

Abstract
Potent antituberculosis aryl oxazoles can be made in an efficient three step process--formation of β-hydroxy amides with serine benzyl ester; cyclization to afford oxazolines; and then dehydration to give the corresponding oxazoles. Furthermore, incorporation of an appropriate aryl halide allows utilization of the Suzuki cross coupling reaction to access new chemical space and more elaborate analogs. The compounds prepared by this method were shown to possess improved activity against M. tuberculosis, extremely low toxicity toward VERO cells and, as a result, high therapeutic indexes.

Keywords: ANTI TUBERCULOSIS AGENTS, SUZUKI COUPLING, ARYL OXAZOLES
INTRODUCTION
Tuberculosis (TB) is a serious global health risk resulting in an estimated 1,700,000 deaths in 2006 (1.5 million in HIV-negative people and 0.2 million in HIV-positive people).1 Moreover, there were a staggering 14,400,000 prevalent cases estimated worldwide in 2006 and 83% of the total cases were located in the African, South-East Asia and Western Pacific regions.1 Mycobacterium tuberculosis, the causative agent of TB, is an airborne pathogen that can be spread when infected people come in close contact with others such as in trans-Atlantic flights. Because it can and will often lie dormant in a latent state for many years, it is a silent killer, especially among the poor, HIV-infected, immune compromised and elderly. To make matters worse, multiple-drug resistant TB (MDR-TB, strains that are resistant to first line drugs) cases and extensively drug resistant TB (XDR-TB, strains that are resistant to the best second line drugs) are a growing concern.2 The current treatment for active drug sensitive TB involves the administration of isoniazid, pyrazinamide, ethambutol and rifampin for two months followed by four additional months of rifampin and isoniazid.3 The treatment of latent TB involves strict administration of isoniazid over nine months.3
Given that the current TB drug treatment utilizes various small heterocyclic compounds there is an opportunity as well as a challenge to replace them with more therapeutically effective and cost-effective alternatives. Our laboratory was the first to synthesize Mycobactins S and T, iron-chelating siderophores of Mycobacterium smegmatis and Mycobacterium tuberculosis (respectively) that structurally differ at only one stereogenic center, and show that a simple controlled variation of the siderophore structure results in either promotion or inhibition of the growth of tuberculosis.4–6 This work led to the design and synthesis of an unnatural siderophore anti-TB agent 1 (ND-005825)7 and, through the broad screening of protected and unprotected intermediates, the discovery that the bis-benzyl protected oxazoline, 2 (ND-005859), had notable activity against M. tuberculosis H37Rv (Scheme 2). We recently described fundamental structure-activity-relationship (SAR) studies of these oxazoline (2) / oxazole (3) scaffolds8 and herein we describe how further elaboration through the use of the Suzuki coupling reactions has generated bi-aryl oxazole derivatives (4) with improved anti-TB activity.
Scheme 2.

SAR of analogs 1–4 indicate that further optimization by Suzuki cross coupling is warranted.
RESULTS AND DISCUSSION
As illustrated in Scheme 3, the process for the preparation of oxazole-based anti-TB inhibitors, such as 4, is both straightforward and scalable, important attributes since any useful new anti-TB therapeutic agent will need to be made more cost effectively than drugs that target most other diseases. Thus, conversion of the starting carboxylic acid (5) to an acid chloride (6), subsequent coupling with L-serine benzyl ester to give β-hydroxy amide (7) followed by dehydrative cyclizaton to oxazoline (8) with bis(2-methoxyethyl)amino-sulfur trifluoride (DAST) as reported by Wipf et. al.9 and then a final oxidation with bromine trichloride and DBU produced the desired the oxazole (4) in 69% yield over the entire sequence.
Scheme 3.

Reagents and conditions: (a) Oxalyl chloride, DCM, DMF (catalytic), room temp, 4 h; (b) L-Serine-OBn, Et3N, DCM, 50 °C, 14 h,; (c) DAST, DCM, K2CO3, −78 °C, 1 h; (d) DBU, BrCCl3, DCM, 0 °C to room temp, 1 h.
While it is possible to generate many analogs through this four step procedure and screen all intermediates, our previously generated SAR suggested that the oxazole scaffold was, on average, more active than the oxazoline scaffold and an optimal place to focus efforts. Therefore, we decided to utilize the Suzuki cross coupling reaction10 on a common intermediate instead of using the process shown in Scheme 3 which would require running thirty-six reactions to produce nine new oxazoles useful for SAR studies. In effect, use the Suzuki reaction to generate the SAR also was “green” and in the spirit of continued sustainability of our resources as it not only eliminated an additional 24 chemical reactions if the analogs were made one by one according to Scheme 3 but saved all the energy, solvents and waste generated from each step! Scheme 4 shows the synthesis of the penultimate iodo intermediate (12) which was used in the Suzuki chemistry to make the panel of di-aryl oxazoles (14 – 22) for subsequent determination of anti-TB activity. The synthesis involved an EDC coupling of 4-iodobenzoic acid (9) and L-serine benzyl ester, then cyclization of the β-hydroxy amide (10) with DAST to give oxazoline (11). Next, the iodo-phenyloxazole (12) was produced through a mild oxidation with bromotrichloromethane and DBU and then the oxaozle and requisite boronic acids were coupled using palladium tetrakis(triphenylphosphine)palladium(0) and potassium carbonate in 1,4-dioxane to give the desired compounds for SAR studies.
Scheme 4.

Reagents and conditions: (a) L-Serine-OBn, EDC, DMAP, ACN, room temp, 14 h; (b) DAST, K2CO3, −78°C, 1 h; (c) DBU, BrCCl3, DCM, 0°C to RT, 1 h; (d) R1-B(OH)2, K2CO3, Pd(PPh3)4, 1,4-Dioxanes, reflux, 16 h.
Nine analogs were prepared by the Suzuki chemistry and their activity (MIC = minimum inhibitory concentration effecting a reduction in metabolic activity of ≥90%) was determined against M. tuberculosis H37Rv and their toxicity assessed by determination of IC50 values (inhibitory concentration effecting a decrease in tetrazolium dye reduction of 50%) in a VERO11 (African Green Monkey kidney epithelial) cell viability assay. The results are shown in Table 1 along with the unsubstituted di-phenyl oxazole (4), first line TB drug rifampin and clinical candidate PA-82412 as points of reference.
Table 1.
Antituberculosis structure-activity-relationships (SAR) of di-aryl oxazole compounds.
| Compound ID | R1 | GAS (µM)a | GAST (µM)b | VERO (µM)c |
|---|---|---|---|---|
| ND-006759 (4) | 1.86 | 3.36 | >128 | |
| ND-007183 (14) | 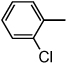 |
7.60 | 26.6 | >128 |
| ND-007185 (15) | 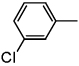 |
4.10 | 6.80 | >128 |
| ND-007225 (16) | 3.71 | 2.45 | >128 | |
| ND-007148 (17) | 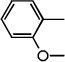 |
3.05 | 13.2 | >128 |
| ND-007181 (18) | 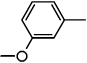 |
1.61 | 3.06 | >128 |
| ND-007182 (19) | 51.3 | 62.4 | >128 | |
| ND-007143 (20) | 3.60 | 7.37 | >128 | |
|
|
0.60 | 0.95 | >128 | |
| ND-007227 (22) | 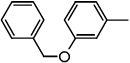 |
3.84 | 1.59 | 3.63 |
| Rifampin (control) |  |
0.06 | 0.01 | 124.7 |
| PA-824 (control) |  |
0.31 | 0.12 | >128 |
H37Rv TB assay using glycerol-alanine-salts medium (GAS).
H37Rv TB assay using glycerol-alanine-salts medium without added iron but with Tween 80 (GAST).
Toxicity assay using kidney epithelial cells of the African Green Monkey (VERO).
In whole cell assays such as the ones reported here it is not unexpected to see about a 3-fold range in the MIC values as demonstrated by the MIC range of control Rifampin (from 0.04 to 0.11 µM) when screened over 100 times. Taking that into consideration the following SAR trends can be inferred. First, with the exception of 22, the compounds were found to be non-toxic for VERO cells. Second, that the activities of these compounds are not dependant upon either iron or the detergent Tween as the potency of the compounds is within error in both the GAS and GAST assays (see Table 1). Lastly, 21 (ND-007186) is a substantially more potent compound and 19 (ND-007182) is the most inactive compound with the others being of consistently moderate activity despite the vast differences in substituents on the pendant aryl group.
CONCLUSION
Utilization of the Suzuki cross coupling reaction facilitated the rapid synthesis of various potent anti-TB analogs based on a general oxazole benzyl ester core. While, the majority of analogs produced have anti-TB activity slightly less than that of the current clinical candidate PA-824, one analog 21 is similarly potent to PA-824 with the added benefit of having no stereogenic centers and can be produced in only one step from the iodo intermediate 12 which, in turn is readily made in only four steps from readily available starting materials. If further studies demonstrate continued efficacy of this new class of anti-TB agents, additional SAR done by simple Suzuki couplings might produce new analogs of equal or improved potency against TB. This “green” approach to anti-TB analog generation further demonstrates the power and value of the Suzuki coupling to medicinal chemistry.
EXPERIMENTAL
All anhydrous solvents, reagent grade solvents for chromatography and starting materials were purchased from either Aldrich Chemical Co. (Milwaulkee, WI) or Fisher Scientific (Suwanee, GA). Water was distilled and purified through a Milli-Q water system (Millipore Corp., Bedford, MA). General methods of purification of compounds involved the use of silica cartridges purchased from AnaLogix, Inc. (Burlington, WI; www.varianinc.com) and/or recrystallization. The reactions were monitored by thin-layer chromatography (TLC) on precoated Merck 60 F254 silica gel plates and visualized using UV light (254 nm). All compounds were analyzed for purity and characterized by 1H and 13C NMR using Varian 300MHz NMR and/or Varian 500 MHz NMR spectrometers. Chemical shifts are reported in ppm (δ) relative to the residual solvent peak in the corresponding spectra; chloroform 7.26 ppm and 77.23 ppm, methanol 3.31 ppm and 49.00 ppm and coupling constants (J) are reported in hertz (Hz) (where, s = singlet, bs = broad singlet, d = doublet, dd = double doublet, bd = broad doublet, ddd = double doublet of dublet, t = triplet, tt – triple triplet, q = quartet, m = multiplet) and analyzed using MestReC NMR data processing. Mass spectra values are reported as m/z. Melting points were measured on a Thomas-Hoover capillary melting point apparatus and are uncorrected. The LC/HRMS analyses were carried out on a Dionex RSLC Ultimate 3000 instrument consisting of a photodiode array detector, using the Acclaim RSLC 120 C18 2.2 µm particle size, 2.1 × 100 mm column (Dionex Corporation, Sunnyvale, CA, www.dionex.com). Column Temp: 50°C, Post-column Temp: 40 °C. Mobile phases: Millipore purified water with 0.1% formic acid (A) HPLC grade acetonitrile with 0.1% formic acid (B). The column was eluted with 30% A and 70% B from T = 0 to T = 3 minutes at 0.5 mL/min. Then T = 6 to T = 8 minutes the column was eluted with 10% A and 90% B at 0.5 mL/min. Lastly, T = 8.1 to T = 10 minutes the column was eluted with 30% A and 70% B at 0.5 mL/min. The MS electrospray source operated at capillary voltage 3.5 kV and a desolvation temperature 300 °C. All reactions were conducted under argon unless otherwise noted. Solvents were removed in vacuo on a rotary evaporator. Abbreviations: DCM = dichloromethane; DMF = dimethylformamide; ACN = acetonitrile; EtOAc = ethyl acetate; HOAc = acetic acid; DAST = Bis(2-methoxyethyl)amino-sulfur trifluoride; EDC = N-(3-Dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride.
Benzyl 2-(biphenyl-4-yl)oxazole-4-carboxylate (4)
Biphenyl-4-carboxylic acid (1.00 g, 5.05 mmol) was dissolved in 25 mL of anhydrous DCM and then oxalyl chloride (0.97 mL, 11.4 mmol) was added drop wise followed by the addition of 20 µL of DMF to catalyze the reaction. The reaction mixture bubbled and was stirred at room temperature for 4h. During this time it became a homogeneous solution. The reaction mixture was concentrated to give a yellow paste which was subsequently concentrated in vacuo. Toluene was added to the residue and then evaporated under reduced pressure repeatedly to facilitate removal of excess reagent. Biphenyl-4-carbonyl chloride (6) was thus obtained as a crude yellow solid (1.09 g, quantitative) which was used immediately in the subsequent reaction.
Biphenyl-4-carbonyl chloride (1.09 g, 5.05 mmol) was dissolved in 25 mL of DCM and then L-serine benzyl ester hydrochloride (1.23 g, 5.30 mmol) and triethylamine (1.45 mL, 10.3 mmol) were added while the reaction was chilled in an ice bath (0 °C bath temperature). The reaction mixture was warmed to room temperature and then heated to reflux, 50 °C (bath temperature) for 14 h. The reaction mixture was cooled, washed with 10% aqueous NaHCO3 solution (2×), water, then washed with 0.5 N citric acid (2×) and then brine. The organic phase was collected and dried over Na2SO4, filtered and then concentrated in vacuo to give (S)-benzyl 2-biphenyl-4-ylcarboxamido-3-hydroxypropanoate (7, 5.04 g, 99%) as an off white solid which was used without further purification. 1H NMR (300 MHz, CDCl3) δ 7.94-7.87 (2 H, m), 7.71-7.59 (4 H, m), 7.52-7.34 (6 H, m), 7.26 (2 H, s), 7.14 (1 H, s, NH), 5.28 (2 H, d, J = 1.5 Hz), 4.99-4.91 (1 H, m), 4.11 (2 H, d, J = 4.1 Hz).
(S)-benzyl 2-biphenyl-4-ylcarboxamido-3-hydroxypropanoate (1.89 g, 5.04 mmol) was dissolved in DCM (25 mL) and cooled to −78 °C (bath temp, dry ice-acetone). DAST (0.77 mL, 5.80 mmol) was added slowly to the cold mixture under argon and the reaction was stirred for 30 min. Then K2CO3 (1.74 g, 12.6 mmol) was added and the reaction was allowed to warm to room temperature. Once the reaction appeared complete by TLC analysis, the mixture was poured into 30 mL of 10% aqueous NaHCO3 and extracted with DCM (2×). The organic layers were combined and washed again with 10% aqueous NaHCO3 solution, brine, and then dried over Na2SO4, filtered and concentrated in vacuo. Crude material obtained was titurated by dissolving it in DCM and adding small amounts of hexanes until the mixture became slightly turbid. The mixture was then cooled in a freezer (< 0 °C) overnight to give 1.03 g (57%) of (S)-benzyl 2-(biphenyl-4-yl)-4,5-dihydrooxazole-4-carboxylate (8) as a white solid. Mp 105–106 °C; 1H NMR (300 MHz, CDCl3) δ 8.05 (2 H, d, J = 8.21 Hz), 7.64 (4 H, dd, J = 10.7, 4.1 Hz), 7.53-7.21 (8 H, m), 5.27 (2 H, q, J = 12.3, 12.3, 12.3 Hz), 5.01 (1 H, dd, J = 10.6, 7.9 Hz), 4.77-4.53 (2 H, m). HRMS (EI), M+1 calcd. for C23H20NO3, 358.1443; found 358.1442.
(S)-benzyl 2-(biphenyl-4-yl)-4,5-dihydrooxazole-4-carboxylate (8, 853 mg, 2.39 mmol) was dissolved in 15 mL of dry DCM and chilled in an ice bath (0 °C bath temperature). Next, DBU (1.07 mL, 7.17 mmol) was added followed by slow addition of bromotrichloromethane (0.71 mL, 7.17 mmol). The reaction was stirred for 1 h and then allowed to warm to room temperature. Once the reaction appeared complete by TLC analysis, the reaction mixture was poured into 20 mL of 10% aqueous NaHCO3 solution and extracted with DCM (2×). The combined organic layers were washed again with the NaHCO3 solution, brine, dried over Na2SO4, filtered and then concentrated in vacuo. Crude material obtained was purified by silica gel column chromatography with DCM to give 588 mg (69%) of 4 as a white solid. Mp 110–111 °C; 1H NMR (300 MHz, CDCl3) δ 8.30 (1 H, s), 8.20-8.16 (2 H, m), 7.78-7.57 (4 H, m), 7.55-7.21 (8 H, m), 5.43 (2 H, s). HRMS (EI), M+1 calcd. for C23H18NO3, 356.1281; found 356.1275. HPLC retention time = 2.4 – 2.6 min.
Benzyl 2-(4-iodophenyl)oxazole-4-carboxylate (12)
4-Iodobenzoic acid (5.18 g, 20.9 mmol) and L-serine benzyl ester hydrochloride (4.61 g, 19.9 mmol) were dissolved in 70 mL of DCM and treated with EDC (4.58 g, 23.9 mmol) and triethylamine (5.5 mL, 39.8 mmol). The resulting solution was stirred for 12 h at room temperature. The reaction mixture was diluted with DCM, washed with 0.5 N citric acid (2×), water and 10% aqueous NaHCO3 solution (2×). The organic phase was collected, dried over sodium sulfate (Na2SO4), filtered and then concentrated in vacuo. Crude material obtained was purified by silica gel column chromatography using 10% EtOAc in DCM as the eluent to give 5.58 g (66%) of (S)-benzyl 3-hydroxy-2-(4-iodobenzamido)propanoate (10) as a white solid. 1H NMR (300 MHz, CDCl3) δ 7.84 (2 H, d, J = 8.1 Hz), 7.58 (2 H, d, J = 8.1 Hz), 7.40 (4 H, s), 7.30 (1 H, s), 7.10 (1H, s, NH), 5.30 (2 H, d, J = 1.3 Hz), 4.97-4.88 (1 H, m), 4.13 (2 H, m).
(S)-benzyl 3-hydroxy-2-(4-iodobenzamido)propanoate (10, 5.58 g, 13.1 mmol) was dissolved in DCM (200 mL) and cooled to −78 °C (bath temp, dry ice-acetone). DAST (2.0 mL, 15.1 mmol) was added slowly to the cold mixture under argon and the reaction was stirred for 30 min. Then K2CO3 (4.90 g, 35.4 mmol) was added and the reaction was allowed to warm to room temperature. Once the reaction appeared complete by TLC analysis, the mixture was poured into 220 mL of 10% aqueous NaHCO3 and extracted with DCM (2×). The combined organic layers were washed again with the NaHCO3 solution, brine, and then dried over Na2SO4, filtered and concentrated in vacuo. Crude material obtained was purified by silica gel column chromatography with 10% EtOAc in DCM as the eluent to give 5.01 g (94%) of (S)-benzyl 2-(4-iodophenyl)-4,5-dihydrooxazole-4-carboxylate (11) as a white solid. Mp 75–76 °C; 1H NMR (300 MHz, CDCl3) δ 7.84-7.63 (4 H, m), 7.46-7.19 (5 H, m), 5.31-5.19 (2 H, m), 4.96 (1 H, dd, J = 10.6, 7.9 Hz), 4.74-4.50 (2 H, m). HRMS (EI), M+1 calcd. for C17H15INO3, 408.0091 found 408.0087. HPLC retention time = 1.7 – 1.8 min.
(S)-benzyl 2-(4-iodophenyl)-4,5-dihydrooxazole-4-carboxylate (11, 4.20 g, 10.3 mmol) was dissolved in 90 mL of dry DCM and chilled in an ice bath (0 °C bath temperature). Next, DBU (3.85 mL, 25.8 mmol) was added followed by slow addition of bromotrichloromethane (2.56 mL, 25.8 mmol). The reaction was stirred for 1 h cold and then allowed to warm to room temperature. Once the reaction appeared complete by TLC analysis, the reaction mixture was poured into a 10% aqueous NaHCO3 solution and extracted with DCM (2×). The combined organic layers were washed again with the NaHCO3 solution, brine, dried over Na2SO4, filtered and then concentrated in vacuo. Crude material obtained was purified by silica gel column chromatography with 10% EtOAc in DCM as the eluent to give 3.51 mg (84%) of 12 as a white solid. Mp 130–131 °C; 1H NMR (300 MHz, CDCl3) δ 8.27 (1 H, s), 7.91-7.74 (4 H, m), 7.51-7.21 (5 H, m), 5.40 (2H, s). HRMS (EI), M+1 calcd. for C17H13INO3, 405.9935 found 405.9944 (difference of 0.9 amu). HPLC retention time = 4.8 – 4.9 min.
General procedure for Suzki coupling
Benzyl 2-(4-iodophenyl)oxazole-4-carboxylate (12, 225 mg, 0.56 mmol) was dissolved in 1,4-dioxane (2 mL, which was then degassed for 10 min with argon) and then 4-methoxyphenylboronic acid (96 mg, 0.61 mmol, 1.1 equiv.) and K2CO3 (153 mg, 1.11 mmol, 2 equiv.) were added followed by the canary yellow palladium tetrakis(triphenylphosphine)palladium(0) (122 mg, 0.17 mmol, 0.3 equiv.). The reaction was heated to 110°C under argon for 16 h. The reaction mixture was then cooled to room temperature, diluted with DCM and filtered in vacuo through a pad of Celite®. The filtrate was washed with 0.5 N citric acid (2×), water and 10% aqueous NaHCO3 (2×). The organic phase was collected, dried over sodium sulfate (Na2SO4), filtered and then concentrated in vacuo. Crude material obtained was purified by silica gel column chromatography with a 30% to 100% hexanes : DCM solvent gradient to give desired product an a colored solid.
Benzyl 2-(2'-chlorobiphenyl-4-yl)oxazole-4-carboxylate (14)
Yield: 135 mg (62%), light orange oil; 1H NMR (300 MHz, CDCl3) δ 8.33 (1 H, s), 8.24-8.19 (2 H, m), 7.63-7.57 (2 H, m), 7.56-7.29 (9 H, m), 5.45 (2 H, s). HRMS (EI), M+1 calcd. for C23H17ClNO3, 390.0891 found 390.0894. HPLC retention time = 3.0 – 3.1 min.
Benzyl 2-(3'-chlorobiphenyl-4-yl)oxazole-4-carboxylate (15)
Yield: 46 mg (21%), light orange solid; mp 92–93 °C; 1H NMR (300 MHz, CDCl3) δ 8.33 (1 H, s), 8.22 (2 H, dd, J = 8.6, 2.1 Hz), 7.70 (4 H, td, J = 12.6, 5.3, 5.3 Hz), 7.53-7.40 (7 H, m), 5.45 (2 H, s). HRMS (EI), M+1 calcd. for C23H17ClNO3, 390.0891 found 390.0912. HPLC retention time = 2.4 – 2.6 min.
Benzyl 2-(4'-chlorobiphenyl-4-yl)oxazole-4-carboxylate (16)
Yield: 47 mg (58%), light orange solid; mp 120–121 °C; 1H NMR (300 MHz, CDCl3) δ 1H NMR (300 MHz, CDCl3) δ ppm 8.33 (1 H, s), 8.21-8.16 (2 H, m), 7.68-7.65 (2 H, m), 7.60-7.54 (2 H, m), 7.47-7.36 (7 H, m), 5.45 (2 H, s). HRMS (EI), M+1 calcd. for C23H17ClNO3, 390.0891 found 390.0890. HPLC retention time = 3.6 – 3.7 min.
Benzyl 2-(2'-methoxybiphenyl-4-yl)oxazole-4-carboxylate (17)
Yield: 57 mg (24%), light orange solid; mp 84–85 °C; 1H NMR (300 MHz, CDCl3) δ 8.28 (1 H, s), 8.14 (2 H, d, J = 8.2 Hz), 7.65 (2 H, d, J = 8.4 Hz), 7.51-7.32 (7 H, m), 7.04 (2 H, d, J = 8.5 Hz), 5.42 (2 H, s), 3.83 (3 H, s). HRMS (EI), M+1 calcd. for C24H20NO4, 386.1387 found 386.1392. HPLC retention time = 2.3 – 2.4 min.
Benzyl 2-(3'-methoxybiphenyl-4-yl)oxazole-4-carboxylate (18)
Yield: 68 mg (36%), off white solid; mp 133–134 °C; 1H NMR (300 MHz, CDCl3) δ 8.29 (1 H, s), 8.17 (2 H, m), 7.70 (2 H, m), 7.42 (6 H, m), 7.20 (2 H, ddd, J = 14.7, 5.8, 1.4 Hz), 6.94 (1 H, m), 5.42 (2 H, s), 3.88 (3 H, s). HRMS (EI), M+1 calcd. for C24H20NO4, 386.1387 found 386.1389. HPLC retention time = 2.4 – 2.6 min.
Benzyl 2-(4'-methoxybiphenyl-4-yl)oxazole-4-carboxylate (19)
Yield: 64 mg (30%), off white solid; mp 161–162 °C; 1H NMR (300 MHz, CDCl3) δ 8.28 (1 H, s), 8.15 (2 H, d, J = 8.24 Hz), 7.61 (4 H, m), 7.42 (5 H, m), 7.00 (2 H, d, J = 8.76 Hz), 5.42 (2 H, s), 3.87 (3 H, s). HRMS (EI), M+1 calcd. for C24H20NO4, 386.1387 found 386.1382. HPLC retention time = 2.4 – 2.5 min.
Benzyl 2-(4'-(trifluoromethoxy)biphenyl-4-yl)oxazole-4-carboxylate (20)
Yield: 43 mg (31%), light orange solid; mp 104–105 °C; 1H NMR (300 MHz, CDCl3) δ 8.31 (1 H, s), 8.24-8.16 (2 H, m), 7.85-7.29 (11 H, m), 5.41 (2 H, s). HRMS (EI), M+1 calcd. for C24H17F3NO4, 440.1104 found 440.1111. HPLC retention time = 3.9 – 4.0 min.
Benzyl 2-(4'-cyanobiphenyl-4-yl)oxazole-4-carboxylate (21)
Yield: 50 mg (24%), tan solid; mp 156–157 °C; 1H NMR (300 MHz, CDCl3) δ 8.33 (1 H, s), 8.28-8.24 (2 H, m), 7.80-7.71 (6 H, m), 7.46 (5 H, tdd, J = 15.3, 7.7, 2.0, 2.0 Hz), 5.45 (2 H, s). HRMS (EI), M+1 calcd. for C24H17N2O3, 381.1234 found 381.1228. HPLC retention time = 1.7 – 1.8 min.
Benzyl 2-(3'-(benzyloxy)biphenyl-4-yl)oxazole-4-carboxylate (22)
Yield: 84 mg (70%), off white solid; mp 109–110 °C; 1H NMR (300 MHz, CDCl3) δ 8.29 (1 H, s), 8.22-8.14 (2 H, m), 7.76-7.61 (3 H, m), 7.53-7.30 (10 H, m), 7.24 (2 H, t, J = 4.4, 4.4 Hz), 7.04-6.98 (1 H, m), 5.42 (2 H, s), 5.14 (2 H, s). HRMS (EI), M+1 calcd. for C30H24F3NO4, 462.1700 found 462.1706. HPLC retention time = 4.7 – 4.9 min.
Anti-tuberculosis and Cytotoxicity Assays
Activity against replicating Mycobacterium tuberculosis H37Rv (ATCC 27294, American Type Culture Collection, Rockville, MD) was determined using a fluorescence readout in the Microplate Alamar Blue Assay (MABA)13–16 following incubation for one week with test compounds in glycerol-alanine-salts medium (GAS) as well as in medium without added iron but with Tween 80 (GAST).16 The MIC was defined as the minimum concentration inhibiting fluorescence by 90% relative to bacteria-only controls. Toxicity to African green monkey kidney cells (VERO) was determined using a colorimetric assay as previously described.8
Scheme 1.

First line anti-TB drugs for the treatment of active and latent forms of TB.
ACKNOWLEDGEMENTS
This work was supported by Grant AI054193 from the National Institutes of Health. We would like to thank the University of Notre Dame especially the Mass Spectrometry & Proteomics Facility (Bill Boggess, Nonka Sevova, Michelle Joyce) which is supported by the by Grant CHE-0741793 from the National Science Foundation and Prof. Jennifer DuBois for profound scientific discussion. The excellent technical assistance of Baojie Wan and Yuehong Wang with anti-TB assays at UIC is greatly appreciated.
REFERENCES
- 1.WHO report 2008. WHO/HTM/TB/2008
- 2.Sacchettini JC, Rubin EJ, Freundlich JS. Nat. Rev. Microbiol. 2008;6:41. doi: 10.1038/nrmicro1816. [DOI] [PubMed] [Google Scholar]
- 3.Maher D, Blanc L, Raviglione M. The Lancet. 2004;363:1911. doi: 10.1016/S0140-6736(04)16377-0. [DOI] [PubMed] [Google Scholar]
- 4.Miller M, Zhu H, Xu Y, Wu C, Walz A, Vergne A, Roosenberg J, Moraski G, Minnick A, McKee-Dolence J, Hu J, Fennell K, Dolence K, Dong L, Franzblau S, Malouin F, Mollmann U. Biometals. 2009;22:61. doi: 10.1007/s10534-008-9185-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walz AJ, Moellmann U, Miller MJ. Org. Biomol. Chem. 2007;5:1621. doi: 10.1039/b703116e. [DOI] [PubMed] [Google Scholar]
- 6.Hu JD, Miller MJ. J. Am. Chem. Soc. 1997;119:3462. [Google Scholar]
- 7.Xu Y, Miller MJ. J. Org. Chem. 1998;63:4314. [Google Scholar]
- 8.Moraski GC, Chang M, Villegas-Estrada, A A, Franzblau SG, Mollmann U, Miller MJ. 2009 doi: 10.1016/j.ejmech.2009.12.074. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Phillips AJ, Uto Y, Wipf P, Reno MJ, Williams DR. Org. Lett. 2000;2:1165. doi: 10.1021/ol005777b. [DOI] [PubMed] [Google Scholar]
- 10.Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457. [Google Scholar]
- 11.Gommer AM. Dev. Biol. Stand. 1996;86:217. [PubMed] [Google Scholar]
- 12.Stover CK, Warrener P, VanDevanter DR, Sherman DR, Arain TM, Langhorne MH, Anderson SW, Towell JA, Yuan Y, McMurray DN, Kreiswirth BN, Barry CE, Baker WR. Nature. 2000;405:962. doi: 10.1038/35016103. [DOI] [PubMed] [Google Scholar]
- 13.Collins LA, Franzblau SG. Antimicrob Agents Chemother. 1997;41:1004–1009. doi: 10.1128/aac.41.5.1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Changsen C, Franzblau SG, Palittapongarnpim P. Antimicrob Agents Chemother. 2003;47:3682. doi: 10.1128/AAC.47.12.3682-3687.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Voss JJ, Rutter K, Schroeder BG, Su H, Zhu YQ, Barry CE. Proc. Natl. Acad. Sci. U. S. A. 2000;97:1252. doi: 10.1073/pnas.97.3.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cho SH, Warit S, Wan B, Hwang CH, Pauli GF, Franzblau SG. Antimicrob. Agents Chemother. 2007;51:1380. doi: 10.1128/AAC.00055-06. [DOI] [PMC free article] [PubMed] [Google Scholar]