

Summary
The centromere-specific histone variant CENP-A (CID in Drosophila) is a structural and functional foundation for kinetochore formation and chromosome segregation. Here, we show that overexpressed CID is mislocalized into normally noncentromeric regions in Drosophila tissue culture cells and animals. Analysis of mitoses in living and fixed cells reveals that mitotic delays, anaphase bridges, chromosome fragmentation, and cell and organismal lethality are all direct consequences of CID mislocalization. In addition, proteins that are normally restricted to endogenous kinetochores assemble at a subset of ectopic CID incorporation regions. The presence of microtubule motors and binding proteins, spindle attachments, and aberrant chromosome morphologies demonstrate that these ectopic kinetochores are functional. We conclude that CID mislocalization promotes formation of ectopic centromeres and multicentric chromosomes, which causes chromosome missegregation, aneuploidy, and growth defects. Thus, CENP-A mislocalization is one possible mechanism for genome instability during cancer progression, as well as centromere plasticity during evolution.
Introduction
Genome instability plays a key role in birth defects and cancer progression (Balmain et al., 2003). The centromeric DNA and chromatin are the most important chromosomal elements required for segregation in mitosis and meiosis (Cleveland et al., 2003; Sullivan et al., 2001). In most eukaryotes, there is only one centromere per chromosome, which is usually embedded in heterochromatin. The centromere and associated kinetochore are essential for microtubule spindle attachments, congression to the metaphase plate, anaphase segregation to the poles, and the function of the mitotic checkpoint (or spindle assembly checkpoint [SAC]). Centromere dysfunction results in chromosome loss due to the absence of spindle attachments, and chromosomes with more than one kinetochore (di- or multicentrics) frequently fragment and missegregate, due to attachments of the same chromatid to both poles (McClintock, 1939).
Specification of only one site for centromere function (centromere identity) is regulated by epigenetic mechanisms in most eukaryotes (Cleveland et al., 2003; Sullivan et al., 2001). Transmissible dicentric chromosomes exist in which a kinetochore forms on only one of two regions of centromeric DNA, demonstrating that centromeric DNA is not sufficient for kinetochore formation (Agudo et al., 2000; Sullivan and Willard, 1998). Furthermore, centromeric DNA is not necessary for kinetochore formation, since noncentromeric DNA can acquire and faithfully propagate centromere proteins and functions (neocentromeres) without any change to the DNA sequences (Lo et al., 2001; Maggert and Karpen, 2001; Satinover et al., 2001). Finally, chromosome rearrangements are a hallmark of evolution and speciation, and they are accompanied by centromere gains, losses, and movements with respect to genome sequences (Ferreri et al., 2005; Murphy and Karpen, 1998).
Members of the CENP-A family of centromere-specific histone H3-like proteins serve as both structural and functional foundations for the kinetochore, and they are excellent candidates for an epigenetic mark that establishes and propagates centromere identity (Cleveland et al., 2003; Sullivan et al., 2001). CENP-A proteins are constitutive chromatin components that are assembled into a cylindrical 3D structure on mitotic chromosomes, around which the inner and outer kinetochore proteins are wrapped (Blower et al., 2002). They are essential for recruitment of kinetochore proteins, establishment of spindle attachments, and normal chromosome segregation in many eukaryotes (Blower and Karpen, 2001; Buchwitz et al., 1999; Chen et al., 2003; Howman et al., 2000; Stoler et al., 1995). In addition, reciprocal epistasis experiments have shown that CENP-A proteins are very high in the kinetochore assembly pathway (Cleveland et al., 2003; Sullivan et al., 2001), consistent with CENP-A’s location in chromatin at the base of the kinetochore (Blower et al., 2002).
CENP-A depletion provides one mechanism for generating aneuploidy in mitosis and meiosis, as well as centromere loss during evolution. In Drosophila, neocentromeres are generated due to their proximity in cis to a functional, endogenous centromere, suggesting that spreading of key centromere chromatin proteins such as CENP-A is one molecular mechanism for centromere gain (Maggert and Karpen, 2001). However, the types of rearrangements observed during evolution or in human neocentromeric chromosomes indicate that cis spreading cannot account for all cases of centromere gains.
Alternatively, centromere gain could also occur in response to CENP-A incorporation into normally noncentromeric regions, resulting in the formation of ectopic kinetochores. Studies testing this hypothesis in mammals have produced ambiguous and contradictory results. In human tissue culture cells, overexpressed CENP-A was incorporated into noncentromeric regions, but it did not appear to produce functional ectopic kinetochores (Van Hooser et al., 2001). In another study, 11 out of 11 human primary colorectal tumors sampled displayed CENP-A overexpression and mistargeting to normally noncentromeric regions, suggesting a potential link between CENP-A mislocalization and genome instability in cancer (Tomonaga et al., 2003).
Here, we directly test the hypothesis that CENP-A mislocalization into normally noncentromeric regions results in ectopic kinetochore formation. The effects of elevated levels of the Drosophila CENP-A homolog (CID) (Blower and Karpen, 2001; Henikoff et al., 2000) on cell and organismal proliferation and chromosome behavior were evaluated in both tissue culture cells and developing flies. Our results demonstrate that CID mislocalization can nucleate the formation of functional kinetochores at ectopic sites, which results in chromosome missegregation, aneuploidy, and growth defects.
Results
CID Mislocalization Results in Growth Defects in Cells and Animals
Stable Drosophila S2 cell lines were established that expressed either CID-GFP or histone H3-GFP fusion proteins, under the control of the inducible metallothionein promoter. For studies in flies, we induced expression of transgenic CID-V5 or H3-V5 constructs by using the GAL4-UAS system (Brand and Perrimon, 1993). Uninduced S2 cells or animals displayed leaky expression of the tagged CID proteins, which was exclusively targeted to endogenous centromeres (labeled “control,” Figure 1A), whereas leaky expression of tagged H3 was more broadly distributed in chromatin. Quantitative Western analysis indicated that CID-GFP induction in the entire population was 70-fold over endogenous CID levels (Figure S1; see the Supplemental Data available with this article online). However, subsequent functional analyses were performed on individual cells, which exhibited very different amounts of H3 and CID after induction (Figures 1A and 1B). Therefore, we quantitated protein levels in cells by using GFP fluorescence; low, medium, and high levels of CID-GFP expression corresponded to ~10-, 20-, and 30-fold induction, respectively, over control levels (see Experimental Procedures). Consistent with previous work (Collins et al., 2004; Henikoff et al., 2000; Van Hooser et al., 2001), overexpressed CID exhibited broad mislocalization in mitotic and polytene chromosomes, but it was incorporated preferentially into euchromatin; little, if any, signal was visible in the pericentric heterochromatin (Figures 1C and 1D). Epitope-tagged H3 was also incorporated into chromatin, but it predominantly localized to the heterochromatin.
Figure 1. CID-GFP- or H3-GFP-Expressing S2 Cells Display Different Protein Levels and Localizations upon Induction.
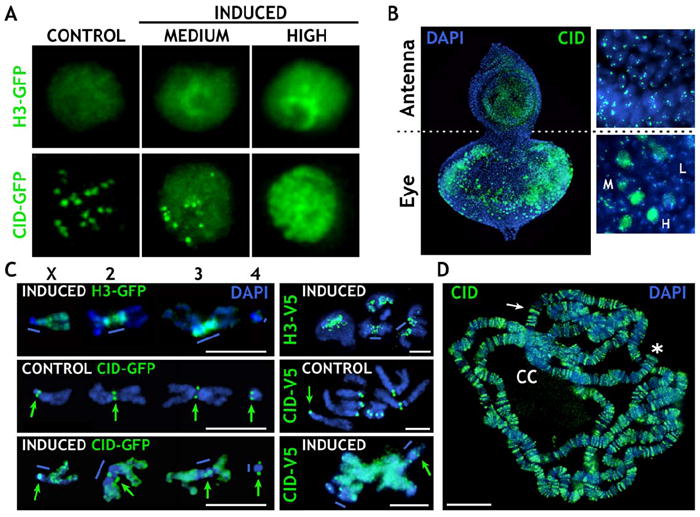
(A) H3-GFP staining was widely distributed in the nucleus at all levels of expression, whereas CID-GFP staining patterns depended on expression levels: centromere only, control; diffuse plus centromere, medium; and diffuse, high.
(B) CID overexpression in animals carrying UAS-CID-V5. IF with anti-CID antibodies (green) on eye-antenna discs shows that the EY-GAL4 driver is very specific to the eye disc. The levels of CID varied from low (L), to medium (M), to high (H). Cells in the antenna disc display endogenous CID levels.
(C) Distribution of H3-GFP/H3-V5 (top images) and low and high levels of CID-GFP/CID-V5 (green: GFP or V5 antibodies) on metaphase chromosomes from S2 cells (left), and from larval discs (right). High-CID-GFP/CID-V5 staining is predominantly euchromatic and at endogenous centromeres (green arrows), and it is not extensively incorporated into pericentric heterochromatin (blue bars), unlike H3-GFP/H3-V5.
(D) CID (green) was abnormally incorporated into polytene chromosome arms, bands (white arrow), and telomeres (asterisk) after heat shock induction of larvae containing HSP70-GAL4 and UAS-CID-V5. Only low levels of CID-V5 were incorporated into the heterochromatic chromocenter (CC).
The scale bars are 5 μm in (C) and 15 μm in (D).
Uninduced H3-GFP, CID-GFP, and untransfected S2 cells exhibited normal exponential growth, and untransfected S2 cell and H3-GFP cell growth was only slightly reduced after induction (Figure 2A). In contrast, overall cell growth was significantly reduced after CID-GFP induction; specifically, cells expressing low, medium, and high levels of CID-GFP were quickly eliminated during culture, and cells with no or leaky CID-GFP levels eventually took over the population (Figure 2B). Ubiquitous and strong CID-V5 expression starting in early embryos (TUB-GAL-4 driver) resulted in abnormal development and lethality (Figure 2C). Another severe phenotype was observed with the EY-GAL4 driver; most of the adults displayed a reduced eye phenotype (85%) (Figure 2D), as observed previously (Jager et al., 2005). Analysis of mitotic indices and acridine orange staining showed that these phenotypes were caused by cell death or severe retardation of cell division (Figure S2). None of these phenotypes were observed with overexpression of H3 or control levels of CID (Figure 2).
Figure 2. CID Overexpression Results in Cellular and Organismal Phenotypes.
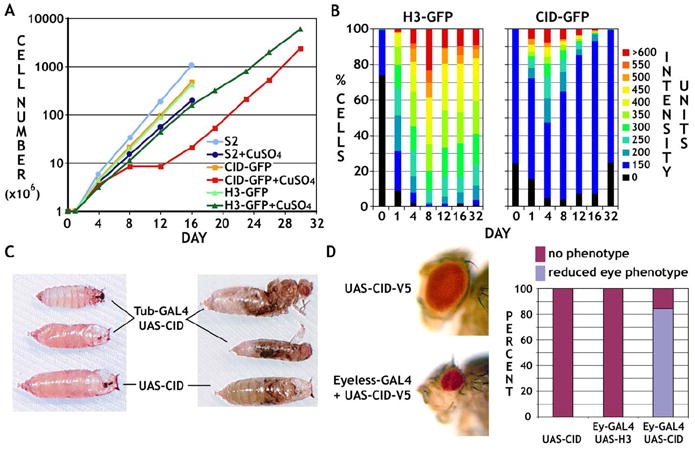
(A) Growth curves are shown for untransfected or stably transfected S2 cells carrying CID-GFP or H3-GFP, with and without CuSO4 induction. Induced CID-GFP cells displayed significant growth defects from day 8 to 12.
(B) H3-GFP-expressing cells maintained a similar distribution of protein levels once fully induced (~day 4), whereas the percentage of cells with CID-GFP levels above 150 decreased significantly after day 4.
(C) Ubiquitous CID induction resulted in organismal lethality (96%, n = 305). The majority (76%) stopped developing before pupariation (left panel), and 20% metamorphosed but were unable to hatch (right panel). The few flies (4%) that hatched, as well as the unhatched pupae, were substantially smaller than control flies.
(D) Eye-specific CID induction resulted in strongly reduced eye size (lower panel) in comparison to controls (upper panel). Approximately 85% of induced flies (n = 391) showed a severe visible phenotype (histogram).
We conclude that CID mislocalization leads to mitotic arrest or severe delay, and cell death in S2 cells and animals, consistent with the observed phenotypes. In addition, the normal growth of control cells and animals demonstrates that the CID fusion proteins did not interfere with endogenous centromere function.
CID Mislocalization Causes Severe Chromosome Segregation Defects
Cytological analysis of mitosis was used to explore the cellular basis for the growth defects and lethality associated with CID overexpression and mislocalization. In fixed cells from S2 cultures, embryos, larval brains, and imaginal discs, CID incorporation into noncentromeric regions was associated with a variety of mitotic defects, including anaphase bridges, chromosomes stretched along the spindle axis, and chromosome fragmentation (Figure S3). Similar mitotic abnormalities were observed in S2 cells overexpressing untagged CID, demonstrating that the defects were not due to the GFP fusion (data not shown). None of these defects were observed in controls, or in response to high levels of H3 expression.
Time-lapse analysis of mitosis in live S2 cells extended our understanding of the chromosomal defects displayed by cells with CID mislocalization. Most control cells (control CID-GFP or induced H3-GFP) displayed normal chromosome segregation during anaphase (Figure 3A, rows 1 and 2; quantitation in Figure 3C; Movies S1 and S2). CID-overexpressing cells displayed significantly higher frequencies of lagging and abnormally stretched chromosomes (Figure 3A, row 3; quantitation in Figure 3C; Movie S3). Stretched chromosomes also produced fragments that failed to be incorporated into daughter nuclei (Figure 3A, row 4; Movie S4). Finally, a substantially higher number of cells with induced CID-GFP expression displayed “cut” phenotypes, where chromosomes abnormally positioned near the spindle midzone in late anaphase were severed by the cleavage furrow during cytokinesis (Figure 3A, row 5; quantitation in Figure 3C; Movie S5).
Figure 3. Time-Lapse Analysis of Live S2 Cells Reveals Mitotic Defects after CID Induction.
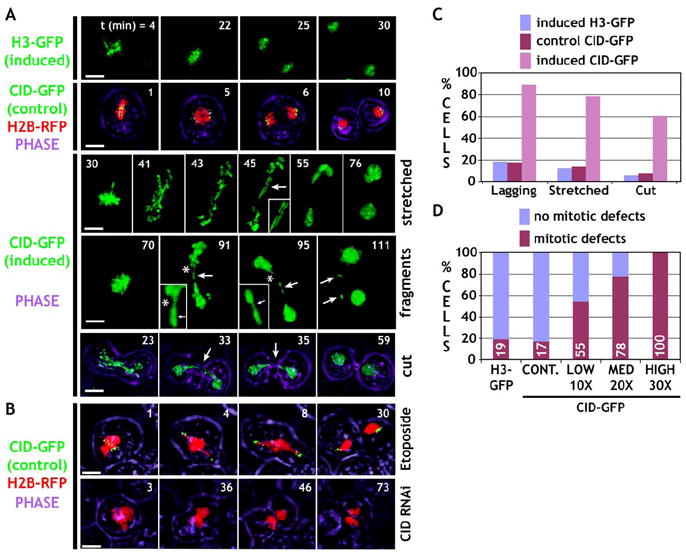
See the Supplemental Movies.
(A) Frames from time-lapse microscopy are shown for induced H3-GFP, control CID-GFP coexpressed with H2B-RFP (chromosome counter stain), and induced CID-GFP. Phase indicates cell outlines. Control chromosomes segregate normally. In contrast, cells with induced CID-GFP expression displayed stretched chromosomes, fragmentation (asterisk, inset), and “cutting” of the unsegregated chromosome mass by cytokinesis (see [C] for quantitation).
(B) Cells were treated with Etoposide to mimic problems in resolving sister chromatids (row 6), or they were depleted for CID by RNAi (row 7), and displayed distinct segregation defects in comparison to CID mislocalization. The scale bars are 5 μm.
(C) Quantitation of defects observed in the time-lapse analysis. Cells induced for CID-GFP displayed higher percentages of all types of mitotic defects (induced nH3-GFP = 32, nCID-GFP control = 50, ninduced CID-GFP = 27). For all phenotypes, the differences were highly significant (p < 0.001, Chi square test).
(D) Levels of CID-GFP expression correlated with a highly elevated overall frequency of defective mitoses, and all are significantly different from the controls (p < 0.01, in comparison to controls; H3-GFP: n = 32, control: n = 50, CID-GFP, low: n = 11, medium: n = 9 and high: n = 7).
Overall, time-lapse analysis revealed that mitotic defects were observed in 75% of cells with induced CID-GFP, compared to only ~18% of control cells (Figure 3D). Furthermore, even the lowest levels of induction produced significantly higher frequencies of mitotic abnormalities (55%) than controls, which were further elevated in cells with medium and high levels of expression (78% and 100%, respectively; Figure 3D). Thus, the frequency of mitotic abnormalities correlated with the amount of CID-GFP expression, suggesting that the defects are a direct result of CID mislocalization to normally noncentromeric regions. In addition, we frequently observed chromosomes that did not move in the typical V-form, as expected for a single spindle-chromosome attachment. Instead, chromosomes appeared to be stretched parallel to the spindle axis (Figure 3A, rows 3 and 4; quantitation in Figure 3C), a morphology consistent with spindle forces being applied to both chromosome arms, as observed for dicentric chromosomes (Ahmad and Golic, 1998). Finally, cells with induced CID-GFP displayed a 3- to 4-fold increase in the transition time between metaphase and anaphase onset (30.7 min) relative to control CID-GFP (7.5 min) or induced H3-GFP (10.8 min, p < 0.001). These results demonstrate that CID mislocalization leads to significant mitotic segregation defects, aneuploidy, and a delay in progression from metaphase to anaphase, which likely caused the observed growth defects and cell death.
CID Mislocalization Causes Mitotic Defects that Differ from Loss of Endogenous Centromere Function or Failure to Separate Sister Chromatids
Although the chromosome morphologies and mitotic phenotypes imply that multicentric chromosomes form upon CID mislocalization, anaphase bridges and stretched chromosomes could also arise from defects in sister chromatid separation. In addition, lagging chromosomes could result from interference with endogenous centromere function. To address these possibilities, we performed time-lapse analysis of S2 cells with defective sister separation by using the topoisomerase II inhibitor etoposide, and with cells depleted for CID by RNAi.
Etoposide treatment resulted in defects expected for inhibiting sister chromatid separation during anaphase (Chang et al., 2003; Coelho et al., 2003). Although centromeres moved normally and synchronously to the poles during anaphase, arms remained near the metaphase plate, producing massive bridges that were eventually “cut” by cytokinesis (Figure 3B, row 1; Movie S6). In addition, CID signals were stretched, consistent with previous observations suggesting that interlocking of sister chromatids elevates forces at kinetochores (Coelho et al., 2003).
The anaphase bridges formed after CID mislocalization were qualitatively and quantitatively different from failures in sister chromatid separation. After CID mislocalization, asynchronous poleward movement of centromeres and individual lagging chromosomes were observed, with no obvious centromere stretching (compare Movies S3 and S6). Consistent with these differences, PROD, which binds a satellite DNA near the centromeres of chromosomes 2 and 3 (Torok et al., 1997), was located predominantly near the poles after etoposide treatment, and telomeric HOAP antibody signals (Cenci et al., 2003) were enriched near the middle (Figures 4A and 4B). After CID mislocalization, PROD signals were less frequently observed near the poles (p < 0.01), and HOAP signals were less abundant near the middle (p < 0.025). Finally, we only observed chromosomes stretched parallel to the spindle and endogenous centromeres at the center and telomeres oriented toward opposite poles after CID mislocalization, and not after etoposide treatment (Figure 4A, inset).
Figure 4. CID Mislocalization Causes Mitotic Defects that Are Different from Failure to Separate Sister Chromatids or the Loss of Endogenous Centromere Function.
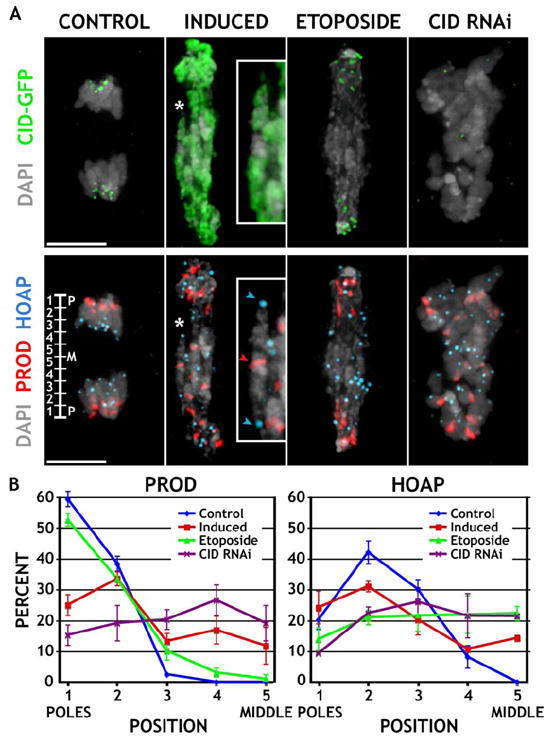
(A) Induced S2 cells, and uninduced cells treated with Etoposide or depleted for CID (CID RNAi), were fixed and stained for CID (green), PROD (red), and HOAP (blue). The inset shows a frequently observed phenotype of a chromosome (asterisk) being stretched along its length axis, with the centromeres (PROD) positioned in the middle, and the telomeres (HOAP) facing opposite poles. The scale bars are 5 μm.
(B) The distance of all PROD (endogenous centromere) and HOAP (telomere) foci from the two poles was measured, normalized to the length of the cell, and expressed as percent distance from the pole (see ruler, bottom left in [A]). Cells with induced CID expression had PROD spots positioned less frequently near the poles and fewer HOAP spots in the middle, compared to Etoposide treatment. Cells depleted for CID display random staining for both. Note that two of ten PROD spots per cell colocalized with HOAP and were omitted from the quantitation. Error bars are ±1 standard deviation from the average.
Similarly, the phenotypes observed after CID depletion versus CID induction were significantly different. After CID RNAi, chromosomes did not congress to the metaphase plate, and they displayed little movement toward the poles; no bridges were formed, and the chromosome mass that remained at the center was asymmetrically severed by cytokinesis (Figure 3B, row 2; Movie S7). These differences were substantiated by the observation that PROD and HOAP were randomly distributed across the spindle after CID depletion—localizations that were distinct from those observed after CID mislocalization (Figures 4A and 4B; p < 0.05 and p < 0.01, respectively).
We conclude that the CID mislocalization mitotic defects are phenotypically distinct from failure to resolve sister chromatid cohesion or loss of endogenous centromere function. Furthermore, phenotypes consistent with the presence of multiple kinetochores, such as stretched chromosomes with endogenous centromeres in the middle, were only observed after CID mislocalization.
Inner and Outer Kinetochore Proteins Are Recruited to Sites of Ectopic CID Incorporation
The hypothesis that ectopic kinetochores form in response to CID mislocalization was further tested by examining the distributions of proteins involved in centromeric chromatin and kinetochore functions. CENP-C is an inner kinetochore protein located between CENP-A and outer kinetochore proteins in mitotic chromosomes (Blower et al., 2002), and it is required for normal kinetochore formation (Sullivan et al., 2001). The Drosophila CENP-C homolog was recently shown to localize to centromeres in a CID-dependent manner (Jager et al., 2005), consistent with previous results in other organisms (Sullivan et al., 2001). Another protein associated with centromeric chromatin is MEI-S332 (Blower and Karpen, 2001; Kerrebrock et al., 1995), whose homologs (shugoshins) have recently been demonstrated to block degradation of centromeric cohesins (Kitajima et al., 2004; Salic et al., 2004). CID is required for normal MEI-S332 localization to the centromere region, whereas CID localization does not depend on MEI-S332 (Blower and Karpen, 2001). In order to examine the effects on localization of an outer kinetochore protein, we stained cells for BUBR1, which is an outer kinetochore protein required for the mitotic checkpoint (Basu et al., 1998).
CENP-C, MEI-S332, and BUBR1 were localized exclusively to centromeres in S2 and fly imaginal disc cell controls (Figure 5); the number of spots was close to the amount expected for two sister kinetochores on the 13 ± 2 chromosomes present in S2 cells, and the 8 chromosomes in flies (Tables S1 and S2). In contrast, significantly more sites (1.7- to 3.3-fold) were observed for all three proteins after CID induction, and the additional signals were located in normally noncentromeric (ectopic) regions (all p values < 0.01). These differences were not due to elevated numbers of chromosomes in induced CID cells (data not shown).
Figure 5. Induction of Ectopic CID Results in Aberrant Localization of Centromere and Kinetochore Proteins to Normally Noncentromeric Regions.
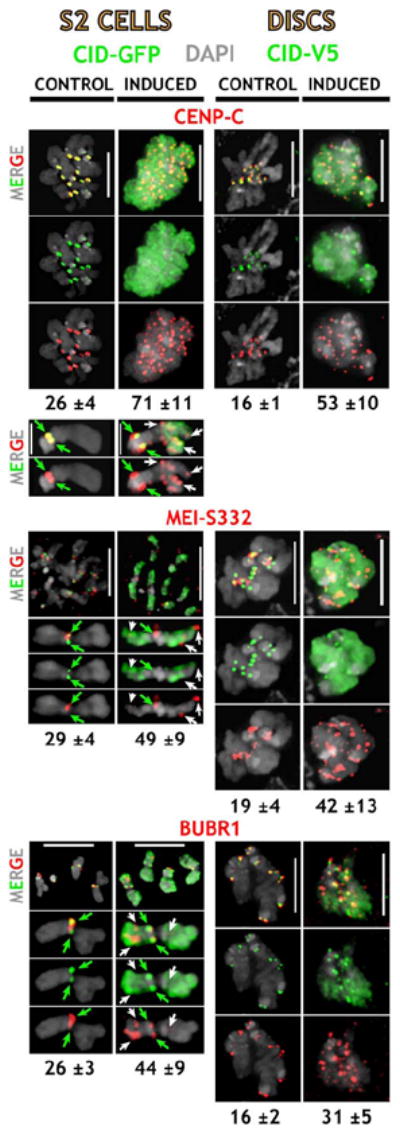
Localization of the inner kinetochore protein CENP-C, the sister cohesion protein MEI-S332, and BUBR1. CENP-C, MEI-S332, and BUBR1 are in red; CID-GFP in S2 cells and CID-V5 in larval disc cells are in green. Enlargements of individual chromosomes of S2 cells are shown below; green arrows = endogenous centromeres, white arrows = ectopic sites. Normal centromeric localization of all three proteins was observed in controls. Upon CID induction, these proteins were abnormally localized to many noncentromeric sites, and the average number of sites (below) was significantly higher in induced versus control cells (p < 0.01, Tables S1 and S2). The scale bars are 5 μm.
To address whether individual ectopic sites of CID incorporation recruit multiple kinetochore proteins, we simultaneously stained control and induced cells with combinations of antibodies to different inner and outer kinetochore proteins. Specifically, we quantitated colocalization of CENP-C/MEI-S332, CENP-C/POLO, POLO/ROD, and MEI-S332/BUBR1 (Figure 6). POLO kinase strongly associates with the outer kinetochore from prometaphase through anaphase, and it is required for spindle integrity and chromosome segregation (Logarinho and Sunkel, 1998). ROD is also localized to the outer kinetochore, and it is required for recruitment of microtubule motors and normal segregation (Basto et al., 2000; Starr et al., 1998; Williams et al., 2003).
Figure 6. Inner and Outer Kinetochore Proteins Colocalize at Sites of Ectopic CID Incorporation.
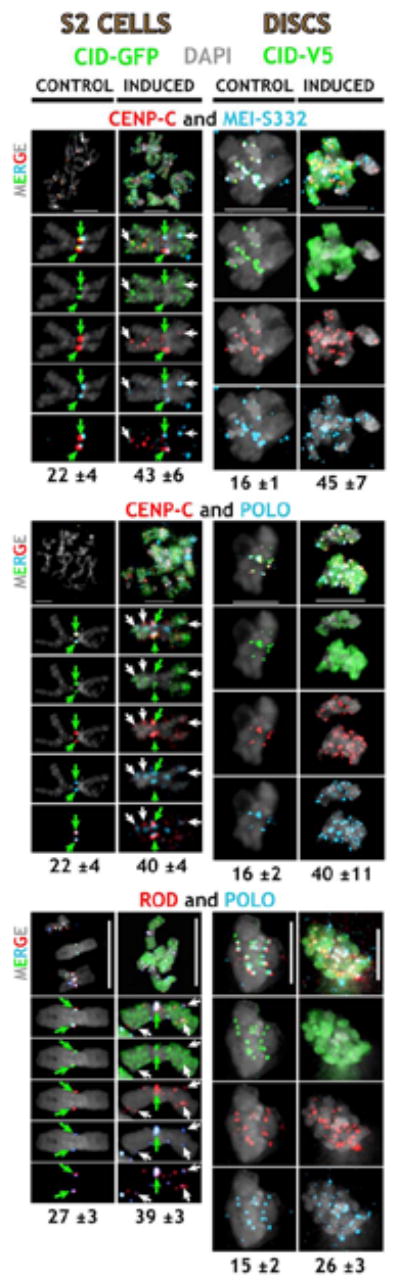
Control and induced cells were simultaneously stained with CENPC/MEI-S332, CENP-C/POLO, POLO/ROD, or MEI-S332/BUBR1 antibodies. CID-GFP in S2 cells and CID-V5 in larval disc cells are in green, CENP-C and ROD are in red, and POLO and MEI-S332 are in blue. Enlargements of individual chromosomes of S2 cells are shown below. Inner and outer kinetochore proteins were often colocalized at ectopic sites (white arrows), in addition to the endogenous centromeres (green arrows). The average number of colocalization sites (below) was significantly higher in induced versus control cells (p < 0.01, Tables S1 and S2). The scale bars are 5 μm.
Normal CENP-C, MEI-S332, ROD, POLO, and BUBR1 kinetochore localization was observed in control S2 and imaginal disc cells (Figure 6), and in cells with induced H3 (data not shown). In contrast, all centromere/kinetochore proteins were mislocalized to significantly more ectopic sites in cells with CID mislocalization, in addition to endogenous centromeres (Figure 6; Tables S1 and S2; all p values < 0.01). As observed previously in single-label experiments for CENP-C, MEI-S332, and BUBR1 (see above), ROD and POLO were present at 1.4- to 3.6-fold more sites after induction. Most importantly, all combinations of inner/inner, inner/outer, and outer/outer kinetochore proteins colocalized at significantly more sites after CID induction, compared to controls (1.4- to 2.8-fold; all p values < 0.01). The percentage of centromere/kinetochore sites that contained both proteins after induction ranged from 39% to 85% in S2 cells and from 72% to 97% in disc cells, supporting the idea that most kinetochore proteins are recruited to the same ectopic sites.
These results demonstrate that CID mislocalization is associated with recruitment of multiple centromeric cohesion, inner kinetochore, and outer kinetochore proteins to normally noncentromeric sites, suggesting at least partial formation of ectopic kinetochores. Note that these proteins were not recruited to all sites of ectopic CID incorporation (see Discussion).
CID Mislocalization Results in the Formation of Functional Ectopic Kinetochores
The presence of ectopic centromere and kinetochore proteins, and the types of segregation defects, strongly suggests that ectopic kinetochores are formed after CID mislocalization. In order to evaluate the functionality of these ectopic kinetochores, we determined whether kinetochore-associated microtubule motors and binding proteins, stable microtubule connections, and spindle-mediated forces were present at normally noncentromeric regions after CID mislocalization.
Chromosome movement is mediated by microtubule assembly and disassembly and the functions of microtubule motors. The kinesin KLP59C is a microtubule-depolymerizing protein located in the outer kinetochore, and it is required for chromosome movement along spindles (Rogers et al., 2004). Dynein is a minus-end motor that links kinetochores to microtubules and contributes to poleward chromosome movement during anaphase A (Howell et al., 2001; Rogers et al., 2004). In mitosis, both Dynein and KLP59C were specifically associated with endogenous centromeres (and for Dynein on microtubules) in control CID-GFP cells, but they were colocalized at significantly more chromosomal sites after CID mislocalization, (2.3-fold increase, Table S1, p < 0.02).
Plus-end microtubule binding proteins, including Drosophila MAST, are concentrated at the interface between microtubules and functional kinetochores (Maiato et al., 2002, 2004), providing an independent test for the presence of ectopic spindle attachments. Control S2 cells contained on average 26 MAST spots in metaphase, whereas cells with induced CID contained significantly more MAST spots (42, p < 0.001; Figures 7B and 7C). Similar results were obtained in flies; control imaginal disc cells contained 16 MAST spots in metaphase, compared to 35 spots after induction. In contrast, the number of MAST spots observed in etoposide-treated control S2 cells (28) was equivalent to the results from untreated control cells, and cells depleted for CID by RNAi contained only 6 MAST spots (Figure 7C). These results demonstrate that the number of functional ectopic kinetochore-microtubule attachments increases significantly, specifically in cells with extensive CID mislocalization.
Figure 7. Distributions of Microtubule Motors and the Plus-End Binding Protein MAST Suggest the Presence of Functional Ectopic Kinetochores.
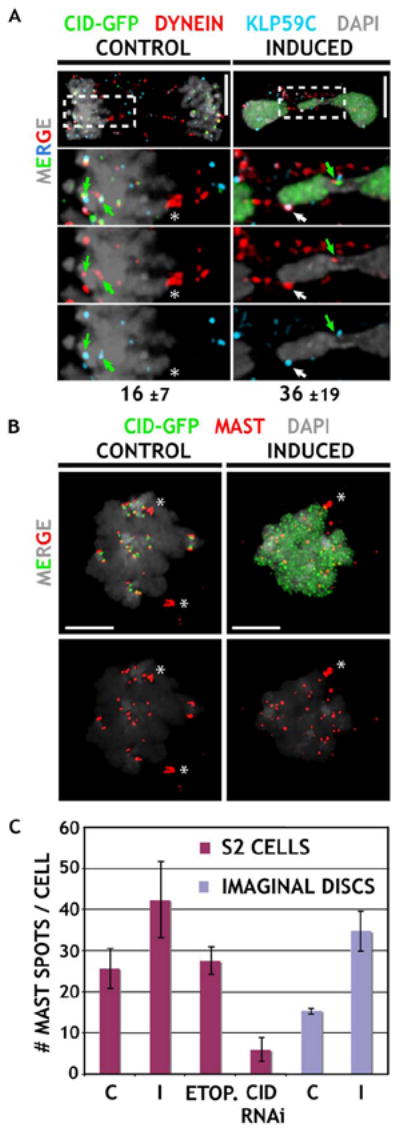
(A) Localization of the motor proteins Dynein and kinesin KLP59C. Merged images for anaphase figures are shown on top, and enlargements are shown below. CID-GFP-expressing cells were stained for CID (green), Dynein, and KLP59C (blue). Control cells display close association of both proteins only at endogenous centromeres (green arrows), whereas they frequently colocalize at ectopic chromosomal sites (white arrow) after induction (average number of sites indicated below, see Table S1). Note that Dynein also decorates underlying spindle microtubules (asterisk). The scale bars are 5 μm.
(B) Localization of the microtubule plus-end binding protein MAST. Metaphase figures from induced cells contain more MAST (red) spots than observed in controls. The scale bars are 5 μm.
(C) Quantitation of MAST spots in S2 and imaginal disc cells. MAST signals associated with spindle poles (asterisk in [B]) were not included in the quantitation. Induced cells contained significantly elevated numbers of MAST spots compared to controls (p values in text). Etoposide treatment did not alter the number of MAST spots from control values, and CID depletion by RNAi resulted in significantly fewer MAST spots than controls (p < 0.001 for I or CID RNAi versus C; S2 cells: nC = 19, nI = 17, nETOP = 16, nCID RNAi = 14, imaginal disks: nC = 12, nI = 13). Error bars are ±1 standard deviation from the average.
To visualize microtubule attachments, we exposed cells to cold treatment, which preferentially depolymerizes spindle microtubules that are not attached to kinetochores (Rieder, 1981). After CID induction, cells displayed cold-stable microtubule connections at ectopic sites that also contained CID and the outer kinetochore protein ROD (Figure 8A). Three-dimensional modeling demonstrated that microtubules ended at sites in the chromosome arms, far from the endogenous centromere (Movies S8 and S9). In order to visualize ectopic microtubule attachments, we had to examine missegregating chromosomes (e.g., stretched) during late anaphase, when pole-to-pole microtubules are also retained after cold-treatment, which made quantitative comparisons difficult. However, such ectopic microtubule connections were never observed in S2 or imaginal disc cells overexpressing H3 or control levels of CID, or in S2 cells perturbed by CID depletion or etoposide treatment. In addition, the idea that these ectopic microtubule attachments exert force on the chromosome arms was supported by observations of stretched or bent chromosome arms with microtubule attachments to ectopic sites (Figure 8B, also see Figure 4A). Finally, the presence of significantly more ectopic sites containing the key kinetochore motors Dynein and KLP59C, and the plus-end binding protein MAST, provided quantitative evidence that ectopic spindle attachments were significantly increased after CID induction.
Figure 8. Ectopic Microtubule Attachments and Aberrant Chromosome Morphologies Are Observed after CID Induction.
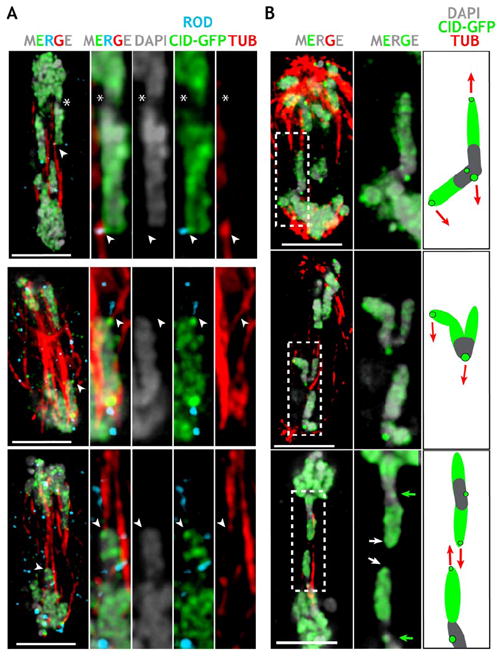
(A) Anaphase figure from CID-GFP-expressing S2 cells (higher magnification to the right) displaying prominent ectopic CID-GFP foci (white arrows) connected to microtubule bundles (red), and colocalized with ROD (light blue), far from the endogenous centromere (asterisk).
(B) Chromosome morphologies suggest poleward forces applied to chromosome arms after CID induction. The panel to the right shows enlargements of chromosomes, plus a schematic interpretation of the forces (red arrows) responsible for the observed chromosome morphologies.
The scale bars are 5 μm.
We conclude that CID induction results in ectopic chromosomal sites containing proteins implicated in forming functional microtubule attachments at kinetochores, as well as attachments similar to those found at endogenous kinetochores.
Discussion
Sites of Ectopic CID Incorporation Are Associated with Kinetochore Formation and Function
CENP-A has been demonstrated to provide a structural and functional foundation for the kinetochore in a variety of organisms (Cleveland et al., 2003; Sullivan et al., 2001). This study shows that ectopic incorporation of Drosophila CID into normally noncentromeric chromatin occurs in response to overexpression in S2 and animal cells, as observed previously in tissue culture cells and yeast (Collins et al., 2004; Henikoff et al., 2000; Van Hooser et al., 2001). Here, we show that CID mislocalization results in defective cell growth, cell and organismal death, and abnormal development.
Our results strongly support the conclusion that these mitotic abnormalities and growth defects are caused by formation of ectopic kinetochores and multiple spindle attachments on individual chromatids. First, studies of fixed and live cells demonstrated that CID overexpression caused significant mitotic defects, including increased mitotic index and stretched, fragmented, and lagging chromosomes during anaphase. Time-lapse analysis in S2 cells revealed that CID overexpression also caused mitotic delays, as well as cut phenotypes, chromosome loss, and abnormal chromosome morphology during anaphase segregation.
Second, proteins that are normally associated with endogenous centromeres were present at ectopic sites in response to CID mislocalization. We examined the distribution and colocalization of proteins involved in different centromere/kinetochore structures and functions, extending from the centromeric chromatin to the outer kinetochore. Proteins associated with centromeric chromatin (CENP-C), centromeric cohesion (MEI-S332, BUBR1), outer kinetochore formation and motor protein recruitment (ROD, POLO), and the SAC (BUBR1) were mislocalized and colocalized to normally noncentromeric regions in S2 and animal cells with mislocalized CID. Thus, proteins involved in a wide spectrum of centromere and kinetochore functions are recruited together to ectopic sites after CID mislocalization.
Third, CID mislocalization resulted in significantly elevated numbers of sites containing the kinetochore-associated KLP59C and Dynein motor proteins, and the microtubule plus-end binding protein MAST. KLP59C and Dynein were frequently colocalized at normally noncentromeric regions of chromosomes that were displaying aberrant anaphase chromosome morphology. This data strongly suggests that ectopic CID incorporation can seed kinetochores that are able to form stable microtubule attachments, and that these attachments are able to transmit forces to chromosomes during mitosis. MAST localization in metaphase is likely to provide the best estimate for the number of ectopic functional kinetochores formed after CID induction, approximately twice the number observed in controls.
Finally, CID mislocalization resulted in the appearance of cold-stable microtubule attachments at normally noncentromeric regions, in addition to endogenous centromeres. The presence of ectopic spindle forces was confirmed in fixed preparations and time-lapse analysis by observing chromosomes with bent or stretched chromosome arms, which can only result from forces directing different sites on a single chromatid to the same pole. Likewise, in fixed cells and time-lapse analysis, we observed chromosomes stretched along their longitudinal axes with endogenous centromeres in the middle, indicating that arms are under tension from opposite poles.
It is possible that other chromosome defects are caused by mislocalization of CID, in addition to ectopic kinetochore formation and multicentric chromosomes. However, inhibition of sister chromatid separation with the topoisomerase II inhibitor etoposide, and CID depletion by RNAi, produced mitotic defects that were qualitatively and quantitatively distinct from those observed after CID mislocalization. Thus, loss of endogenous centromere function or sister separation defects alone cannot account for the predominant chromosome phenotypes observed after CID mislocalization. Determining if other chromosome segregation defects in addition to ectopic kinetochores occur in response to CID mislocalization warrants further study.
We conclude that CID induction results in broad incorporation into normally noncentromeric, predominantly euchromatic regions, a subset of which recruit key kinetochore proteins and exhibit kinetochore function. We propose that the mitotic, cellular, and organismal phenotypes are caused by the presence of more than one functional kinetochore and spindle attachment per chromatid. These results also provide further evidence that CENP-A is a key epigenetic mark for centromere identity (Sullivan et al., 2001).
Multiple Factors Regulate Kinetochore Formation at Sites of CENP-A Incorporation
Although CENP-A is currently the highest protein in the kinetochore assembly pathway, previous studies have not addressed whether CENP-A is also sufficient for kinetochore formation. The fact that most ectopic sites of CID incorporation were not associated with kinetochore proteins and spindle attachments indicates that CENP-A is not absolutely sufficient for kinetochore formation. However, there are several reasons why it is unlikely that a strict correlation between CENP-A incorporation and kinetochore formation would be observed in this system. First, it seems unlikely that all kinetochore proteins are present in the vast excess required for kinetochore formation at all ectopic CID sites. Interestingly, the inner kinetochore protein CENP-C was recruited more efficiently to ectopic sites in comparison to all of the outer kinetochore proteins (Figures 5 and 6), suggesting that kinetochore formation may be limited by processes downstream from centromeric chromatin formation. Second, it is possible that only regions with CID incorporation above a threshold level, perhaps equivalent to the density at the endogenous centromere, are capable of establishing a functional kinetochore. This hypothesis is supported by the observation that the severity of chromosome segregation defects is closely correlated with CID expression levels. Lower levels of CENP-A induction are the most likely explanation for why ectopic kinetochores were not detected in the human study (Van Hooser et al., 2001). Alternatively, human cells may possess a more efficient clearing mechanism for eliminating CENP-A from noncentromeric regions, as has been reported for S. cerevisiae (Collins et al., 2004). Third, other broadly distributed chromatin factors may contribute to functional kinetochore formation in combination with CID. Centromeric chromatin in flies and humans contains histone modification patterns that are distinct from euchromatin and the flanking heterochromatin, which may also be required for the formation of ectopic, functional kinetochores (Sullivan and Karpen, 2004).
Although centromere function and kinetochore assembly may require flanking heterochromatin, the presence of heterochromatin can also inhibit CID incorporation and kinetochore formation. In Drosophila, neocentromeres are produced when noncentromeric DNA and an endogenous centromere are juxtaposed, but not when heterochromatin separates these regions (Maggert and Karpen, 2001). The lack of CID incorporation into heterochromatin after induction is consistent with the hypothesis that heterochromatin antagonizes the spread of centromeric chromatin and normally acts to limit the size and distribution of centromeric chromatin (Maggert and Karpen, 2000, 2001). Thus, differences in the distribution of heterochromatin may also limit CID incorporation into ectopic sites, or the ability of ectopic sites to form functional kinetochores. Further studies are needed to determine exactly what factors limit kinetochore formation at ectopic sites, and to examine the sufficiency of CENP-A in more detail.
CENP-A and Genome Instability
Studies in mammals, insects, and other lineages have shown that centromere gains and losses are a hallmark of chromosome evolution (Ferreri et al., 2005). Loss of an epigenetic mark such as CENP-A provides one mechanism for centromere inactivation without deletion of centromeric DNA. Identifying mechanisms for centromere gain is more challenging, as it requires acquisition of an epigenetic mark in the absence of DNA sequence changes. Studies of experimentally induced neocentromeres in flies suggest one molecular mechanism for centromere gain, specifically cis spreading of key centromere chromatin proteins such as CENP-A (Maggert and Karpen, 2001). However, this model cannot account for human neocentromere formation or most examples of centromere gain during evolution. The results presented here suggest a more appropriate mechanism for these cases of centromere acquisition, specifically CENP-A mislocalization, perhaps in response to transient overexpression. In addition, our results demonstrate that similar levels of CENP-A overexpression in Drosophila and human tumors (Tomonaga et al., 2003) can produce a spectrum of mitotic phenotypes consistent with the chromosome abnormalities observed in cancers (Balmain et al., 2003). Further investigations into the prevalence of CENP-A mislocalization in different types of human cancers, and its timing during cancer initiation and progression, are required to directly test this hypothesis.
Experimental Procedures
Cloning and DNA Constructs
Full-length H3 and CID were cloned into a modified pMT/V5 vector (Invitrogen), which contained an in-frame EGFP, and the hygromycin resistance gene full-length histone H2B was inserted into a modified pIB/V5 vector (Invitrogen), which contained an in-frame mRFP in the XhoI-SacII sites.
Cell Culture
S2 cells were grown under standard conditions, and Ca phosphate-DNA coprecipitation was used for transfection (Cherbas et al., 1994). Stable lines were selected and maintained with 100 μg/ml Hygromycin-B (Invitrogen). Protein expression was induced from the metallothionein promoter (pMT/V5 vectors) by using 250 μM CuSO4 for 24 hr.
Drosophila Culture
Transgenic animals (Ashburner, 1990) were generated from the pP[UAST]CID-V5-6His or pP[UAST]H3-V5-6His constructs, which carry the mini-white gene. Several UAS-CID and UAS-H3 lines were established. Strong phenotypes were only observed at higher temperatures (28°C–29°C). All “UAS-driver lines” were obtained from the Bloomington stock center.
Cytological Preparations and Immunofluorescence
Unless otherwise noted, all S2 cells used for indirect IF were plated on Concanavalin-A (Sigma)-coated slides and processed as described (Henderson, 2004), except that cells stained for MAST were treated with 3 μg/ml colcemid for 30 min. After fixation, all S2 cells were processed for IF as described (Blower and Karpen, 2001), and they were mounted in SlowFade Light (Molecular Probes). Anti-tubulin staining was performed as described (Henderson, 2004), except that cells were cold treated for 30 min at 4°C prior to fixation (Rieder, 1981). Cells were incubated with etoposide for 30 min at a final concentration of 10 μM 2% DMSO. RNAi for CID was performed as described (Blower and Karpen, 2001).
Animal tissues were prepared and IF was performed as described (Henderson, 2004), except that tissues were prefixed for 1 min and incubated in 1 mg/ml dispase/collagenase (Roche) for 5 min prior to fixation. Mitotic chromosome and salivary gland squashes, and IF on embryos, were performed as described (Sullivan et al., 2000). IF for Tubulin was performed as described (Henderson, 2004), except that the tissues were incubated on ice for 1 min and fixation was carried out on ice. Embryos and disc cells were mounted in Vectashield (Vector Laboratories) containing DAPI.
For IF of both S2 cells and animal tissues, dilutions for the CID, ROD, POLO, TUBULIN, BUBR1, PROD, and MEI-S332 primary antibodies were as described (Blower and Karpen, 2001). Dilutions for other primary antibodies were mouse anti-V5, 1:500; rabbit anti-H3 Ser10, 1:250; guinea pig anti-HOAP, 1:100; rabbit anti-KLP59C, 1:500; mouse anti-Dynein, 1:500; rabbit anti-MAST, 1:10; and rabbit anti-CENP-C, 1:5000. Secondary antibodies were coupled to Alexa 488, Alexa 546, and Alexa 647 fluorophores (Molecular Probes) and were used at 1:500 dilutions.
Microscopy
All images were taken on a Deltavision Spectris Microscope and were deconvolved by using softWoRx (Applied Precision). For indirect IF, images were taken as z-stacks of 0.2 μm increments, by using a 100× oil-immersion objective. For the growth rate and protein expression studies, ten z-stacks of 1 μm increments were taken with a 40× oil-immersion objective, quick projected, then quantified for GFP levels by using arbitrary density units. For time-lapse microscopy, 10 μl of exponentially growing cultures was added to a cover slip and processed as described by using the “hanging drop” method (Shields and Sang, 1970). Seven z-stacks of 1 μm increments were captured for each channel (GFP, RFP, and transmission light) at 1 frame/min by using a 60× oil-immersion objective. Movies are displayed as quick projections, and GFP levels of metaphase plates were quantified by using softWoRx. Fold induction of CID-GFP expression in S2 cells was quantified by using the sum of pixel intensity in the different z-sections for the first picture of each time-lapse movie for both induced and uninduced cells, and levels were categorized as control (uninduced) ≤ 14, low = 90–190, medium = 190–290, and high ≥ 290. Images of flies, heads, and pupae cases were taken on a dissecting microscope with a Polaroid CCD camera, and images analyzed with the DMC direct software. All statistical comparisons of the numbers of localization sites utilized the Mann-Whitney U Test.
Supplementary Material
Acknowledgments
We thank Roger Tsien for the mRFP construct, and we thank the following people for antibodies: Roger Karess, Claudio Sunkel, David Sharp and Daniel Buster, Terry Orr-Weaver, Stefan Heideman, Tibor Török, and Jamy Peng. We also thank Abby Dernburg, Arshad Desai, Aaron Straight, and many lab members for discussions and critical reading of the manuscript; Pete Carlton for help with 3D modeling; and Katie Freeman, David Acevedo, Cameron Kennedy, and Tomas Bogardus for general assistance. Funding for this research was provided by the Wellcome Trust to S.E. (072016), Deutsche Forschungsgemeinschaft (HE 3434/1-1) and Human Frontier Science Program (LT 00174/2002) to P.H., and LBNL-LDRD (366987) and National Institutes of Health R01 (GM 66272) grants to G.H.K.
Footnotes
Supplemental Data Supplemental Data including time-lapse movies, figures, tables, and Experimental Procedures are available at http://www.developmentalcell.com/cgi/content/full/10/3/303/DC1/.
References
- Agudo M, Abad JP, Molina I, Losada A, Ripoll P, Villasante A. A dicentric chromosome of Drosophila melanogaster showing alternate centromere inactivation. Chromosoma. 2000;109:190–196. doi: 10.1007/s004120050427. [DOI] [PubMed] [Google Scholar]
- Ahmad K, Golic KG. The transmission of fragmented chromosomes in Drosophila melanogaster. Genetics. 1998;148:775–792. doi: 10.1093/genetics/148.2.775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M. Drosophila: A Laboratory Handbook. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1990. [Google Scholar]
- Balmain A, Gray J, Ponder B. The genetics and genomics of cancer. Nat Genet. 2003;33(Suppl):238–244. doi: 10.1038/ng1107. [DOI] [PubMed] [Google Scholar]
- Basto R, Gomes R, Karess RE. Rough deal and Zw10 are required for the metaphase checkpoint in Drosophila. Nat Cell Biol. 2000;2:939–943. doi: 10.1038/35046592. [DOI] [PubMed] [Google Scholar]
- Basu J, Logarinho E, Herrmann S, Bousbaa H, Li Z, Chan GK, Yen TJ, Sunkel CE, Goldberg ML. Localization of the Drosophila checkpoint control protein Bub3 to the kinetochore requires Bub1 but not Zw10 or Rod. Chromosoma. 1998;107:376–385. doi: 10.1007/s004120050321. [DOI] [PubMed] [Google Scholar]
- Blower MD, Karpen GH. The role of Drosophila CID in kinetochore formation, cell-cycle progression and heterochromatin interactions. Nat Cell Biol. 2001;3:730–739. doi: 10.1038/35087045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blower MD, Sullivan BA, Karpen GH. Conserved organization of centromeric chromatin in flies and humans. Dev Cell. 2002;2:319–330. doi: 10.1016/s1534-5807(02)00135-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Buchwitz BJ, Ahmad K, Moore LL, Roth MB, Henikoff S. A histone H3-like protein in C. elegans. Nature. 1999;401:547–548. doi: 10.1038/44062. [DOI] [PubMed] [Google Scholar]
- Cenci G, Siriaco G, Raffa GD, Kellum R, Gatti M. The Drosophila HOAP protein is required for telomere capping. Nat Cell Biol. 2003;5:82–84. doi: 10.1038/ncb902. [DOI] [PubMed] [Google Scholar]
- Chang CJ, Goulding S, Earnshaw WC, Carmena M. RNAi analysis reveals an unexpected role for topoisomerase II in chromosome arm congression to a metaphase plate. J Cell Sci. 2003;116:4715–4726. doi: 10.1242/jcs.00797. [DOI] [PubMed] [Google Scholar]
- Chen ES, Saitoh S, Yanagida M, Takahashi K. A cell cycle-regulated GATA factor promotes centromeric localization of CENP-A in fission yeast. Mol Cell. 2003;11:175–187. doi: 10.1016/s1097-2765(03)00011-x. [DOI] [PubMed] [Google Scholar]
- Cherbas L, Moss R, Cherbas P. Transformation techniques for Drosophila cell lines. Methods Cell Biol. 1994;44:161–179. doi: 10.1016/s0091-679x(08)60912-7. [DOI] [PubMed] [Google Scholar]
- Cleveland DW, Mao Y, Sullivan KF. Centromeres and kinetochores: from epigenetics to mitotic checkpoint signaling. Cell. 2003;112:407–421. doi: 10.1016/s0092-8674(03)00115-6. [DOI] [PubMed] [Google Scholar]
- Coelho PA, Queiroz-Machado J, Sunkel CE. Condensin-dependent localisation of topoisomerase II to an axial chromosomal structure is required for sister chromatid resolution during mitosis. J Cell Sci. 2003;116:4763–4776. doi: 10.1242/jcs.00799. [DOI] [PubMed] [Google Scholar]
- Collins KA, Furuyama S, Biggins S. Proteolysis contributes to the exclusive centromere localization of the yeast Cse4/CENP-A histone H3 variant. Curr Biol. 2004;14:1968–1972. doi: 10.1016/j.cub.2004.10.024. [DOI] [PubMed] [Google Scholar]
- Ferreri GC, Liscinsky DM, Mack JA, Eldridge MD, O’Neill RJ. Retention of latent centromeres in the Mammalian genome. J Hered. 2005;96:217–224. doi: 10.1093/jhered/esi029. [DOI] [PubMed] [Google Scholar]
- Henderson DS. Visualizing mitosis in whole-mount larval brains. In: Henderson DS, editor. Drosophila Cytogenetics Protocols. Totowa, NJ: Humana Press; 2004. pp. 363–371. [DOI] [PubMed] [Google Scholar]
- Henikoff S, Ahmad K, Platero JS, van Steensel B. Heterochromatic deposition of centromeric histone H3-like proteins. Proc Natl Acad Sci USA. 2000;97:716–721. doi: 10.1073/pnas.97.2.716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howell BJ, McEwen BF, Canman JC, Hoffman DB, Farrar EM, Rieder CL, Salmon ED. Cytoplasmic dynein/dynactin drives kinetochore protein transport to the spindle poles and has a role in mitotic spindle checkpoint inactivation. J Cell Biol. 2001;155:1159–1172. doi: 10.1083/jcb.200105093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howman EV, Fowler KJ, Newson AJ, Redward S, MacDonald AC, Kalitsis P, Choo KH. Early disruption of centromeric chromatin organization in centromere protein A (Cenpa) null mice. Proc Natl Acad Sci USA. 2000;97:1148–1153. doi: 10.1073/pnas.97.3.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager H, Rauch M, Heidmann S. The Drosophila melanogaster condensin subunit Cap-G interacts with the centromere-specific histone H3 variant CID. Chromosoma. 2005;113:350–361. doi: 10.1007/s00412-004-0322-4. [DOI] [PubMed] [Google Scholar]
- Kerrebrock AW, Moore DP, Wu JS, Orr-Weaver TL. Mei-S332, a Drosophila protein required for sister-chromatid cohesion, can localize to meiotic centromere regions. Cell. 1995;83:247–256. doi: 10.1016/0092-8674(95)90166-3. [DOI] [PubMed] [Google Scholar]
- Kitajima TS, Kawashima SA, Watanabe Y. The conserved kinetochore protein shugoshin protects centromeric cohesion during meiosis. Nature. 2004;427:510–517. doi: 10.1038/nature02312. [DOI] [PubMed] [Google Scholar]
- Lo AWI, Magliano DJ, Sibson MC, Kalitsis P, Craig JM, Choo KHA. A novel chromatin immunoprecipitation and array (CIA) analysis identifies a 460-kb CENP-A-binding neocentromere DNA. Genome Res. 2001;11:448–457. doi: 10.1101/gr.167601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logarinho E, Sunkel CE. The Drosophila POLO kinase localises to multiple compartments of the mitotic apparatus and is required for the phosphorylation of MPM2 reactive epitopes. J Cell Sci. 1998;111:2897–2909. doi: 10.1242/jcs.111.19.2897. [DOI] [PubMed] [Google Scholar]
- Maggert KA, Karpen GH. Acquisition and metastability of centromere identity and function: sequence analysis of a human neocentromere. Genome Res. 2000;10:725–728. doi: 10.1101/gr.10.6.725. [DOI] [PubMed] [Google Scholar]
- Maggert KA, Karpen GH. Neocentromere formation occurs by an activation mechanism that requires proximity to a functional centromere. Genetics. 2001;158:1615–1628. doi: 10.1093/genetics/158.4.1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiato H, Sampaio P, Lemos CL, Findlay J, Carmena M, Earnshaw WC, Sunkel CE. MAST/Orbit has a role inmicrotubule-kinetochore attachment and is essential for chromosome alignment and maintenance of spindle bipolarity. J Cell Biol. 2002;157:749–760. doi: 10.1083/jcb.200201101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiato H, DeLuca J, Salmon ED, Earnshaw WC. The dynamic kinetochore-microtubule interface. J Cell Sci. 2004;117:5461–5477. doi: 10.1242/jcs.01536. [DOI] [PubMed] [Google Scholar]
- McClintock B. The behaviour of successive nuclear divisions of a chromosome broken at meiosis. Proc Natl Acad Sci USA. 1939;25:405–416. doi: 10.1073/pnas.25.8.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TD, Karpen GH. Centromeres take flight: alpha satellite and the quest for the human centromere. Cell. 1998;93:317–320. doi: 10.1016/s0092-8674(00)81158-7. [DOI] [PubMed] [Google Scholar]
- Rieder CL. The structure of the cold-stable kinetochore fiber in metaphase PtK1 cells. Chromosoma. 1981;84:145–158. doi: 10.1007/BF00293368. [DOI] [PubMed] [Google Scholar]
- Rogers GC, Rogers SL, Schwimmer TA, Ems-McClung SC, Walczak CE, Vale RD, Scholey JM, Sharp DJ. Two mitotic kinesins cooperate to drive sister chromatid separation during anaphase. Nature. 2004;427:364–370. doi: 10.1038/nature02256. [DOI] [PubMed] [Google Scholar]
- Salic A, Waters JC, Mitchison TJ. Vertebrate shugoshin links sister centromere cohesion and kinetochore microtubule stability in mitosis. Cell. 2004;118:567–578. doi: 10.1016/j.cell.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Satinover DL, Vance GH, Van Dyke DL, Schwartz S. Cytogenetic analysis and construction of a BAC contig across a common neocentromeric region from 9p. Chromosoma. 2001;110:275–283. doi: 10.1007/s004120100143. [DOI] [PubMed] [Google Scholar]
- Shields G, Sang JH. Characteristics of five cell types appearing during in vitro culture of embryonic material from Drosophila melanogaster. J Embryol Exp Morphol. 1970;23:53–69. [PubMed] [Google Scholar]
- Starr DA, Williams BC, Hays TS, Goldberg ML. ZW10 helps recruit dynactin and dynein to the kinetochore. J Cell Biol. 1998;142:763–774. doi: 10.1083/jcb.142.3.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoler S, Keith KC, Curnick KE, Fitzgerald-Hayes M. A mutation in CSE4, an essential gene encoding a novel chromatinassociated protein in yeast, causes chromosome nondisjunction and cell cycle arrest at mitosis. Genes Dev. 1995;9:573–586. doi: 10.1101/gad.9.5.573. [DOI] [PubMed] [Google Scholar]
- Sullivan BA, Karpen GH. Centromeric chromatin exhibits a histone modification pattern that is distinct from both euchromatin and heterochromatin. Nat Struct Mol Biol. 2004;11:1076–1083. doi: 10.1038/nsmb845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan BA, Willard HF. Stable dicentric X chromosomes with two functional centromeres. Nat Genet. 1998;20:227–228. doi: 10.1038/3024. [DOI] [PubMed] [Google Scholar]
- Sullivan W, Ashburner M, Hawley RS. Drosophila Protocols. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 2000. [Google Scholar]
- Sullivan BA, Blower MD, Karpen GH. Determining centromere identity: cyclical stories and forking paths. Nat Rev Genet. 2001;2:584–596. doi: 10.1038/35084512. [DOI] [PubMed] [Google Scholar]
- Tomonaga T, Matsushita K, Yamaguchi S, Oohashi T, Shimada H, Ochiai T, Yoda K, Nomura F. Overexpression and mistargeting of centromere protein-A in human primary colorectal cancer. Cancer Res. 2003;63:3511–3516. [PubMed] [Google Scholar]
- Torok T, Harvie PD, Buratovich M, Bryant PJ. The product of proliferation disrupter is concentrated at centromeres and required for mitotic chromosome condensation and cell proliferation in Drosophila. Genes Dev. 1997;11:213–225. doi: 10.1101/gad.11.2.213. [DOI] [PubMed] [Google Scholar]
- Van Hooser AA, Ouspenski II, Gregson HC, Starr DA, Yen TJ, Goldberg ML, Yokomori K, Earnshaw WC, Sullivan KF, Brinkley BR. Specification of kinetochore-forming chromatin by the histone H3 variant CENP-A. J Cell Sci. 2001;114:3529–3542. doi: 10.1242/jcs.114.19.3529. [DOI] [PubMed] [Google Scholar]
- Williams BC, Li Z, Liu S, Williams EV, Leung G, Yen TJ, Goldberg ML. Zwilch, a new component of the ZW10/ROD complex required for kinetochore functions. Mol Biol Cell. 2003;14:1379–1391. doi: 10.1091/mbc.E02-09-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.