig WJ. J Biol Chem. 1999;274:36505. doi: 10.1074/jbc.274.51.36505. [DOI] [PubMed] [Google Scholar]
- 401.Rennex D, Hemmings BA, Hofsteenge J, Stone SR. Biochemistry. 1991;30:2195. doi: 10.1021/bi00222a025. [DOI] [PubMed] [Google Scholar]
- 402.Fulop V, Bocskei Z, Polgar L. Cell. 1998;94:161. doi: 10.1016/s0092-8674(00)81416-6. [DOI] [PubMed] [Google Scholar]
- 403.Wilk S. Life Sci. 1983;33:2149. doi: 10.1016/0024-3205(83)90285-0. [DOI] [PubMed] [Google Scholar]
- 404.Toide K, Iwamoto Y, Fujiwara T, Abe H. J Pharmacol Exp Ther. 1995;274:1370. [PubMed] [Google Scholar]
- 405.Toide K, Fujiwara T, Iwamoto Y, Shinoda M, Okamiya K, Kato T. Naunyn Schmiedebergs Arch Pharmacol. 1996;353:355. doi: 10.1007/BF00168640. [DOI] [PubMed] [Google Scholar]
- 406.Barelli H, Petit A, Hirsch E, Wilk S, De Nanteuil G, Morain P, Checler F. Biochem Biophys Res Commun. 1999;257:657. doi: 10.1006/bbrc.1999.0366. [DOI] [PubMed] [Google Scholar]
- 407.Nolte WM, Tagore DM, Lane WS, Saghatelian A. Biochemistry. 2009;48:11971. doi: 10.1021/bi901637c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Bellemere G, Morain P, Vaudry H, Jegou S. J Neurochem. 2003;84:919. doi: 10.1046/j.1471-4159.2003.01536.x. [DOI] [PubMed] [Google Scholar]
- 409.Huston JP, Hasenohrl RU. Behav Brain Res. 1995;66:117. doi: 10.1016/0166-4328(94)00132-y. [DOI] [PubMed] [Google Scholar]
- 410.Shinoda M, Okamiya K, Toide K. Jpn J Pharmacol. 1995;69:273. doi: 10.1254/jjp.69.273. [DOI] [PubMed] [Google Scholar]
- 411.Schneider JS, Giardiniere M, Morain P. Neuropsychopharmacology. 2002;26:176. doi: 10.1016/S0893-133X(01)00307-4. [DOI] [PubMed] [Google Scholar]
- 412.Williams RS, Harwood AJ. Trends Pharmacol Sci. 2000;21:61. doi: 10.1016/s0165-6147(99)01428-5. [DOI] [PubMed] [Google Scholar]
- 413.Williams RS, Cheng L, Mudge AW, Harwood AJ. Nature. 2002;417:292. doi: 10.1038/417292a. [DOI] [PubMed] [Google Scholar]
- 414.Morain P, Lestage P, De Nanteuil G, Jochemsen R, Robin JL, Guez D, Boyer PA. CNS Drug Rev. 2002;8:31. doi: 10.1111/j.1527-3458.2002.tb00214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Parvari R, Gonen Y, Alshafee I, Buriakovsky S, Regev K, Hershkovitz E. Genomics. 2005;86:195. doi: 10.1016/j.ygeno.2005.04.001. [DOI] [PubMed] [Google Scholar]
- 416.Szeltner Z, Alshafee I, Juhasz T, Parvari R, Polgar L. Cell Mol Life Sci. 2005;62:2376. doi: 10.1007/s00018-005-5262-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Jaeken J, Martens K, Francois I, Eyskens F, Lecointre C, Derua R, Meulemans S, Slootstra JW, Waelkens E, de Zegher F, Creemers JW, Matthijs G. Am J Hum Genet. 2006;78:38. doi: 10.1086/498852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Martens K, Derua R, Meulemans S, Waelkens E, Jaeken J, Matthijs G, Creemers JW. Biol Chem. 2006;387:879. doi: 10.1515/BC.2006.111. [DOI] [PubMed] [Google Scholar]
- 419.Mentlein R. Regul Pept. 1999;85:9. doi: 10.1016/s0167-0115(99)00089-0. [DOI] [PubMed] [Google Scholar]
- 420.Tanaka T, Camerini D, Seed B, Torimoto Y, Dang NH, Kameoka J, Dahlberg HN, Schlossman SF, Morimoto C. J Immunol. 1992;149:481. [PubMed] [Google Scholar]
- 421.Kettmann U, Humbel B, Holzhausen HJ. Acta Histochem. 1992;92:225. doi: 10.1016/S0065-1281(11)80085-1. [DOI] [PubMed] [Google Scholar]
- 422.Heymann E, Mentlein R, Nausch I, Erlanson-Albertsson C, Yoshimoto T, Feller AC. Biomed Biochim Acta. 1986;45:575. [PubMed] [Google Scholar]
- 423.Mitro A, Lojda Z. Histochemistry. 1988;88:645. doi: 10.1007/BF00570337. [DOI] [PubMed] [Google Scholar]
- 424.Mentlein R, Gallwitz B, Schmidt WE. Eur J Biochem. 1993;214:829. doi: 10.1111/j.1432-1033.1993.tb17986.x. [DOI] [PubMed] [Google Scholar]
- 425.Engel M, Hoffmann T, Wagner L, Wermann M, Heiser U, Kiefersauer R, Huber R, Bode W, Demuth HU, Brandstetter H. Proc Natl Acad Sci U S A. 2003;100:5063. doi: 10.1073/pnas.0230620100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Rasmussen HB, Branner S, Wiberg FC, Wagtmann N. Nat Struct Biol. 2003;10:19. doi: 10.1038/nsb882. [DOI] [PubMed] [Google Scholar]
- 427.Thoma R, Loffler B, Stihle M, Huber W, Ruf A, Hennig M. Structure. 2003;11:947. doi: 10.1016/s0969-2126(03)00160-6. [DOI] [PubMed] [Google Scholar]
- 428.Nausch I, Mentlein R, Heymann E. Biol Chem Hoppe-Seyler. 1990;371:1113. doi: 10.1515/bchm3.1990.371.2.1113. [DOI] [PubMed] [Google Scholar]
- 429.Thorens B. Proc Natl Acad Sci U S A. 1992;89:8641. doi: 10.1073/pnas.89.18.8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Drucker DJ, Philippe J, Mojsov S, Chick WL, Habener JF. Proc Natl Acad Sci U S A. 1987;84:3434. doi: 10.1073/pnas.84.10.3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Zander M, Madsbad S, Madsen JL, Holst JJ. Lancet. 2002;359:824. doi: 10.1016/S0140-6736(02)07952-7. [DOI] [PubMed] [Google Scholar]
- 432.Marguet D, Baggio L, Kobayashi T, Bernard AM, Pierres M, Nielsen PF, Ribel U, Watanabe T, Drucker DJ, Wagtmann N. Proc Natl Acad Sci U S A. 2000;97:6874. doi: 10.1073/pnas.120069197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Green BD, Flatt PR, Bailey CJ. Diab Vasc Dis Res. 2006;3:159. doi: 10.3132/dvdr.2006.024. [DOI] [PubMed] [Google Scholar]
- 434.Kim D, Wang L, Beconi M, Eiermann GJ, Fisher MH, He H, Hickey GJ, Kowalchick JE, Leiting B, Lyons K, Marsilio F, McCann ME, Patel RA, Petrov A, Scapin G, Patel SB, Roy RS, Wu JK, Wyvratt MJ, Zhang BB, Zhu L, Thornberry NA, Weber AE. J Med Chem. 2005;48:141. doi: 10.1021/jm0493156. [DOI] [PubMed] [Google Scholar]
- 435.Villhauer EB, Brinkman JA, Naderi GB, Burkey BF, Dunning BE, Prasad K, Mangold BL, Russell ME, Hughes TE. J Med Chem. 2003;46:2774. doi: 10.1021/jm030091l. [DOI] [PubMed] [Google Scholar]
- 436.Augeri DJ, Robl JA, Betebenner DA, Magnin DR, Khanna A, Robertson JG, Wang A, Simpkins LM, Taunk P, Huang Q, Han SP, Abboa-Offei B, Cap M, Xin L, Tao L, Tozzo E, Welzel GE, Egan DM, Marcinkeviciene J, Chang SY, Biller SA, Kirby MS, Parker RA, Hamann LG. J Med Chem. 2005;48:5025. doi: 10.1021/jm050261p. [DOI] [PubMed] [Google Scholar]
- 437.Ahren B, Landin-Olsson M, Jansson PA, Svensson M, Holmes D, Schweizer A. J Clin Endocrinol Metab. 2004;89:2078. doi: 10.1210/jc.2003-031907. [DOI] [PubMed] [Google Scholar]
- 438.Drucker D, Easley C, Kirkpatrick P. Nat Rev Drug Discov. 2007;6:109. doi: 10.1038/nrd2245. [DOI] [PubMed] [Google Scholar]
- 439.Aschner P, Kipnes MS, Lunceford JK, Sanchez M, Mickel C, Williams-Herman DE. Diabetes Care. 2006;29:2632. doi: 10.2337/dc06-0703. [DOI] [PubMed] [Google Scholar]
- 440.Ahren B, Pacini G, Foley JE, Schweizer A. Diabetes Care. 2005;28:1936. doi: 10.2337/diacare.28.8.1936. [DOI] [PubMed] [Google Scholar]
- 441.Ohnuma K, Dang NH, Morimoto C. Trends Immunol. 2008;29:295. doi: 10.1016/j.it.2008.02.010. [DOI] [PubMed] [Google Scholar]
- 442.Tagore DM, Nolte WM, Neveu JM, Rangel R, Guzman-Rojas L, Pasqualini R, Arap W, Lane WS, Saghatelian A. Nat Chem Biol. 2009;5:23. doi: 10.1038/nchembio.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 443.Tinoco AD, Tagore DM, Saghatelian A. J Am Chem Soc. 2010;132:3819. doi: 10.1021/ja909524e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.O’Brien P, O’Connor BF. Biochim Biophys Acta. 2008;1784:1130. doi: 10.1016/j.bbapap.2008.01.006. [DOI] [PubMed] [Google Scholar]
- 445.Scanlan MJ, Raj BK, Calvo B, Garin-Chesa P, Sanz-Moncasi MP, Healey JH, Old LJ, Rettig WJ. Proc Natl Acad Sci U S A. 1994;91:5657. doi: 10.1073/pnas.91.12.5657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Niedermeyer J, Garin-Chesa P, Kriz M, Hilberg F, Mueller E, Bamberger U, Rettig WJ, Schnapp A. Int J Dev Biol. 2001;45:445. [PubMed] [Google Scholar]
- 447.Garin-Chesa P, Old LJ, Rettig WJ. Proc Natl Acad Sci U S A. 1990;87:7235. doi: 10.1073/pnas.87.18.7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 448.Collins PJ, McMahon G, O’Brien P, O’Connor B. Int J Biochem Cell Biol. 2004;36:2320. doi: 10.1016/j.biocel.2004.05.006. [DOI] [PubMed] [Google Scholar]
- 449.Aertgeerts K, Levin I, Shi L, Snell GP, Jennings A, Prasad GS, Zhang Y, Kraus ML, Salakian S, Sridhar V, Wijnands R, Tennant MG. J Biol Chem. 2005;280:19441. doi: 10.1074/jbc.C500092200. [DOI] [PubMed] [Google Scholar]
- 450.Lee KN, Jackson KW, Christiansen VJ, Lee CS, Chun JG, McKee PA. Blood. 2006;107:1397. doi: 10.1182/blood-2005-08-3452. [DOI] [PubMed] [Google Scholar]
- 451.Pineiro-Sanchez ML, Goldstein LA, Dodt J, Howard L, Yeh Y, Tran H, Argraves WS, Chen WT. J Biol Chem. 1997;272:7595. doi: 10.1074/jbc.272.12.7595. [DOI] [PubMed] [Google Scholar]
- 452.Adams S, Miller GT, Jesson MI, Watanabe T, Jones B, Wallner BP. Cancer Res. 2004;64:5471. doi: 10.1158/0008-5472.CAN-04-0447. [DOI] [PubMed] [Google Scholar]
- 453.Eager RM, Cunningham CC, Senzer NN, Stephenson J, Jr, Anthony SP, O’Day SJ, Frenette G, Pavlick AC, Jones B, Uprichard M, Nemunaitis J. BMC Cancer. 2009;9:263. doi: 10.1186/1471-2407-9-263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 454.Narra K, Mullins SR, Lee HO, Strzemkowski-Brun B, Magalong K, Christiansen VJ, McKee PA, Egleston B, Cohen SJ, Weiner LM, Meropol NJ, Cheng JD. Cancer Biol Ther. 2007;6:1691. doi: 10.4161/cbt.6.11.4874. [DOI] [PubMed] [Google Scholar]
- 455.Hofheinz RD, al-Batran SE, Hartmann F, Hartung G, Jager D, Renner C, Tanswell P, Kunz U, Amelsberg A, Kuthan H, Stehle G. Onkologie. 2003;26:44. doi: 10.1159/000069863. [DOI] [PubMed] [Google Scholar]
- 456.Niedermeyer J, Kriz M, Hilberg F, Garin-Chesa P, Bamberger U, Lenter MC, Park J, Viertel B, Puschner H, Mauz M, Rettig WJ, Schnapp A. Mol Cell Biol. 2000;20:1089. doi: 10.1128/mcb.20.3.1089-1094.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457.Abbott CA, Yu DM, Woollatt E, Sutherland GR, McCaughan GW, Gorrell MD. Eur J Biochem. 2000;267:6140. doi: 10.1046/j.1432-1327.2000.01617.x. [DOI] [PubMed] [Google Scholar]
- 458.Olsen C, Wagtmann N. Gene. 2002;299:185. doi: 10.1016/s0378-1119(02)01059-4. [DOI] [PubMed] [Google Scholar]
- 459.Bjelke JR, Christensen J, Nielsen PF, Branner S, Kanstrup AB, Wagtmann N, Rasmussen HB. Biochem J. 2006;396:391. doi: 10.1042/BJ20060079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 460.Kobayashi K, Lin LW, Yeadon JE, Klickstein LB, Smith JA. J Biol Chem. 1989;264:8892. [PubMed] [Google Scholar]
- 461.Bartlam M, Wang G, Yang H, Gao R, Zhao X, Xie G, Cao S, Feng Y, Rao Z. Structure. 2004;12:1481. doi: 10.1016/j.str.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 462.Yamin R, Bagchi S, Hildebrant R, Scaloni A, Widom RL, Abraham CR. J Neurochem. 2007;100:458. doi: 10.1111/j.1471-4159.2006.04251.x. [DOI] [PubMed] [Google Scholar]
- 463.Jones WM, Manning JM. Biochem Biophys Res Commun. 1985;126:933. doi: 10.1016/0006-291x(85)90275-x. [DOI] [PubMed] [Google Scholar]
- 464.Jones WM, Manning JM. Biochim Biophys Acta. 1988;953:357. doi: 10.1016/0167-4838(88)90045-3. [DOI] [PubMed] [Google Scholar]
- 465.Jones WM, Manning LR, Manning JM. Biochem Biophys Res Commun. 1986;139:244. doi: 10.1016/s0006-291x(86)80105-x. [DOI] [PubMed] [Google Scholar]
- 466.Scaloni A, Jones WM, Barra D, Pospischil M, Sassa S, Popowicz A, Manning LR, Schneewind O, Manning JM. J Biol Chem. 1992;267:3811. [PubMed] [Google Scholar]
- 467.Fujino T, Watanabe K, Beppu M, Kikugawa K, Yasuda H. Biochim Biophys Acta. 2000;1478:102. doi: 10.1016/s0167-4838(00)00004-2. [DOI] [PubMed] [Google Scholar]
- 468.Galjart NJ, Gillemans N, Meijer D, d’Azzo A. J Biol Chem. 1990;265:4678. [PubMed] [Google Scholar]
- 469.Hiraiwa M. Cell Mol Life Sci. 1999;56:894. doi: 10.1007/s000180050482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Galjart NJ, Gillemans N, Harris A, van der Horst GT, Verheijen FW, Galjaard H, d’Azzo A. Cell. 1988;54:755. doi: 10.1016/s0092-8674(88)90999-3. [DOI] [PubMed] [Google Scholar]
- 471.Jackman HL, Tan FL, Tamei H, Beurling-Harbury C, Li XY, Skidgel RA, Erdos EG. J Biol Chem. 1990;265:11265. [PubMed] [Google Scholar]
- 472.Rudenko G, Bonten E, d’Azzo A, Hol WG. Structure. 1995;3:1249. doi: 10.1016/s0969-2126(01)00260-x. [DOI] [PubMed] [Google Scholar]
- 473.Bonten EJ, Galjart NJ, Willemsen R, Usmany M, Vlak JM, d’Azzo A. J Biol Chem. 1995;270:26441. doi: 10.1074/jbc.270.44.26441. [DOI] [PubMed] [Google Scholar]
- 474.Jackman HL, Morris PW, Deddish PA, Skidgel RA, Erdos EG. J Biol Chem. 1992;267:2872. [PubMed] [Google Scholar]
- 475.D’Azzo A, Hoogeveen A, Reuser AJ, Robinson D, Galjaard H. Proc Natl Acad Sci U S A. 1982;79:4535. doi: 10.1073/pnas.79.15.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 476.Kyllerman M, Mansson JE, Westphal O, Conradi N, Nellstrom H. Pediatr Neurol. 1993;9:318. doi: 10.1016/0887-8994(93)90073-l. [DOI] [PubMed] [Google Scholar]
- 477.Vinogradova MV, Michaud L, Mezentsev AV, Lukong KE, El-Alfy M, Morales CR, Potier M, Pshezhetsky AV. Biochem J. 1998;330(Pt 2):641. doi: 10.1042/bj3300641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Morreau H, Galjart NJ, Willemsen R, Gillemans N, Zhou XY, d’Azzo A. J Biol Chem. 1992;267:17949. [PubMed] [Google Scholar]
- 479.Galjart NJ, Morreau H, Willemsen R, Gillemans N, Bonten EJ, d’Azzo A. J Biol Chem. 1991;266:14754. [PubMed] [Google Scholar]
- 480.Zhou XY, Morreau H, Rottier R, Davis D, Bonten E, Gillemans N, Wenger D, Grosveld FG, Doherty P, Suzuki K, Grosveld GC, d’Azzo A. Genes Dev. 1995;9:2623. doi: 10.1101/gad.9.21.2623. [DOI] [PubMed] [Google Scholar]
- 481.Seyrantepe V, Hinek A, Peng J, Fedjaev M, Ernest S, Kadota Y, Canuel M, Itoh K, Morales CR, Lavoie J, Tremblay J, Pshezhetsky AV. Circulation. 2008;117:1973. doi: 10.1161/CIRCULATIONAHA.107.733212. [DOI] [PubMed] [Google Scholar]
- 482.Haynes WG, Webb DJ. J Hypertens. 1998;16:1081. doi: 10.1097/00004872-199816080-00001. [DOI] [PubMed] [Google Scholar]
- 483.Tan F, Morris PW, Skidgel RA, Erdos EG. J Biol Chem. 1993;268:16631. [PubMed] [Google Scholar]
- 484.Kumamoto K, Stewart TA, Johnson AR, Erdos EG. J Clin Invest. 1981;67:210. doi: 10.1172/JCI110015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 485.Miller JJ, Changaris DG, Levy RS. Life Sci. 1991;48:1529. doi: 10.1016/0024-3205(91)90277-i. [DOI] [PubMed] [Google Scholar]
- 486.Odya CE, Marinkovic DV, Hammon KJ, Stewart TA, Erdos EG. J Biol Chem. 1978;253:5927. [PubMed] [Google Scholar]
- 487.Soisson SM, Patel SB, Abeywickrema PD, Byrne NJ, Diehl RE, Hall DL, Ford RE, Reid JC, Rickert KW, Shipman JM, Sharma S, Lumb KJ. BMC Struct Biol. 2010;10:16. doi: 10.1186/1472-6807-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Wallingford N, Perroud B, Gao Q, Coppola A, Gyengesi E, Liu ZW, Gao XB, Diament A, Haus KA, Shariat-Madar Z, Mahdi F, Wardlaw SL, Schmaier AH, Warden CH, Diano S. J Clin Invest. 2009;119:2291. doi: 10.1172/JCI37209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 489.Zhou C, Garcia-Calvo M, Pinto S, Lombardo M, Feng Z, Bender K, Pryor KD, Bhatt UR, Chabin RM, Geissler WM, Shen Z, Tong X, Zhang Z, Wong KK, Roy RS, Chapman KT, Yang L, Xiong Y. J Med Chem. 2010;53:7251. doi: 10.1021/jm101013m. [DOI] [PubMed] [Google Scholar]
- 490.Zhao X, Southwick K, Cardasis HL, Du Y, Lassman ME, Xie D, El-Sherbeini M, Geissler WM, Pryor KD, Verras A, Garcia-Calvo M, Shen DM, Yates NA, Pinto S, Hendrickon RC. Proteomics. 2010;10:2882. doi: 10.1002/pmic.201000145. [DOI] [PubMed] [Google Scholar]
- 491.Araki H, Li Y, Yamamoto Y, Haneda M, Nishi K, Kikkawa R, Ohkubo I. J Biochem. 2001;129:279. doi: 10.1093/oxfordjournals.jbchem.a002855. [DOI] [PubMed] [Google Scholar]
- 492.Chiravuri M, Agarraberes F, Mathieu SL, Lee H, Huber BT. J Immunol. 2000;165:5695. doi: 10.4049/jimmunol.165.10.5695. [DOI] [PubMed] [Google Scholar]
- 493.Maes MB, Lambeir AM, Gilany K, Senten K, Van der Veken P, Leiting B, Augustyns K, Scharpe S, De Meester I. Biochem J. 2005;386:315. doi: 10.1042/BJ20041156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Leiting B, Pryor KD, Wu JK, Marsilio F, Patel RA, Craik CS, Ellman JA, Cummings RT, Thornberry NA. Biochem J. 2003;371:525. doi: 10.1042/BJ20021643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Chiravuri M, Schmitz T, Yardley K, Underwood R, Dayal Y, Huber BT. J Immunol. 1999;163:3092. [PubMed] [Google Scholar]
- 496.Danilova OV, Tai AK, Mele DA, Beinborn M, Leiter AB, Greenberg AS, Perfield JW, 2nd, Defuria J, Singru PS, Lechan RM, Huber BT. Endocrinology. 2009;150:5240. doi: 10.1210/en.2009-0386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497.Senten K, Van der Veken P, Bal G, De Meester I, Lambeir AM, Scharpe S, Bauvois B, Haemers A, Augustyns K. Bioorg Med Chem Lett. 2002;12:2825. doi: 10.1016/s0960-894x(02)00603-0. [DOI] [PubMed] [Google Scholar]
- 498.Senten K, Van Der Veken P, De Meester I, Lambeir AM, Scharpe S, Haemers A, Augustyns K. J Med Chem. 2004;47:2906. doi: 10.1021/jm031122f. [DOI] [PubMed] [Google Scholar]
- 499.Danilova O, Li B, Szardenings AK, Huber BT, Rosenblum JS. Bioorg Med Chem Lett. 2007;17:507. doi: 10.1016/j.bmcl.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Xing Y, Li Z, Chen Y, Stock JB, Jeffrey PD, Shi Y. Cell. 2008;133:154. doi: 10.1016/j.cell.2008.02.041. [DOI] [PubMed] [Google Scholar]
- 501.Devedjiev Y, Dauter Z, Kuznetsov SR, Jones TL, Derewenda ZS. Structure. 2000;8:1137. doi: 10.1016/s0969-2126(00)00529-3. [DOI] [PubMed] [Google Scholar]
- 502.Bellizzi JJ, 3rd, Widom J, Kemp C, Lu JY, Das AK, Hofmann SL, Clardy J. Proc Natl Acad Sci U S A. 2000;97:4573. doi: 10.1073/pnas.080508097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Ortega-Gutierrez S, Leung D, Ficarro S, Peters EC, Cravatt BF. PLoS One. 2008;3:e2486. doi: 10.1371/journal.pone.0002486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Longin S, Zwaenepoel K, Martens E, Louis JV, Rondelez E, Goris J, Janssens V. Exp Cell Res. 2008;314:68. doi: 10.1016/j.yexcr.2007.07.030. [DOI] [PubMed] [Google Scholar]
- 505.Ogris E, Du X, Nelson KC, Mak EK, Yu XX, Lane WS, Pallas DC. J Biol Chem. 1999;274:14382. doi: 10.1074/jbc.274.20.14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Oliver CJ, Shenolikar S. Front Biosci. 1998;3:D961. doi: 10.2741/a336. [DOI] [PubMed] [Google Scholar]
- 507.Shi Y. Cell. 2009;139:468. doi: 10.1016/j.cell.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 508.Tolstykh T, Lee J, Vafai S, Stock JB. EMBO J. 2000;19:5682. doi: 10.1093/emboj/19.21.5682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Puustinen P, Junttila MR, Vanhatupa S, Sablina AA, Hector ME, Teittinen K, Raheem O, Ketola K, Lin S, Kast J, Haapasalo H, Hahn WC, Westermarck J. Cancer Res. 2009;69:2870. doi: 10.1158/0008-5472.CAN-08-2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Wu J, Tolstykh T, Lee J, Boyd K, Stock JB, Broach JR. EMBO J. 2000;19:5672. doi: 10.1093/emboj/19.21.5672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 511.Longin S, Jordens J, Martens E, Stevens I, Janssens V, Rondelez E, De Baere I, Derua R, Waelkens E, Goris J, Van Hoof C. Biochem J. 2004;380:111. doi: 10.1042/BJ20031643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 512.Sontag JM, Nunbhakdi-Craig V, Mitterhuber M, Ogris E, Sontag E. J Neurochem. 2010;115:1455. doi: 10.1111/j.1471-4159.2010.07049.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Bachovchin DA, Mohr JT, Speers AE, Wang C, Berlin JM, Spicer TP, Fernandez-Vega V, Chase P, Hodder PS, Schurer SC, Nomura DK, Rosen H, Fu GC, Cravatt BF. Proc Natl Acad Sci U S A. 2011;108:6811. doi: 10.1073/pnas.1015248108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 514.Wang A, Yang HC, Friedman P, Johnson CA, Dennis EA. Biochim Biophys Acta. 1999;1437:157. doi: 10.1016/s1388-1981(99)00012-8. [DOI] [PubMed] [Google Scholar]
- 515.Hirano T, Kishi M, Sugimoto H, Taguchi R, Obinata H, Ohshima N, Tatei K, Izumi T. Biochim Biophys Acta. 2009;1791:797. doi: 10.1016/j.bbalip.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 516.Sugimoto H, Hayashi H, Yamashita S. J Biol Chem. 1996;271:7705. doi: 10.1074/jbc.271.13.7705. [DOI] [PubMed] [Google Scholar]
- 517.Shanado Y, Kometani M, Uchiyama H, Koizumi S, Teno N. Biochem Biophys Res Commun. 2004;325:1487. doi: 10.1016/j.bbrc.2004.10.193. [DOI] [PubMed] [Google Scholar]
- 518.Duncan JA, Gilman AG. J Biol Chem. 1998;273:15830. doi: 10.1074/jbc.273.25.15830. [DOI] [PubMed] [Google Scholar]
- 519.Yeh DC, Duncan JA, Yamashita S, Michel T. J Biol Chem. 1999;274:33148. doi: 10.1074/jbc.274.46.33148. [DOI] [PubMed] [Google Scholar]
- 520.Dekker FJ, Rocks O, Vartak N, Menninger S, Hedberg C, Balamurugan R, Wetzel S, Renner S, Gerauer M, Scholermann B, Rusch M, Kramer JW, Rauh D, Coates GW, Brunsveld L, Bastiaens PI, Waldmann H. Nat Chem Biol. 2010;6:449. doi: 10.1038/nchembio.362. [DOI] [PubMed] [Google Scholar]
- 521.Siegel G, Obernosterer G, Fiore R, Oehmen M, Bicker S, Christensen M, Khudayberdiev S, Leuschner PF, Busch CJ, Kane C, Hubel K, Dekker F, Hedberg C, Rengarajan B, Drepper C, Waldmann H, Kauppinen S, Greenberg ME, Draguhn A, Rehmsmeier M, Martinez J, Schratt GM. Nat Cell Biol. 2009;11:705. doi: 10.1038/ncb1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Toyoda T, Sugimoto H, Yamashita S. Biochim Biophys Acta. 1999;1437:182. doi: 10.1016/s1388-1981(99)00007-4. [DOI] [PubMed] [Google Scholar]
- 523.Tomatis VM, Trenchi A, Gomez GA, Daniotti JL. PLoS One. 2010;5:e15045. doi: 10.1371/journal.pone.0015045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Camp LA, Hofmann SL. J Biol Chem. 1993;268:22566. [PubMed] [Google Scholar]
- 525.Camp LA, Verkruyse LA, Afendis SJ, Slaughter CA, Hofmann SL. J Biol Chem. 1994;269:23212. [PubMed] [Google Scholar]
- 526.Hellsten E, Vesa J, Olkkonen VM, Jalanko A, Peltonen L. EMBO J. 1996;15:5240. [PMC free article] [PubMed] [Google Scholar]
- 527.Verkruyse LA, Hofmann SL. J Biol Chem. 1996;271:15831. doi: 10.1074/jbc.271.26.15831. [DOI] [PubMed] [Google Scholar]
- 528.Das AK, Bellizzi JJ, 3rd, Tandel S, Biehl E, Clardy J, Hofmann SL. J Biol Chem. 2000;275:23847. doi: 10.1074/jbc.M002758200. [DOI] [PubMed] [Google Scholar]
- 529.Sleat DE, Donnelly RJ, Lackland H, Liu CG, Sohar I, Pullarkat RK, Lobel P. Science. 1997;277:1802. doi: 10.1126/science.277.5333.1802. [DOI] [PubMed] [Google Scholar]
- 530.Vesa J, Hellsten E, Verkruyse LA, Camp LA, Rapola J, Santavuori P, Hofmann SL, Peltonen L. Nature. 1995;376:584. doi: 10.1038/376584a0. [DOI] [PubMed] [Google Scholar]
- 531.Gupta P, Soyombo AA, Atashband A, Wisniewski KE, Shelton JM, Richardson JA, Hammer RE, Hofmann SL. Proc Natl Acad Sci U S A. 2001;98:13566. doi: 10.1073/pnas.251485198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 532.Soyombo AA, Hofmann SL. J Biol Chem. 1997;272:27456. doi: 10.1074/jbc.272.43.27456. [DOI] [PubMed] [Google Scholar]
- 533.Gupta P, Soyombo AA, Shelton JM, Wilkofsky IG, Wisniewski KE, Richardson JA, Hofmann SL. Proc Natl Acad Sci U S A. 2003;100:12325. doi: 10.1073/pnas.2033229100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 534.Raetz CR, Reynolds CM, Trent MS, Bishop RE. Annu Rev Biochem. 2007;76:295. doi: 10.1146/annurev.biochem.76.010307.145803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Raetz CR, Whitfield C. Annu Rev Biochem. 2002;71:635. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 536.Nagai Y, Kobayashi T, Motoi Y, Ishiguro K, Akashi S, Saitoh S, Kusumoto Y, Kaisho T, Akira S, Matsumoto M, Takatsu K, Miyake K. J Immunol. 2005;174:7043. doi: 10.4049/jimmunol.174.11.7043. [DOI] [PubMed] [Google Scholar]
- 537.Munford RS, Hall CL. Science. 1986;234:203. doi: 10.1126/science.3529396. [DOI] [PubMed] [Google Scholar]
- 538.Munford RS, Varley AW. PLoS Pathog. 2006;2:e67. doi: 10.1371/journal.ppat.0020067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Ojogun N, Kuang TY, Shao B, Greaves DR, Munford RS, Varley AW. J Infect Dis. 2009;200:1685. doi: 10.1086/646616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Munford RS, Hall CL. J Biol Chem. 1989;264:15613. [PubMed] [Google Scholar]
- 541.Hagen FS, Grant FJ, Kuijper JL, Slaughter CA, Moomaw CR, Orth K, O’Hara PJ, Munford RS. Biochemistry. 1991;30:8415. doi: 10.1021/bi00098a020. [DOI] [PubMed] [Google Scholar]
- 542.Munford RS, Hunter JP. J Biol Chem. 1992;267:10116. [PubMed] [Google Scholar]
- 543.Lu M, Zhang M, Kitchens RL, Fosmire S, Takashima A, Munford RS. J Exp Med. 2003;197:1745. doi: 10.1084/jem.20030420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 544.Shao B, Lu M, Katz SC, Varley AW, Hardwick J, Rogers TE, Ojogun N, Rockey DC, Dematteo RP, Munford RS. J Biol Chem. 2007;282:13726. doi: 10.1074/jbc.M609462200. [DOI] [PubMed] [Google Scholar]
- 545.Lu M, Zhang M, Takashima A, Weiss J, Apicella MA, Li XH, Yuan D, Munford RS. Nat Immunol. 2005;6:989. doi: 10.1038/ni1246. [DOI] [PubMed] [Google Scholar]
- 546.Tanaka S, Maeda Y, Tashima Y, Kinoshita T. J Biol Chem. 2004;279:14256. doi: 10.1074/jbc.M313755200. [DOI] [PubMed] [Google Scholar]
- 547.Ueda Y, Yamaguchi R, Ikawa M, Okabe M, Morii E, Maeda Y, Kinoshita T. J Biol Chem. 2007;282:30373. doi: 10.1074/jbc.M705601200. [DOI] [PubMed] [Google Scholar]
- 548.Higa HH, Manzi A, Varki A. J Biol Chem. 1989;264:19435. [PubMed] [Google Scholar]
- 549.Takematsu H, Diaz S, Stoddart A, Zhang Y, Varki A. J Biol Chem. 1999;274:25623. doi: 10.1074/jbc.274.36.25623. [DOI] [PubMed] [Google Scholar]
- 550.Stoddart A, Zhang Y, Paige CJ. Nucleic Acids Res. 1996;24:4003. doi: 10.1093/nar/24.20.4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551.Butor C, Higa HH, Varki A. J Biol Chem. 1993;268:10207. [PubMed] [Google Scholar]
- 552.Cariappa A, Takematsu H, Liu H, Diaz S, Haider K, Boboila C, Kalloo G, Connole M, Shi HN, Varki N, Varki A, Pillai S. J Exp Med. 2009;206:125. doi: 10.1084/jem.20081399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 553.Surolia I, Pirnie SP, Chellappa V, Taylor KN, Cariappa A, Moya J, Liu H, Bell DW, Driscoll DR, Diederichs S, Haider K, Netravali I, Le S, Elia R, Dow E, Lee A, Freudenberg J, De Jager PL, Chretien Y, Varki A, Macdonald ME, Gillis T, Behrens TW, Bloch D, Collier D, Korzenik J, Podolsky DK, Hafler D, Murali M, Sands B, Stone JH, Gregersen PK, Pillai S. Nature. 2010;466:243. doi: 10.1038/nature09115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 554.Pillai S, Cariappa A, Pirnie SP. Trends Immunol. 2009;30:488. doi: 10.1016/j.it.2009.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555.Karasik A, Aschner P, Katzeff H, Davies MJ, Stein PP. Curr Med Res Opin. 2008;24:489. doi: 10.1185/030079908x261069. [DOI] [PubMed] [Google Scholar]
- 556.Probst MR, Beer M, Beer D, Jeno P, Meyer UA, Gasser R. J Biol Chem. 1994;269:21650. [PubMed] [Google Scholar]
- 557.Probst MR, Jeno P, Meyer UA. Biochem Biophys Res Commun. 1991;177:453. doi: 10.1016/0006-291x(91)92005-5. [DOI] [PubMed] [Google Scholar]
- 558.Trickett JI, Patel DD, Knight BL, Saggerson ED, Gibbons GF, Pease RJ. J Biol Chem. 2001;276:39522. doi: 10.1074/jbc.M101764200. [DOI] [PubMed] [Google Scholar]
- 559.Watanabe A, Fukami T, Takahashi S, Kobayashi Y, Nakagawa N, Nakajima M, Yokoi T. Drug Metab Dispos. 2010;38:1532. doi: 10.1124/dmd.110.033720. [DOI] [PubMed] [Google Scholar]
- 560.Watanabe A, Fukami T, Nakajima M, Takamiya M, Aoki Y, Yokoi T. Drug Metab Dispos. 2009;37:1513. doi: 10.1124/dmd.109.026567. [DOI] [PubMed] [Google Scholar]
- 561.Lo V, Erickson B, Thomason-Hughes M, Ko KW, Dolinsky VW, Nelson R, Lehner R. J Lipid Res. 2010;51:368. doi: 10.1194/jlr.M000596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 562.Edgar AJ, Polak JM. Biochem Biophys Res Commun. 2002;292:617. doi: 10.1006/bbrc.2002.6692. [DOI] [PubMed] [Google Scholar]
- 563.Miyata K, Oike Y, Hoshii T, Maekawa H, Ogawa H, Suda T, Araki K, Yamamura K. Biochem Biophys Res Commun. 2005;329:296. doi: 10.1016/j.bbrc.2005.01.127. [DOI] [PubMed] [Google Scholar]
- 564.Jin S, Zhao G, Li Z, Nishimoto Y, Isohama Y, Shen J, Ito T, Takeya M, Araki K, He P, Yamamura K. Biochem Biophys Res Commun. 2009;380:419. doi: 10.1016/j.bbrc.2009.01.098. [DOI] [PubMed] [Google Scholar]
- 565.Stoelting M, Geyer M, Reuter S, Reichelt R, Bek MJ, Pavenstadt H. Biochem Biophys Res Commun. 2009;379:81. doi: 10.1016/j.bbrc.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 566.Forner F, Foster LJ, Campanaro S, Valle G, Mann M. Mol Cell Proteomics. 2006;5:608. doi: 10.1074/mcp.M500298-MCP200. [DOI] [PubMed] [Google Scholar]
- 567.Merla G, Ucla C, Guipponi M, Reymond A. Hum Genet. 2002;110:429. doi: 10.1007/s00439-002-0710-x. [DOI] [PubMed] [Google Scholar]
- 568.Padmanabhan B, Kuzuhara T, Adachi N, Horikoshi M. J Biol Chem. 2004;279:9615. doi: 10.1074/jbc.M312165200. [DOI] [PubMed] [Google Scholar]
- 569.Kim I, Chu XY, Kim S, Provoda CJ, Lee KD, Amidon GL. J Biol Chem. 2003;278:25348. doi: 10.1074/jbc.M302055200. [DOI] [PubMed] [Google Scholar]
- 570.Lai L, Xu Z, Zhou J, Lee KD, Amidon GL. J Biol Chem. 2008;283:9318. doi: 10.1074/jbc.M709530200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571.Puente XS, Lopez-Otin C. J Biol Chem. 1995;270:12926. doi: 10.1074/jbc.270.21.12926. [DOI] [PubMed] [Google Scholar]
- 572.Kim I, Song X, Vig BS, Mittal S, Shin HC, Lorenzi PJ, Amidon GL. Mol Pharm. 2004;1:117. doi: 10.1021/mp0499757. [DOI] [PubMed] [Google Scholar]
- 573.Nakajima K, Sonoda H, Mizoguchi T, Aoki J, Arai H, Nagahama M, Tagaya M, Tani K. J Biol Chem. 2002;277:11329. doi: 10.1074/jbc.M111092200. [DOI] [PubMed] [Google Scholar]
- 574.Tani K, Mizoguchi T, Iwamatsu A, Hatsuzawa K, Tagaya M. J Biol Chem. 1999;274:20505. doi: 10.1074/jbc.274.29.20505. [DOI] [PubMed] [Google Scholar]
- 575.Shimoi W, Ezawa I, Nakamoto K, Uesaki S, Gabreski G, Aridor M, Yamamoto A, Nagahama M, Tagaya M, Tani K. J Biol Chem. 2005;280:10141. doi: 10.1074/jbc.M409673200. [DOI] [PubMed] [Google Scholar]
- 576.Ong YS, Tang BL, Loo LS, Hong W. J Cell Biol. 2010;190:331. doi: 10.1083/jcb.201003005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577.Mizoguchi T, Nakajima K, Hatsuzawa K, Nagahama M, Hauri HP, Tagaya M, Tani K. Biochem Biophys Res Commun. 2000;279:144. doi: 10.1006/bbrc.2000.3846. [DOI] [PubMed] [Google Scholar]
- 578.Schultz DC, Vanderveer L, Berman DB, Hamilton TC, Wong AJ, Godwin AK. Cancer Res. 1996;56:1997. [PubMed] [Google Scholar]
- 579.Phillips N, Ziegler M, Saha B, Xynos F. Int J Cancer. 1993;54:85. doi: 10.1002/ijc.2910540115. [DOI] [PubMed] [Google Scholar]
- 580.Vorobiev SM, Su M, Seetharaman J, Huang YJ, Chen CX, Maglaqui M, Janjua H, Proudfoot M, Yakunin A, Xiao R, Acton TB, Montelione GT, Tong L. Proteins. 2009;74:526. doi: 10.1002/prot.22278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 581.Woitach JT, Zhang M, Niu CH, Thorgeirsson SS. Nat Genet. 1998;19:371. doi: 10.1038/1258. [DOI] [PubMed] [Google Scholar]
- 582.Shields DJ, Niessen S, Murphy EA, Mielgo A, Desgrosellier JS, Lau SK, Barnes LA, Lesperance J, Bouvet M, Tarin D, Cravatt BF, Cheresh DA. Proc Natl Acad Sci U S A. 2010;107:2189. doi: 10.1073/pnas.0911646107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 583.Bachovchin DA, Brown SJ, Rosen H, Cravatt BF. Nat Biotechnol. 2009;27:387. doi: 10.1038/nbt.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 584.Bachovchin DA, Wolfe MR, Masuda K, Brown SJ, Spicer TP, Fernandez-Vega V, Chase P, Hodder PS, Rosen H, Cravatt BF. Bioorg Med Chem Lett. 2010;20:2254. doi: 10.1016/j.bmcl.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585.Crenon I, Foglizzo E, Kerfelec B, Verine A, Pignol D, Hermoso J, Bonicel J, Chapus C. Protein Eng. 1998;11:135. doi: 10.1093/protein/11.2.135. [DOI] [PubMed] [Google Scholar]
- 586.Ren J, Chen Z, Zhang W, Li L, Sun R, Deng C, Fei Z, Sheng Z, Wang L, Sun X, Wang Z, Fei J. J Nutr Biochem. 2010 doi: 10.1016/j.jnutbio.2010.06.002. [DOI] [PubMed] [Google Scholar]
- 587.Smith TS, Southan C, Ellington K, Campbell D, Tew DG, Debouck C. Genomics. 2001;78:12. doi: 10.1006/geno.2001.6643. [DOI] [PubMed] [Google Scholar]
- 588.Polianskyte Z, Peitsaro N, Dapkunas A, Liobikas J, Soliymani R, Lalowski M, Speer O, Seitsonen J, Butcher S, Cereghetti GM, Linder MD, Merckel M, Thompson J, Eriksson O. Proc Natl Acad Sci U S A. 2009;106:18960. doi: 10.1073/pnas.0906734106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589.Wilson PA, Gardner SD, Lambie NM, Commans SA, Crowther DJ. J Lipid Res. 2006;47:1940. doi: 10.1194/jlr.M600185-JLR200. [DOI] [PubMed] [Google Scholar]
- 590.Lee TH, Streb JW, Georger MA, Miano JM. J Histochem Cytochem. 2006;54:701. doi: 10.1369/jhc.5A6894.2006. [DOI] [PubMed] [Google Scholar]
- 591.Lee TH, Chen J, Miano JM. Circ Res. 2009;105:271. doi: 10.1161/CIRCRESAHA.109.199869. [DOI] [PubMed] [Google Scholar]
- 592.Kollmann K, Damme M, Deuschl F, Kahle J, D’Hooge R, Lullmann-Rauch R, Lubke T. FEBS J. 2009;276:1356. doi: 10.1111/j.1742-4658.2009.06877.x. [DOI] [PubMed] [Google Scholar]
- 593.Chen J, Streb JW, Maltby KM, Kitchen CM, Miano JM. J Biol Chem. 2001;276:34175. doi: 10.1074/jbc.M104162200. [DOI] [PubMed] [Google Scholar]
- 594.Mahoney JA, Ntolosi B, DaSilva RP, Gordon S, McKnight AJ. Genomics. 2001;72:243. doi: 10.1006/geno.2000.6484. [DOI] [PubMed] [Google Scholar]
- 595.Harris J, Schwinn N, Mahoney JA, Lin HH, Shaw M, Howard CJ, da Silva RP, Gordon S. Int J Exp Pathol. 2006;87:29. doi: 10.1111/j.0959-9673.2006.00450.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596.Borgstrom B. Biochim Biophys Acta. 1977;488:381. doi: 10.1016/0005-2760(77)90197-7. [DOI] [PubMed] [Google Scholar]
- 597.Jessani N, Humphrey M, McDonald WH, Niessen S, Masuda K, Gangadharan B, Yates JR, 3rd, Mueller BM, Cravatt BF. Proc Natl Acad Sci U S A. 2004;101:13756. doi: 10.1073/pnas.0404727101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598.Jessani N, Young JA, Diaz SL, Patricelli MP, Varki A, Cravatt BF. Angew Chem Int Ed Engl. 2005;44:2400. doi: 10.1002/anie.200463098. [DOI] [PubMed] [Google Scholar]
- 599.Jessani N, Cravatt BF. Curr Opin Chem Biol. 2004;8:54. doi: 10.1016/j.cbpa.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 600.Leung D, Hardouin C, Boger DL, Cravatt BF. Nat Biotechnol. 2003;21:687. doi: 10.1038/nbt826. [DOI] [PubMed] [Google Scholar]
- 601.Li W, Blankman JL, Cravatt BF. J Am Chem Soc. 2007;129:9594. doi: 10.1021/ja073650c. [DOI] [PubMed] [Google Scholar]
- 602.Tully SE, Cravatt BF. J Am Chem Soc. 2010;132:3264. doi: 10.1021/ja1000505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 603.Speers AE, Adam GC, Cravatt BF. J Am Chem Soc. 2003;125:4686. doi: 10.1021/ja034490h. [DOI] [PubMed] [Google Scholar]
- 604.Khidekel N, Ficarro SB, Peters EC, Hsieh-Wilson LC. Proc Natl Acad Sci U S A. 2004;101:13132. doi: 10.1073/pnas.0403471101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.Mahal LK, Yarema KJ, Bertozzi CR. Science. 1997;276:1125. doi: 10.1126/science.276.5315.1125. [DOI] [PubMed] [Google Scholar]
- 606.Martin BR, Cravatt BF. Nat Methods. 2009;6:135. doi: 10.1038/nmeth.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 607.Heal WP, Tate EW. Org Biomol Chem. 2010;8:731. doi: 10.1039/b917894e. [DOI] [PubMed] [Google Scholar]