

Abstract
Neural stem cells (NSCs) continually produce new neurons in postnatal brains. However, the majority of these cells stay in a nondividing, inactive state. The molecular mechanism that is required for these cells to enter proliferation still remains largely unknown. Here, we show that nuclear receptor TLX (NR2E1) controls the activation status of postnatal NSCs in mice. Lineage tracing indicates that TLX-expressing cells give rise to both activated and inactive postnatal NSCs. Surprisingly, loss of TLX function does not result in spontaneous glial differentiation, but rather leads to a precipitous age-dependent increase of inactive cells with marker expression and radial morphology for NSCs. These inactive cells are mispositioned throughout the granular cell layer of the dentate gyrus during development and can proliferate again after reintroduction of ectopic TLX. RNA-seq analysis of sorted NSCs revealed a TLX-dependent global expression signature, which includes the p53 signaling pathway. TLX regulates p21 expression in a p53-dependent manner, and acute removal of p53 can rescue the proliferation defect of TLX-null NSCs in culture. Together, these findings suggest that TLX acts as an essential regulator that ensures the proliferative ability of postnatal NSCs by controlling their activation through genetic interaction with p53 and other signaling pathways.
Introduction
Postnatal neural stem cells (NSCs) exist normally in the subgranular zone (SGZ) of the dentate gyrus (DG) and the subventricular zone (SVZ) of the lateral ventricles (LVs) (Lois and Alvarez-Buylla, 1993; Kuhn et al., 1996; Lie et al., 2004; Zhao et al., 2008). These cells may play a critical role in certain forms of learning and memory and may significantly contribute to the maintenance of brain homeostasis (Imayoshi et al., 2008; Zhao et al., 2008). In the SGZ, type-1 NSCs have long radial glia-like processes spanning the entire granule cell layer. They express Nestin, GFAP, Sox2, and basic lipid binding protein (BLBP). While the majority of them remain in an inactive state, some of these NSCs slowly divide and give rise to transiently amplifying type-2 cells with short processes and tangential orientation. These type-2 cells rapidly proliferate and generate type-3 cells, which resemble immature neuroblasts and express doublecortin (DCX). They eventually mature into granule neurons that functionally integrate into the existing neural networks. In the LV, glia-like, GFAP+Nestin+ NSCs (type B cells) are located adjacent to the ependyma, a thin layer of cells lining the ventricle. These slow-dividing type B cells give rise to transiently amplifying Dlx2+ type C cells, which produce type A (DCX+PSA-NCAM+) neuroblasts. Newly generated neuroblasts migrate into the olfactory bulbs and become granule or periglomerular interneurons. Neurogenesis in both the SVZ and the SGZ continues throughout the adult life but decreases dramatically with age (Seki and Arai, 1995; Kuhn et al., 1996; Tropepe et al., 1997).
Despite advances in understanding adult NSCs, it still remains unclear how their activity is molecularly regulated and what signals are responsible for the age-dependent decline in replication. Previously, we and others have identified that TLX (NR2E1) is expressed in the neurogenic niche and is required for adult neurogenesis in the SGZ and the SVZ (Shi et al., 2004; Liu et al., 2008; Zhang et al., 2008). Furthermore, TLX-dependent NSCs and neurogenesis play a role in spatial learning and memory (Zhang et al., 2008). TLX is a member of the nuclear hormone receptor superfamily and functions as a transcriptional repressor by recruiting corepressors (Yu et al., 1994; Monaghan et al., 1995; Wang et al., 2006; Zhang et al., 2006; Sun et al., 2007; Yokoyama et al., 2008). The function of TLX is largely thought to prevent precocious differentiation of NSCs into mature neurons or glial cells during development (Roy et al., 2004; Shi et al., 2004; Li et al., 2008). This notwithstanding, the role of TLX in NSCs is far from clear, since our detailed analysis using genetic tracers revealed that cells expressing markers for NSCs still exist in postnatal TIx-null brains. These NSCs are mispositioned and rapidly lose their ability to proliferate in an age-dependent manner. Interestingly, these inert cells can be reactivated by reintroducing TLX. Furthermore, our RNA-seq analysis and in vitro cell culture studies showed a genetic interaction of TLX with the p53 signaling pathway. Together, these findings suggest that TLX function is essential in postnatal NSCs by controlling the switch from quiescence to activation.
Materials and Methods
Animals.
pTlx-CreERT2 mice were generated through recombineering technology (http://web.ncifcrf.gov/research/brb/recombineeringInformation.aspx). Briefly, a tamoxifen-inducible CreERT2 gene (Feil et al., 1997) was inserted through homologous recombination into the first exon of the Tlx locus in a BAC clone (RP24-344A4). The correctly recombined BAC clones were confirmed by restriction digestion and sequencing. The genomic DNA was released by sequential digestion with BsiWI and AscI and separated from the vector backbone through a CL-4B Sepharose column. Transgenic animals were then produced by pronuclear injection of fertilized mouse eggs by the Transgenic Core Facility at University of Texas (UT) Southwestern. Twenty-three founders were identified after genotyping and were further screened for inducible expression of enhanced yellow fluorescent protein (EYFP) after crossing into Rosa-EYFP reporter mice and treatment with tamoxifen. The pTlx-CreERT2 mice were kept in a mixed background of FVB, C57BL/6J, and 129S1/SvImJ.
The strategies and methods for generating TlxLacZ/LacZ mice or mice with conditional alleles of Tlx have been described (Yu et al., 2000; Zhang et al., 2008). In short, TlxLacZ/LacZ mice were generated by replacing exons 3–5 with LacZ and Neo genes. Thus, expression of LacZ is under the direct regulation of the endogenous Tlx promoter and enhancers. To create a conditional allele of Tlx, exon 2 was flanked by two loxP sites through homologous recombination. Cre-mediated recombination resulted in a null allele of Tlx by creating stop codons after reading frame shifts. Detailed information has also been provided for the generation and characterization of transgenic Nes-GFP mice (Yamaguchi et al., 2000) and a conditional allele of Trp53 (Marino et al., 2000). All mice were housed under a 12 h light/dark cycle and had ad libitum access to food and water in a controlled animal facility. No significant phenotypic differences were observed between male and female mice; thus, both genders were included in the analysis. Experimental protocols were approved by the Institutional Animal Care and Use Committee at UT Southwestern.
Administration of tamoxifen, bromodeoxyuridine, chlorodeoxyuridine, and iododeoxyuridine.
Tamoxifen (Sigma) was dissolved through bursts of sonication in sesame oil with a final stock concentration of 20 mg/ml. Mice were injected intraperitoneally once daily with 4 mg of tamoxifen per 20 g of body weight or sterile sesame oil (vehicle) for 5 consecutive days. The mice were killed 24 h after the last injection or at the indicated time points. Dividing cells in vivo were labeled by intraperitoneal injection of bromodeoxyuridine (BrdU), chloro-deoxyuridine (CldU), or iodo-deoxyuridine (IdU) (in PBS) at the indicated dose for the specified duration.
Lentivirus production and stereotactic brain injections.
Tlx cDNA was subcloned into either CMV-ires-GFP or hGfap-GFP-T2A lentiviral vector in which the expression of the transgene was driven by the CMV or hGfap promoter (Lee et al., 2008). HEK293T cells were cotransfected with lentiviral and packaging plasmids (pMDL, VSV-G, and pREV) by the CaPO4 method. Virus-containing supernatants were collected at 24, 48, and 72 h posttransfection, pooled, and filtered through a 0.22 μm filter to remove cellular debris. Viral particles were then concentrated by centrifugation at 25,000 rpm for 2 h at 4°C. Lentiviral titers were measured in either HEK293 cells (CMV promoter) or primary astrocytes (hGfap promoter). One microliter of viruses (1 × 108 colony-forming units/ml) were stereotactically injected into the DG. We used the following coordinates from bregma for Tlx-null mice: anterior/posterior, −1.3 mm; medial/lateral, ±2 mm; and dorsal/ventral from skull (DV), −1.5 mm.
Tissue preparation and immunohistochemistry.
The adult mice were killed via CO2 overdose and perfused with 1× PBS followed by ice-cold 4% paraformaldehyde (PFA) in PBS. Brains were dissected and postfixed overnight with 4% PFA at 4°C, followed by cryoprotection with 30% sucrose solution in PBS for another 24 h. Frozen brains were sectioned at 40 μm with a sliding microtome (Leica), and free-floating sections were collected and stored in antifreeze solution at 20°C. For immunostaining, sections were washed three times with PBS and blocked for 1 h at room temperature with blocking solution (3% BSA/0.2% Triton X-100 in PBS). The sections were then incubated overnight at 4°C with primary antibodies diluted in blocking solution. After three rinses with PBST buffer (0.2% Triton X-100 in PBS), the sections were further incubated with the corresponding secondary antibodies in blocking solution for 2 h at room temperature. When necessary, nuclei were stained with Hoechst 33342 (Hst, Sigma) (1 μg/ml in PBS). Sections were washed and mounted onto Superfrost glass slides (Fisher Scientific) with mounting medium containing diazabicyclo-octane (DABCO, Sigma). For BrdU, CldU, and IdU detection, before incubation with primary antibodies the sections were treated with 50% formamide in 2× SSC buffer for 2 h at 65°C, followed by further treatment with 2 m HCl for 30 min at 37°C and equilibration with 0.1 m boric acid (pH 8.5). Sequential detection of CldU and IdU were conducted essentially as described previously (Tuttle et al., 2011). Briefly, HCl-treated brain sections were first incubated overnight with antibody against IdU/BrdU (mouse clone 3D4) at 4°C, followed by high stringency wash with fresh TBST buffer (36 mm Tris, 50 mm NaCl, 0.5% Tween 20, pH 8.0) at 37°C. The sections were then incubated overnight with antibody against CldU/BrdU (rat BU1/75) at 4°C, washed with PBST, and detected with corresponding secondary antibodies.
The following primary antibodies were used: GFP (rabbit, 1:500, Invitrogen; chick, 1:1000, Aves Labs); GFAP (mouse, 1:500, Sigma; guinea pig, 1:1000, Advanced ImmunoChemical); BrdU/CldU (rat BU1/75, 1:500, Accurate Chemical); BrdU/IdU (mouse clone 3D4, 1:1000, BD Pharmingen); NeuN (rabbit, 1:500, Millipore); Sox2 (rabbit, 1:500, Millipore); BLBP (rabbit, 1:500, Millipore); Nestin (mouse, 1:200, BD Pharmingen); Ki67 (rabbit, 1:500, Novocastra); MCM2 (rabbit, 1:500,Cell Signaling Technology); DCX (goat, 1:150, Santa Cruz Biotechnology); Olig2 (rabbit, 1:500, Millipore), pSMAD1/5/8 (rabbit, 1:500, Cell Signaling Technology); GSTπ (mouse, 1:200, BD Biosciences); S100β (rabbit, 1:1000, Swant); RIP (mouse, 1:250, Hybridoma Bank, University of Iowa, Iowa City, IA); and Histone H3 (rabbit, 1:500, Cell Signaling Technology). Alexa Fluor 488-, 594-, or 647-conjugated secondary antibodies produced in goat or donkey (Invitrogen) were used for indirect fluorescence. Images were taken using a Zeiss LSM510 confocal microscope. A Cell Counter software plugin in the ImageJ program was used to count cells. Data were obtained from 12 random sections from three to five mice in each group.
NSC culture, immunocytochemistry, and flow cytometry.
NSCs from 6-to-8-week-old Tlx+/LacZ;CreER (CZ) or Tlxflox/LacZ;CreER (FCZ) mice were isolated and cultured in growth medium [DMEM/F12 medium supplemented with N2 (Invitrogen), heparin (5 μg/ml, Sigma), EGF (20 ng/ml, Peprotech), and bFGF (20 ng/ml, Peprotech)] as described previously (Zhang et al., 2008). Tlx+/LacZ NSCs were sorted based on LacZ expression using FluoReporter lacZ Flow Cytometry Kits according to the user's manual (Invitrogen). To acutely delete Tlx, FCZ cells were treated with 10 nm 4-hydroxytamoxifen (Sigma) for the indicated duration. Similarly, Tlx-null NSCs were isolated from embryonic day (E)18.5 cortices and cultured in growth medium. These cells were labeled the following day with 1 μm 1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI, Invitrogen) for 15 min at room temperature, followed by lentiviral transduction. Four hours later, the cells were washed with growth medium and continuously cultured for another 14 d with medium change in every other day. DiI and GFP were used for gating in flow cytometry. For immunocytochemistry, cultured cells on chamber slides (BD Biosciences) were fixed with 4% PFA, washed with PBS, blocked for 30 min at room temperature, and followed by overnight incubation with primary antibodies in blocking solution at 4°C. For detection of BrdU-labeled cells, the fixed cells on slides were treated with 2 m HCl at 37°C for 30 min, followed by washing with PBS and incubation with primary antibodies.
Fluorescence-activated cell sorting of Nes-GFP cells and RNA-Seq.
Three-week-old mice (n = 5–9 for each genotype) were used. Brains were cut into 1 mm coronal sections with a brain matrix, and fresh tissues surrounding the lateral ventricles were microdissected on ice. Tissues were enzymatically digested for 30 min at 37°C with PPD solution that consists of papain (2.5 u/ml), DNase I (250 u/ml), and dispase II (1.0 u/ml) in DMEM culture medium supplemented with 4.5 g/L glucose. After trituration with a 5 ml pipette, cells were sequentially washed three times with DMEM/10% FBS and once with PBS/4% FBS. They were then filtered through a 40 μm cell strainer for live cell sorting based on GFP expression. GFP+ cells were directly sorted into TRIzol LS reagent (Invitrogen), and the total RNAs were isolated using RNeasy Mini Kit (Qiagen). RNA quality was determined by Bioanalyzer (Agilent). cDNAs were synthesized from 50 ng of total RNA and further amplified with an RNA-seq Ovation system (NuGEN). A cDNA library was prepared and subjected to parallel sequencing using Genome Analyzer II (Illumina). Sequence tags were analyzed as described previously (Masui et al., 2010). A total of 20.8 million unique tags were obtained for wild-type cells and 19.7 million with RNAs from Tlx−/− cells. Based on the RPKM (reads per kilobase of exon model per million mapped reads) value for Mcm2, which is an essential gene for licensing DNA replication, a cutoff value of 2.0 was used for either wild-type or Tlx−/−. Gene expression changes were calculated as the ratio of the RPKM for wild-type to that of Tlx−/− and further analyzed with DAVID program for KEGG (Kyoto Encyclopedia of Genes and Genomes) signaling pathways.
Quantitative RT-PCR analysis.
Total RNAs from cultured cells or cells sorted by fluorescence-activated cell sorting (FACS) were isolated using TRIzol reagent (Invitrogen) and RNeasy Mini Kit (Qiagen). The Superscript III system (Invitrogen) and random primers were used to synthesize cDNA from 1.0 μg of total RNA from cultured cells. For sorted cells, 20 ng of total RNA was used to make the first-strand cDNAs, which were further amplified using the RNA-seq Ovation system according to manufacturer's protocol (NuGEN). Gene expression was analyzed using the SYBR Greener system (Invitrogen) on a 384-well ABI 7900HT thermocycler (Applied Biosystems). Primer sequences for PCRs are available upon request.
Statistical analysis.
Differences between groups were determined for significance using the two-tailed Student's t test with equal variance or ANOVA. A p value of <0.05 was considered significant.
Results
Tlx-expressing cells generate both activated and inactive adult NSCs
We used an inducible approach to specifically examine the identity and lineage of Tlx-expressing cells in the adult mouse brain. Such a strategy has been used to show that TLX-expressing cells are type B NSCs in the SVZ (Liu et al., 2008). However, the activation status of those cells and the identity of the cells in the SGZ are not known. Through homologous recombination, the synthetic gene CreERT2 was inserted into the first exon of the Tlx locus in a 148-kb BAC genomic clone, which covers the entire Tlx gene and the flanking genomic sequences (Fig. 1A). Such a recombination strategy presumably preserved all of the regulatory elements required for Tlx expression. The resulting mouse strain was termed pTlx-CreERT2. After breeding to the reporter Rosa-GFP mouse line, tamoxifen (TM)-induced expression of GFP was identified in the SVZ and the SGZ in the adult mouse brains (Fig. 1B). Some of these GFP+ cells exhibit radial glia-like morphology and coexpress the marker GFAP, two hallmarks of adult NSCs (Fig. 1C, arrows). These cells resemble type-1 or type B stem cells within the SGZ or SVZ, respectively. In addition, GFP expression can also be identified in GFAP-negative cells, with a tangential orientation in the SGZ. These cells morphologically resemble those of type-2 transient amplifying cells (Fig. 1C, asterisks).
Figure 1.
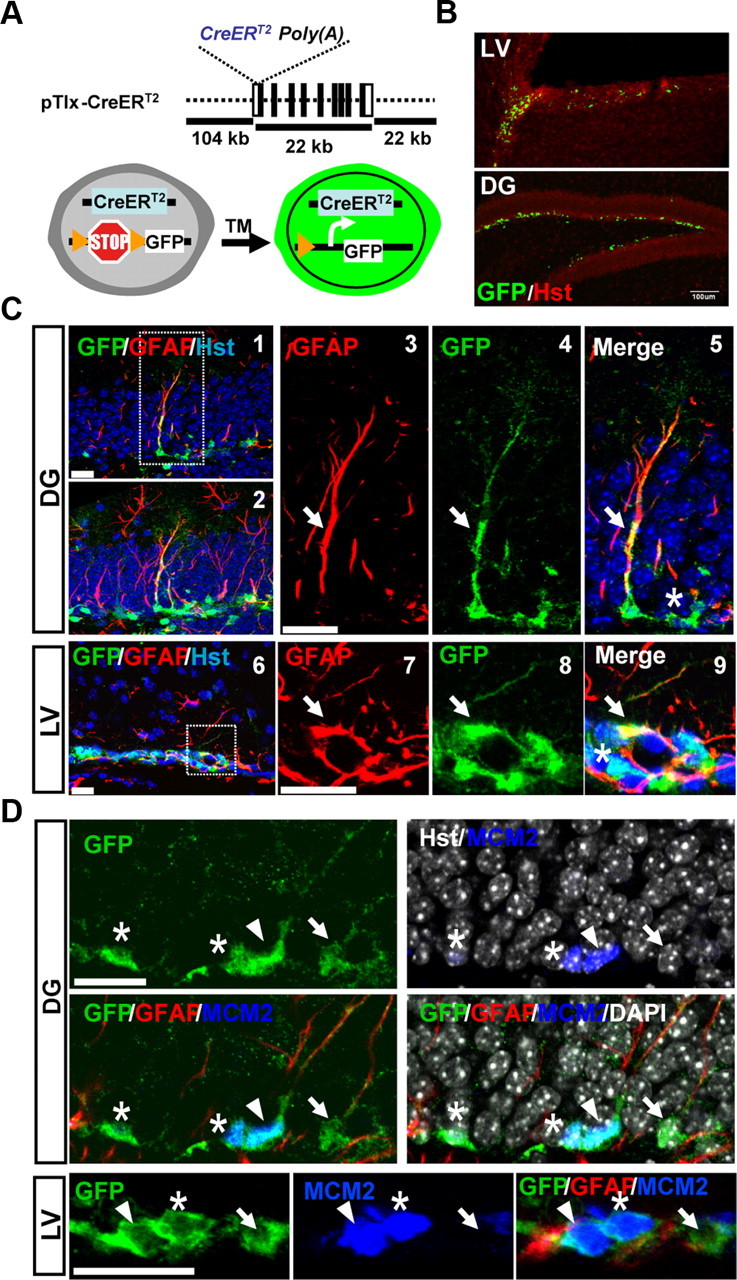
Tracing TLX lineage in inactive and activated adult NSCs. A, The CreERT2 gene was knocked into the first exon of the Tlx gene in a BAC clone through homologous recombination. Transgenic mice were produced through pronuclear injection of fertilized eggs. B, Tamoxifen-induced expression of GFP in adult neurogenic niches. C, Induced expression of GFP in type-1 (C1–C5) or type B (C6–C9) stem cells (marked by colocalization with GFAP expression and long processes, arrows) in the DG or the LV, respectively. Asterisks indicate tangentially oriented type-2 cells. C2 is a projection view of C1. C2–C5 and C7–C9 are enlarged views of boxed regions in C1 and C6, respectively. D, Induced GFP expression in inactive (arrow, GFAP+MCM2−) and activated (arrowhead, GFAP+MCM2+) stem cells. Transient amplifying cells (asterisk, GFP+GFAP−MCM2+) were also observed. Scale bars: B, 100 μm; C, D, 20 μm.
NSCs exist in either an inactive or activated state. We used the marker MCM2 to distinguish these two states. MCM2, together with MCM4, 6 and 7, forms the pre-replication complex, which is essential for the initiation of eukaryotic genome replication. Its expression is present throughout all phases of the proliferative cell cycle but downregulated when cells exit from cycle into quiescent, fully differentiated or replicative senescent states (Stoeber et al., 2001; Wharton et al., 2001; Blow and Hodgson, 2002; Maslov et al., 2004; Williams and Stoeber, 2007). Downregulation of MCM2 in intestinal stem cells marks their quiescent state (Kingsbury et al., 2005). Careful analysis through confocal microscopy revealed that MCM2 is expressed in ∼20% (20 ± 2.5%) of GFP+GFAP+ cells that have a radial morphology, showing that Tlx-expressing cells generate both dormant and activated stem cells (Fig. 1D, indicated by arrows and arrowheads, respectively). Furthermore, MCM2 was also detected in all of the GFP+GFAP− cells with short processes and tangential orientation in the SGZ. These cells resemble type-2 transient amplifying cells in this neurogenic niche (Fig. 1D, denoted by asterisks). Interestingly, some of the MCM2+GFP+GFAP− type-2 cells can be detected in close contact with MCM2+GFP+GFAP+ type-1 cells (Fig. 1D, denoted by asterisk), suggesting that TLX-expressing activated type-1 cells give rise to rapidly dividing type-2 cells. Together, these results suggest that TLX lies at the top level of a hierarchy that is required for postnatal neurogenesis.
Persistence of cells expressing markers for NSCs in postnatal brains after Tlx deletion
TLX is essential for NSC proliferation and neurogenesis in the postnatal brain (Shi et al., 2004; Liu et al., 2008; Zhang et al., 2008). The fate of stem cells lacking Tlx is not clear, although there is an indication that they undergo spontaneous differentiation into mature astrocytes and thus deplete NSCs (Shi et al., 2004). Taking advantage of the LacZ marker that was knocked into the Tlx locus, we examined the fate of Tlx-expressing cells. Surprisingly, β-galactosidase staining for LacZ gene expression can be robustly detected in the neurogenic niche even in the adult Tlx-null brain, suggesting that deletion of Tlx does not cause cell death or elimination of those Tlx-expressing cells (Fig. 2A). Importantly, Nestin+ or Sox2+GFAP+ cells can still be detected in the neurogenic niche in the adult Tlx-null brains (Fig. 2B).
Figure 2.
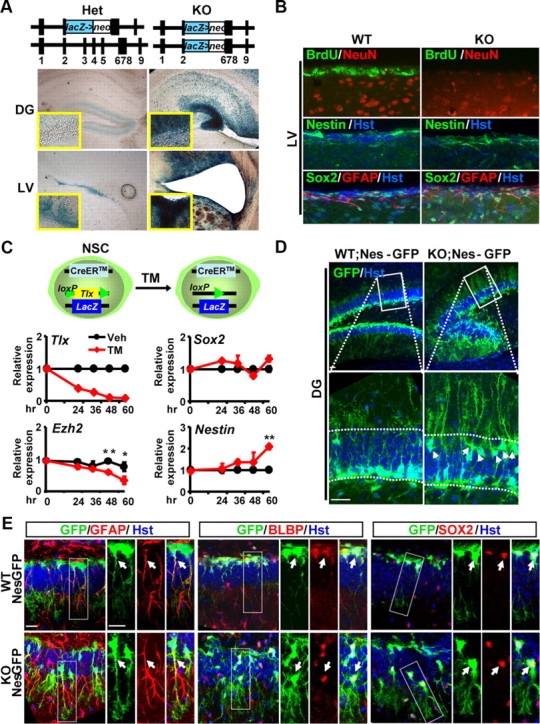
Persistence of postnatal NSCs in Tlx-null brains. A, Existence of Tlx-expressing cells, indicated by blue staining for β-galactosidase in the neurogenic niches of Tlx-null mice. Deletion of Tlx leads to a hypomorphic DG and enlarged ventricles. B, Although lacking proliferating cells (indicated by BrdU staining), Tlx-null mice continue to express stem cell markers (Nestin, Sox2, and GFAP) in the neurogenic niche. Images were taken from the LV. WT, Wild type; KO, knockout. C, Inducible deletion of Tlx does not result in downregulation of stem cell markers (Sox2 or Nestin) in culture. Tlx+ stem cells were isolated based on LacZ expression. The Tlx gene was then deleted through Cre/loxP-mediated recombination after TM treatment. Vehicle (Veh, ethanol) was used as a control. Gene expression was determined by qRT-PCR at the indicated time points (n = 3; *p = 0.014; **p = 0.0001). D, Persistence of Nes-GFP-expressing cells in Tlx-null mice. Expression of Nes-GFP in the dentate gyrus of 30-d-old mice was detected by confocal analysis. Enlarged views of the boxed regions are also shown. Arrows denote mispositioned GFP+ cells. Hoechst 33342 staining, designated by Hst, was used to reveal the nuclei. Scale bar, 50 μm. E, NSCs in adult Tlx-null brains. Enlarged views are taken from the boxed regions. NSCs have long, radial processes and coexpress GFAP, BLBP, and Sox2. Arrows indicate colocalization. Please also note the displacement of Tlx-null NSCs from the subgranule zone of the dentate gyrus. Scale bars, 20 μm.
To examine the early fate of stem cells lacking Tlx, we isolated TLX-positive cells based on LacZ expression and cultured them in monolayer. These cells also contained a floxed allele of Tlx and a CreER transgene (Fig. 2C). TM treatment can induce robust deletion of the conditional allele of Tlx in a time-dependent manner. However, quantitative RT-PCR (qRT-PCR) analysis showed that the markers for stem cells, such as Sox2 and Nestin, are still expressed. In fact, the expression of Nestin is rather enhanced after inducible deletion of Tlx in these cultured cells. In contrast, deletion of Tlx results in a significant downregulation of Ezh2, a member of the polycomb group that regulates the methylation status of core histones and embryonic stem cell development.
To further investigate the identity of those Tlx-null stem cells, we crossed the Tlx-null mice into the pNestin-GFP (Nes-GFP) transgenic background. Expression of gfp is under a neural-specific Nestin enhancer, which marks NSCs and transient amplifying cells in the neurogenic niche (Yamaguchi et al., 2000). In agreement with our immunostaining for endogenous Nestin expression, Nes-GFP+ cells with radial morphology can be observed in both of the neurogenic niches (the SVZ and the SGZ) in adult Tlx-null brains (Fig. 2D and data not shown). Interestingly, many GFP+ cells in Tlx-null brains are mislocalized in the outer layer of the dentate blades (see below, Label-retaining Nes-GFP+ cells are mispositioned in the DG of Tlx-null brains), although they still maintain their normal outward orientation. Through confocal microscopy, we examined in detail the identity of these Nes-GFP+ cells and found that they coexpress GFAP, BLBP, and Sox2 and exhibit radial glia morphology in the DG (Fig. 2E). These are characteristics of postnatal NSCs (Ihrie and Alvarez-Buylla, 2008; Zhao et al., 2008; Kriegstein and Alvarez-Buylla, 2009). Furthermore, these GFP+ cells lack marker staining for astrocytes [S100β and glutamine synthetase (GS); Fig. 3A,B], oligodendrocytes (Olig2, GSTπ, and RIP; Fig. 3C,D), or immature (DCX) or mature (NeuN) neurons (Fig. 3E,F). In contrast to a previous report (Shi et al., 2004), these data clearly indicate that deletion of Tlx during embryogenesis does not lead to a depletion of cells that have characteristics of NSCs or result in spontaneous differentiation of NSCs into mature astrocytes at the time points examined.
Figure 3.
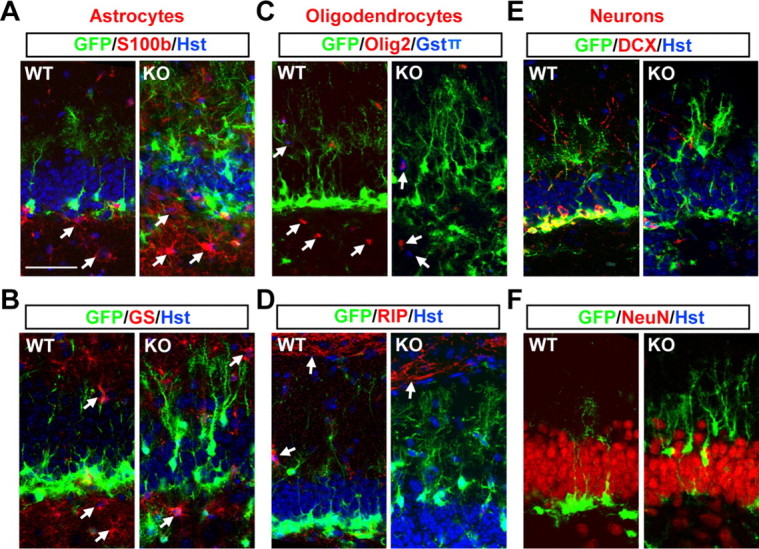
Deletion of Tlx does not lead to spontaneous differentiation of postnatal Nes-GFP+ stem cells. A–F, The identity of Nes-GFP+ cells in 30-d-old mice was analyzed by colabeling with markers for astrocytes (A, B), oligodentrocytes (C, D), and neurons (E, F). Some of the glial cells are indicated by arrows. WT, Wild type; KO, knockout. Scale bar (in A) A–F, 50 μm.
Deletion of Tlx leads to nonproliferative NSCs
The above data demonstrates that cells with morphology and marker expression resembling that of NSCs persist in the adult Tlx-null brains, raising the question about the status of these cells. By 4 weeks of age, Nes-GFP+ cells in Tlx-null brains are no longer able to generate DCX+ immature neurons (Fig. 3E). In addition, they do not proliferate. This is indicated by a lack of BrdU incorporation (Fig. 2B) or staining for Ki67, an endogenous marker for cell proliferation. Importantly, these cells also lack MCM2 expression, suggesting that they are in a nonlicensed state for replication (see below, last paragraph in this section).
Next, we examined the time course during which TLX is required for stem cell activation. Ki67, PCNA (proliferating cell nuclear antigen), and MCM2 are endogenous markers that were used to label the status of Nes-GFP+ NSCs. Expression of Ki67 is present throughout the cell cycle, but not in the G0 or early G1 phase, and thus is tightly associated with active cell proliferation (Gerdes et al., 1984; Scholzen and Gerdes, 2000). At 7 d of age [postnatal day (P)7], ∼20% of Nes-GFP+ cells in the DG were labeled by Ki67-staining in both the wild-type and the mutant brains (Fig. 4A). However, 7 d later the number of Ki67+GFP+ cells dropped >8-fold to 2.4% in the mutant brains, while Ki67+GFP+ cells from their wild-type littermates continued to proliferate at a rate of 14.2%. By 3 weeks of age (P21), only 0.7% of GFP+ cells in Tlx-null mice expressed Ki67, which was in sharp contrast to 10.8% in the control brains (Fig. 4B). It is known that the proliferation rate of stem cells drops with age, which was manifested in the control brains where we observed a 40% decrease in proliferating stem cells between 7 and 21 d of age. However, this rate of age-dependent decline was dramatically increased to 96% in Tlx-null NSCs.
Figure 4.
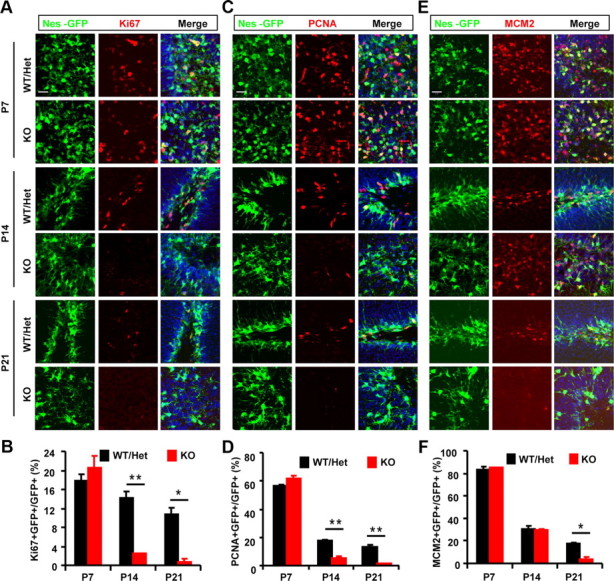
Age-dependent inactivation of postnatal NSCs due to loss of TLX function. A–D, Endogenous marker Ki67 (A, B) or PCNA (C, D) was used to label actively proliferating NSCs in the DG during early postnatal stages (postnatal days 7, 14, and 21). Stem cells were identified by expression of Nes-GFP. E–F, Activated stem cells were examined by expression of MCM2. Please note that only a fraction of MCM2+GFP+ cells were undergoing active cell cycling (Ki67+GFP+ or PCNA+GFP+) (n = 12 sections from 3 mice for each genotype; *p < 0.005; **p < 0.002). Scale bars: A, C, E, 20 μm. WT, Wild type; KO, knockout; Het, heterozygote.
Similar to Ki67, PCNA expression identifies cells in late G1, S, and G2-M phase; thus, is associated with active cell proliferation (Giordano et al., 1991). The percentage of PCNA-labeled cells was much higher than those labeled by Ki67 (compare Fig. 4D to Fig. 4B). This may be due to differential sensitivity of the antibodies used and/or an additional role of PCNA in DNA repair (Stoimenov and Helleday, 2009). This notwithstanding, deletion of Tlx resulted in a 72 and 90% reduction of PCNA+ cells among the total Nes-GFP+ cells at P14 and P21, respectively (Fig. 4D), which is consistent with the results obtained with Ki67-staining (Fig. 4B).
Formation of the preinitiation complex for DNA replication and its chromosomal loading are the prerequisite steps for cell to proliferate (Geng et al., 2003; Lea et al., 2003; Chuang et al., 2009). One of the key components of this complex is the MCM2 protein, which regulates the helicase activity of the Mcm4/6/7 hexamer (Ishimi et al., 2001). Unlike Ki67 or PCNA, expression of MCM2 marks all activated cells, including those leaving the G0 to the G1 phase of the cell cycle; thus, it also identifies noncycling cells with proliferative potential (Kayes et al., 2009; Torres-Rendon et al., 2009). At 7 d of age, 83% of Nes-GFP+ cells express MCM2 in both the Tlx-null mice and their littermate controls. This number dropped to 30% 7 days later, suggesting a sharp increase with age in the number of stem cells that are in an inactive stage. Interestingly, we did not observe any difference between Tlx mutants and their wild-type controls at this early postnatal stage (Fig. 4E,F). However, by 3 weeks MCM2+GFP+ cells decreased by 80% in Tlx-null compared to their littermate controls, resulting in a mere 3.5% of stem cells in proliferation-competent state. Because the decrease of Ki67+ or PCNA+ cells (at 2 weeks) precedes that of MCM2+ cells (at 3 weeks), these data indicate that a loss of TLX function first results in an age-dependent decrease of active proliferation, followed by an exit of cell cycle indicated by a nonlicensed state.
Label-retaining Nes-GFP+ cells are mispositioned in the DG of Tlx-null brains
The granule cell layer of the DG is established in an outside–in layering pattern: the early generated cells form the outer layer, whereas later born cells progressively populate the deeper inner layer. The SGZ within the inner layer constitutes a neurogenic niche for postnatal NSCs (Zhao et al., 2008). The cell bodies of these cells are localized in this microenvironment while their long, radial processes pass through the dentate granular cell layer and protrude outward. We found that many of the Nes-GFP+ cells in the adult Tlx-null brains are localized in the outer layer of the dentate blade yet still maintain the correct orientation of their radial processes (Fig. 2D,E). Such mispositioning of stem cells in the DG of Tlx-null mice can be detected as early as postnatal day 7 and becomes most obvious by the adult stage, when nearly 50% of these Nes-GFP+ cells are localized in the outer half of the granular cell layer in the Tlx-null mice (Fig. 5A,B).
Figure 5.
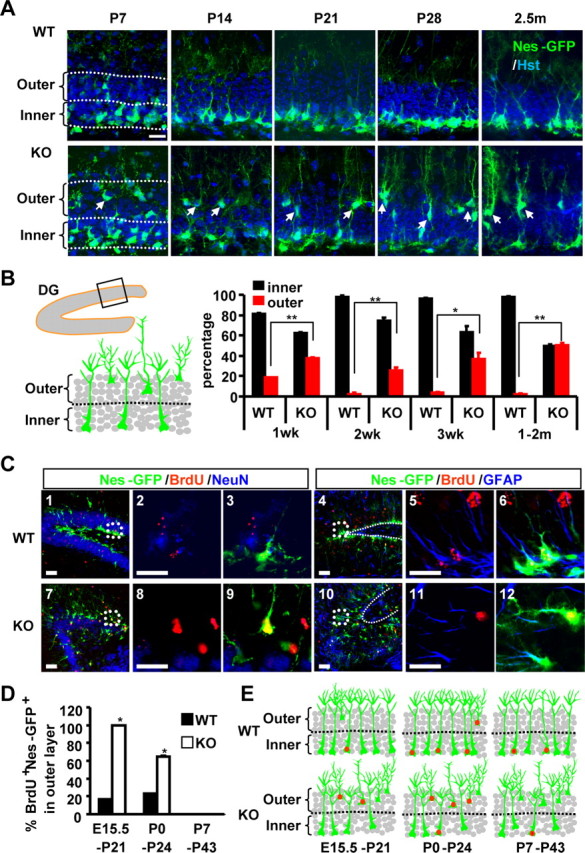
Mispositioning of label-retaining Nes-GFP cells in Tlx-null mice. A, NSCs are indicated by Nes-GFP expression in the DG during the early postnatal and adult stages. To quantify the position of NSCs, the DG was divided into the outer and the inner halves. Scale bar, 20 μm. B, Quantification of the positions of NSCs in the DG. Values are presented as percentage of GFP+ cells in either the inner or the outer half of dentate blade (n = 3; *p < 0.005; **p < 0.002). C, Confocal images of BrdU-retaining cells colabeled with a mature neuronal marker (NeuN) or a radial glia marker (GFAP). Cells were pulse labeled with BrdU at P0 and examined at P24. Scale bar, 20 μm. D, Quantification of BrdU-retaining Nes-GFP cells in the outer half of the granule cell layer. Cells were pulse-labeled with BrdU at E15.5, P0, or P7 and examined at postnatal days P21, P24, or P43, respectively (n = 3; *p < 0.002). E, Schematic representation of BrdU-retaining Nes-GFP+ cells (indicated by red dots) in the DG of either wild-type or Tlx-null mice. WT, Wild type; KO, knockout.
To trace the origin of these mispositioned Nes-GFP+ cells, we birth dated cells born in embryonic or paranatal stages with BrdU. After 3–5 weeks, the cellular identity of label-retaining cells and their positions in the DG were analyzed. Although the majority of BrdU-retaining cells are mature NeuN+ neurons, a small subset of these cells displays radial morphology and coexpresses Nes-GFP and GFAP, which are characteristics of NSCs (Fig. 5C). Close to 100 and 62% of these cells are mispositioned in the outer layer of the Tlx-null DG when cells are labeled at E15.5 and P0, respectively. In contrast, none of the label-retaining cells can be detected in the outer layer when cells are pulse labeled at P7 (Fig. 5D,E). Such mispositioned BrdU-retaining cells are rarely observed in wild-type mice. These data suggest that the cells that are mispositioned in the outer layer of the Tlx-null DG originate from the embryonic or paranatal stages. Although dysregulated migration of label-retaining cells from inner layer to outer layer cannot be fully excluded, such results are consistent with an outside–in layering pattern of the DG and further indicate that deletion of TLX leads to inactive NSCs that are temporally “frozen” in their original birthplace.
Activation of label-retaining NSCs by reintroducing TLX
Our results demonstrate that deletion of TLX leads to rapid age-dependent inactivation of NSCs. Since they do not yet spontaneously differentiate into either glial or neuronal cells, these inactive cells may become senescent or quiescent. We performed staining for senescence-associated β-galactosidase at pH 6.0. Unexpectedly, both wild-type and mutant brains showed staining in the DG and other regions of the hippocampus, although the staining in the mutant brains were stronger. We also conducted the same staining using in vitro cultured cells and observed similar robust staining in both wild-type or mutant cells in which Tlx is conditionally deleted. Thus, such staining cannot clearly differentiate whether the cells in the Tlx-null brains are senescent. Reasoning that quiescent but not senescent stem cells can be reactivated, we then performed rescue experiments by reintroducing Tlx. We initially tried an in vitro culture system. For this, Tlx-null cells from E18.5 cortices were isolated and labeled with DiI, followed by transduction with lentivirus expressing either GFP or GFP-T2A-TLX (Fig. 6A). Fourteen days later, flow cytometry showed that TLX-reintroduction caused a dramatic reduction of label-retaining cells when compared to that of GFP-transduced cells (GFP, 19.7 ± 1.7%; GFP-T2A-TLX, 1.9 ± 0.93%; n = 3, p < 0.0001). This data suggests that re-expression of TLX rescued and promoted proliferation of the cultured Tlx-null cells.
Figure 6.

Activation of label-retaining Tlx-null NSCs by reintroducing exogenous TLX. A, Ectopic TLX rescued proliferation of Tlx-null cells, indicated by a reduction of DiI label. Isolated Tlx-null NSCs were transduced by the indicated lentiviruses. Flow cytometry was performed after 14 d in culture. B, Inactive adult type-1 NSCs induced by Tlx deletion. Proliferating type-1 cells in the DG of 9 weeks old mice were labeled by BrdU-pulse (100 mg/kg, twice a day for 5 d) and staining for Nes-GFP and GFAP. A BrdU-labeled type-1 cell with radial morphology and an adjacent one with semitangential orientation in wild-type (WT) mice are indicated by an arrow and an asterisk, respectively. C, Enhancing proliferation of Tlx-null NSCs by exogenous TLX in vivo. Six-week-old Tlx-null;Nes-GFP mice were injected with control lentiviruses or viruses expressing TLX. Three weeks later, proliferating cells were labeled by BrdU pulse. A proliferating type-1 cell induced by re-expression of TLX is indicated by an arrow. D, Reactivation of label-retaining cells with exogenous TLX. Tlx-null mice were pulsed with CldU (50 mg/kg) at E18.5 and E19.5, chased for 6 weeks, stereotactically injected with lentiviruses expressing GFP or GFP-T2A-TLX, and then pulsed again with IdU (100 mg/kg, twice a day for 7 or 10 d). A few examples of TLX-induced GFP+CldU+IdU+ cells are shown. Scale bars (in B–D): images at lower magnification, 50 μm; images at higher magnification, 20 μm.
To examine the in vivo role of TLX in inactive NSCs, we first checked the basal level of proliferating type-1 NSCs in both 9-week-old Tlx-null mice and their littermate controls after BrdU-labeling (100 mg/kg, twice a day for 5 d). Whereas 9 of 215 Nes-GFP+;GFAP+ cells were also labeled by BrdU in control mice, only 1 cell was BrdU+ after counting 255 Nes-GFP+;GFAP+ cells in Tlx-null mice, indicating a very low level of basal proliferation of type-1 cells after deletion of Tlx (Fig. 6B). Through lentiviral delivery, Tlx was then reintroduced into Tlx-null;Nes-GFP brains at 6 weeks of age. Three weeks later, proliferating cells were labeled by BrdU incorporation (100 mg/kg, twice a day for 5 d). We observed a significant increase in the number of BrdU+;Nes-GFP+;GFAP+ cells in the Tlx-null brains after re-expressing Tlx (6.25 vs 0.25 cell/injection site when comparing Tlx-virus-transduced mutant mice to those transduced with control viruses). Such an increase is most likely due to reactivation of originally nondividing stem cells rather than a mere enhancement of the rate/frequency of those already proliferating cells, since the latter case would result in clusters of BrdU-labeled cells (Encinas et al., 2011), which were not detected in this study (Fig. 6C).
To further directly examine the role of TLX in label-retaining NSCs, we performed a pulse–chase–pulse experiment to investigate whether exogenous TLX could enable Tlx-null NSCs to proliferate in adult mice. For this, we first labeled proliferating NSCs with one pulse of CldU (50 mg/kg) each at E18.5 and E19.5. After 6 weeks of chase, the DG of Tlx-null mice was stereotactically injected with lentiviruses expressing GFP or GFP-T2A-TLX under the control of hGfap promoter (Lee et al., 2008). Proliferating cells were then pulsed again with IdU (100 mg/kg, twice a day for 7 or 10 d). By immunohistochemistry and confocal microscopy, Tlx-induced GFP+CldU+IdU+ cells could be observed as early as 7 d postinjection (dpi) (Fig. 6D). Analysis at 10 dpi showed more triple-labeled cells in mice injected with viruses expressing GFP-T2A-TLX versus control GFP (18.3 vs 3.75 cells/section). Together, these data demonstrate that re-expression of TLX enables label-retaining cells to proliferate.
TLX controls multiple pathways in postnatal NSCs
How does TLX regulate stem cell activation? Previously, we used cultured Tlx+ NSCs for global gene expression analysis after acute deletion of Tlx and found that many genes involved in cell proliferation showed significant changes (Zhang et al., 2008). Considering the potential side effects of long-term culture and the exogenous growth factors on stem cells, we wished to determine the function of TLX in endogenous NSCs. For this purpose, we took advantage of the recently developed RNA-seq technology. Lateral ventricles were dissected from 3-week-old mice (5–9 mice for each genotype). After enzymatic dissociation, Nes-GFP+ cells were isolated by FACS based on GFP expression, which resulted in >97% purity. Total RNA was purified from these sorted cells and converted into cDNA, which was then amplified using the Ovation RNA-seq system from NuGEN.
After sequencing of the prepared cDNA libraries, around 20 million sequence tags for each genotype were uniquely mapped onto the mouse genome. The expression for MCM2, which is 2 RPKM, was chosen as the cutoff. Such analysis identified 1721 genes with twofold changes. Importantly, we did not detect any significant changes of many housekeeping genes (such as Hprt, Rpl23, and Rpp30). Additionally, the expression of several stem cell markers, such as Nestin, Sox2, or Blbp/Fabp7, was not altered, suggesting that we indeed isolated and used the same population of cells for RNA-seq analysis (Fig. 7A). Together, these results demonstrated the robustness of our RNA-seq data.
Figure 7.
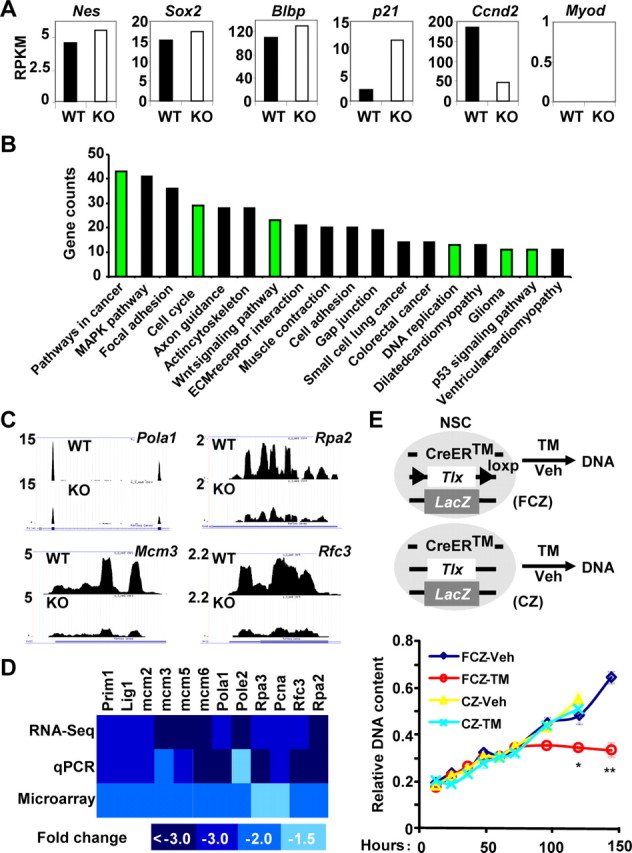
TLX-dependent signaling in postnatal NSCs. A, Whole-genome expression was examined by sequencing of the RNAs isolated from sorted Nes-GFP+ cells from 3-week-old wild-type or Tlx-null brains. Representative genes for four categories: no expression (Myod1); no change on expression (Nes, Sox2, and Blbp); upregulated (p21); and downregulated (Ccnd2). B, KEGG pathway analysis. Please note the significant changes on genes involved in cancer/glioma, cell cycle, Wnt signaling, DNA replication and p53 signaling. C, D, Expression changes on genes in DNA replication. C, Sequencing data. D, Heat map representation of fold changes on gene expression, which was determined by sequencing or qRT-PCR analysis of RNA from Tlx-null NSCs. Data from microarray analysis of adult NSCs after inducible deletion of Tlx (Zhang et al., 2008) were also included. E, Inducible deletion of Tlx leads to inactive stem cells. Tlx+/LacZ adult NSCs with a CreER transgene and/or a floxed allele of Tlx (FCZ and CZ, respectively) were treated with 4-hydroxytamoxifen, TM, or vehicle (Veh, ethanol). Genomic DNA was isolated and measured at the indicated time points after treatment (n = 3. *p = 0.02; **p = 0.001). WT, Wild type; KO, knockout; Veh, vehicle.
KEGG analysis of RNA-seq data showed that several signaling pathways were significantly altered upon deletion of Tlx in stem cells (Fig. 7B). Among these, changes of genes involved in DNA replication, cell cycle, and p53 signaling may contribute to the inactivation of NSCs upon deletion of Tlx. Using independently FACS-isolated Nes-GFP+ cells and qRT-PCR, we analyzed and confirmed the genes involved in DNA replication. Significantly, these genes were also identified in our previous microarray analysis using cultured cells (Zhang et al., 2008) (Fig. 7C,D). To further analyze the role of TLX in DNA replication, we isolated and cultured Tlx+ NSCs and found that the DNA content reached a plateau by 60 h after TM-induced deletion of Tlx, whereas the control cells continued their growth (Fig. 7E).
TLX genetically interacts with p53 signaling pathway in postnatal NSCs
Of the genes involved in cell cycle control and p53 signaling, p21/Cdkn1a is significantly upregulated in Tlx-null stem cells (Fig. 7A). This upregulation of p21 is confirmed by use of an inducible deletion system in cultured NSCs by qRT-PCR (Fig. 8A), indicating a direct link between p21 expression and TLX function. This is further supported by data showing that TLX directly binds to the promoter region of p21 (Sun et al., 2007). Because of its demonstrated role in maintaining stem cell quiescence (Cheng et al., 2000; Perucca et al., 2009), these data strongly raised the possibility that p21 may be a critical component in mediating TLX-deletion-induced NSC inactivation. We tested this possibility by ectopic expression of p21 in cultured NSCs and indeed observed a significant reduction of Ki67+ cells (Fig. 8B).
Figure 8.
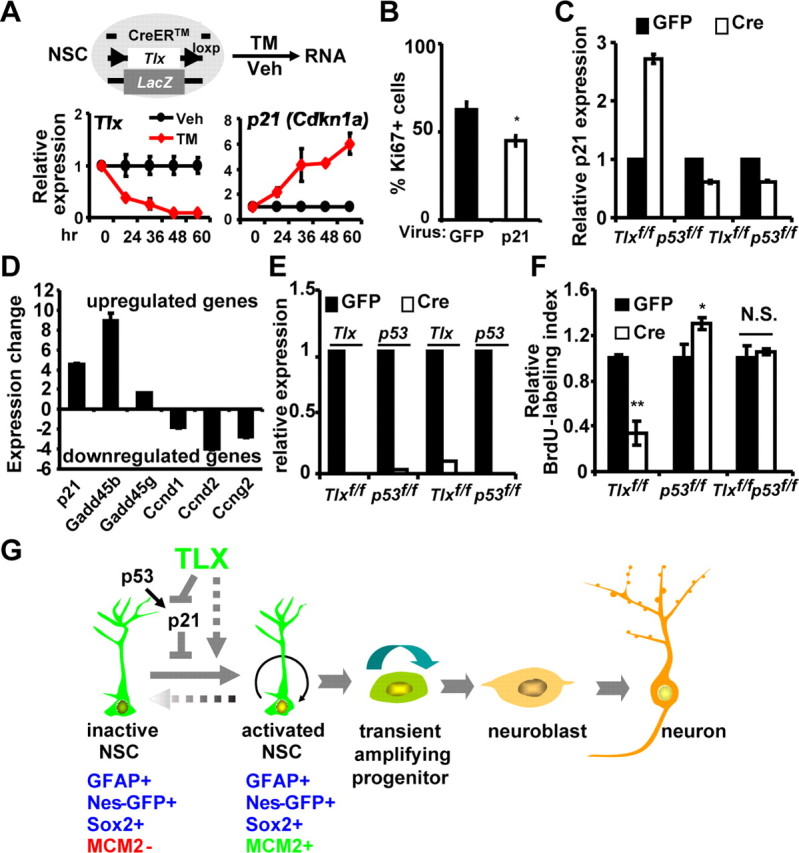
Role of p53 signaling in TLX-dependent regulation of NSC proliferation. A, Increased p21 expression upon inducible deletion of Tlx by 4-hydroxytamoxifen (TM) treatment of cultured adult Tlxflox/LacZ NSCs. The expression of Tlx and p21 were determined by qRT-PCR. Veh indicates ethanol as vehicle control. B, Transient ectopic expression of p21 in cultured NSCs inhibited their proliferation (n = 4; *p = 0.019). C, p53-dependent regulation of p21 expression by TLX. Cultured NSCs with the indicated genotypes were transduced with adenoviruses expressing either GFP or GFP-Cre. D, Confirmation of altered p53 signaling. Gene expression was determined by qRT-PCR using sorted Nes-GFP+ NSCs from 3-week-old wild-type or Tlx-null mice. E, Efficiency of Cre-mediated deletion of either Tlx or p53 in cultured NSCs determined by qRT-PCR. F, Concomitant deletion of p53 in cultured NSCs rescued their proliferation defects that were induced by Cre-mediated deletion of Tlx (n = 4; *p = 0.023, **p < 0.0001). G, A model showing the role of TLX in postnatal NSCs. Tlx-expressing cells generate both activated and inactive postnatal NSCs, which are identified by marker expression. TLX is required for inactive NSCs to proliferate by modulating p21 expression in a p53-dependent manner. Besides p53 signaling, TLX also modulates many other signaling pathways, which may contribute to the regulation of NSC activation.
It is well established that the expression of p21 is under the direct control of the p53 signaling pathway in most cellular contexts. We examined whether p53 is also involved in Tlx deletion-induced upregulation of p21 in postnatal NSCs. We isolated these cells from 2-month-old mice harboring floxed alleles of Tlx and/or p53. Consistent with our previous observation (Zhang et al., 2008), Cre-mediated deletion of Tlx results in upregulation of p21. Such upregulation is completely abolished upon acute removal of p53, suggesting that TLX controls p21 expression in a p53-dependent manner (Fig. 8C). Besides p21, our RNA-seq also identified altered expression of several other genes that are involved in the p53 signaling pathway. These changes can be further confirmed by qRT-PCR analysis of independently isolated RNA samples from sorted Nes-GFP+ cells (Fig. 8D). These results suggest that p53 may play a role in controlling the activation status of Tlx-null NSCs. To test this possibility, we acutely deleted Tlx with or without concomitant removal of p53 expression in cultured adult NSCs. As demonstrated previously (Zhang et al., 2008), Cre-mediated deletion of Tlx led to a significant reduction of BrdU+ cells. Interestingly, this proliferation defect of Tlx-null NSCs was completely rescued by concomitant deletion of p53 (Fig. 8E,F). It should also be noted that deletion of p53 alone resulted in a small but significant increase of proliferating cells, which is consistent with a demonstrated role of p53 in adult NSCs (Meletis et al., 2006). Together, these data show that TLX genetically interacts with the p53 signaling pathway to tightly regulate the activity of postnatal NSCs.
Discussion
Postnatal neurogenesis depends on the continued presence of infrequently dividing NSCs in the SGZ and the SVZ. However, the underlying driving force for NSCs to become activated remains largely unknown. Using inducible lineage tracing and genetic markers for adult NSCs, we show here for the first time that Tlx-expressing cells generate both activated and nonproliferative postnatal NSCs and that TLX is required for NSC activation and positioning in the neurogenic niche. Our whole-genome RNA-seq data reveals that TLX acts as a key coordinator for multiple signaling pathways in the regulation of NSC behavior. Specifically, TLX genetically interacts with the p53 pathway to control NSC activation (Fig. 8G). Together, these data uncover a key role of TLX in controlling the proliferative capacity of postnatal neural stem cells, but not their radial glia morphology and the expression of multiple NSC markers.
TLX function in postnatal NSCs
TLX was recently shown to be expressed in type B cells in the LV (Liu et al., 2010). In agreement with this finding, our inducible lineage tracing further demonstrated that Tlx-expressing cells give rise to both type-1 cells in the SGZ and type B cells in the SVZ. This implies a much broader role of TLX in the regulation of neurogenic stem cells in the adult brain. Consequently, deletion of Tlx leads to completely abolished neurogenesis in both of the neurogenic regions (Shi et al., 2004; Liu et al., 2008; Zhang et al., 2008). Type-1/B cells are slowly dividing stem cells that give rise to transiently but rapidly amplifying type-2/C cells (Kriegstein and Alvarez-Buylla, 2009). Interestingly, both active and inactive NSCs are derived from Tlx-expressing cells. We cannot unambiguously differentiate which of the two types of NSCs express Tlx due to the lack of a sensitive antibody that recognizes endogenous TLX in the adult neurogenic regions. Nevertheless, three pieces of data indicate that Tlx is expressed in inactive NSCs. First, the LacZ reporter, which was knocked into the endogenous Tlx locus, can still be robustly detected in both the SVZ and the SGZ in Tlx-null mice. Second, whole-genome sequencing of total RNAs clearly shows the expression of undeleted exons in sorted nonproliferative Nes-GFP+ cells from 3-week-old Tlx-null brains. Third, it is known that aging is accompanied by a significant decrease of proliferating stem cells; however, our qRT-PCR analysis cannot detect a significant change in Tlx expression between young (1 month old) and aged mice (26 months old) (data not shown). Expression of Tlx in nonproliferative NSCs raised the possibility that the activity of TLX itself in NSCs may be under regulation through either posttranslational modifications or ligand binding. The ligand for TLX (a member of the nuclear hormone receptor superfamily) remains unknown. Future studies are clearly needed to identify this ligand, which may provide a unique opportunity to control TLX-dependent postnatal neurogenesis.
The function of TLX during development is to prevent precocious cell cycle exit and premature differentiation (Roy et al., 2004; Zhang et al., 2006; Li et al., 2008). By contrast, the role of TLX in postnatal NSCs is not clear, although it has been proposed to inhibit spontaneous differentiation of NSCs into astrocytes (Shi et al., 2004). We were able to detect significantly more astrocytes in postnatal Tlx-null mice. However, these cells are largely localized to the gliogenic regions with typical stellate morphology and the expression of S100β and glutamine synthetase, which are characteristics of differentiated astrocytes. Since our RNA-seq showed that genes involved in bone morphogenetic protein (BMP) and leukemia inhibitory factor (LIF) signaling, such as Bmpr1b (2.1-fold increase), Bmper (2.08-fold decrease), Bmp4 (8.3-fold increase), and Lifr (2.25-fold increase), are significantly altered upon deletion of Tlx, we suspect that enhanced response of neural progenitor cells to BMP and/or LIF signaling may be the underlying mechanism for the observed increase of astrocytes in gliogenic regions. On the other hand, cells within the neurogenic niche of Tlx-null brains exhibit radial glia morphology, a characteristic of adult NSCs. Although they are GFAP+, they do not express markers for mature glia (Fig. 3); rather, they still express markers for stem cells such as Nestin, Sox2, and BLBP (Fig. 2E). Our whole-genome RNA-seq data further support this conclusion by showing unaltered expression of these stem cell markers (Fig. 7A). These data clearly demonstrate that loss of TLX function does not lead to spontaneous differentiation of NSCs directly into mature glia, but rather results in nonproliferative cells that genetically and morphologically resemble NSCs.
Age-dependent inactivation of postnatal NSCs
Neurogenesis continues into adulthood but is reduced considerably with the aging process (Seki and Arai, 1995; Kuhn et al., 1996; Tropepe et al., 1997). Such age-dependent reduction may be attributed to both decreased proliferation and/or division-coupled astrocytic differentiation (Hattiangady and Shetty, 2008; Dranovsky et al., 2011; Encinas et al., 2011). By using MCM2 as a marker for all activated stem cells and Ki67 and PCNA to label actively proliferating cells, our detailed analysis of postnatal NSCs revealed a previously unappreciated role of TLX in stem cell biology. Loss of TLX function results in an age-dependent, dramatic reduction of proliferating cells with a compensatory increase in the number of MCM− inactive NSCs. Remarkably, ectopic expression of TLX can reactivate these inactive NSCs in vivo. Such a result is consistent with a recent report showing that overexpression of Tlx is sufficient to enhance neurogenesis even in aged animals (Liu et al., 2010). Increased expression of the tumor suppressors INK4A/ARF has been linked to NSC aging (Molofsky et al., 2006; Levi and Morrison, 2008). Yet, we did not detect any expression change on these two genes through our extensive genome-wide analysis using either cultured adult NSCs or sorted live NSCs from wild-type or Tlx-null animals. This suggests that TLX controls stem cell aging in an INK4A/ARF-independent manner. Our unbiased global expression analysis revealed that many genes in p53 signaling (such as p21, Gadd45, Btg2, Ccnd1, and Ccnd2) are significantly altered. Interestingly, our data show that ectopic expression of p21 is sufficient to inhibit proliferation of cultured NSCs. Conversely, concomitant loss of p53 function rescues the Tlx-deletion-induced proliferation defect of cultured NSCs. Such a role of p21 and p53 in postnatal NSCs is consistent with their demonstrated function in controlling stem cell quiescence (Cheng et al., 2000; Kippin et al., 2005; Gil-Perotin et al., 2006; Meletis et al., 2006; Liu et al., 2009; Perucca et al., 2009). In addition to p53 signaling, however, it should be noted that TLX also controls the expression of a plethora of other genes that may also play important roles in the regulation of NSC activation (Fig. 8G). Future studies are required to tease out the in vivo function of these genes in NSCs and during adult neurogenesis.
Implications in brain tumorigenesis
Emerging evidence indicates that slowly dividing, self-renewable NSC-like cells are the cellular origin for brain tumors (Stiles and Rowitch, 2008). Therefore, our finding that TLX plays a key role in NSC activation may have therapeutic implications in brain tumorigenesis. Recent data demonstrating that increased activity of TLX may initiate brain tumor formation in flies or mice (Kurusu et al., 2009; Liu et al., 2010) support our findings. Overexpression of Tlx also has been associated with certain human brain tumors, including astrocytomas, ependymomas, and glioblastomas (Taylor et al., 2005; Phillips et al., 2006; Sim et al., 2006). Mechanistically, our systematic genome-wide analysis using microarray (Zhang et al, 2008) and RNA-seq (current study) revealed that TLX controls a plethora of genes involved in DNA replication, cell cycle, p53 signaling, and pathways in cancers. These data suggest that TLX lies at a nodal point to control NSC activation and replication by coordinating a complex genetic network. Because of its essential role in controlling postnatal neurogenesis, it will be interesting to examine whether TLX is also required for the development of certain type of brain tumors and to determine the relationship of adult neurogenesis and tumorigenesis. Our finding that the proliferation defect of Tlx-deleted NSCs can be rescued by concomitant removal of p53 suggests that targeting TLX alone may not be sufficient to control tumorigenesis. However, because of its restricted expression in the nervous system and the potential regulation by a ligand, TLX could be an excellent drug target in combination with other therapeutic strategies to treat certain human brain tumors.
Footnotes
This work was supported by the Whitehall Foundation (2009-12-05), the Welch Foundation (I-1724), the American Heart Association (09SDG2260602), the National Institutes of Health (1DP2OD006484 and R01NS070981), and a Startup Fund from University of Texas (UT) Southwestern (to C.-L.Z.). C.-L.Z. is a W. W. Caruth, Jr. Scholar in Biomedical Research. We thank Ronald Evans for sharing Tlx mutant mice, Amelia Eisch for providing Nes-GFP mice, Michael Brenner for providing hGfap enhancer, Pierre Chambon for providing the CreERT2 plasmid, Richard Lu for sharing the Olig2 antibody, Ray MacDonald and Galvin Swift for assistance with RNA-seq, the Transgenic Core Facility at UT Southwestern for generating BAC transgenic mice, and the Flow Cytometry Core for FACS. We also thank Eric Olson, Jane Johnson, Steven Kliewer, and Jenny Hsieh for comments, Ronald Evans and Fred Gage for intellectual support, and Pamela Jackson for administrative assistance.
The authors declare no competing financial interests.
References
- Blow JJ, Hodgson B. Replication licensing–defining the proliferative state? Trends Cell Biol. 2002;12:72–78. doi: 10.1016/s0962-8924(01)02203-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, Scadden DT. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287:1804–1808. doi: 10.1126/science.287.5459.1804. [DOI] [PubMed] [Google Scholar]
- Chuang LC, Teixeira LK, Wohlschlegel JA, Henze M, Yates JR, Méndez J, Reed SI. Phosphorylation of Mcm2 by Cdc7 promotes pre-replication complex assembly during cell-cycle re-entry. Mol Cell. 2009;35:206–216. doi: 10.1016/j.molcel.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranovsky A, Picchini AM, Moadel T, Sisti AC, Yamada A, Kimura S, Leonardo ED, Hen R. Experience dictates stem cell fate in the adult hippocampus. Neuron. 2011;70:908–923. doi: 10.1016/j.neuron.2011.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encinas JM, Michurina TV, Peunova N, Park JH, Tordo J, Peterson DA, Fishell G, Koulakov A, Enikolopov G. Division-coupled astrocytic differentiation and age-related depletion of neural stem cells in the adult hippocampus. Cell Stem Cell. 2011;8:566–579. doi: 10.1016/j.stem.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil R, Wagner J, Metzger D, Chambon P. Regulation of Cre recombinase activity by mutated estrogen receptor ligand-binding domains. Biochem Biophys Res Commun. 1997;237:752–757. doi: 10.1006/bbrc.1997.7124. [DOI] [PubMed] [Google Scholar]
- Geng Y, Yu Q, Sicinska E, Das M, Schneider JE, Bhattacharya S, Rideout WM, Bronson RT, Gardner H, Sicinski P. Cyclin E ablation in the mouse. Cell. 2003;114:431–443. doi: 10.1016/s0092-8674(03)00645-7. [DOI] [PubMed] [Google Scholar]
- Gerdes J, Lemke H, Baisch H, Wacker HH, Schwab U, Stein H. Cell cycle analysis of a cell proliferation-associated human nuclear antigen defined by the monoclonal antibody Ki-67. J Immunol. 1984;133:1710–1715. [PubMed] [Google Scholar]
- Gil-Perotin S, Marin-Husstege M, Li J, Soriano-Navarro M, Zindy F, Roussel MF, Garcia-Verdugo JM, Casaccia-Bonnefil P. Loss of p53 induces changes in the behavior of subventricular zone cells: implication for the genesis of glial tumors. J Neurosci. 2006;26:1107–1116. doi: 10.1523/JNEUROSCI.3970-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano M, Danova M, Pellicciari C, Wilson GD, Mazzini G, Conti AM, Franchini G, Riccardi A, Romanini MG. Proliferating cell nuclear antigen (PCNA)/cyclin expression during the cell cycle in normal and leukemic cells. Leuk Res. 1991;15:965–974. doi: 10.1016/0145-2126(91)90101-x. [DOI] [PubMed] [Google Scholar]
- Hattiangady B, Shetty AK. Aging does not alter the number or phenotype of putative stem/progenitor cells in the neurogenic region of the hippocampus. Neurobiol Aging. 2008;29:129–147. doi: 10.1016/j.neurobiolaging.2006.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihrie RA, Alvarez-Buylla A. Cells in the astroglial lineage are neural stem cells. Cell Tissue Res. 2008;331:179–191. doi: 10.1007/s00441-007-0461-z. [DOI] [PubMed] [Google Scholar]
- Imayoshi I, Sakamoto M, Ohtsuka T, Takao K, Miyakawa T, Yamaguchi M, Mori K, Ikeda T, Itohara S, Kageyama R. Roles of continuous neurogenesis in the structural and functional integrity of the adult forebrain. Nat Neurosci. 2008;11:1153–1161. doi: 10.1038/nn.2185. [DOI] [PubMed] [Google Scholar]
- Ishimi Y, Komamura-Kohno Y, Arai K, Masai H. Biochemical activities associated with mouse Mcm2 protein. J Biol Chem. 2001;276:42744–42752. doi: 10.1074/jbc.M106861200. [DOI] [PubMed] [Google Scholar]
- Kayes OJ, Loddo M, Patel N, Patel P, Minhas S, Ambler G, Freeman A, Wollenschlaeger A, Ralph DJ, Stoeber K, Williams GH. DNA replication licensing factors and aneuploidy are linked to tumor cell cycle state and clinical outcome in penile carcinoma. Clin Cancer Res. 2009;15:7335–7344. doi: 10.1158/1078-0432.CCR-09-0882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingsbury SR, Loddo M, Fanshawe T, Obermann EC, Prevost AT, Stoeber K, Williams GH. Repression of DNA replication licensing in quiescence is independent of geminin and may define the cell cycle state of progenitor cells. Exp Cell Res. 2005;309:56–67. doi: 10.1016/j.yexcr.2005.05.027. [DOI] [PubMed] [Google Scholar]
- Kippin TE, Martens DJ, van der Kooy D. p21 loss compromises the relative quiescence of forebrain stem cell proliferation leading to exhaustion of their proliferation capacity. Genes Dev. 2005;19:756–767. doi: 10.1101/gad.1272305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegstein A, Alvarez-Buylla A. The glial nature of embryonic and adult neural stem cells. Annu Rev Neurosci. 2009;32:149–184. doi: 10.1146/annurev.neuro.051508.135600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn HG, Dickinson-Anson H, Gage FH. Neurogenesis in the dentate gyrus of the adult rat: age-related decrease of neuronal progenitor proliferation. J Neurosci. 1996;16:2027–2033. doi: 10.1523/JNEUROSCI.16-06-02027.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurusu M, Maruyama Y, Adachi Y, Okabe M, Suzuki E, Furukubo-Tokunaga K. A conserved nuclear receptor, Tailless, is required for efficient proliferation and prolonged maintenance of mushroom body progenitors in the Drosophila brain. Dev Biol. 2009;326:224–236. doi: 10.1016/j.ydbio.2008.11.013. [DOI] [PubMed] [Google Scholar]
- Lea NC, Orr SJ, Stoeber K, Williams GH, Lam EW, Ibrahim MA, Mufti GJ, Thomas NS. Commitment point during G0 →G1 that controls entry into the cell cycle. Mol Cell Biol. 2003;23:2351–2361. doi: 10.1128/MCB.23.7.2351-2361.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Messing A, Su M, Brenner M. GFAP promoter elements required for region-specific and astrocyte-specific expression. Glia. 2008;56:481–493. doi: 10.1002/glia.20622. [DOI] [PubMed] [Google Scholar]
- Levi BP, Morrison SJ. Stem cells use distinct self-renewal programs at different ages. Cold Spring Harb Symp Quant Biol. 2008;73:539–553. doi: 10.1101/sqb.2008.73.049. [DOI] [PubMed] [Google Scholar]
- Li W, Sun G, Yang S, Qu Q, Nakashima K, Shi Y. Nuclear receptor TLX regulates cell cycle progression in neural stem cells of the developing brain. Mol Endocrinol. 2008;22:56–64. doi: 10.1210/me.2007-0290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lie DC, Song H, Colamarino SA, Ming GL, Gage FH. Neurogenesis in the adult brain: new strategies for central nervous system diseases. Annu Rev Pharmacol Toxicol. 2004;44:399–421. doi: 10.1146/annurev.pharmtox.44.101802.121631. [DOI] [PubMed] [Google Scholar]
- Liu HK, Belz T, Bock D, Takacs A, Wu H, Lichter P, Chai M, Schütz G. The nuclear receptor tailless is required for neurogenesis in the adult subventricular zone. Genes Dev. 2008;22:2473–2478. doi: 10.1101/gad.479308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu HK, Wang Y, Belz T, Bock D, Takacs A, Radlwimmer B, Barbus S, Reifenberger G, Lichter P, Schütz G. The nuclear receptor tailless induces long-term neural stem cell expansion and brain tumor initiation. Genes Dev. 2010;24:683–695. doi: 10.1101/gad.560310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Elf SE, Miyata Y, Sashida G, Liu Y, Huang G, Di Giandomenico S, Lee JM, Deblasio A, Menendez S, Antipin J, Reva B, Koff A, Nimer SD. p53 regulates hematopoietic stem cell quiescence. Cell Stem Cell. 2009;4:37–48. doi: 10.1016/j.stem.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois C, Alvarez-Buylla A. Proliferating subventricular zone cells in the adult mammalian forebrain can differentiate into neurons and glia. Proc Natl Acad Sci U S A. 1993;90:2074–2077. doi: 10.1073/pnas.90.5.2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino S, Vooijs M, van Der Gulden H, Jonkers J, Berns A. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev. 2000;14:994–1004. [PMC free article] [PubMed] [Google Scholar]
- Maslov AY, Barone TA, Plunkett RJ, Pruitt SC. Neural stem cell detection, characterization, and age-related changes in the subventricular zone of mice. J Neurosci. 2004;24:1726–1733. doi: 10.1523/JNEUROSCI.4608-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masui T, Swift GH, Deering T, Shen C, Coats WS, Long Q, Elsässer HP, Magnuson MA, MacDonald RJ. Replacement of Rbpj with Rbpjl in the PTF1 complex controls the final maturation of pancreatic acinar cells. Gastroenterology. 2010;139:270–280. doi: 10.1053/j.gastro.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meletis K, Wirta V, Hede SM, Nistér M, Lundeberg J, Frisén J. p53 suppresses the self-renewal of adult neural stem cells. Development. 2006;133:363–369. doi: 10.1242/dev.02208. [DOI] [PubMed] [Google Scholar]
- Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ. Increasing p16INK4a expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448–452. doi: 10.1038/nature05091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan AP, Grau E, Bock D, Schütz G. The mouse homolog of the orphan nuclear receptor tailless is expressed in the developing forebrain. Development. 1995;121:839–853. doi: 10.1242/dev.121.3.839. [DOI] [PubMed] [Google Scholar]
- Perucca P, Cazzalini O, Madine M, Savio M, Laskey RA, Vannini V, Prosperi E, Stivala LA. Loss of p21 CDKN1A impairs entry to quiescence and activates a DNA damage response in normal fibroblasts induced to quiescence. Cell Cycle. 2009;8:105–114. doi: 10.4161/cc.8.1.7507. [DOI] [PubMed] [Google Scholar]
- Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, Misra A, Nigro JM, Colman H, Soroceanu L, Williams PM, Modrusan Z, Feuerstein BG, Aldape K. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. 2006;9:157–173. doi: 10.1016/j.ccr.2006.02.019. [DOI] [PubMed] [Google Scholar]
- Roy K, Kuznicki K, Wu Q, Sun Z, Bock D, Schutz G, Vranich N, Monaghan AP. The Tlx gene regulates the timing of neurogenesis in the cortex. J Neurosci. 2004;24:8333–8345. doi: 10.1523/JNEUROSCI.1148-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholzen T, Gerdes J. The Ki-67 protein: from the known and the unknown. J Cell Physiol. 2000;182:311–322. doi: 10.1002/(SICI)1097-4652(200003)182:3<311::AID-JCP1>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Seki T, Arai Y. Age-related production of new granule cells in the adult dentate gyrus. Neuroreport. 1995;6:2479–2482. doi: 10.1097/00001756-199512150-00010. [DOI] [PubMed] [Google Scholar]
- Shi Y, Chichung Lie D, Taupin P, Nakashima K, Ray J, Yu RT, Gage FH, Evans RM. Expression and function of orphan nuclear receptor TLX in adult neural stem cells. Nature. 2004;427:78–83. doi: 10.1038/nature02211. [DOI] [PubMed] [Google Scholar]
- Sim FJ, Keyoung HM, Goldman JE, Kim DK, Jung HW, Roy NS, Goldman SA. Neurocytoma is a tumor of adult neuronal progenitor cells. J Neurosci. 2006;26:12544–12555. doi: 10.1523/JNEUROSCI.0829-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiles CD, Rowitch DH. Glioma stem cells: a midterm exam. Neuron. 2008;58:832–846. doi: 10.1016/j.neuron.2008.05.031. [DOI] [PubMed] [Google Scholar]
- Stoeber K, Tlsty TD, Happerfield L, Thomas GA, Romanov S, Bobrow L, Williams ED, Williams GH. DNA replication licensing and human cell proliferation. J Cell Sci. 2001;114:2027–2041. doi: 10.1242/jcs.114.11.2027. [DOI] [PubMed] [Google Scholar]
- Stoimenov I, Helleday T. PCNA on the crossroad of cancer. Biochem Soc Trans. 2009;37:605–613. doi: 10.1042/BST0370605. [DOI] [PubMed] [Google Scholar]
- Sun G, Yu RT, Evans RM, Shi Y. Orphan nuclear receptor TLX recruits histone deacetylases to repress transcription and regulate neural stem cell proliferation. Proc Natl Acad Sci U S A. 2007;104:15282–15287. doi: 10.1073/pnas.0704089104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor MD, Poppleton H, Fuller C, Su X, Liu Y, Jensen P, Magdaleno S, Dalton J, Calabrese C, Board J, Macdonald T, Rutka J, Guha A, Gajjar A, Curran T, Gilbertson RJ. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell. 2005;8:323–335. doi: 10.1016/j.ccr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Torres-Rendon A, Roy S, Craig GT, Speight PM. Expression of Mcm2, geminin and Ki67 in normal oral mucosa, oral epithelial dysplasias and their corresponding squamous-cell carcinomas. Br J Cancer. 2009;100:1128–1134. doi: 10.1038/sj.bjc.6604967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropepe V, Craig CG, Morshead CM, van der Kooy D. Transforming growth factor-alpha null and senescent mice show decreased neural progenitor cell proliferation in the forebrain subependyma. J Neurosci. 1997;17:7850–7859. doi: 10.1523/JNEUROSCI.17-20-07850.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuttle AH, Rankin MM, Teta M, Sartori DJ, Stein GM, Kim GJ, Virgilio C, Granger A, Zhou D, Long SH, Schiffman AB, Kushner JA. Immunofluorescent detection of two thymidine analogues (CldU and IdU) in primary tissue. J Vis Exp. 2011 doi: 10.3791/2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Rajan H, Pitman JL, McKeown M, Tsai CC. Histone deacetylase-associating Atrophin proteins are nuclear receptor corepressors. Genes Dev. 2006;20:525–530. doi: 10.1101/gad.1393506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wharton SB, Chan KK, Anderson JR, Stoeber K, Williams GH. Replicative Mcm2 protein as a novel proliferation marker in oligodendrogliomas and its relationship to Ki67 labelling index, histological grade and prognosis. Neuropathol Appl Neurobiol. 2001;27:305–313. doi: 10.1046/j.0305-1846.2001.00333.x. [DOI] [PubMed] [Google Scholar]
- Williams GH, Stoeber K. Cell cycle markers in clinical oncology. Curr Opin Cell Biol. 2007;19:672–679. doi: 10.1016/j.ceb.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Yamaguchi M, Saito H, Suzuki M, Mori K. Visualization of neurogenesis in the central nervous system using nestin promoter-GFP transgenic mice. Neuroreport. 2000;11:1991–1996. doi: 10.1097/00001756-200006260-00037. [DOI] [PubMed] [Google Scholar]
- Yokoyama A, Takezawa S, Schüle R, Kitagawa H, Kato S. Transrepressive function of TLX requires the histone demethylase LSD1. Mol Cell Biol. 2008;28:3995–4003. doi: 10.1128/MCB.02030-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu RT, McKeown M, Evans RM, Umesono K. Relationship between Drosophila gap gene tailless and a vertebrate nuclear receptor Tlx. Nature. 1994;370:375–379. doi: 10.1038/370375a0. [DOI] [PubMed] [Google Scholar]
- Yu RT, Chiang MY, Tanabe T, Kobayashi M, Yasuda K, Evans RM, Umesono K. The orphan nuclear receptor Tlx regulates Pax2 and is essential for vision. Proc Natl Acad Sci U S A. 2000;97:2621–2625. doi: 10.1073/pnas.050566897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CL, Zou Y, Yu RT, Gage FH, Evans RM. Nuclear receptor TLX prevents retinal dystrophy and recruits the corepressor atrophin1. Genes Dev. 2006;20:1308–1320. doi: 10.1101/gad.1413606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CL, Zou Y, He W, Gage FH, Evans RM. A role for adult TLX-positive neural stem cells in learning and behaviour. Nature. 2008;451:1004–1007. doi: 10.1038/nature06562. [DOI] [PubMed] [Google Scholar]
- Zhao C, Deng W, Gage FH. Mechanisms and functional implications of adult neurogenesis. Cell. 2008;132:645–660. doi: 10.1016/j.cell.2008.01.033. [DOI] [PubMed] [Google Scholar]