

Abstract
The repeated use of drugs that directly or indirectly stimulate dopamine transmission carry addiction liability and produce enduring pathological changes in the brain circuitry that normally regulates adaptive behavioral responding to a changing environment. This circuitry is rich in glutamatergic projections, and addiction-related behaviors in animal models have been linked to impairments in excitatory synaptic plasticity. Among the best-characterized glutamatergic projection in this circuit is the prefrontal efferent to the nucleus accumbens. A variety of molecular adaptations have been identified in the prefrontal glutamate synapses in the accumbens, many of which are induced by different classes of addictive drugs. Based largely on work with cocaine, we hypothesize that the drug-induced adaptations impair synaptic plasticity in the cortico-accumbens projection, and thereby dysregulate the ability of addicts to control their drug-taking habits. Accordingly, we go on to describe the literature implicating the drug-induced changes in protein content or function that impinge upon synaptic plasticity and have been targeted in preclinical models of relapse and, in some cases, in pilot clinical trials. Based upon modeling drug-induced impairments in neuroplasticity in the cortico-accumbens pathway, we argue for a concerted effort to clinically evaluate the hypothesis that targeting glial and neuronal proteins regulating excitatory synaptic plasticity may prove beneficial in treating addiction.
Keywords: accumbens, addiction, glutamate, prefrontal cortex, relapse, synaptic plasticity
The prevalence and impact of drug abuse and addiction (dependence as per Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition (DSM-IV)) to our society are enormous. It is estimated that substance use disorders (term proposed by DSM-V) affect 20.4 million Americans or 8.3% of the population aged ≥ 12 years.1 The economic costs associated with substance use disorders in the United States are calculated to exceed half a trillion dollars annually, which include costs from adverse effects of drugs to health, judicial/incarceration-related costs, productivity losses and accidents.2 Substance use disorders also have a major negative public health impact including family disintegration, domestic violence, school dropout, loss of employment, accidents and crimes. Despite the impact of substance use disorders, there are very few medications currently available for the treatment of drug addiction, highlighting the urgent need for research and development in this area.
Legal (for example, alcohol and nicotine) and illegal (for example, cocaine, methamphetamine, heroin and marijuana) drugs are used for various reasons, including their rewarding effects, alteration of mental state, to improve performance and for self-medication. However, repeated drug use can result in addiction, which is manifested as an intense desire for the drug with an impaired ability to control the urges to take that drug, even at the expense of serious adverse consequences.3 Although the aberrant behavioral manifestations of addiction have been viewed for many years as bad ‘choices’, preclinical studies in animal models of drug self-administration and clinical studies using imaging technology are giving us a better understanding of the neuropathologies underlying addiction.
In this review, we bring focus to the circuitry and cellular adaptations produced by repeated drug use in animal models and how this may relate to the neuropathology of addiction in humans. Accordingly, we will not present in detail the better-understood neurobiology of reward and how the binding sites for different addictive drugs can directly or indirectly augment dopamine release in cortical and limbic brain regions to reinforce drug-seeking behavior. Excellent reviews along these lines have recently been published.4 Rather, we will describe the brain circuitry underlying the retrieval and execution of drug-seeking behaviors. We will argue that the circuitry normally responsible for adaptively molding behavior to fit environmental contingencies develops drug-induced neuropathologies in synaptic plasticity that creates the repeated, maladaptive relapse to drug taking that characterizes addiction. Moreover, we will suggest targets for potential pharmacotherapeutic treatments for addiction based upon the long-lasting drug-induced molecular neuroadaptations harbored within the affected brain circuits.
Addiction usurps brain circuits regulating the execution of behavior
Although it can be assumed that the brain functions as a single organ to guide complex behaviors such as reward retrieval or avoiding negative consequences, specific circuits have been identified as sites of pathology in addiction. In particular, neuroimaging of drug addicts in a variety of cognitive and motivated tasks consistently reveals differences in patterns of brain activity in regions of the prefrontal cortex, areas receiving prefrontal cortical projections, such as the amygdala, limbic areas of the basal ganglia (nucleus accumbens) and the dopamine-rich ventral tegmental area (VTA) in the ventromedial mesencephalon.5-8 Additionally, activity in the insular cortex and cerebellum is often elevated with states of drug craving.5,9 Interestingly, these circuits are shared with other neuropsychiatric diseases that are characterized in part by maladaptive motivational states, such as depression, mania and post-traumatic stress disorder, a fact that points to endophenotypes shared between diseases, such as impulsivity, poor insight and impaired decision making.5,10 Although it seems likely that the underlying molecular mechanisms contributing to these diseases will be distinct, the apparent overlap in endophenotypes reflects impairments in the capacity of affected individuals to mobilize specific brain circuits needed for the homeostatic regulation of cognitive and emotional functions, and for the execution of adaptive behavioral responses. Thus, while environmental or genetic vulnerabilities may be shared, a fact reflected in part by the high comorbidity between addiction and certain other neuropsychiatric diseases,11-13 in contrast to most other neuropsychiatric diseases the precipitant of molecular changes that ultimately impair circuitry function in addiction is well known; the repeated use of addictive drugs. The fact that addiction is precipitated and maintained by drug use gives preclinical biomedical researchers a unique avenue to explore the molecular underpinnings of addiction that may be buried within the circuits showing impairments in human addicts.
The brain circuits identified in neuroimaging studies with addicts can be amalgamated into an integrated circuit shown in Figure 1. This circuit provides a map to guide analysis in experimental animals of the drug-induced cellular and molecular neuropathologies that might underlie relapse and constitute neuropathologies that are induced by repeated drug use in combination with genetic or epigenetic vulnerabilities. The interconnected nature of the brain permits a great deal of artistic license in drawing the nucleus accumbens as a point of integration in the circuit in Figure 1. Nonetheless, the nucleus accumbens has been identified as a site important for both the acquisition (learning) and expression (relapse) of addictive behavior.4 This region has been further reduced into shell and core subcompartments, each with its own topographic organization of efferent and afferent connections.14 In Figure 1, we illustrate in particular the different regions of the prefrontal cortex projecting to the shell and core. Thus, the anterior cingulate cortex (prelimbic, in rat) projects largely to the core, whereas the subgenual cingulate (infralimbic, in rat) and ventromedial orbital frontal cortex project more densely to the shell (see Box 1 for detailed discussion of distinct regions of prefrontal cortex). As well, the projection from the basolateral amygdala and hippocampus is denser in the shell (although substantial projections from these areas to the core are also present). Outputs from the core and shell to the ventral pallidum are topographically organized, such that the dorsal ventral pallidum receives input from the core, whereas the ventromedial ventral pallidum is innervated by the shell. It is notable that although the well-established distinction between D2 dopamine receptor expressing projections to the pallidum and D1 receptor containing projections to the dopamine-rich ventral mesencephalon is largely true for the core efferents,15 this relationship breaks down for the shell projections.16 Thus, the shell projections to the ventral pallidum are mixed, whereas the projections to the VTA are only from D1-containing neurons. The implications of this difference remain unknown and largely ignored experimentally. Finally, projections from the VTA are to both the core and shell, although reciprocal projections from the accumbens back to the VTA arise largely from the shell. Glutamatergic inputs to dopamine neurons, likely largely from the pedunculo-pontine nucleus, undergo significant synaptic plasticity in response to repeated use of addictive drugs. Initially, this plasticity was thought to be a temporary, but critical step in the development of addiction into a chronic relapsing disorder. However, recent studies have identified enduring changes in excitatory plasticity in dopamine neurons that are necessary for relapse in animal models of addiction (see Box 2 for discussion).
Figure 1.
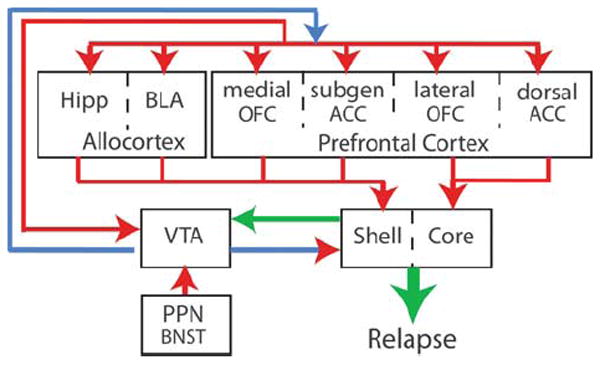
Model of the circuitry contributing to relapse in animal models and altered in neuroimaging studies in addicts, with emphasis on the key position of the cortico-accumbens glutamatergic projection, and the interconnected glutamatergic projections between allocortical and prefrontal cortical regions. This review focuses on drug-induced pathologies in the glutamatergic projections from prefrontal and allocortex into the nucleus accumbens. Glutamatergic pathologies are relatively less known for the cortico-cortical projections, but any glutamatergic-based pharmacological therapy will likely affect these projections in a manner either synergistic or countermanding the therapeutic effects on the cortico-accumbens pathologies. Arrows indicate direction of the projection and are color coded as follows: blue, dopamine; blue/red, dopamine/glutamate co-released; green, GABA/peptides co-released; red, glutamate. Abbreviations: BLA, basolateral amygdala; BNST, bed nucleus of the stria terminalis; core- nucleus accumbens core; dorsal ACC, dorsal anterior cingulate cortex; Hipp, hippocampus; OFC, orbital frontal cortex; PPN, pedunculopontine nucleus; shell, nucleus accumbens shell; subgen ACC, subgenual anterior cingulate cortex; VTA, ventral tegmental area.
Box 1 Role of prefrontal cortex in addiction.
The prefrontal cortex (PFC) plays a central role in executive function, which allows the individual to plan and execute goal-directed behaviors as a function of changing internal needs and environmental contingencies. The PFC occupies a large portion of the human brain, accounting for 29% of the total cortex, and is extensively connected with other cortical regions and with subcortical and limbic structures.134 The PFC is constituted of several regions including Brodmann areas (BAs) BA 8, BA 9, BA 10, BA 11, anterior BA 13, BA 44, BA 45, BA 46, BA 47 and anterior regions in the cingulate gyrus (ACC) including BA 24, BA 25 and BA 32.134 Although there is significant overlap and connectivity between PFC regions, there are also distinct connections to subcortical and limbic regions that determine the functional involvement of a given PFC region.135 Imaging studies, and in particular functional magnetic resonance imaging (fMRI), have started to map the functional involvement of specific PFC regions in the human brain and in addiction.136 Overall, a pattern has emerged that distinguishes between ventral and dorsal PFC. The ventral PFC (BA 11, BA 24, BA 25) is more involved with reward- and emotion-associated behaviors, whereas the dorsal PFC is involved with appraisal of errors, working memory and inhibitory control (BA 32, BA 9, BA 44). Similarly, a medial versus lateral distinction also favors medial areas to greater external influence (BA 10) and with salience attribution (medial BA 11, BA 25). However, because PFC functions reflect the dynamic interaction between neuronal networks, any given PFC region is involved in more than one functional network. Imaging studies in drug addicts have implicated disruption of PFC activity in ventral, dorsal medial and lateral regions that are believed to underlie the inflexible behavior that addicted subjects express when faced with conditioned cues (likely mediated by impaired corticofugal neuroplasticity, see text). Thus, the ventromedial PFC (BA 11, BA 25) is implicated in the disruption in inhibitory control that results in impulsivity and poor control over behavior in addicts, the ACC (BA 32) and inferior PFC (BA 44) are implicated in impaired decision making that also implicates dorsolateral PFC (BA 9, BA 10) and the impairment in internal awareness and error detection that is associated with altered activity in ACC and anterior BA 13.4,137,138 Thus, neuroplastic changes triggered by repeated drug use leading to long-term potentiation (LTP) are likely to result in distinct behaviors depending on where the plasticity occurred within the PFC. Although most preclinical studies investigating the effects of chronic drugs on excitatory synaptic plasticity have focused on the PFC-accumbens pathway and conditioned drug associations, further studies on the effects of long-term drug exposures in other PFC pathways are needed to understand how they influence the behavior of the animal beyond conditioned associations.
Box 2 Role of dopamine neurons in the VTA in relapse.
For over a decade it has been known that phasic activity in ventral tegmental area (VTA) dopamine neurons is induced by reward-predictive cues.139 More recently, it was shown that natural rewards and stressors, and acute or chronic cocaine administration, induces a long-term potentiation (LTP)-like increase in excitatory synaptic strength on dopamine neurons that is mediated by increased GluR2-lacking AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptors.140-142 Thus, these treatments potentiate dopamine involvement in learning reward-predictive cues by potentiating excitatory inputs to dopamine neurons and thereby, dopamine release into cortical and striatal terminal fields. However, the fact that this synaptic potentiation endured for less than a week after discontinuing drug treatment indicated that although LTP in the VTA was important for developing enduring addiction-related behaviors like behavioral sensitization and relapse, it probably did not mediate the expression of these behaviors. This view is reinforced by findings that genetic deletion of GluR1 or NR1 in dopamine neurons does not consistently affect the expression of chronic cocaine-induced conditioned place preference (CPP) or behavioral sensitization.143,144 Also, drug-induced LTP in dopamine neurons is gated by mGluR1, and over-riding mGluR1 inhibition of LTP resulted in long-lasting synaptic potentiation in the nucleus accumbens (a cardinal feature of the enduring synaptic plasticity thought to contribute to relapse, see text).145 However, it was recently shown that if cocaine was self-administered, LTP in VTA dopamine neurons endures for at least 3 months.146 Considering that increased dopamine release in areas like the prefrontal cortex, amygdala and nucleus accumbens reinstates drug seeking,147-150 this LTP would be predicted to make animals more susceptible to relapse. Thus, although we focus on drug-induced neuroadaptations in the prefrontal to accumbens pathway as a potential source of pharmacotherapeutic targets for treating relapse, it is important to note that continuing research on other glutamatergic projections may reveal enduring neuroadaptations that can synergize with or countermand the effect of glutamatergic drugs designed to offset drug-induced plasticity in the prefrontal to accumbens pathway.151-153 Finally, a complete understanding on how glutamate may be targeted therapeutically to manipulate dopamine plasticity will require an integrated understanding of how glutamatergic input interacts with the well-established tonic inhibition of dopamine neurons by GABAergic interneurons.154
Nucleus accumbens: a site of integration and glutamatergic pathology in addiction
The nucleus accumbens comprises medium spiny neurons (MSNs) that constitute >90% of all neurons in both the shell and core.14,17 These neurons are GABAergic and colocalized with various neuropeptides according to their projection patterns.14,16 Thus, projections to the ventral mesencephalon are D1 containing and coexpress dynorphin and neurotensin, whereas the D2-containing neurons projecting to the ventral pallidum coexpress enkephalin. In the case of the shell projections to the ventromedial ventral pallidum, the MSNs also contain D1 receptors, and neuropeptides including substance P, dynorphin and neurotensin. The remaining 5–10% of neurons in the accumbens is split between cholinergic aspiny cells and GABAergic interneurons. These two cell types innervate the MSNs providing excitatory (acetylcholine) and inhibitory (GABA) tone.18 All three cell types are thought to be innervated by glutamatergic afferents from the prefrontal cortex, amygdala and thalamus, as well as dopamine innervation from the VTA. Interestingly, innervation from the VTA is partially GABAergic and glutamatergic, the latter thought to be partly colocalized with dopamine.19 This raises potential complications in interpreting the physiological and pathophysiological changes classically attributed to ‘dopaminergic’ innervation from the VTA.20
Figure 2 illustrates a canonical dendritic spine from an MSN that shows the relative location of glutamatergic input on the spine head and the dopamine input to the shaft of the spine. This orientation provides the morphological underpinning of the classic relationship between these two afferents that is described as dopamine ‘gating’ excitatory input.21 The gating function of dopamine is further supported by the fact that dopamine receptors are G-protein coupled to signaling pathways that generally produce slow, relatively long-lasting biasing on membrane conductances.22 Thus, dopamine does not by itself induce large changes in MSN current, but potentiates or inhibits the capacity of glutamate to depolarize neurons. Many of these intracellular mechanisms are well characterized and involve modulation of calcium (D1) and potassium (D2) channels. Also, gating of glutamatergic activity can be very long lasting because of the capacity of dopamine to regulate enduring excitatory synaptic plasticity (see below). Although the gating function of dopamine is well embedded in our perspectives on accumbens physiology, a number of caveats to this relationship need to be kept in mind. First, as mentioned above, dopamine is at least partly colocalized with glutamate, and hence these afferents are capable of inducing fast changes in membrane conductance.19 Second, the extent to which specific glutamatergic afferents are gated by dopamine is unclear. Glutamatergic inputs can arise from the prefrontal cortex, basolateral amygdala, hippocampus or medial thalamus,18 and as mentioned above, many of these afferents are at least partly topographically segregated between the core and shell.
Figure 2.
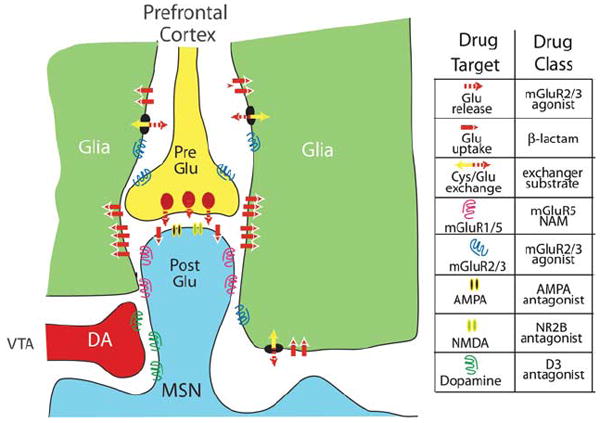
Canonical glutamatergic and dopaminergic synapse on a dendritic spine of a medium spiny cell illustrating proteins important for regulating excitatory synaptic plasticity and that can be readily targeted for treating addiction. The accompanying legend for specific proteins also indicates the category of drug that is hypothesized to be therapeutically useful based upon the type of glutamatergic neuropathologies produced in prefrontal-accumbens synapses that are described in the text. Abbreviations: AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; Cys/Glu exchange, cystine/glutamate exchange; mGluR, metabotropic glutamate receptor; MSN, medium spiny neuron; NAM, negative allosteric modulator; NMDA, N-methyl d-aspartate; NR2B, N-methyl d-aspartate receptor subtype 2B; VTA, ventral tegmental area.
Finally, in Figure 2 it is clear that the proximity of glia creates a ‘tripartite’ glutamatergic synapse, and in considering synaptic homeostasis and plasticity it is critical to include the role of glia. Over the last decade, it has become clear that the relationship goes beyond a classic metabolic relationship. In particular, glutamate released by glia directly regulates metabotropic glutamate receptors (mGluRs) that strongly influence the ability of glutamatergic synapses to increase or decrease in strength (that is, to undergo synaptic plasticity).23-26 Although detailed studies are not available for the accumbens, based on studies in the hippocampus, it is thought that the patterned expression of glutamate uptake transporters in the vicinity of excitatory synapses is responsible for limiting the effect of synaptic glutamate largely to the synaptic cleft at the site of release.27,28 More recently, it has become clear that these transporters also protect the synaptic cleft from glutamate released from glia via calcium-dependent or cystine–glutamate exchange mechanisms.29-31 This is important, given that glial release of glutamate maintains ~1–5 μM glutamate in the extracellular, nonsynaptic space, a concentration capable of stimulating N-methyl d-aspartate (NMDA) receptors.32 This concentration of glutamate also ensures tonic stimulation of mGluRs that are located largely outside of the synaptic cleft.33,34 This balance between synaptic and nonsynaptic glutamate, termed ‘glutamate homeostasis’, plays a critical role in excitatory synaptic plasticity and is an important site of neuropathology in animal models of relapse.35
Corticostriatal synaptic plasticity and addiction
Classic forms of excitatory synaptic plasticity include long-term potentiation (LTP) and long-term depression (LTD), both of which can be demonstrated in the cortico-accumbens projection.36 As well, LTP has been shown in the amygdala and hippocampus afferents to the accumbens.37 Synaptic plasticity should not be viewed as two states, but rather as a continuum along which synaptic strength can increase or decrease to a new, relatively stable state. Synaptic strength can be controlled presynaptically by regulating glutamate release and postsynaptically by the insertion or removal of the ionotropic AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) or NMDA glutamate receptors.38 Although many intracellular mechanisms have been implicated in affecting glutamate release and glutamate receptor trafficking,39,40 in this review we bring focus to cell surface receptors that provide more ready targets for pharmacotherapies that may be useful in treating addiction.
Presynaptic glutamate release in the accumbens and striatum can be modulated by a variety of receptors, including being decreased by activation of mGluR2/3, adenosine A1, dopamine D2 or cannabinoid CB1 receptors, and increased by activation of mGluR1 or possibly presynaptic ionotropic glutamate receptors.41-45 The trafficking of AMPA and NMDA receptors can also be regulated by receptor activation, including mGluR5 to decrease and D1 receptors to promote AMPA surface expression.46,47 Accordingly, the stimulation or inhibition of these receptors has been implicated in regulating the induction of LTP or LTD.18 As well, many of these proteins, or their immediate signaling partners, are altered by chronic administration of addictive drugs. For example, withdrawal from chronic cocaine, heroin or ethanol reduces Gi signaling by upregulating activator of G-protein signaling 3 (AGS3), and pharmacologically decreasing AGS3 reduces relapse to these drugs in animal models.48-50 Given that all of the inhibitory glutamate release-regulating presynaptic receptors listed above are Gi coupled, upregulated AGS3 should reduce the efficacy of inhibitory modulation by ligand binding; although this has been shown directly only for D2 and mGluR2/3.42,48 Moreover, the capacity of mGluR1 stimulation to promote glutamate release (presumably synaptic) is downregulated after withdrawal from chronic cocaine.44,51 The insertion of high calcium permeability AMPA receptors (lacking GluR2 subunit) is increased after withdrawal from cocaine, which contributes to a well-characterized increase in an LTP-like increase in the AMPA/NMDA ratio.52-54 There is also an increase in NMDA receptors containing the NR2B (N-methyl d-aspartate receptor subtype 2B) subunit after chronic cocaine or heroin that is thought to be perisynaptic and localized to silent synapses, perhaps as a prelude to developing mature synapses and an LTP-like increase in synaptic strength.55,56
Recently, it has been shown that glial regulation of nonsynaptic, extracellular glutamate strongly regulates synaptic plasticity by providing tone onto mGluRs and extrasynaptic NMDA receptors.25,26,35 Glial regulation is twofold: first by the patterned expression of glutamate transporters in the vicinity of the synaptic cleft, and second by the release of glial glutamate via calcium-dependent mechanisms or cystine–glutamate exchange. The glial glutamate transporter 1 (GLT1) and the catalytic subunit of the cystine–glutamate exchanger xCT are downregulated after chronic cocaine or nicotine self-administration. 57-59 Importantly, downregulated xCT results in decreased tone on release-regulating mGluR2/3 and postsynaptic mGluR5.29,36,60
In addition to pre- and post-synaptic proteins, it is important to consider that morphological changes occur that parallel the relative strength of excitatory synapses.61 Thus, LTP is associated with larger and LTD with smaller synapses and dendritic spine density. These morphological adaptations are thought to contribute to long-lasting synaptic plasticity by forming the protein and metabolic infrastructure needed for an altered state of synaptic activity, but it should be noted that this association is largely derived from in vitro studies. Importantly, considering cell surface or extracellular proteins involved in regulating synaptic architecture provides additional potential targets for drug development in treating addiction, including tyrosine kinase receptors (for example, Trk2 brain-derived neurotrophic factor (BDNF) receptor),62 integrins (extracellular matrix cell surface receptor)63 and matrix metalloproteases (proteases that cleave extracellular matrix).64 Preclinical data support a role for β-3 integrins and metalloproteases in regulating reinstated drug seeking and conditioned place preference (A Wiggins, unpublished observation),65 and both are being developed as drug targets in cancer and vascular disease.66,67 However, to date, these targets have not been examined clinically in drug addiction. Similarly, BDNF application into the cortico-accumbens pathway is well established in preclinical models to alter the reinstated cocaine seeking, although the direction of change is mixed. Thus, if BDNF is administered acutely into the prefrontal cortex and transported into the nucleus accumbens, reinstated drug seeking is inhibited and glutamate homeostasis normalized,68,69 whereas viral overexpression of BDNF directly into the accumbens promotes reinstated cocaine seeking.70 Although these data pose the possibility that BDNF may directly influence cocaine-induced neuroplasticity, perhaps by an action on synaptic morphology, this remains to be determined.
Addiction as a pathology in cortico-accumbens synaptic plasticity
Over the past 4 years, a number of studies have shown a defect in classic forms of synaptic plasticity in MSNs from the accumbens of rats withdrawn from cocaine self-administration. Martin et al.71 first demonstrated a loss of LTD in slices from core, but not shell, MSNs. This finding was replicated in vivo,36 although the loss of in vivo LTD in the corticoaccumbens pathway appeared only in animals undergoing extinction training during withdrawal.72 Most recently, using an extended access model of drug self-administration, it was shown that the cocaine-induced loss of LTD endured only in the sub-population of rats that developed uncontrolled intake and resistance to modulation by drug-associated negative environmental contingencies (that is, in rats considered to more closely parallel diagnostic criteria for addiction in humans).73 In this latter study, slices of the core were made 24 h after the last cocaine injection, indicating that although an interaction between extinction training and cocaine self-administration may be necessary to create enduring loss of LTD in all cocaine-trained animals, a sub-population of rats possessing an unidentified vulnerability to developing addiction lose the ability to undergo LTD after chronic cocaine without accompanying extinction training. Alternatively, deafferenting core MSNs by making a tissue slice alters glutamate homeostasis, 29 and thereby may unmask the cocaine-induced deficit in LTD regardless of the neuroplasticity produced in the cortico-accumbens circuit by extinction learning.72
In contrast to LTD, LTP is variably induced in striatal slices.18 However, LTP can be induced in vivo by stimulating prelimbic afferents to the core, and this is abolished after withdrawal from cocaine self-administration in a manner independent of extinction training.36,72 The loss of both LTP and LTD in vivo after cocaine self-administration has led to the hypothesis that the inability of the MSNs in the core to undergo bidirectional prefrontal-induced neuroplasticity may reduce the capacity of drug addicts to adaptively organize a behavioral response that incorporates environmental contingencies that mediate against relapse.35
Potential targets for restoring cortico-accumbens plasticity and treating addiction
The remainder of this review combines the possibility that reduced cortico-accumbens plasticity is an important neuropathology in addiction with knowledge about the various surface proteins that are involved in glutamate homeostasis and regulating synaptic plasticity in order to consider potential targets for developing a pharmacotherapy that restores or countermands the drug-induced loss of neuroplasticity. In this section we will examine each protein in Figure 2, both in terms of how addictive drugs alter the protein itself or signaling related to the protein, how targeting these proteins affects relapse in animals models and, when available, clinical trials that have tested the viability of each target. The discussion is organized first to discuss glial proteins that have the capacity to alter extracellular, nonsynaptic glutamate, followed by extrasynaptic mGluRs that will homeostatically respond to changes in extracellular glutamate, to proteins involved in regulating presynaptic glutamate release and the capacity of the postsynapse to detect synaptically released glutamate. Table 1 outlines the drugs that have the potential to be used to modulate glutamate synaptic plasticity in treating addiction.
Table 1.
Potential drugable targets in treating addiction
| Target | Effects on animal modes of drug abuse | Clinically available |
|---|---|---|
| AMPA | Antagonists inhibit relapse | Talampanel |
| Tezampanel | ||
| Perampanel | ||
| Positive allosteric modulators increase BDNF | ||
| NMDA | Partial agonists facilitate extinction | d-Cycloserinea |
| mGluR2/3 | Agonists inhibit relapse | |
| mGluR5 | Negative allosteric modulators inhibit with drug administration and relapse Positive allosteric modulators facilitate extinction, but promote relapse | Fenobam |
| Cystine–glutamate exchanger | Upregulation prevents relapse and facilitates extinction | N-acetylcysteinea |
| GLT1 | Upregulation prevents relapse | Ceftriaxone |
| Modulators | Acamprosateb | |
| Modafinila |
Abbreviations: AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; BDNF, brain-derived neurotrophic factor; GLT1, glutamate transporter 1; mGluR, metabotropic glutamate receptor; NMDA, N-methyl d-aspartate.
Have been tested in clinical trials.
Approved for the treatment of alcoholism.
Proteins regulating glial glutamate release and uptake
As outlined above, cocaine and nicotine self-administration downregulates the content and function of the cystine–glutamate exchanger and GLT1 in the accumbens core. Heroin also reduces GLT1 content (H Shen, unpublished observation). For cocaine and heroin, the net result of these changes is reduced extracellular levels of glutamate as measured by microdialysis, and reduced tone on mGluRs.60 Pharmacological mechanisms have been developed to restore both cystine–glutamate exchange and GLT1. The administration of N-acetylcysteine activates the exchanger, presumably by being deacetylated and dimerizing to provide the extracellular cystine substrate. Ceftriaxone is a β-lactam antibiotic that increases the synthesis and membrane insertion of GLT1. Interestingly, chronically treating animals trained to self-administer cocaine with either drug simultaneously restores both GLT1 and xCT expression, indicating that these two proteins may be homeostatically regulated in tandem.74,75 Importantly, restoring GLT1 with ceftriaxone prevents reinstated cocaine seeking,75,76 and N-acetylcysteine inhibits cocaine, nicotine and heroin seeking.77 Also, N-acetylcysteine restored the ability of prefrontal stimulation to induce LTP and LTD in the core of animals withdrawn from cocaine self-administration. 36 Finally, chronic administration of N-acetylcysteine provides enduring protection from reinstated heroin and cocaine seeking,57,60,78 and recent clinical trials indicate that it reduces cigarette smoking and the desire to use cocaine.59,79 Limitations on the use of these drugs in an outpatient setting to treat addiction includes N-acetylcysteine having poor bioavailability to brain,80 and ceftriaxone requiring intramuscular administration.
A final drug that is potentially in this class is modafinil, a drug used to treat narcolepsy and as a cognitive enhancer.81 Although also characterized as an inhibitor of dopamine and norepinephrine transporters,81 modafinil increases extracellular glutamate82,83 and in preclinical models inhibits reinstated morphine conditioned place preference (CPP) and cocaine- and methamphetamine-induced drug seeking. 84,85 Importantly, akin to glutamate released by N-acetylcysteine, the glutamate released by modafinil acts on mGluR2/3 to inhibit reinstated CPP.84 Clinically, the effects of modafinil on cocaine use have been mixed.86,87 Note also that modafinil blocks dopamine transporters, which may contribute to its potential therapeutic effects in cocaine addiction.88
mGluR2/3
The reduced levels of nonsynaptic, extracellular glutamate after chronic cocaine or heroin decreases tonic stimulation of presynaptic, release-regulating mGluR2/3 (see above). The reduced tone is compounded by an increase in AGS3 and corresponding decrease in G-protein coupling by mGluR2/3 (see above), as well as downregulation of mGluR2/3 after chronic cocaine or nicotine.42,89 Accordingly, restoring tone onto mGluR2/3 decreases glutamate release probability and inhibits reinstated cocaine, nicotine, ethanol and heroin seeking.90-95 Moreover, blocking mGluR2/3 prevents the capacity of N-acetylcysteine to inhibit cocaine seeking or to restore LTP.29,36 In addition to glutamate release, heterosynaptic mGluR2/3 are located on dopamine terminals and decrease dopamine release when stimulated. This probably contributes to why, in addition to reducing reinstated drug seeking, mGluR2/3 agonists also reduce cocaine, ethanol and nicotine self-administration,90 as well as reinstated food seeking at higher doses.94,95 In contrast with agonists, mGluR2/3 antagonists increase glutamate and dopamine release, a property that poses these drugs as potential treatment for the dysphoria experienced by addicted subjects during withdrawal.96 Indeed, in rodent models of chronic nicotine administration, treatment with an mGlu2/3 receptor antagonist attenuated reward deficits associated with nicotine withdrawal.97
mGluR5
mGluR5s are Gq coupled and bind with Homer proteins in the postsynaptic density that facilitate mGluR5 trafficking and function.98 The loss of LTD associated with animals extinguished from self-administered cocaine results from increased Homer levels and a sequestering of mGluR5 off the membrane surface.72 The increase in Homer and reduced surface expression of mGluR5 appears to be a compensatory adaptation that inhibits relapse, as genetically reducing Homer promotes cocaine self-administration, and elevating Homer reduces reinstated cocaine seeking.72,98 Similarly, mGluR5-negative allosteric modulators or mGluR5 gene deletion reduce the self-administration and reinstated drug seeking of most drugs of abuse (with the apparent exception of μ-opioids), whereas increased stimulation of mGluR5 with a positive allosteric modulator prevents N-acetylcysteine from reducing cocaine seeking.36,99,100 Moreover, the capacity of N-acetylcysteine to restore LTD depends on stimulation of mGluR5, affirming the presence of mGluR5-dependent LTD in accumbens MSNs.36 Although there are no reported clinical studies in addiction, there are several ongoing clinical trials with mGluR5-negative allosteric modulators for the treatment of other diseases including Fragile X syndrome, anxiety, depression, gastroesophageal reflux disease and dyskinesias in Parkinson’s disease.101,102 Thus far, these drugs are largely well tolerated and result in few serious adverse cognitive effects.
One caveat when considering pharmacological regulation of mGluR5 is the high density of this receptor on glia.103 Indeed, regulation of glial mGluR5 stimulates calcium-dependent release of glutamate from glia that modulate synaptic plasticity via extrasynaptic NR2B-containing NMDA receptors.103 Thus, it is unclear whether the ameliorative effect of mGluR5-negative allosteric modulators on relapse results from a neuronal or glial action or both. Along these lines, it is clear that the proposed mechanism for N-acetylcysteine to increase nonsynaptic glutamate will stimulate both mGluR5 to promote and mGluR2/3 to reduce drug seeking. Although questions such as why the indirect stimulation of mGluR2/3 by N-acetylcysteine is prepotent over mGluR5 stimulation remain to be answered, these data point to the possibility that combining an mGluR5-negative allosteric modulator and N-acetylcysteine may improve the capacity of either mechanism to inhibit relapse.
AMPA ionotropic glutamate receptors
As discussed above, after chronic cocaine withdrawal there is an LTP-like upregulation in the expression of GluR1-containing AMPA receptors.104 This fact indicates that excessive AMPA receptor signaling may contribute to relapse. Indeed, by restoring AMPA/NMDA ratio and the LTP-like increase field potentials elicited in the accumbens core by prefrontal stimulation, N-acetylcysteine inhibits relapse.60 Moreover, administration of AMPA receptor antagonists directly into the accumbens inhibits reinstated ethanol, cocaine and heroin seeking.105-109 Several clinical trials with AMPA antagonists are ongoing to evaluate their efficacy in several neurological disorders, although none is being tested of addiction. Tezampanel (AMPA/kainate receptor antagonist) is being tested for acute migraine and is of particular relevance, as in preclinical studies it reduced cocaine self-administration.110 Other potentially interesting compounds are Talampanel (AMPA/kainate receptor antagonist) and Perampanel (noncompetitive AMPA antagonist), which are being evaluated for Parkinson’s disease and partial seizures in other neurological conditions.111
A variety of compounds with mixed actions that include AMPA receptor antagonism have been studied in addiction clinical trials. Topiramate is an antiepileptic drug with AMPA antagonist and GABA-enhancing effects, and has shown positive results in the treatment of alcoholism, and a pilot clinical study indicates benefit in the treatment of cocaine addiction. 112,113 Similarly, Lamotrigine, another antiepileptic drug with AMPA antagonist properties that also reduces glutamate release, was shown to reduce cocaine intake and craving in cocaine abusers.114 However, it is important to note that the actions of lamotrigine on AMPA receptors are likely secondary to the well-established inhibitory effect on voltage-dependent sodium and calcium conductances that is thought to underlie its efficacy as an anticonvulsant.
Ampakines are positive allosteric modulators of AMPA receptors that increase ion channel flux in the presence of agonist by suppressing desensitization and/or deactivation of the AMPA receptors.115 Ampakines have shown positive effects in rodent models of memory and cognition and have antidepressant properties.115,116 They have also been shown to increase BDNF,117,118 which modulates drug seeking and glutamate synaptic plasticity (see above). Very few studies have evaluated the effects of ampakines in animal models of drug self-administration. One such study reported that ampakines reverse methamphetamine-induced hyperactivity,119 and another reported that ampakines prevent circling behavior induced by methamphetamine in unilaterally 6-hydroxydopamine-lesioned rats.120 In a rodent model of acute alcohol administration, an ampakine (LY404187) reversed the effects of acute ethanol intoxication on locomotor and operant behavior, and on brain activity (reversed decreases in blood oxygen level-dependent magnetic resonance signal after 0.6 g kg−1 ethanol and attenuated the decreases in glucose metabolism after 2.0 g kg−1 ethanol).121 Common among these three studies is the inhibition of some effects associated with the acute drug administration, thereby posing ampakines as potential treatments for acute intoxication in emergency room settings. Ampakines also have neuroprotective effects, which are mediated in part by inducing BDNF, and LY503430 and LY404187 were neuroprotective in an animal model of Parkinson’s disease (6-hydroxydopamine and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) toxicity), even when the treatment was delayed for 14 days after the lesion was established.117,122 Thus, AMPA receptor potentiators are potential medications for reversing neurotoxic effects of drugs, including a possible enhanced risk for Parkinson’s disease in methamphetamine abusers.123
NMDA ionotropic glutamate receptors
Pharmacological blockade of NMDA receptors inhibits acquisition and expression of CPP, which most likely reflect the involvement of NMDA receptors in memory consolidation.124 In contrast, the effect of NMDA receptor blockade on reinstated cocaine seeking is mixed.108,125 There is also extensive evidence that implicates NMDA receptor in extinction. 126 For example, enhancement of NMDA receptor function by the NMDA receptor partial agonist d-cycloserine (DCS) facilitates extinction of cocaine CPP and naloxone-induced conditioned place aversion in morphine-dependent rats, and retards subsequent reconditioning to ethanol and cocaine. Thus, DCS may be beneficial for preventing cue-induced relapse in addicted subjects. Two clinical studies have evaluated the effects of DCS in extinction. One study in smokers undergoing extinction exposure showed that DCS facilitated extinction in contrast to subjects receiving placebo.127 The other study conducted in cocaine abusers evaluated the effects of DCS on extinction of cocaine cue-induced craving and showed a trend toward a decrease in craving.128 Further clinical studies should evaluate long-term outcomes to determine if DCS when given during an extinction intervention can prevent relapse.
The NR2B-containing NMDA receptors are particularly important in extinction, and repeated drug exposures modify the expression of these subunits in the accumbens.55,56,129 Selective blockade of NR2B-containing NMDA receptors with ifenprodil in accumbens can inhibit conditioned drug craving and reinstated cocaine- and heroin-induced drug seeking. 56,124 Thus, selective targeting of NR2B-containing NMDA receptors may be useful in treating addiction. Notably, there is substantial interest from the pharmaceutical industry for developing NR2B subtype-selective antagonists for neurodegenerative disorders for chronic pain states130 that will likely result in novel compounds that could be of value in addiction.
Finally, acamprosate is a nonspecific glutamate receptor modulator that weakly activates NMDA receptors and shows some efficacy to reduce cocaine reinforcement and prevent alcohol and morphine in preclinical models.131 Moreover, acamprosate is clinically beneficial in supporting continuous abstinence after detoxification in alcohol-dependent patients.132 However, given the relatively weak effect of acamprosate on glutamate, it remains likely that additional, non-glutamatergic mechanisms contribute to its effects on alcohol-dependent patients.
Conclusions
A drug-induced pathology that impairs synaptic plasticity in the glutamatergic projection from the prefrontal cortex to the nucleus accumbens has been identified in preclinical models of addiction. These findings are consistent with the imaging literature that identified engagement of the prefrontal cortex in craving, compulsive drug administration, impaired inhibitory control and impaired decision making in addiction (see Box 1). Drug-induced neuroplasticity include changes in glial and synaptic proteins that are known to regulate glutamatergic synaptic plasticity (Figure 2). Many of the proteins have been pharmacologically targeted in animal models of relapse and successfully shown to inhibit relapse. Moreover, some treatments have been shown to affect drug-induced neuroadaptations in glutamate synaptic transmission and plasticity. Notably, this has been shown for N-acetylcysteine treatment that not only normalizes its molecular target, the cystine–glutamate exchanger, but when given chronically also produces an enduring restoration of many measures of cocaine-induced adaptations in glutamate homeostasis and neuronal plasticity.60 Whether other compounds found effective at preventing relapse in animal models will also prove capable of normalizing these measures remains to be determined. Nonetheless, early clinical trials with N-acetylcysteine in both cocaine and nicotine addiction have been positive. For example, double-blind, placebo-controlled reductions in the number of cigarettes smoked and cue-induced desire to use cocaine have been published.59,79 As well, cocaine-induced craving was also shown to be reduced by N-acetylcysteine.133 Accordingly, we propose that targeting other proteins shown in Figure 2 that are downstream from N-acetylcysteine-induced stimulation of cystine–glutamate exchange to release glutamate could be beneficial in treating addiction. This includes mGluR2/3 agonists, mGluR5-negative allosteric modulators, stimulators of glutamate transport and perhaps inhibitors of either AMPA or NMDA receptors. Importantly, most of these targets are active in preclinical models of relapse or withdrawal. Finally, given that all of these targets can be functionally linked via the concept of glutamate homeostasis, it is possible that combinations of drugs acting at different proteins may be more clinically effective and permit lower, more tolerable doses to be used.
Footnotes
Conflict of interest The authors declare no conflict of interest.
References
- 1.Substance Abuse and Mental Health Services Administration. Results from the 2009 National Survey on Drug Use and Health: Volume I. Summary of National Findings (Office of Applied Studies. NSDUH Series H-38A, HHS Publication No. SMA 10-4586Findings; Rockville, MD: 2010. [Google Scholar]
- 2.Policy OoNDC. The economic costs of drug abuse in the United States: 1992-2002, vol. Publication number (207303) Executive Office of the President; Washington, DC: 2004. [Google Scholar]
- 3.O’Brien C. Drug addiction and drug abuse. In: Hardman J, Limbird L, Gilman AG, editors. The Pharmacological Basis of Therapeutics. McGraw-Hill; New York: 2001. pp. 621–642. [Google Scholar]
- 4.Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goldstein RZ, Craig AD, Bechara A, Garavan H, Childress AR, Paulus MP, et al. The neurocircuitry of impaired insight in drug addiction. Trends Cogn Sci. 2009;13:372–380. doi: 10.1016/j.tics.2009.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verdejo-Garcia A, Bechara A. A somatic marker theory of addiction. Neuropharmacology. 2009;56(Suppl 1):48–62. doi: 10.1016/j.neuropharm.2008.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li CS, Sinha R. Inhibitory control and emotional stress regulation: neuroimaging evidence for frontal-limbic dysfunction in psycho-stimulant addiction. Neurosci Biobehav Rev. 2008;32:581–597. doi: 10.1016/j.neubiorev.2007.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goldstein RZ, Volkow ND. Drug addiction and its underlying neurobiological basis: neuroimaging evidence for the involvement of the frontal cortex. Am J Psychiatry. 2002;159:1642–1652. doi: 10.1176/appi.ajp.159.10.1642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strick PL, Dum RP, Fiez JA. Cerebellum and nonmotor function. Annu Rev Neurosci. 2009;32:413–434. doi: 10.1146/annurev.neuro.31.060407.125606. [DOI] [PubMed] [Google Scholar]
- 10.Fineberg NA, Potenza MN, Chamberlain SR, Berlin HA, Menzies L, Bechara A, et al. Probing compulsive and impulsive behaviors, from animal models to endophenotypes: a narrative review. Neuropsychopharmacology. 2010;35:591–604. doi: 10.1038/npp.2009.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brady KT, Verduin ML, Tolliver BK. Treatment of patients comorbid for addiction and other psychiatric disorders. Curr Psychiatry Rep. 2007;9:374–380. doi: 10.1007/s11920-007-0048-0. [DOI] [PubMed] [Google Scholar]
- 12.Back SE, Sonne SC, Killeen T, Dansky BS, Brady KT. Comparative profiles of women with PTSD and comorbid cocaine or alcohol dependence. Am J Drug Alcohol Abuse. 2003;29:169–189. doi: 10.1081/ada-120018845. [DOI] [PubMed] [Google Scholar]
- 13.Lising-Enriquez K, George TP. Treatment of comorbid tobacco use in people with serious mental illness. J Psychiatry Neurosci. 2009;34:E1–E2. [PMC free article] [PubMed] [Google Scholar]
- 14.Zahm DS. An integrative neuroanatomical perspective on some subcortical substrates of adaptive responding with emphasis on the nucleus accumbens. Neurosci Biobehav Rev. 2000;24:85–105. doi: 10.1016/s0149-7634(99)00065-2. [DOI] [PubMed] [Google Scholar]
- 15.Gerfen CR. The neostriatal mosaic: multiple levels of compartmental organization in the basal ganglia. Annu Rev Neurosci. 1993;15:285–320. doi: 10.1146/annurev.ne.15.030192.001441. [DOI] [PubMed] [Google Scholar]
- 16.Lu X-Y, Ghasemzadeh MB, Kalivas PW. Expression of D1 receptor, D2 receptor, substance P and enkephalin messenger RNAs in the neurons projecting from the nucleus accumbens. Neuroscience. 1998;82:767–780. doi: 10.1016/s0306-4522(97)00327-8. [DOI] [PubMed] [Google Scholar]
- 17.Meredith GE, Pennartz CMA, Groenewegen HJ. The cellular framework for chemical signalling in the nucleus accumbens. Prog Brain Res. 1993;99:3–24. doi: 10.1016/s0079-6123(08)61335-7. [DOI] [PubMed] [Google Scholar]
- 18.Kreitzer AC, Malenka RC. Striatal plasticity and basal ganglia circuit function. Neuron. 2008;60:543–554. doi: 10.1016/j.neuron.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stuber GD, Hnasko TS, Britt JP, Edwards RH, Bonci A. Dopaminergic terminals in the nucleus accumbens but not the dorsal striatum corelease glutamate. J Neurosci. 2010;30:8229–8233. doi: 10.1523/JNEUROSCI.1754-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lapish CC, Seamans JK, Chandler LJ. Glutamate-dopamine cotransmission and reward processing in addiction. Alcohol Clin Exp Res. 2006;30:1451–1465. doi: 10.1111/j.1530-0277.2006.00176.x. [DOI] [PubMed] [Google Scholar]
- 21.Surmeier DJ, Ding J, Day M, Wang Z, Shen W. D1 and D2 dopamine-receptor modulation of striatal glutamatergic signaling in striatal medium spiny neurons. Trends Neurosci. 2007;30:228–235. doi: 10.1016/j.tins.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 22.Lavin A, Nogueira L, Lapish CC, Wightman RM, Phillips PE, Seamans JK. Mesocortical dopamine neurons operate in distinct temporal domains using multimodal signaling. J Neurosci. 2005;25:5013–5023. doi: 10.1523/JNEUROSCI.0557-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Anwyl R. Metabotropic glutamate receptors: electrophysiological properties and role in plasticity. Brain Res Rev. 1999;29:83–120. doi: 10.1016/s0165-0173(98)00050-2. [DOI] [PubMed] [Google Scholar]
- 24.Gladding CM, Fitzjohn SM, Molnar E. Metabotropic glutamate receptor-mediated long-term depression: molecular mechanisms. Pharmacol Rev. 2009;61:395–412. doi: 10.1124/pr.109.001735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fellin T. Communication between neurons and astrocytes: relevance to the modulation of synaptic and network activity. J Neurochem. 2009;108:533–544. doi: 10.1111/j.1471-4159.2008.05830.x. [DOI] [PubMed] [Google Scholar]
- 26.Haydon PG, Blendy J, Moss SJ, Rob Jackson F. Astrocytic control of synaptic transmission and plasticity: a target for drugs of abuse? Neuropharmacology. 2009;56(Suppl 1):83–90. doi: 10.1016/j.neuropharm.2008.06.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bergles DE, Diamond JS, Jahr CE. Clearance of glutamate inside the synapse and beyond. Curr Opin Neurobiol. 1999;9:293–298. doi: 10.1016/s0959-4388(99)80043-9. [DOI] [PubMed] [Google Scholar]
- 28.Diamond JS, Jahr CE. Synaptically released glutamate does not overwhelm transporters on hippocampal astrocytes during high-frequency stimulation. J Neurophysiol. 2000;83:2835–2843. doi: 10.1152/jn.2000.83.5.2835. [DOI] [PubMed] [Google Scholar]
- 29.Moran MM, McFarland K, Melendez RI, Kalivas PW, Seamans JK. Cystine/glutamate exchange regulates metabotropic glutamate receptor presynaptic inhibition of excitatory transmission and vulnerability to cocaine seeking. J Neurosci. 2005;25:6389–6393. doi: 10.1523/JNEUROSCI.1007-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cavelier P, Attwell D. Tonic release of glutamate by a DIDS-sensitive mechanism in rat hippocampal slices. J Physiol. 2005;564(Part 2):397–410. doi: 10.1113/jphysiol.2004.082131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Herman MA, Jahr CE. Extracellular glutamate concentration in hippocampal slice. J Neurosci. 2007;27:9736–9741. doi: 10.1523/JNEUROSCI.3009-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van der Zeyden M, Oldenziel WH, Rea K, Cremers TI, Westerink BH. Microdialysis of GABA and glutamate: analysis, interpretation and comparison with microsensors. Pharmacol Biochem Behav. 2008;90:135–147. doi: 10.1016/j.pbb.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 33.Tamaru Y, Nomura S, Mizuno N, Shigemoto R. Distribution of metabotropic glutamate receptor mGluR3 in the mouse CNS: differential location relative to pre- and postsynaptic sites. Neuroscience. 2001;106:481–503. doi: 10.1016/s0306-4522(01)00305-0. [DOI] [PubMed] [Google Scholar]
- 34.Mitrano DA, Arnold C, Smith Y. Subcellular and subsynaptic localization of group I metabotropic glutamate receptors in the nucleus accumbens of cocaine-treated rats. Neuroscience. 2008;154:653–666. doi: 10.1016/j.neuroscience.2008.03.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nat Rev Neurosci. 2009;10:561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- 36.Moussawi K, Pacchioni A, Moran M, Olive MF, Gass JT, Lavin A, et al. N-Acetylcysteine reverses cocaine-induced metaplasticity. Nat Neurosci. 2009;12:182–189. doi: 10.1038/nn.2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Goto Y, Grace AA. Limbic and cortical information processing in the nucleus accumbens. Trends Neurosci. 2008;31:552–558. doi: 10.1016/j.tins.2008.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 39.Russo SJ, Dietz DM, Dumitriu D, Morrison JH, Malenka RC, Nestler EJ. The addicted synapse: mechanisms of synaptic and structural plasticity in nucleus accumbens. Trends Neurosci. 2010;33:267–276. doi: 10.1016/j.tins.2010.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- 41.Lafourcade M, Elezgarai I, Mato S, Bakiri Y, Grandes P, Manzoni OJ. Molecular components and functions of the endocannabinoid system in mouse prefrontal cortex. PLoS ONE. 2007;2:e709. doi: 10.1371/journal.pone.0000709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xi ZX, Ramamoorthy S, Baker DA, Shen H, Samuvel DJ, Kalivas PW. Modulation of group II metabotropic glutamate receptor signaling by chronic cocaine. J Pharmacol Exp Ther. 2002;303:608–615. doi: 10.1124/jpet.102.039735. [DOI] [PubMed] [Google Scholar]
- 43.Pistis M, Muntoni AL, Pillolla G, Gessa GL. Cannabinoids inhibit excitatory inputs to neurons in the shell of the nucleus accumbens: an in vivo electrophysiological study. Eur J Neurosci. 2002;15:1795–1802. doi: 10.1046/j.1460-9568.2002.02019.x. [DOI] [PubMed] [Google Scholar]
- 44.Swanson C, Baker D, Carson D, Worley P, Kalivas P. Repeated cocaine administration attenuates group I metabotropic glutamate receptor-mediated glutamate release and behavioral activation: a potential role for Homer 1b/c. J Neurosci. 2001;21:9043–9052. doi: 10.1523/JNEUROSCI.21-22-09043.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Solinas M, Ferre S, You ZB, Karcz-Kubicha M, Popoli P, Goldberg SR. Caffeine induces dopamine and glutamate release in the shell of the nucleus accumbens. J Neurosci. 2002;22:6321–6324. doi: 10.1523/JNEUROSCI.22-15-06321.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Venkitaramani DV, Gladding CM, Kurup P, Molnar E, Collingridge GL, et al. The tyrosine phosphatase STEP mediates AMPA receptor endocytosis after metabotropic glutamate receptor stimulation. J Neurosci. 2008;28:10561–10566. doi: 10.1523/JNEUROSCI.2666-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao C, Sun X, Wolf ME. Activation of D1 dopamine receptors increases surface expression of AMPA receptors and facilitates their synaptic incorporation in cultured hippocampal neurons. J Neurochem. 2006;98:1664–1677. doi: 10.1111/j.1471-4159.2006.03999.x. [DOI] [PubMed] [Google Scholar]
- 48.Bowers MS, McFarland K, Lake RW, Peterson YK, Lapish CC, Gregory ML, et al. Activator of G-protein signaling 3: a gatekeeper of cocaine sensitization and drug-seeking. Neuron. 2004;42:269–281. doi: 10.1016/s0896-6273(04)00159-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yao L, McFarland K, Fan P, Jiang Z, Inoue Y, Diamond I. Activator of G protein signaling 3 regulates opiate activation of protein kinase A signaling and relapse of heroin-seeking behavior. Proc Natl Acad Sci USA. 2005;102:8746–8751. doi: 10.1073/pnas.0503419102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bowers MS, Hopf FW, Chou JK, Guillory AM, Chang SJ, Janak PH, et al. Nucleus accumbens AGS3 expression drives ethanol seeking through G betagamma. Proc Natl Acad Sci USA. 2008;105:12533–12538. doi: 10.1073/pnas.0706999105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Szumlinski KK, Dehoff MH, Kang SH, Frys KA, Lominac KD, Klugmann M, et al. Homer proteins regulate sensitivity to cocaine. Neuron. 2004;43:401–413. doi: 10.1016/j.neuron.2004.07.019. [DOI] [PubMed] [Google Scholar]
- 52.Boudreau AC, Reimers JM, Milovanovic M, Wolf ME. Cell surface AMPA receptors in the rat nucleus accumbens increase during cocaine withdrawal but internalize after cocaine challenge in association with altered activation of mitogen-activated protein kinases. J Neurosci. 2007;27:10621–10635. doi: 10.1523/JNEUROSCI.2163-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kourrich S, Rothwell PE, Klug JR, Thomas MJ. Cocaine experience controls bidirectional synaptic plasticity in the nucleus accumbens. J Neurosci. 2007;27:7921–7928. doi: 10.1523/JNEUROSCI.1859-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Conrad KL, Tseng KY, Uejima JL, Reimers JM, Heng LJ, Shaham Y, et al. Formation of accumbens GluR2-lacking AMPA receptors mediates incubation of cocaine craving. Nature. 2008;454:118–121. doi: 10.1038/nature06995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang YH, Lin Y, Mu P, Lee BR, Brown TE, Wayman G, et al. In vivo cocaine experience generates silent synapses. Neuron. 2009;63:40–47. doi: 10.1016/j.neuron.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shen H, Moussawi K, Zhou W, Toda S, Kalivas P. Heroin relapse requires LTP-like plasticity mediated by NR2B-containing receptors. Nat Neurosci. 2011 doi: 10.1073/pnas.1112052108. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Madayag A, Lobner D, Kau KS, Mantsch JR, Abdulhameed O, Hearing M, et al. Repeated N-acetylcysteine administration alters plasticity-dependent effects of cocaine. J Neurosci. 2007;27:13968–13976. doi: 10.1523/JNEUROSCI.2808-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kau KS, Madayag A, Mantsch JR, Grier MD, Abdulhameed O, Baker DA. Blunted cystine-glutamate antiporter function in the nucleus accumbens promotes cocaine-induced drug seeking. Neuroscience. 2008;155:530–537. doi: 10.1016/j.neuroscience.2008.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Knackstedt LA, LaRowe S, Mardikian P, Malcolm R, Upadhyaya H, Hedden S, et al. The role of cystine-glutamate exchange in nicotine dependence in rats and humans. Biol Psychiatry. 2009;65:841–845. doi: 10.1016/j.biopsych.2008.10.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moussawi K, Zhou W, Shen H, Reichel CM, See R, Carr D, et al. Reversing cocaine-induced synaptic potentiation provides enduring protection from relapse. Proc Natl Acad Sci USA. 2011;108:385–390. doi: 10.1073/pnas.1011265108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.De Roo M, Klauser P, Garcia PM, Poglia L, Muller D. Spine dynamics and synapse remodeling during LTP and memory processes. Prog Brain Res. 2008;169:199–207. doi: 10.1016/S0079-6123(07)00011-8. [DOI] [PubMed] [Google Scholar]
- 62.Soule J, Messaoudi E, Bramham CR. Brain-derived neurotrophic factor and control of synaptic consolidation in the adult brain. Biochem Soc Trans. 2006;34(Part 4):600–604. doi: 10.1042/BST0340600. [DOI] [PubMed] [Google Scholar]
- 63.Shi Y, Ethell IM. Integrins control dendritic spine plasticity in hippocampal neurons through NMDA receptor and Ca2 +/ calmodulin-dependent protein kinase II-mediated actin reorganization. J Neurosci. 2006;26:1813–1822. doi: 10.1523/JNEUROSCI.4091-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bozdagi O, Nagy V, Kwei KT, Huntley GW. In vivo roles for matrix metalloproteinase-9 in mature hippocampal synaptic physiology and plasticity. J Neurophysiol. 2007;98:334–344. doi: 10.1152/jn.00202.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brown TE, Forquer MR, Cocking DL, Jansen HT, Harding JW, Sorg BA. Role of matrix metalloproteinases in the acquisition and reconsolidation of cocaine-induced conditioned place preference. Learn Mem. 2007;14:214–223. doi: 10.1101/lm.476207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Rodriguez D, Morrison CJ, Overall CM. Matrix metalloproteinases: what do they not do? New substrates and biological roles identified by murine models and proteomics. Biochim Biophys Acta. 2010;1803:39–54. doi: 10.1016/j.bbamcr.2009.09.015. [DOI] [PubMed] [Google Scholar]
- 67.Desgrosellier JS, Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010;10:9–22. doi: 10.1038/nrc2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Berglind WJ, See RE, Fuchs RA, Ghee SM, Whitfield TW, Jr, Miller SW, et al. A BDNF infusion into the medial prefrontal cortex suppresses cocaine seeking in rats. Eur J Neurosci. 2007;26:757–766. doi: 10.1111/j.1460-9568.2007.05692.x. [DOI] [PubMed] [Google Scholar]
- 69.Berglind WJ, Whitfield TW, Jr, LaLmiere RT, Kalivas PW, McGinty JF. A single intra-PFC infusion of BDNF prevents cocaine-induced alterations in extracellular glutamate within the nucleus accumbens. J Neurosci. 2009;29:3715–3719. doi: 10.1523/JNEUROSCI.5457-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Graham DL, Edwards S, Bachtell RK, Dileone RJ, Rios M, Self DW. Dynamic BDNF activity in nucleus accumbens with cocaine use increases self-administration and relapse. Nat Neurosci. 2007;10:1029–1037. doi: 10.1038/nn1929. [DOI] [PubMed] [Google Scholar]
- 71.Martin M, Chen BT, Hopf FW, Bowers MS, Bonci A. Cocaine self-administration selectively abolishes LTD in the core of the nucleus accumbens. Nat Neurosci. 2006;9:868–869. doi: 10.1038/nn1713. [DOI] [PubMed] [Google Scholar]
- 72.Knackstedt LA, Moussawi K, Lalumiere R, Schwendt M, Klugmann M, Kalivas PW. Extinction training after cocaine self-administration induces glutamatergic plasticity to inhibit cocaine seeking. J Neurosci. 2010;30:7984–7992. doi: 10.1523/JNEUROSCI.1244-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kasanetz F, Deroche-Gamonet V, Berson N, Balado E, Lafourcade M, Manzoni O, et al. Transition to addiction is associated with a persistent impairment in synaptic plasticity. Science. 2010;328:1709–1712. doi: 10.1126/science.1187801. [DOI] [PubMed] [Google Scholar]
- 74.Lewerenz J, Albrecht P, Tien ML, Henke N, Karumbayaram S, Kornblum HI, et al. Induction of Nrf2 and xCT are involved in the action of the neuroprotective antibiotic ceftriaxone in vitro. J Neurochem. 2009;111:332–343. doi: 10.1111/j.1471-4159.2009.06347.x. [DOI] [PubMed] [Google Scholar]
- 75.Knackstedt LA, Melendez RI, Kalivas PW. Ceftriaxone restores glutamate homeostasis and prevents relapse to cocaine seeking. Biol Psychiatry. 2010;67:81–84. doi: 10.1016/j.biopsych.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sari Y, Smith KD, Ali PK, Rebec GV. Upregulation of GLT1 attenuates cue-induced reinstatement of cocaine-seeking behavior in rats. J Neurosci. 2009;29:9239–9243. doi: 10.1523/JNEUROSCI.1746-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, et al. Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci. 2003;6:743–749. doi: 10.1038/nn1069. [DOI] [PubMed] [Google Scholar]
- 78.Zhou W, Kalivas PW. N-Acetylcysteine reduces extinction responding and induces enduring reductions in cue- and heroin-induced drug-seeking. Biol Psychiatry. 2007;63:338–340. doi: 10.1016/j.biopsych.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.LaRowe SD, Myrick H, Hedden S, Mardikian P, Saladin M, McRae A, et al. Is cocaine desire reduced by N-acetylcysteine? Am J Psychiatry. 2007;164:1115–1117. doi: 10.1176/ajp.2007.164.7.1115. [DOI] [PubMed] [Google Scholar]
- 80.Holdiness MR. Clinical pharmacokinetics of N-acetylcysteine. Clin Pharmacokinet. 1991;20:123–134. doi: 10.2165/00003088-199120020-00004. [DOI] [PubMed] [Google Scholar]
- 81.Ballon JS, Feifel D. A systematic review of modafinil: potential clinical uses and mechanisms of action. J Clin Psychiatry. 2006;67:554–566. doi: 10.4088/jcp.v67n0406. [DOI] [PubMed] [Google Scholar]
- 82.Ferraro L, Antonelli T, O’Connor WT, Tanganelli S, Rambert FA, Fuxe K. The effects of modafinil on striatal, pallidal and nigral GABA and glutamate release in the conscious rat: evidence for a preferential inhibition of striato-pallidal GABA transmission. Neurosci Lett. 1998;253:135–138. doi: 10.1016/s0304-3940(98)00629-6. [DOI] [PubMed] [Google Scholar]
- 83.Ferraro L, Antonelli T, Tanganelli S, O’Connor WT, Perez de la Mora M, Mendez-Franco J, et al. The vigilance promoting drug modafinil increases extracellular glutamate levels in the medial preoptic area and the posterior hypothalamus of the conscious rat: prevention by local GABAA receptor blockade. Neuropsychopharmacology. 1999;20:346–356. doi: 10.1016/S0893-133X(98)00085-2. [DOI] [PubMed] [Google Scholar]
- 84.Tahsili-Fahadan P, Carr GV, Harris GC, Aston-Jones G. Modafinil blocks reinstatement of extinguished opiate-seeking in rats: mediation by a glutamate mechanism. Neuropsychopharmacology. 2010;35:2203–2210. doi: 10.1038/npp.2010.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Reichel CM, See RE. Modafinil effects on reinstatement of methamphetamine seeking in a rat model of relapse. Psychopharmacology (Berl) 2010;210:337–346. doi: 10.1007/s00213-010-1828-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tahsili-Fahadan P, Malcolm R, Aston-Jones G. Modafinil: an anti-relapse medication. Neuropsychopharmacology. 2010;35:343–344. doi: 10.1038/npp.2009.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dackis CA, Kampman KM, Lynch KG, Pettinati HM, O’Brien CP. A double-blind, placebo-controlled trial of modafinil for cocaine dependence. Neuropsychopharmacology. 2005;30:205–211. doi: 10.1038/sj.npp.1300600. [DOI] [PubMed] [Google Scholar]
- 88.Volkow ND, Fowler JS, Logan J, Alexoff D, Zhu W, Telang F, et al. Effects of modafinil on dopamine and dopamine transporters in the male human brain: clinical implications. JAMA. 2009;301:1148–1154. doi: 10.1001/jama.2009.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liechti ME, Lhuillier L, Kaupmann K, Markou A. Metabotropic glutamate 2/3 receptors in the ventral tegmental area and the nucleus accumbens shell are involved in behaviors relating to nicotine dependence. J Neurosci. 2007;27:9077–9085. doi: 10.1523/JNEUROSCI.1766-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gass JT, Olive MF. Glutamatergic substrates of drug addiction and alcoholism. Biochem Pharmacol. 2008;75:218–265. doi: 10.1016/j.bcp.2007.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Adewale AS, Platt DM, Spealman RD. Pharmacological stimulation of group ii metabotropic glutamate receptors reduces cocaine self-administration and cocaine-induced reinstatement of drug seeking in squirrel monkeys. J Pharmacol Exp Ther. 2006;318:922–931. doi: 10.1124/jpet.106.105387. [DOI] [PubMed] [Google Scholar]
- 92.Xi ZX, Li X, Peng XQ, Li J, Chun L, Gardner EL, et al. Inhibition of NAALADase by 2-PMPA attenuates cocaine-induced relapse in rats: a NAAG-mGluR2/3-mediated mechanism. J Neurochem. 2010;112:564–576. doi: 10.1111/j.1471-4159.2009.06478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bossert JM, Gray SM, Lu L, Shaham Y. Activation of group II metabotropic glutamate receptors in the nucleus accumbens shell attenuates context-induced relapse to heroin seeking. Neuropsychopharmacology. 2006;31:2197–2209. doi: 10.1038/sj.npp.1300977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Baptista MA, Martin-Fardon R, Weiss F. Preferential effects of the metabotropic glutamate 2/3 receptor agonist LY379268 on conditioned reinstatement versus primary reinforcement: comparison between cocaine and a potent conventional reinforcer. J Neurosci. 2004;24:4723–4727. doi: 10.1523/JNEUROSCI.0176-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Peters J, Kalivas PW. The group II metabotropic glutamate receptor agonist, LY379268, inhibits both cocaine- and food-seeking behavior in rats. Psychopharmacology (Berl) 2006;186:143–149. doi: 10.1007/s00213-006-0372-9. [DOI] [PubMed] [Google Scholar]
- 96.Markou A. Metabotropic glutamate receptor antagonists: novel therapeutics for nicotine dependence and depression? Biol Psychiatry. 2007;61:17–22. doi: 10.1016/j.biopsych.2006.03.053. [DOI] [PubMed] [Google Scholar]
- 97.Kenny PJ, Gasparini F, Markou A. Group II metabotropic and alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA)/kainate glutamate receptors regulate the deficit in brain reward function associated with nicotine withdrawal in rats. J Pharmacol Exp Ther. 2003;306:1068–1076. doi: 10.1124/jpet.103.052027. [DOI] [PubMed] [Google Scholar]
- 98.Szumlinski KK, Kalivas PW, Worley PF. Homer proteins: implications for neuropsychiatric disorders. Curr Opin Neurobiol. 2006;16:251–257. doi: 10.1016/j.conb.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 99.Chiamulera C, Epping-Jordan M, Zocchi A, Marcon C, Cottiny C, Tacconi S, et al. Reinforcing and locomotor stimulant effects of cocaine are absent in mGluR5 null mutant mice. Nat Neurosci. 2001;4:873–874. doi: 10.1038/nn0901-873. [DOI] [PubMed] [Google Scholar]
- 100.Olive MF. Cognitive effects of Group I metabotropic glutamate receptor ligands in the context of drug addiction. Eur J Pharmacol. 2010;639:47–58. doi: 10.1016/j.ejphar.2010.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zerbib F, Keywood C, Strabach G. Efficacy, tolerability and pharmacokinetics of a modified release formulation of ADX10059, a negative allosteric modulator of metabotropic glutamate receptor 5: an esophageal pH-impedance study in healthy subjects. Neurogastroenterol Motil. 2010;22:859–865. e231. doi: 10.1111/j.1365-2982.2010.01484.x. [DOI] [PubMed] [Google Scholar]
- 102.Berry-Kravis E, Hessle D, Coffey S, Nervey C, Scheider A, Yuhas J, et al. A pilot open label, single dose trial of fenobam in adults with fragile X sindrome. J Med Genet. 2009;46:266–271. doi: 10.1136/jmg.2008.063701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.D’Ascenzo M, Fellin T, Terunuma M, Revilla-Sanchez R, Meaney DF, Auberson YP, et al. mGluR5 stimulates gliotransmission in the nucleus accumbens. Proc Natl Acad Sci USA. 2007;104:1995–2000. doi: 10.1073/pnas.0609408104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Wolf ME. The Bermuda Triangle of cocaine-induced neuroadaptations. Trends Neurosci. 2010;33:391–398. doi: 10.1016/j.tins.2010.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.LaLumiere RT, Kalivas PW. Glutamate release in the nucleus accumbens core is necessary for heroin seeking. J Neurosci. 2008;28:3170–3177. doi: 10.1523/JNEUROSCI.5129-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cornish J, Kalivas P. Glutamate transmission in the nucleus accumbens mediates relapse in cocaine addiction. J Neurosci. 2000;20:RC89, 81–85. doi: 10.1523/JNEUROSCI.20-15-j0006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Di Ciano P, Everitt BJ. Dissociable effects of antagonism of NMDA and AMPA/KA receptors in the nucleus accumbens core and shell on cocaine-seeking behavior. Neuropsychopharmacology. 2001;25:341–360. doi: 10.1016/S0893-133X(01)00235-4. [DOI] [PubMed] [Google Scholar]
- 108.Backstrom P, Hyytia P. Involvement of AMPA/kainate, NMDA, and mGlu5 receptors in the nucleus accumbens core in cue-induced reinstatement of cocaine seeking in rats. Psychopharmacology (Berl) 2007;192:571–580. doi: 10.1007/s00213-007-0753-8. [DOI] [PubMed] [Google Scholar]
- 109.Sanchis-Segura C, Borchardt T, Vengeliene V, Zghoul T, Bachteler D, Gass P, et al. Involvement of the AMPA receptor GluR-C subunit in alcohol-seeking behavior and relapse. J Neurosci. 2006;26:1231–1238. doi: 10.1523/JNEUROSCI.4237-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Di Ciano P, Everitt BJ. Direct interactions between the basolateral amygdala and nucleus accumbens core underlie cocaine-seeking behavior by rats. J Neurosci. 2004;24:7167–7173. doi: 10.1523/JNEUROSCI.1581-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bowers MS, Chen BT, Bonci A. AMPA receptor synaptic plasticity induced by psychostimulants: the past, present, and therapeutic future. Neuron. 2010;67:11–24. doi: 10.1016/j.neuron.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Rubio G, Ponce G, Jimenez-Arriero MA, Palomo T, Manzanares J, Ferre F. Effects of topiramate in the treatment of alcohol dependence. Pharmacopsychiatry. 2004;37:37–40. doi: 10.1055/s-2004-815473. [DOI] [PubMed] [Google Scholar]
- 113.Kampman KM, Pettinati H, Lynch KG, Dackis C, Sparkman T, Weigley C, et al. A pilot trial of topiramate for the treatment of cocaine dependence. Drug Alcohol Depend. 2004;75:233–240. doi: 10.1016/j.drugalcdep.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 114.Brown ES, Nejtek VA, Perantie DC, Orsulak PJ, Bobadilla L. Lamotrigine in patients with bipolar disorder and cocaine dependence. J Clin Psychiatry. 2003;64:197–201. doi: 10.4088/jcp.v64n0213. [DOI] [PubMed] [Google Scholar]
- 115.O’Neill MJ, Bleakman D, Zimmerman DM, Nisenbaum ES. AMPA receptor potentiators for the treatment of CNS disorders. Curr Drug Targets CNS Neurol Disord. 2004;3:181–194. doi: 10.2174/1568007043337508. [DOI] [PubMed] [Google Scholar]
- 116.Quirk JC, Nisenbaum ES. LY404187: a novel positive allosteric modulator of AMPA receptors. CNS Drug Rev. 2002;8:255–282. doi: 10.1111/j.1527-3458.2002.tb00228.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.O’Neill MJ, Murray TK, Whalley K, Ward MA, Hicks CA, Woodhouse S, et al. Neurotrophic actions of the novel AMPA receptor potentiator, LY404187, in rodent models of Parkinson’s disease. Eur J Pharmacol. 2004;486:163–174. doi: 10.1016/j.ejphar.2003.12.023. [DOI] [PubMed] [Google Scholar]
- 118.Lauterborn JC, Pineda E, Chen LY, Ramirez EA, Lynch G, Gall CM. Ampakines cause sustained increases in brain-derived neurotrophic factor signaling at excitatory synapses without changes in AMPA receptor subunit expression. Neuroscience. 2009;159:283–295. doi: 10.1016/j.neuroscience.2008.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Larson J, Quach CN, LeDuc BQ, Nguyen A, Rogers GA, Lynch G. Effects of an AMPA receptor modulator on methamphetamine-induced hyperactivity in rats. Brain Res. 1996;738:353–356. doi: 10.1016/s0006-8993(96)01049-9. [DOI] [PubMed] [Google Scholar]
- 120.Hess US, Whalen SP, Sandoval LM, Lynch G, Gall CM. Ampakines reduce methamphetamine-driven rotation and activate neocortex in a regionally selective fashion. Neuroscience. 2003;121:509–521. doi: 10.1016/s0306-4522(03)00423-8. [DOI] [PubMed] [Google Scholar]
- 121.Jones N, Messenger MJ, O’Neill MJ, Oldershaw A, Gilmour G, Simmons RM, et al. AMPA receptor potentiation can prevent ethanol-induced intoxication. Neuropsychopharmacology. 2008;33:1713–1723. doi: 10.1038/sj.npp.1301562. [DOI] [PubMed] [Google Scholar]
- 122.Murray TK, Whalley K, Robinson CS, Ward MA, Hicks CA, Lodge D, et al. LY503430, a novel alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptor potentiator with functional, neuroprotective and neurotrophic effects in rodent models of Parkinson’s disease. J Pharmacol Exp Ther. 2003;306:752–762. doi: 10.1124/jpet.103.049445. [DOI] [PubMed] [Google Scholar]
- 123.Callaghan RC, Cunningham JK, Sajeev G, Kish SJ. Incidence of Parkinson’s disease among hospital patients with methamphetamine-use disorders. Mov Disord. 2010;25:2333–2339. doi: 10.1002/mds.23263. [DOI] [PubMed] [Google Scholar]
- 124.Ma YY, Cepeda C, Cui CL. The role of striatal NMDA receptors in drug addiction. Int Rev Neurobiol. 2009;89:131–146. doi: 10.1016/S0074-7742(09)89006-5. [DOI] [PubMed] [Google Scholar]
- 125.Cornish JL, Duffy P, Kalivas PW. A role of nucleus accumbens glutamate transmission in the relapse to cocaine-seeking behavior. Neuroscience. 1999;93:1359–1368. doi: 10.1016/s0306-4522(99)00214-6. [DOI] [PubMed] [Google Scholar]
- 126.Myers KM, Carlezon WA, Jr, Davis M. Glutamate receptors in extinction and extinction-based therapies for psychiatric illness. Neuropsychopharmacology. 2011;36:274–293. doi: 10.1038/npp.2010.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Santa Ana EJ, Rounsaville BJ, Frankforter TL, Nich C, Babuscio T, Poling J, et al. D-Cycloserine attenuates reactivity to smoking cues in nicotine dependent smokers: a pilot investigation. Drug Alcohol Depend. 2009;104:220–227. doi: 10.1016/j.drugalcdep.2009.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Price KL, McRae-Clark AL, Saladin ME, Maria MM, DeSantis SM, Back SE, et al. D-cycloserine and cocaine cue reactivity: preliminary findings. Am J Drug Alcohol Abuse. 2009;35:434–438. doi: 10.3109/00952990903384332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Mao LM, Wang W, Chu XP, Zhang GC, Liu XY, Yang YJ, et al. Stability of surface NMDA receptors controls synaptic and behavioral adaptations to amphetamine. Nat Neurosci. 2009;12:602–610. doi: 10.1038/nn.2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Gogas KR. Glutamate-based therapeutic approaches: NR2B receptor antagonists. Curr Opin Pharmacol. 2006;6:68–74. doi: 10.1016/j.coph.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 131.Bowers MS, Chen BT, Chou JK, Osborne MP, Gass JT, See RE, et al. Acamprosate attenuates cocaine- and cue-induced reinstatement of cocaine-seeking behavior in rats. Psychopharmacology (Berl) 2007;195:397–406. doi: 10.1007/s00213-007-0904-y. [DOI] [PubMed] [Google Scholar]
- 132.Rosner S, Hackl-Herrwerth A, Leucht S, Lehert P, Vecchi S, Soyka M. Acamprosate for alcohol dependence. Cochrane Database Syst Rev. 2010(9):CD004332. doi: 10.1002/14651858.CD004332.pub2. [DOI] [PubMed] [Google Scholar]
- 133.Amen SL, Piacentine LB, Ahmad ME, Li SJ, Mantsch JR, Risinger RC, et al. Repeated N-acetyl cysteine reduces cocaine seeking in rodents and craving in cocaine-dependent humans. Neuropsychopharmacology. 2011;36:871–878. doi: 10.1038/npp.2010.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fuster JM. The Prefrontal Cortex: Anatomy, Physiology and Neuropsychology of the Frontal Lobe. Lippincott-Raven; Philadelphia: 1997. [Google Scholar]
- 135.Haber SN. The primate basal ganglia: parallel and integrative networks. J Chem Neuroanat. 2003;26:317–330. doi: 10.1016/j.jchemneu.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 136.D’Esposito M, Chen AJ. Neural mechanisms of prefrontal cortical function: implications for cognitive rehabilitation. Prog Brain Res. 2006;157:123–139. doi: 10.1016/s0079-6123(06)57008-6. [DOI] [PubMed] [Google Scholar]
- 137.Volkow ND, Fowler JS, Wang GJ, Telang F, Logan J, Jayne M, et al. Cognitive control of drug craving inhibits brain reward regions in cocaine abusers. Neuroimage. 2010;49:2536–2543. doi: 10.1016/j.neuroimage.2009.10.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Goldstein RZ, Alia-Klein N, Tomasi D, Carrillo JH, Maloney T, Woicik PA, et al. Anterior cingulate cortex hypoactivations to an emotionally salient task in cocaine addiction. Proc Natl Acad Sci USA. 2009;106:9453–9458. doi: 10.1073/pnas.0900491106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Schultz W. Getting formal with dopamine and reward. Neuron. 2002;36:241–263. doi: 10.1016/s0896-6273(02)00967-4. [DOI] [PubMed] [Google Scholar]
- 140.Ungless M, Whistler J, Malenka R, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001;411:583–587. doi: 10.1038/35079077. [DOI] [PubMed] [Google Scholar]
- 141.Bellone C, Luscher C. Cocaine triggered AMPA receptor redistribution is reversed in vivo by mGluR-dependent long-term depression. Nat Neurosci. 2006;9:636–641. doi: 10.1038/nn1682. [DOI] [PubMed] [Google Scholar]
- 142.Stuber GD, Klanker M, de Ridder B, Bowers MS, Joosten RN, Feenstra MG, et al. Reward-predictive cues enhance excitatory synaptic strength onto midbrain dopamine neurons. Science. 2008;321:1690–1692. doi: 10.1126/science.1160873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zweifel LS, Argilli E, Bonci A, Palmiter RD. Role of NMDA receptors in dopamine neurons for plasticity and addictive behaviors. Neuron. 2008;59:486–496. doi: 10.1016/j.neuron.2008.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Engblom D, Bilbao A, Sanchis-Segura C, Dahan L, Perreau-Lenz S, Balland B, et al. Glutamate receptors on dopamine neurons control the persistence of cocaine seeking. Neuron. 2008;59:497–508. doi: 10.1016/j.neuron.2008.07.010. [DOI] [PubMed] [Google Scholar]
- 145.Mameli M, Halbout B, Creton C, Engblom D, Parkitna JR, Spanagel R, et al. Cocaine-evoked synaptic plasticity: persistence in the VTA triggers adaptations in the NAc. Nat Neurosci. 2009;12:1036–1041. doi: 10.1038/nn.2367. [DOI] [PubMed] [Google Scholar]
- 146.Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM, et al. Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron. 2008;59:288–297. doi: 10.1016/j.neuron.2008.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ledford CC, Fuchs RA, See RE. Potentiated reinstatement of cocaine-seeking behavior following D-amphetamine infusion into the basolateral amygdala. Neuropsychopharmacology. 2003;28:1721–1729. doi: 10.1038/sj.npp.1300249. [DOI] [PubMed] [Google Scholar]
- 148.McFarland K, Kalivas PW. The circuitry mediating cocaine-induced reinstatement of drug-seeking behavior. J Neurosci. 2001;21:8655–8663. doi: 10.1523/JNEUROSCI.21-21-08655.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Park WK, Bari AA, Jey AR, Anderson SM, Spealman RD, Rowlett JK, et al. Cocaine administered into the medial prefrontal cortex reinstates cocaine-seeking behavior by increasing AMPA receptor-mediated glutamate transmission in the nucleus accumbens. J Neurosci. 2002;22:2916–2925. doi: 10.1523/JNEUROSCI.22-07-02916.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Anderson SM, Bari AA, Pierce RC. Administration of the D1-like dopamine receptor antagonist SCH-23390 into the medial nucleus accumbens shell attenuates cocaine priming-induced reinstatement of drug-seeking behavior in rats. Psychopharmacology (Berl) 2003;168:132–138. doi: 10.1007/s00213-002-1298-5. [DOI] [PubMed] [Google Scholar]
- 151.Huang CC, Lin HJ, Hsu KS. Repeated cocaine administration promotes long-term potentiation induction in rat medial prefrontal cortex. Cereb Cortex. 2007;17:1877–1888. doi: 10.1093/cercor/bhl096. [DOI] [PubMed] [Google Scholar]
- 152.Fu Y, Pollandt S, Liu J, Krishnan B, Genzer K, Orozco-Cabal L, et al. Long-term potentiation (LTP) in the central amygdala (CeA) is enhanced after prolonged withdrawal from chronic cocaine and requires CRF1 receptors. J Neurophysiol. 2007;97:937–941. doi: 10.1152/jn.00349.2006. [DOI] [PubMed] [Google Scholar]
- 153.Tye KM, Stuber GD, de Ridder B, Bonci A, Janak PH. Rapid strengthening of thalamo-amygdala synapses mediates cue-reward learning. Nature. 2008;453:1253–1257. doi: 10.1038/nature06963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Melis M, Camarini R, Ungless MA, Bonci A. Long-lasting potentiation of GABAergic synapses in dopamine neurons after a single in vivo ethanol exposure. J Neurosci. 2002;22:2074–2082. doi: 10.1523/JNEUROSCI.22-06-02074.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]