

Abstract
With metal carbene access to dipolar intermediates, 3,6-dihydro-1,2-oxazines are produced in high yields by dirhodium(II) carboxylate catalyzed reactions between nitrones and a β-TBSO-substituted vinyldiazoacetate. High enantiocontrol occurs with catalysis by N-phthaloyl-(S)-(amino acid)-ligated dirhodium carboxylates for [3+3]-cycloaddition reactions with both acyclic and cyclic nitrones.
Nitrones are versatile reagents for cycloaddition reactions that produce cyclic hydroxylamine dervatives.1 Although their catalytic [2+3]-dipolar cycloaddition reactions have been studied extensively,2 and highly enantioselective reactions with α,β-unsaturated aldehydes have been reported,3 scarce attention has been given to their formal [3+3]-cycloaddition reactions. Hayashi reported a palladium-catalyzed [3+3]-cycloaddition reaction between an N,α-diarylnitrone and trimethylenemethane generated from [2-(acetoxymethyl)-2-propenyl)trimethylsilane in high yield,4 but nitrones have otherwise been neglected. Greater attention has been given to azomethine imines and their reactions with trimethylenemethane (Pd catalysis),4 enals (N-heterocyclic carbene catalysis),5 and propargyl esters (Au catalysis);6 and other pairings for [3+3] cycloaddition show broad applicability of this annulation transformation.7 However, although there is recognized high potential for [3+3]-cycloaddition between 1,3-dipoles and electrophilic vinylcarbenes, only the recent communication by Toste has described a [3+3]-annulation process that involves a putative vinylcarbene intermediate.6
We reasoned that the absence of examples of [3+3]-cycloaddition reactions by catalytic dinitrogen extrusion from vinyldiazo compounds was due to the limited ability of structure 1b of the vinylcarbene intermediate to capture a dipolar species in a stepwise process that is a formal [3+3]-cycloaddition (Scheme 1). This problem has now been resolved with the use of TBSO-substituted vinyldiazoacetate 58 in catalytic reactions with nitrones to form 3,6-dihydro-1,2-oxazine derivatives exclusively and in high yield, and high enantiocontrol has been achieved from the application of a chiral dirhodium carboxylate catalyst.
Scheme 1.

Treatment of 1.5 equiv. of 5 in the presence of N,α-diphenylnitrone and 1.0 mol % of dirhodium tetraacetate in dichloromethane gave immediate dinitrogen extrusion from 5 and produced 3,6-dihydro-1,2-oxazine 6a within 20 min in 98% isolated yield (eq 1). No reaction occurred in the absence of
 |
(1) |
catalyst. The installation of the electron-donating MeO substituent or an electron-withdrawing Br substituent on the reactant nitrone also formed the [3+3]-cycloaddition products (6b and 6c) in high yields. Structural confirmation for the 3,6-dihydro-1,2-oxazine products was obtained spectrally and from the X-ray diffraction of a single crystal from 6c (Figure 1). The core structure of these compounds, which up to now has been mainly available via nitroso hetero-Diels-Alder reactions9 and from a tandem one-pot process involving organocatalytic α-oxyamination of an enamine with nitrosobenzene followed by reaction with a vinyl phosphonium salt in an intramolecular Wittig process,10 are versatile intermediates for the synthesis of α-substituted β-amino acids and related compounds that are not easily accessible by other methods.9,11 Substituent groups on 6a make this 3,6-dihydro-1,2-oxazine particularly suitable for further functionalization.
Figure 1.

X-ray structure of the formal [3+3]-cycloaddition reaction product (6c) formed from 5 and N-phenyl-α-(p-bromophenyl) nitrone.

Only one direct chiral catalytic methodology has been advanced for the synthesis of 3,6-dihydro-1,2-oxazines, and this process is limited to pyridylnitroso compounds that undergo cycloaddition with conjugated dienes.12 Ley’s two-step organocatalytic route10 provides high enantiocontrol and modest to good yields, but is limited thus far to nitrosobenzene. In view of the limited availability of catalytic enantioselective methods with which to access these compounds we sought to employ chiral dirhodium carboxylate catalysts for the reaction between 5 and N,α-diphenylnitrone depicted in eq 1. As can be seen by the data in Table 1, Rh2(S-DOSP)4 (7)13 catalyzed the formal [3+3]-cycloaddition reaction but did not provide evidence of any enantiocontrol. However, the phthalimide-amino acid ligated dirhodium catalysts 8a–8d of Hashimoto and coworkers14 produced 6a in good to excellent yields and modest to high enantioselectivities. Surprisingly, the least sterically encumbered catalyst (8d) provided the highest level of enantiocontrol; steric interference by R1 (for 8) and R2 (for 915) appears to be the cause for lowering % ee, and the absence of 6a from catalysis by 9b suggests the absolute limitation for dirhodium carboxylates with this class of chiral ligand. Notably, bulky substituents in ligands also decreased the reaction rate, especially for Rh2(S-PTTL)4 (8c) and Rh2(S-PTAD)4 (8e)16, which only converted a small portion of the nitrone into the [3+3]-annulation product 6a resulting in <20% isolated yield. Both solvent and temperature were varied to reach optimum conditions (entry 14) that were use of tert-butyl methyl ether (TBME) as the solvent and minus 30°C as the reaction temperature for 95% yield and 93% ee with only 2.0 mol% of 8d and a reaction time of 2 h.
Table 1.
Optimization of the Enantioselective Formal [3+3] Cycloaddition Reaction between Siloxyvinyldiazoacetate 5 and N,α-Diphenylnitrone Catalyzed by Chiral Dirhodium(II) Carboxylatesa
| Entry | Rh2L4 | Solvent | T (°C) |
time (h) |
yield (%)b |
ee (%)c |
|---|---|---|---|---|---|---|
| 1 | 7 | PhMe | 23 | 1 | 80 | 0 |
| 2 | 8a | CH2Cl2 | 23 | 0.5 | 98 | 30 |
| 3 | 8a | PhMe | 23 | 0.5 | 98 | 70 |
| 4 | 8a | PhMe | −15 | 2 | 76 | 80 |
| 5 | 8b | PhMe | −15 | 2 | 80 | 62 |
| 6 | 8c | PhMe | −15 | 2 | 15 | 60 |
| 7 | 8d | PhMe | −15 | 2 | 85 | 87 |
| 8 | 8e | PhMe | −15 | 2 | 18 | 64 |
| 9 | 9a | PhMe | −15 | 2 | 50 | 53 |
| 10 | 9b | PhMe | −15 | 2 | <3 | - |
| 11 | 8d | PhMe | −30 | 2 | 60 | 89 |
| 12 | 8d | PhMe/hexanes (2 : 1) | −30 | 2 | 70 | 91 |
| 13d | 8d | PhMe/hexanes (2 : 1) | −30 | 2 | 95 | 91 |
| 14d | 8d | TBME | −30 | 2 | 95 | 93 |
Reactions were performed by addition of a 1.0 mL solution of the 5 (0.38 mmol) to the suspension of 0.25 mmol N,α-diphenylnitrone, 0.0025 mmol catalyst (1.0 mol%), and 100 mg 4 Å MS with 1.0 mL solvent.
Isolated yield; the only other nitrone-derived material observed in the reaction mixture was unreacted N,α-diphenylnitrone.
Determined by HPLC (AD-H column).
The diazo compound was added dropwise with a syringe pump over a time period of 1 h, and 2.0 mol% catalyst was used.
With the optimal conditions in hand, the generality of this enantioselective formal [3+3]-cycloaddition reaction (eq 2) was further investigated, and the results of this investigation are given in Table 2. Product yields were high, and 3,6-dihydro-1,2-oxazines 11 were the sole reaction products; however, enantioselectivities of reactions with nitrones having electron-donating or electron-withdrawing groups all occurred with a 3–16% lower enantiomeric excess than did N,α-diphenylnitrone, indicating little dependence on the electronic nature of aryl substituents. Instead, the size of the α-substituent appears to be a determining factor in the enantioselectivity of these reactions. All substituents on the α-phenyl group decreased enantioselecitivity relative to N,α-diphenylnitrone, but 90% ee and 89% ee were achieved with α-2- and α-3-furyl nitrones (entries 9 and 10), albeit in a lower % conversion of 10 and lower isolated yield of 11. When the α-substituent was changed to the aliphatic cyclohexyl group (entry 12), enantioselectivity was also lower than with aryl groups as α-substituents.
Table 2.
Effects of Nitrone Substituents on Enantio-control for the Formal [3+3]-Cycloaddition Reactiona
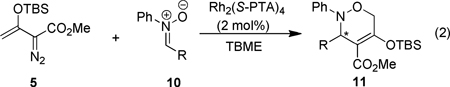 | |||||
|---|---|---|---|---|---|
| Entry | R | T (°C) | time (h) |
yield (%)b |
ee (%)c |
| 1 | C6H5 | −30 | 2 | 95 | 93 |
| 2 | p-MeC6H4 | −30 | 2 | 95 | 87 |
| 3 | p-MeOC6H4 | −15 | 3 | 96 | 78 |
| 4 | p-BrC6H4 | −15 | 4 | 65 | 80 |
| 5 | p-FC6H4 | −15 | 4 | 92 | 77 |
| 6 | m-MeC6H4 | −30 | 2 | 94 | 90 |
| 7 | m-ClC6H4 | −15 | 3 | 89 | 85 |
| 8 | 2-naphthyl | 0 | 3 | 73 | 80 |
| 9 | 2-furyl | 0 | 3 | 53 | 90 |
| 10 | 3-furyl | 0 | 3 | 66 | 89 |
| 11 | 2-thienyl | 0 | 3 | 81 | 80 |
| 12 | cyclohexyl | −30 | 2 | 85 | 77 |
Reactions were performed by slow addition (over 1 hour) of 1.0 mL solution of 5 (0.38 mmol) to the suspension of 0.25 mmol nitrone 10, 0.0050 mmol catalyst (2.0 mol%), and 100 mg 4 Å MS with 1.0 mL TBME.
Isolated yield of 11; the only other nitrone-derived material observed in the reaction mixture was unreacted 10.
Determined by HPLC (AD-H or OD-H column).
Catalytic reactions of 5 with the cyclic nitrone of 3,4-dihydroisoquinoline N-oxide (12), which is the geometric equivalent of a cis-disubstituted nitrone whose stereochemical requirements would be expected to be different from those of trans-disubstituted nitrones such as 10, were also examined (eq 3 and Table 3). In contrast to reactions with 10, catalysis of the reaction between 5 and 12 by Rh2(S-PTA)4 (8d) provided the [3+3]-cycloaddition product in high yield but with only a moderate 54% enantiomeric excess. However, the sterically demanding catalyst Rh2(S-PTTL)4 (8c) improved enantioselectivity to 80% ee without a significant decrease in product yield. That use of Rh2(S-PTAD)4 (8e) had the same degree of enantiocontrol as Rh2(S-PTTL)4, but the yield of 13 in this case was much lower suggests the subtle nature of steric influences in this catalytic process. Solvent tert-butyl methyl ether was not used because of its poor miscibility with 3,4-dihydroisoquinoline N-oxide.
Table 3.
Formal [3+3]-Cycloaddition Reactions of 3,4-Dihydroisoquinoline N-oxide with 5.a
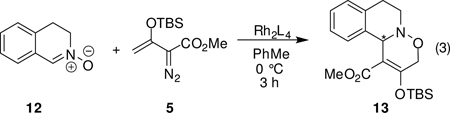 | ||
|---|---|---|
| catalyst | yield (%)b | ee (%)c |
| Rh2(S-PTA)4 (8d) | 97 | 54 |
| Rh2(S-PTTL)4 (8c) | 86 | 80 |
| Rh2(S-PTAD)4 (8e) | 50 | 80 |
| Rh2(S-NTTL)4 (9b) | 75 | 60 |
Reactions were performed by slow addition (over 1 hour) of the diazo compound (0.38 mmol) in 1.0 mL toluene to the suspension of 0.25 mmol 3,4-dihydroisoquinoline N-oxide, 0.0050 mmol catalyst (2.0 mol%), and 100 mg 4 Å MS in 1.0 mL toluene.
Isolated yield; the only other nitrone-derived material observed in the reaction mixture was unreacted 12.
Determined by HPLC (AD-H column).
The mechanism of this formal [3+3]-cycloaddition (Scheme 2) is in accord with the general process given in Scheme 1. Rhodium carboxylate catalysts activate the vinyl carbene for nucleophilic attack by the nitrone at the γ-position of 14 to form adduct 15. Vinylogous reactivity in catalytic reactions of vinyldiazoacetates has ample precedent.17 Intramolecular iminium ion addition to the catalyst-activated vinyl ether functional group of 15 forms, in a stepwise or concerted fashion, the [3+3]-cycloaddition product 11. Iminium ion addition is facilitated by stabilization provided by the TBSO group, and release of the catalyst from 16 is a favorable process. As predicted by Scheme 2, methyl styryldiazoacetate, which undergoes dirhodium(II) carboxylate catalyzed [3+2] annulation reactions of indoles and vinyl ethers,18 failed to produce the corresponding [3+3]-cycloaddition product with N,α-diphenylnitrone with catalysis by either Rh2(OAc)4 or Rh2(S-PTA)4.
Scheme 2.

Mechanism of the formal [3+3]-cycloaddition reaction of siloxyvinyldiazoacetate 5 and nitrone 10
In conclusion, we have developed a general, enantioselective formal [3+3]-cycloaddition process between TBSO-activated vinyldiazoacetate 5 and acyclic (10) and cyclic (12) nitrones that occur in high yields and selectivities. The convenience of this methodology, the absence of a background reaction, and the potential suitability of a spectrum of 1,3-dipoles and β-substituted vinyldiazoacetates for this transformation suggest broad applicability. The high level of dependence of catalyst ligands on enantioselectivity in product formation provides opportunities for new catalyst developments. Further studies exploring these processes are underway.
Supplementary Material
ACKNOWLEDGMENT
Support for this research from the National Institutes of Health (GM 46503) and the National Science Foundation (CHE-0748121) is gratefully acknowledged.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Experimental procedures and compound characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.(a) Kissane M, Maguire AR. Chem. Soc. Rev. 2010;39:845. doi: 10.1039/b909358n. [DOI] [PubMed] [Google Scholar]; (b) Martin JN, Jones RCF. Chapter 1. In: Padwa A, Pearson WH, editors. Synthetic Applications of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products. Hoboken, N. J.: John Wiley & Sons; 2003. [Google Scholar]; (c) Gothelf KV. Chapter 6. In: Kobayashi S, Jørgensen KA, editors. Cycloaddition Reactions in Organic Synthesis. Weinheim, Germany: Wiley-VCH; 2002. [Google Scholar]; (d) Kanemasa S. Chapter 7. In: Kobayashi S, Jørgensen KA, editors. Cycloaddition Reactions in Organic Synthesis. Weinheim, Germany: Wiley-VCH; 2002. [Google Scholar]
- 2.(a) Stanley LM, Sibi MP. Chem. Rev. 2008;108:2887. doi: 10.1021/cr078371m. [DOI] [PubMed] [Google Scholar]; (b) Pellissier H. Tetrahedron. 2007;63:3235. [Google Scholar]; (c) Gothelf KV, Jorgensen KA. Chapter 12. In: Padwa A, Pearson WH, editors. Synthetic Application of 1,3-Dipolar Cycloaddition Chemistry Toward Heterocycles and Natural Products. Hoboken, N. J.: John Wiley & Sons; 2003. [Google Scholar]; (d) Gothelf KV, Jørgensen KA. Chem. Rev. 1998;98:863. doi: 10.1021/cr970324e. [DOI] [PubMed] [Google Scholar]
- 3.(a) Wang X, Weigl C, Doyle MP. J. Am. Chem. Soc. 2011;133:9572. doi: 10.1021/ja202676a. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bädoiu A, Bernardinelli G, Mareda J, Kündig EP, Viton F. Chem. Asian J. 2008;3:1298. doi: 10.1002/asia.200800063. [DOI] [PubMed] [Google Scholar]; (c) Wang Y, Wolf J, Zavalij P, Doyle MP. Angew. Chem., Int. Ed. 2008;47:1439. doi: 10.1002/anie.200704618. [DOI] [PubMed] [Google Scholar]; (d) Carmona D, Lamata MP, Viguri F, Rodriguez R, Fischer T, Lahoz FJ, Dobrinovitch IT, Oro LA. Adv. Synth. Catal. 2007;349:1751. [Google Scholar]; (e) Hashimoto T, Omote M, Kano T, Maruoka K. Org. Lett. 2007;9:4805. doi: 10.1021/ol702123n. [DOI] [PubMed] [Google Scholar]; (f) Kano T, Hashimoto T, Maruoka K. J. Am. Chem. Soc. 2005;127:11926. doi: 10.1021/ja0523284. [DOI] [PubMed] [Google Scholar]; (g) Carmona D, Lamata MP, Viguri F, Rodriguez R, Oro LA, Lahoz FJ, Balana AI, Tejero T, Merino P. J. Am. Chem. Soc. 2005;127:13386. doi: 10.1021/ja0539443. [DOI] [PubMed] [Google Scholar]; (h) Carmona D, Lamata MP, Viguri F, Rodriguez R, Oro LA, Balana AI, Lahoz FJ, Tejero T, Merino P, Franco S, Montesa I. J. Am. Chem. Soc. 2004;126:2716. doi: 10.1021/ja031995z. [DOI] [PubMed] [Google Scholar]; (i) Shirahase M, Kanemasa S, Oderaotoshi Y. Org. Lett. 2004;6:675. doi: 10.1021/ol0361148. [DOI] [PubMed] [Google Scholar]; (j) Viton F, Bernardinelli G, Kündig EP. J. Am. Chem. Soc. 2002;124:4968. doi: 10.1021/ja017814f. [DOI] [PubMed] [Google Scholar]; (k) Mita T, Ohtsuki N, Ikeno T, Yamada T. Org. Lett. 2002;4:2457. doi: 10.1021/ol026079p. [DOI] [PubMed] [Google Scholar]
- 4.Shintani R, Hayashi T. J. Am. Chem. Soc. 2006;128:6330. doi: 10.1021/ja061662c. [DOI] [PubMed] [Google Scholar]
- 5.Chan A, Scheidt KA. J. Am. Chem. Soc. 2007;129:5334. doi: 10.1021/ja0709167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shapiro ND, Shi Y, Toste FD. J. Am. Chem. Soc. 2009;131:11654. doi: 10.1021/ja903863b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Grover HK, Lebold TP, Kerr MA. Org. Lett. 2011;13:220. doi: 10.1021/ol102627e. [DOI] [PubMed] [Google Scholar]; (b) Kanao K, Miyake Y, Nishibayashi Y. Organometal. 2010;29:2126. [Google Scholar]; (c) Al-Harrasi A, Reissig H-U. Angew. Chem. Int. Ed. 2005;44:6227. doi: 10.1002/anie.200501127. [DOI] [PubMed] [Google Scholar]; (c) Helms M, Schade W, Pulz R, Watanabe T, Al-Harrasi A, Fisera L, Hlobilová I, Zahn G, Reissig H-U. Eur. J. Org. Chem. 2005:1003. [Google Scholar]; (d) Gerasyuto AI, Hsung RP, Sydorenko N, Slafer B. J. Org. Chem. 2005;70:4248. doi: 10.1021/jo050171s. [DOI] [PubMed] [Google Scholar]
- 8.Davies HML, Ahmed G, Churchill MR. J. Am. Chem. Soc. 1996;118:10774. [Google Scholar]
- 9.Bodnar BS, Miller MJ. Angew., Chem. Int. Ed. 2011;50:5630. doi: 10.1002/anie.201005764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kumarn S, Shaw DM, Longbottom DA, Ley SV. Org. Lett. 2005;7:4189. doi: 10.1021/ol051577u. [DOI] [PubMed] [Google Scholar]
- 11.(a) Brasholz M, Reissig H-U, Zimmer R. Acc. Chem. Res. 2009;42:45. doi: 10.1021/ar800011h. [DOI] [PubMed] [Google Scholar]; (b) Yamamoto Y, Yamamoto H. Eur. J. Org. Chem. 2006:2031. [Google Scholar]
- 12.(a) Yamamoto Y, Yamamoto H. J. Am. Chem. Soc. 2004;126:4128. doi: 10.1021/ja049849w. [DOI] [PubMed] [Google Scholar]; (b) Yamamoto Y, Yamamoto H. Angew. Chem. Int. Ed. 2005;44:7082. doi: 10.1002/anie.200501345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davies HML, Bruzinski PR, Lake DH, Kong N, Fall MJ. J. Am. Chem. Soc. 1996;118:6897. [Google Scholar]
- 14.(a) Hashimoto S, Watanabe N, Sato T, Shiro M, Ikegami S. Tetrahedron lett. 1993;34:5109. [Google Scholar]; (b) Tsutsui H, Abe T, Nakamura S, Anada M, Hashimoto S. Chem. Pharm. Bull. 2005;53:1366. doi: 10.1248/cpb.53.1366. [DOI] [PubMed] [Google Scholar]
- 15.(a) Liang C, Collet F, Robert-Peillard F, Müller P, Dodd RH, Dauban P. J. Am. Chem. Soc. 2008;130:343. doi: 10.1021/ja076519d. [DOI] [PubMed] [Google Scholar]; (b) Müller P, Allenbach Y, Robert E. Tetrahedron: Asymmetry. 2003;14:779. [Google Scholar]
- 16.Reddy RP, Lee GH, Davies HML. Org. Lett. 2006;8:3437. doi: 10.1021/ol060893l. [DOI] [PubMed] [Google Scholar]
- 17.(a) Davies HML, Saikali E, Young WB. J. Org. Chem. 1991;56:5696. [Google Scholar]; (b) Davies HML, Hu B, Saikali E, Bruzinski PR. J. Org. Chem. 1994;59:4535. [Google Scholar]; (c) Lian Y, Davies HML. Org. Lett. 2010;12:924. doi: 10.1021/ol9028385. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Hansen JH, Davies HML. Chem. Sci. 2011;2:457. [Google Scholar]
- 18.(a) Davies HML, Xiang B, Kong N, Stafford DG. J. Am. Chem. Soc. 2001;123:7461. doi: 10.1021/ja0160546. [DOI] [PubMed] [Google Scholar]; (b) Lian Y, Davies HML. J. Am. Chem. Soc. 2010;132:440. doi: 10.1021/ja9078094. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.