

Abstract
We previously reported that the NH2-terminal variable region of cardiac troponin T (cTnT) is removed by restrictive μ-calpain cleavage in myocardial ischemia-reperfusion (Zhang, Biesiadecki, Jin, Biochemistry 45:11681–94, 2006). Selective removal of the NH2-terminal variable region of cTnT had a compensatory effect on myocardial contractility (Feng, Biesiadecki, Yu, Hossain, Jin, J. Physiol. 586:3537–50, 2008). Here we further studied this posttranslational modification under pathophysiological conditions. Thrombin perfusion of isolated mouse hearts and cardiomyocytes induced the production of NH2-terminal truncated cTnT (cTnT-ND), suggesting a role of calcium overloading. Ouabain treatment of primary cultures of mouse cardiomyocytes in hypokalemic media, another calcium overloading condition, also produced cTnT-ND. Exploring the molecular mechanisms, we found that cTnT phosphorylation was primarily in the NH2-terminal region and the level of cTnT phosphorylation did not change under the calcium overloading conditions. However, alternatively spliced cTnT variants differing in the NH2-terminal primary structure produced significantly different levels of cTnT-ND in vivo in transgenic mouse hearts. The results suggest that stress conditions involving calcium overloading may convey an increased sensitivity of cTnT to the restrictive μ-calpain proteolysis, in which structure of the NH2-terminal variable region may play a determining role.
Keywords: NH2-terminal variable region of troponin T, Restrictive proteolysis, Calcium overloading, Phosphorylation, Cardiac myocytes, Myocardial adaptation
The troponin complex in sarcomeric thin filaments plays a central role in the Ca2+-activation of cardiac and skeletal muscle contraction [1]. Troponin T (TnT) is the tropomyosin-binding subunit of the troponin complex [2]. Three muscle type-specific (fast skeletal, slow skeletal and cardiac) TnT genes have evolved in vertebrates and alternative RNA splicing further produces multiple protein isoforms [3, 4]. The TnT isoforms and alternatively spliced variants differ mainly in their NH2-terminal region of which the alternative splicing is regulated during development [5, 6] and skeletal muscle adaptations to unloading [7] or abnormal neurological stimuli [8]. Aberrant splicing of cardiac TnT (cTnT) in the NH2-terminal region has been found in hypertrophic and failing human hearts [9] and animals with dilated cardiomyopathy [10, 11].
The alternatively spliced NH2-terminal variable region of TnT does not contain binding sites for any known proteins in the myofilaments [12–14], and its removal did not abolish the core function of troponin [15–18]. However, structural variations in the NH2-terminal region of TnT affected the Ca2+-sensitivity in myosin ATPase activation and force development [19, 20]. Protein conformation and binding studies showed that NH2-terminal alterations in TnT altered the molecular conformation of the conserved middle and COOH-terminal regions [21, 22], and the binding affinities for troponin I (TnI), troponin C (TnC) and tropomyosin [21, 23]. Therefore, the NH2-terminal variable region of TnT is a regulatory structure that modulates muscle contractility and cardiac function. Whereas TnT isoform expression and alternative splicing provide chronic regulations, the NH2-terminal variable region of cTnT can also be restrictively cleaved by myofibril-associated μ-calpain in acute responses to stress conditions such as myocardial ischemia-reperfusion [24] and ventricular pressure overload [25].
The NH2-terminal truncated cTnT (cTnT-ND) remains in the myofilaments and is physiologically functional. Studies of cardiac function demonstrated that transgenic mouse hearts over-expressing cTnT-ND exhibited a unique change of ~5–10% slower systolic velocity, which did not decrease but increased cardiac output by elongating the time of ventricular rapid ejection phase [25]. This novel function suggested a plausible adaptive value of the production of cTnT-ND in compensating for cardiac function under energetic crisis due to ischemia and/or pressure overload.
The mechanisms that activate the restrictive NH2-terminal proteolysis of cTnT remain to be investigated. Intracellular calcium overload is known to be a cause of myocardial injury in stress conditions such as ischemia-reperfusion [26]. On the other hand, the rhythm contraction of cardiac muscle is based on Ca2+-induced allosteric changes in the myofilament [1], therefore, cytosolic free Ca2+ in cardiomyocytes normally rises to near micromolar concentration during contraction-relaxation cycles. In the present study, we investigated the induction of cTnT-ND in the heart and in isolated cardiomyocytes for underlying mechanisms. Pathophysiological calcium overloading conditions induced the production of cTnT-ND independent of the phosphorylation states of cTnT. In contrast, the structure of the NH2-terminal variable region exhibited a strong correlation to the production of cTnT-ND in vivo. The results suggest that calcium overload in cardiomyocytes may convey an increased sensitivity of cTnT to the restrictive μ-calpain cleavage, in which the NH2-terminal structure may play a determining role.
Materials and methods
All animal procedures were approved by the Institutional Animal Care and Use Committee and were conducted in accordance with the Guiding Principles in the Care and Use of Animals, as approved by the Council of the American Physiological Society.
Langendorff retrograde perfusion of mouse heart and thrombin treatment
Among the pathological mechanisms that result in myocardial Ca2+ overload, thrombin increases cytosolic [Ca2+] in cardiomyocytes during ischemia-reperfusion injury [27, 28]. Thrombin treatment was tested ex vivo in Langendorff retrograde perfused mouse hearts. Thirty min after intraperitoneal (i.p.) injection of 100 units of heparin, the mouse was anesthetized with pentobarbital sodium (100 mg/kg body weight, i.p.) and the heart was rapidly isolated and cannulated through the aorta as described [25]. The cannulated heart was immediately mounted for Langendorff perfusion at 65 mmHg pressure with Krebs-Henseleit buffer (118 mM NaCl, 4.7 mM KCl, 2.25 mM CaCl2, 2.25 mM MgSO4, 1.2 mM KH2PO4, 0.32 mM EGTA, 25 mM NaHCO3 and 11 mM D-glucose, pH 7.4) bubbled with 95% O2 and 5% CO2 at 37°C. The heart was supraventricularly paced at 480 beats per minute using an isolated stimulator (A365, World Precision Instrument). Aortic pressure was monitored using an MLT844 pressure transducer (Capto, Horten, Norway) and recorded using Chart 5 computer software (AD Instruments) via a Powerlab/16 SP digital data archiving system (AD Instruments). A 0.5 mL air bubble was placed in the aortic compliance chamber to mimic in vivo arterial compliance [25]. After 30 min equilibration, the perfusate was switched to Krebs-Henseleit buffer containing 1 or 10 Unit/mL thrombin (Sigma) [28]. Every heart in the thrombin treatment and control groups was perfused with 100 mL of circulating buffer for 2 hours. After the treatment, cardiac muscle samples were rapidly frozen on dry ice and stored at −80°C for protein analysis.
Primary culture of neonatal mouse cardiomyocytes
Cardiomyocytes from neonatal C57BL/6 mouse hearts were isolated and cultured using a reported protocol [29] with minor modifications. After euthanasia, the pups were soaked in 70% ethanol before dissection. The hearts were removed under sterile conditions and minced in Ca2+-free Joklik modified Eagles medium (MEM) (Sigma) followed by the addition of 1 mg/mL collagenase I (Sigma). The enzymatic digestion was incubated at 37°C for 30 min with mixing by pipetting every 5 min and stopped by adding Joklik MEM containing 10% fetal bovine serum (FBS). The media containing dispersed cardiomyocytes were put through a polypropylene macroporous filter (100 μm mesh opening) and centrifuged at 150 × g at room temperature for 5 min. The cell pellet was gently re-suspended in Joklik MEM supplemented with penicillin-streptomycin-glutamine and 10% FBS. After gradually restoring Ca2+ in the culture media to 1.4 mM by stepwise additions of 0.1 M CaCl2 in 1 h, the cells were collected by centrifugation as above and re-suspended in M199 media (Gibco) supplemented with penicillin-streptomycin-glutamine and 10% FBS. The isolated cells were first plated on a bacterial culture-quality plastic dish and incubated at 37°C for 1 h to remove the highly adherent cells such as fibroblasts. The not-yet-attached cells were then transferred to a 1% gelatin-coated tissue culture-quality dish (Cellstar). After allowing the cardiomyocytes to adhere at 37°C for 2 h, the plating media and non-attached cells were gently removed and fresh serum-free M199 media was added for continuing culture.
Ouabain treatment of mouse primary cardiomyocytes in low K+ media
In classic inotropic treatment of congestive heart failure, cardiac glycosides are used to elevate cytosolic [Ca2+] in cardiac myocytes through the inhibition of plasma membrane Na+-K+-ATPase [30]. We used ouabain, a rapidly effective cardiac glycoside, to treat cardiomyocytes to produce Ca2+ overloading.
On the third day of culture, the neonatal mouse cardiomyocytes were electrically paced at 1 Hz, 5 V/cm with pulse duration of 5 ms for 2 hrs using an IonOptix C-pace stimulator (IonOptix, Milton, MA). Low and high concentrations (50 μM and 200 μM, respectively) of ouabain were then applied in hypokalemic media (Krebs-Henseleit solution minus KCl) to produce Ca2+ overload in the cardiomyocytes [31]. After incubation under continuous pacing for 2 more hrs, the cardiomyocytes were washed with PBS, lysed in SDS-polyacrylamide gel electrophoresis (PAGE) sample buffer, and processed for SDS-PAGE and Western blotting studies.
Cardiac muscle tissue samples
Immediately after euthanasia, hearts were harvested from C57BL/6 mice and transgenic mice that over-express embryonic cTnT or exon 7-deleted cTnT in adult cardiac muscle driven by an α-myosin heavy chain promoter [11] for biochemical analysis of cardiac muscle proteins. Double transgenic mice expressing both embryonic and exon 7-deleted cTnT in the adult heart were produced by cross breeding of the two transgenic mouse lines. Independent Mendelian segregation of the two transgene alleles was observed, indicating their locations in different chromosomes [32].
SDS-PAGE and Western blotting
Fresh ventricular muscle tissues and cardiomyocytes were immediately processed for SDS-PAGE or frozen at −80°C until use. The muscle tissues or myocytes were rapidly homogenized in Laemmli SDS-PAGE sample buffer containing 2% SDS to avoid enzymatic degradation of proteins, heated at 80°C for 5 min, and clarified by centrifugation. The protein extracts were resolved on 14% Laemmli gel with an acrylamide:bisacrylamide ratio of 180:1. The gels were stained with Coomassie Brilliant Blue R250 to reveal the protein bands. Duplicate gels were electrically blotted to polyvinylidene fluoride (PVDF) membranes similarly to that previously described for nitrocellulose membranes [21]. After blocking in Tris-buffered saline (TBS) containing 1% bovine serum albumin (BSA), the membranes were incubated with anti-TnT monoclonal antibodies (mAbs) CT3 recognizing cardiac and slow TnT [24] and 2C8 recognizing all three TnT isoforms with similar affinities [33], an anti-TnI mAb TnI-1 [34], and anti-cardiac myosin heavy chain mAb FA2 [35] diluted in TBS containing 0.1% BSA at 4°C overnight. The membranes were then washed at high stringency with TBS containing 0.5% Triton X-100 and 0.05% SDS, incubated with alkaline phosphatase-conjugated anti-mouse IgG second antibody (Santa Cruz Biotechnology, CA), washed again, and developed in 5-bromo-4-chloro-3-indolylphosphate/nitro blue tetrazolium substrate solution as described previously [21]. The anti-TnT and anti-TnI mAbs were used together as a mixture in some blots.
For effective separation of cTnT-ND and cTnI by SDS-PAGE, we developed a modified 14% Laemmli gel with an acrylamide:bisacrylamide ratio of 180:1 using a high conductivity resolving gel buffer containing 562.5 mM Tris-base and adjusted to pH 9.0 with glycine.
Phosphoprotein staining
To examine protein phosphorylation, SDS-PAGE gels were stained using the Pro-Q Diamond reagents (Invitrogen) following the manufacturer’s instruction. Phosphoprotein markers (PeppermintStick™, Invitrogen) were used as control. Fluorescence imaging was performed using a Typhoon 9210 scanner (GE Healthcare) with excitation at 532 nm and recording of emission at 580 nm to detect and quantify the phosphoprotein bands.
Data analysis
Densitometry analysis of SDS-PAGE gel and Western blot images scanned at 600 dpi was performed using NIH Image program version 1.61. All values are presented as the mean ± SD or mean ± SEM and statistical significance of the differences was tested using unpaired two-tail Student’s t test or ANOVA Fisher test.
Results
Thrombin treatment in perfused mouse hearts induced restrictive NH2-terminal truncation of cTnT
cTnT-ND was first found in ischemia-reperfused heart tissue and cardiomyocytes [24]. Thrombin is one of the factors that increase intracellular Ca2+ concentration in cardiomyocytes during ischemia-reperfusion [28]. Supporting a role of Ca2+ overload in the production of cTnT-ND, the results in Fig. 1 showed that ex vivo perfusion of adult mouse hearts with thrombin induced cTnT-ND production as compared to that in the control hearts perfused without thrombin.
Fig. 1. Thrombin treatment in ex vivo perfused mouse hearts produced cTnT-ND.
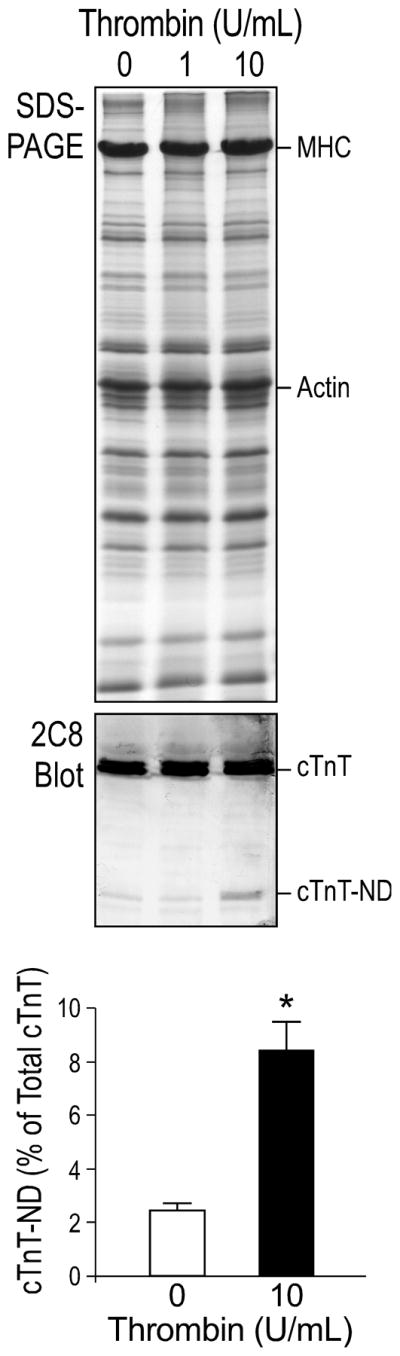
mAb 2C8 Western blots and densitometry quantification showed that Langendorff retrograde perfusion of mouse hearts with 10 U/mL of thrombin increased cTnT-ND production as compared with that in buffer-perfused hearts (*P < 0.05, n = 3 hearts in each group), suggesting a role of myocardial Ca2+ overload in the induction of cTnT-ND. The quantification data are presented as mean ± SD. MHC, myosin heavy chain.
It is worth noting that two variants of cTnT are normally expressed in both embryonic and adult mouse hearts due to the alternative splicing of exon 4 encoding four amino acids in the NH2-terminal variable region (36). These cTnT splicing variants were clearly seen in Fig. 1 (and Western blots in subsequent figures). Consistent with the fact that the restrictive truncation of the NH2-terminal variable region removes this difference, only one band of cTnT-ND was detected.
cTnT-ND produced in cardiomyocytes due to isolation stress was effectively replaced by intact cTnT during culture
We found a significant production of cTnT-ND in neonatal mouse cardiomyocytes due to the perfusion-isolation procedure that simulates the stress in ischemia-reperfusion. This observation indicated that the isolation stress induced the production of cTnT-ND by activating a cellular mechanism in the cardiomyocytes. After removing non-adherent dead cells, a high level of cTnT-ND was detected in the viable cardiomyocytes, demonstrating that the production of cTnT-ND is a response of living cardiomyocytes to stress conditions other than a part of the protein degradation after cell death (Fig. 2A).
Fig. 2. Neonatal mouse cardiomyocytes in primary cultures recovered from isolation stress-induced change of cTnT.

A. The cTnT-ND produced in cardiomyocytes during cell isolation diminished after 3 days of culture. The mAb 2C8 Western blot and densitometry analysis showed that freshly isolated neonatal mouse cardiomyocytes (2 hr attached cells on day 0) had a considerable amount of cTnT-ND. The level of cTnT-ND decreased during culture to nearly non-detectable on days 4 and 5. The results were summarized from four repeated experiments. B. Western blots using mAbs CT3, TnI-1 and FA2 verified the normal expression of cTnT, cardiac TnI (cTnI) and slow TnI (sTnI), and cardiac myosin heavy chain isoforms (MHC) in 3-day cultured neonatal mouse cardiomyocytes, verifying the differentiated state retained during culture.
The isolated neonatal mouse cardiomyocytes were examined in culture at a series of time points up to 5 days. During this short term adherent culture in serum free media, the primary cardiomyocytes retained a differentiated cardiac phenotype as shown by the expression of normal myofilament proteins including cTnT, cardiac TnI, and cardiac myosin (Fig. 2B). No skeletal muscle TnT was detected in the cultured cardiomyocytes and the presence of slow skeletal muscle TnI reflected the normal developmental stage of the neonatal cardiac muscle [37]. The slight decreases in the level of intact cTnT in the 4–5 day cultures shown in the Western blots relative to the level of total cellular actin may reflect an expansion of fibroblast population that contains non-muscle actin. Since we quantified the level of cTnT-ND versus that of intact cTnT in the same sample, the presence of some fibroblasts would not affect our quantification of cTnT modification. However, it is possible that de-differentiation of neonatal cardiomyocytes after 4–5 days of culture also contributed to the decreased level of intact cTnT. Therefore, we chose to use 3-day cultures for further studies.
The mAb 2C8 Western blots in Fig. 2A showed that cTnT-ND gradually diminished during culture and was replaced by intact cTnT with a time frame consistent with the previously reported 3.5-day half-life of cTnT in the heart [38]. The effective synthesis of intact cTnT further demonstrated the viability, differentiated state, and protein synthesis capacity of the cardiomyocytes in culture [39].
Ouabain-generated Ca2+ overload in cardiomyocytes induced cTnT-ND
The primary culture of isolated neonatal mouse cardiomyocytes provided a cellular system to verify the effect of calcium overload on the production of cTnT-ND. As shown in Fig. 3, the background level of cTnT-ND in the day-3 cultures was sufficiently low, allowing the detection of changes in cTnT-ND upon acute treatment of ouabain-hypokalemia, an established condition of calcium overload independent of thrombin treatment. Considering that normal mouse hearts contain cTnT-ND at the level of ~2% of total cTnT (Table 1), this low level of cTnT-ND in the cultured cardiomyocytes represented a nearly in vivo background state for use as an experimental system in our study.
Fig. 3. Calcium agonist induced cTnT-ND in cardiomyocytes.

Primary cultures of neonatal mouse cardiomyocytes were treated on the third day of culture with 50 μM and 200 μM ouabain in low K+ Krebs-Henseleit media under 1 Hz pacing. Western blots using mAb 2C8 and densitometry analysis demonstrated that this calcium overloading condition induced the production of cTnT-ND in a dose dependent manner. MHC, myosin heavy chain. The data were summarized from three repeated experiments.
Table 1.
Quantitative correlations between cTnT NH2-terminal variants and cTnT-ND production in vivo in wild type and transgenic mouse hearts
| cTnT Variants |
Mouse Lines | |||
|---|---|---|---|---|
| Wild Type n = 6 |
Embryonic TG n = 9 |
Exon 7-Deleted TG n = 5 |
Double TG n = 5 |
|
| Adult | 98.0 ± 0.2% | 34.8 ± 1.6% | 19.4 ± 4.2% | 17.7 ± 1.4% |
| Embryonic | N/A | 60.9 ± 1.8% | N/A | 36.6 ± 2.3% |
| Exon 7-Deleted | N/A | N/A | 71.2 ± 5.0% | 39.7 ± 1.4% |
| cTnT-ND | 2.0 ± 0.23% | 4.3 ± 0.6% | 9.4 ± 1.3% | 6.0 ± 0.8% |
The relative amounts of four cTnT variants expressed and cTnT-ND produced in the hearts of adult wild type mice and three lines of single or double transgenic (TG) mice were calculated from densitometry analysis of mAb 2C8 Western blots of total protein extracted from left ventricular muscle samples (Fig. 5). The dada are presented as the percentage of each cTnT variant or cTnT-ND in total cTnT (mean ± SEM). N/A, not applicable. The results demonstrated a positive correlation in vivo between NH2-terminal primary structure of cTnT, especially the deletion of the exon 7-encoded segment, and cTnT-ND production.
The Western blot in Fig. 3 showed that treatment of 50 μM and 200 μM ouabain in hypokalemic media under pacing resulted in increased cTnT-ND in cardiomyocytes in a concentration dependent manner. The effect of ouabain, a classic calcium agonist, on the production of cTnT-ND in isolated cardiomyocytes provides direct evidence for a role of calcium overload in inducing cTnT-ND.
Production of cTnT-ND is independent of cTnT phosphorylation
Cardiac TnT was reported being phosphorylated by several protein kinases, among which protein kinase C (PKC) was the most commonly observed [40–42]. Ca2+ overload in cardiomyocytes may activate conventional PKC [43] and PKC phosphorylation could consequently alter the molecular conformation of cTnT to promote the restrictive μ-calpain cleavage of the NH2-terminal segment. Therefore, we examined cTnT phosphorylation for the correlation to cTnT-ND production. Pro-Q phosphoprotein stain of SDS-gels showed that whereas the mouse neonatal cardiomyocytes in culture had weak phosphorylation of various proteins including myosin binding protein C and cTnI in comparison to that in adult cardiac muscle in vivo, there was no difference in cTnT phosphorylation before and after ouabain treatment (Fig. 4).
Fig. 4. Ouabain treatment did not affect phosphorylation of cTnT in cultured cardiomyocytes.

Total protein extracts from the primary cultures of neonatal mouse cardiomyocytes were examined by SDS-PAGE and Pro-Q phosphoprotein staining. Ouabain treatment did not produce significant changes in the phosphorylation of cTnT, cardiac TnI (cTnI) and myosin binding protein C (MyBP-C). MHC, myosin heavy chain.
Structural variation in the NH2-terminal region of cTnT correlated to the different levels of cTnT-ND in vivo
The overall molecular conformation of TnT is modulated by the structure of the NH2-terminal region [10, 11, 21, 22]. To investigate the effect of substrate conformation on the μ-calpain-catalyzed restrictive proteolysis of the NH2-terminal segment of cTnT, we examined transgenic mouse cardiac muscles co-expressing cTnT variants differing in the NH2-terminal region (Fig. 5A) for the production of cTnT-ND in vivo.
Fig. 5. Alternative splicing-generated cTnT NH2-terminal variation affected the production of cTnT-ND in vivo.

A. Structural comparison of three mouse cTnT variants differing in the alternatively spliced NH2-terminal region. The segments encoded by different exons of the cTnT gene are outlined on the top map. The embryonic exon 5 is normally spliced out from adult cTnT. The abnormal deletion of exon 7 was found in adult cTnT of turkeys [10] and dogs [11] with dilated cardiomyopathy. Exon 4 encoding four amino acids is alternatively spliced to generate two cTnT variants in both embryonic and adult mouse hearts [36]. The dotted line between the NH2-terminal variable region and the conserved region of cTnT indicates the μ-calpain cleavage site for the restrictive deletion of the NH2-terminal segment [24]. The positions of several reported phosphorylated residues in cTnT are indicated with arrowheads. The sequences of the exon 5-encoded segment containing an additional Ser (S25) residue and the exon 7-encoded segment are shown on the map. The identified primary phosphorylation sites S2 and S25 are highlighted in bold font. B. mAb 2C8 Western blots of fresh ventricular muscle samples from 5 to 7 months old mice showed that transgenic (TG) mouse hearts over-expressing embryonic (Emb) cTnT contained higher levels of cTnT-ND than that in wild type hearts. Single or double transgenic mouse hearts over-expressing exon 7 (E7)-deleted adult cTnT exhibited a more increased level of cTnT-ND. As a control, mAb TnI-1 showed no change of cTnI in these groups. C. Densitometry analysis of the Western blots determined that while all three lines of transgenic mice had higher cTnT-ND in the cardiac muscle than the wild type controls, the levels of cTnT-ND in the exon 7-deleted (ΔE7) adult cTnT single transgenic mouse hearts and exon 7-deleted adult cTnT/embryonic cTnT double transgenic mouse hearts were significantly higher than that in the embryonic cTnT single transgenic hearts, indicating a stronger correlation with the levels of exon 7-deleted cTnT. Data are shown as mean ± SEM. *, P<0.05, **, P<0.01, and ***, P<0.001 versus WT group; ###, P<0.001 versus Emb group; &, P<0.05 versus ΔE7 group by ANOVA Fisher test.
The Western blots in Fig. 5B demonstrated that adult transgenic mouse cardiac muscle over-expressing embryonic cTnT with the exon 5 segment included or adult cTnT with the exon 7 segment deleted contained higher levels of cTnT-ND in vivo than that in wild type mouse hearts. The exon 7-deleted cTnT transgenic mouse hearts showed much higher levels of cTnT-ND than that in the embryonic cTnT transgenic hearts. Normalized to total cTnT, densitometry quantification of the Western blots determined that wild type mouse hearts had ~2.0% cTnT-ND, embryonic cTnT transgenic mouse hearts had ~4.3% cTnT-ND, and exon 7-deleted cTnT hearts contained ~9.4% cTnT-ND. Corresponding to the level of exon 7-delted cTnT, the level of cTnT-ND in the double transgenic mouse hearts was ~6.0% (Fig. 5C).
Table 1 summarized the expression levels of the cTnT isoforms different in the NH2-terminal region in the transgenic and wild type mouse hearts together with the corresponding levels of cTnT-ND produced in vivo. The data showed a positive correlation between the level of exon 7-deleted cTnT expressed in the single or double transgenic mouse hearts. This correlation indicates that structure of the NH2-terminal variable region of cTnT affected the restrictive proteolytic removal of itself. This hypothesis is consistent with the established role of the NH2-terminal region in modulating the molecular conformation of TnT, which may alter the substrate conformation at the cleavage site and increase the sensitivity to μ-calpain.
The Western blots in Fig. 5B showed that the transgenic mouse cardiac muscle contained similar levels of intact cardiac TnI (cTnI) without apparent fragmentation. cTnI is also a substrate of μ-calpain [44]. The specific modification of cTnT but not cTnI, two functionally and specially connected subunits of the troponin complex, by calpain proteolysis suggests that the production of cTnT-ND was unlikely due to an increase in overall μ-calpain activity. Therefore, the result supports the hypothesis that the NH2-terminal truncation of cTnT is regulated by modification of substrate conformation and sensitivity to μ-calpain proteolysis.
Pro-Q phosphoprotein staining of the transgenic mouse cardiac muscle found that the embryonic cTnT over-expressed in the adult transgenic mouse hearts had a significantly higher level of phosphorylation in comparison to that of wild type adult cTnT and exon 7-deleted adult cTnT (Fig. 6A). Densitometry analysis of the Pro-Q stained gels quantitatively confirmed the different levels of phosphorylation (Fig. 6B). The phosphorylation levels of these cTnT variants showed no correlation with the levels of cTnT-ND produced in the different transgenic mouse hearts (Fig. 6C). Consistent with no change of cTnT phosphorylation in the ouabain-treated cardiomyocytes in which cTnT-ND was induced (Fig. 4), the results further support the hypothesis that cTnT-ND production is regulated independently of cTnT phosphorylation.
Fig. 6. The production of cTnT-ND did not correlate to cTnT phosphorylation.
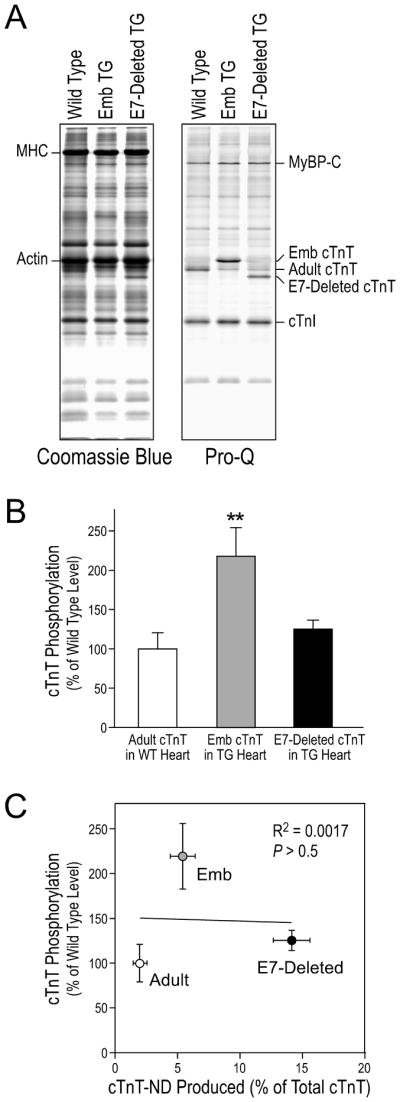
Freshly isolated wild type (WT) and transgenic (TG) mouse hearts were examined using SDS-PAGE and Pro-Q phosphoprotein staining. A. Pro-Q stained SDS-gel showed that embryonic (Emb) cTnT had a significantly higher level of phosphorylation than that of wild type adult cTnT and exon 7 (E7)-deleted adult cTnT. Coomassie Blue staining was used for total protein control. MHC, myosin heavy chain; MyBP-C, myosin binding protein C. B. Normalized to the actin bands, densitometry quantification of Pro-Q-stained gels demonstrated that embryonic cTnT had a level of phosphorylation approximately 2-folds of that of wild type adult and exon 7-deleted adult cTnT in vivo (**P < 0.001). C. The levels of cTnT phosphorylation were plotted against the levels of cTnT-ND using densitometry data from the Pro-Q gel and mAb 2C8 Western blots (Fig. 5). Linear regression showed that there was no significant correlation between the levels of cTnT phosphorylation and cTnT-ND production.
cTnT phosphorylation in vivo is mainly in the NH2-terminal region
The results in Fig. 6 demonstrated in vivo in transgenic mouse hearts similar levels of phosphorylation for the wild type adult cTnT and the exon 7-deleted adult cTnT but twice as much phosphorylation for the embryonic cTnT that contains an additional Ser25 residue due to the inclusion of the exon 5-encoded segment (Fig. 5A). In contrast, Pro-Q phosphoprotein staining did not detect significant phosphorylation of cTnT-ND in vivo in transgenic mouse heart (Fig. 7A). The Western blot in Fig. 7B demonstrated that our modified SDS-PAGE successfully separated the cTnT-ND band from intact cTnI that is normally phosphorylated in cardiac muscle. The absence of significant phosphorylation of cTnT-ND in vivo indicated that the phosphorylation of cTnT under physiological conditions was primarily in the NH2-terminal segment, presumably at Ser2 of all of the alternatively spliced variants and Ser25 in embryonic cTnT (Fig. 5A).
Fig. 7. Phosphorylation of cTnT was primarily in the NH2-terminal region.

A. The Pro-Q stained gel demonstrated the lack of detectable phosphorylation of cTnT-ND in vivo in transgenic (TG) mouse heart in contrast to that of intact adult cTnT, embryonic (Emb) cTnT and exon 7 (E7)-deleted adult cTnT. Peppermint Stick markers were used as control for phosphorylated proteins and non-specific background. A parallel gel was stained with Coomassie Blue R250 for protein loading control. MHC, myosin heavy chain; MBP-C, myosin-binding protein C. B. Using a double transgenic mouse heart that expresses both cTnT-ND and an NH2-terminal truncated cardiac TnI (cTnI-ND) [54] together with controls of wild type and transgenic mouse hearts containing normal level of intact cardiac TnI (cTnI), the Western blot using mixed anti-cTnT mAb 2C8 and anti-TnI mAb TnI-1 demonstrated the effective separation of cTnT-ND and intact cardiac TnI using the modified SDS-PAGE gel.
Discussion
The restrictive proteolysis to selectively remove the NH2-terminal variable region of cTnT is an adaptation mechanism with a compensatory, other than deleterious, impact on cardiac function. To understand the molecular mechanism that induces the production of cTnT-ND will help to explore the possibility of activating this posttranslational modification as a potential treatment for heart failure. In the present study, we demonstrated experimental evidence that calcium overload in cardiomyocytes induces the restrictive proteolysis of the NH2-terminal variable region of cTnT and this cleavage was affected by the NH2-terminal structure of cTnT.
cTnT-ND was first found in myocardial ischemia-reperfusion [24] and ventricular pressure overload [25]. It is known that myocardial ischemia-reperfusion has profound effects on the function and viability of cardiac myocytes by elevation of intracellular calcium concentration [45, 46]. Pressure overload is also known to affect calcium handling in cardiomyocytes [46]. These observations lead to the hypothesis that calcium overload may trigger the production of cTnT-ND. Using established conditions that cause Ca2+ overloading such as thrombin [27, 28] or ouabain-hypokalemia treatment, the present study reproduced cTnT-ND in ex vivo working hearts and isolated cardiomyocytes.
The isolation of cardiomyocytes involves ischemic perfusion with a Ca2+-free digestion media followed by restoring a physiological level of extracellular Ca2+ in oxygenized media. This procedure mimicking ischemia-reperfusion induced cTnT-ND in the cardiomyocytes (Fig. 2A). In addition to suggesting a precaution for functional studies using cardiomyocytes freshly isolated with similar procedures, our results demonstrated that cTnT-ND in the isolated cardiomyocytes diminished during culture in 4 to 5 days (Fig. 2A) and, therefore, the production of cTnT-ND is a reversible transient mechanism to acutely regulate cardiac function.
μ-Calpain is a calcium-activated protease that requires only micromolar concentrations of calcium. Different from that in most non-muscle cells, cytosolic Ca2+ in normal cardiomyocytes periodically rises to the micromolar range during contraction. It is logical to hypothesize that μ-calpain in cardiomyocytes would be largely primed by Ca2+ and in an activated state under physiological conditions. Supporting the hypothesis that the induction of cTnT NH2-terminal truncation in calcium overload is not due to elevated overall activity of μ-calpain in cardiomyocytes, our experimental conditions that produced cTnT-ND did not result in any detectable degradation of cTnI, another subunit of the troponin complex in direct association with cTnT and a known substrate of μ-calpain [44].
Therefore, the induction of cTnT-ND production by calcium overloading that increases the basal level of Ca2+ in cardiomyocytes would require additional mechanism(s). We proposed a hypothesis that the selective cleavage of cTnT by μ-calpain is based on increased sensitivity or accessibility of the specific substrate site due to altered molecular conformation of cTnT. The most extensively studied posttranslational modification of myofilament proteins is phosphorylation that can cause local and overall conformational changes. cTnT was reported to be phosphorylated by PKC [40, 48] that is retargeted or activated in cardiomyocytes under Ca2+ overloading conditions [49, 50]. However, no correlation was found between the levels of cTnT phosphorylation and cTnT-ND production in ouabain-hypokalemia-treated cardiomyocytes or the transgenic mouse hearts.
On the other hand, the NH2-terminal structure of cTnT had a profound effect on the production of cTnT-ND. It has been established that the NH2-terminal structure variations modulate the overall conformation of TnT [3, 4, 11, 21, 22]. Our results showed that cTnT splicing variants with different NH2-terminal structure differed significantly in the production of cTnT-ND in vivo in transgenic mouse hearts. The data in Fig. 5C and Table 1 demonstrated that exon 7-deleted adult cTnT is the most sensitive substrate among the three cTnT variants tested for the production of cTnT-ND.
It was cautiously considered that the transgenic mouse hearts expressing the myopathic cTnT variants had negative effects on cardiac function [10, 11, 32, 51] and could produce cTnT-ND as a secondary response. However, when two or three cTnT variants with NH2-terminal differences co-existed in vivo in the hearts of single and double transgenic mice the exon 7-deleted adult cTnT remained the most sensitive in the production of cTnT-ND. Therefore, it is not the cellular environment adapted to the different transgenic mouse hearts but the structure of cTnT per se determines the production of cTnT-ND.
Our data indicated that cTnT phosphorylation in vivo was primarily in the NH2-terminal region (Fig. 7). The lack of significant phosphorylation in cTnT-ND confirms the observation of recent mass spectrometry studies, which identified 100% monophosphorylation of cTnT at Ser2 in adult rat heart in vivo [52, 53]. Rat and mouse cTnTs have almost identical amino acid sequences in the relevant regions. When embryonic cTnT containing the additional Ser25 in the NH2-terminal variable region encoded by exon 5 was expressed in the adult transgenic mouse heart, the intensity of phosphorylation signal approximately doubled, indicating an effective phosphorylation of this residue. This observation strongly supports the notion that phosphorylation of cTnT in vivo is primarily in the NH2-terminal region. The NH2-terminal specific phosphorylation of cTnT in vivo implicates function significance and is worth further investigation.
The NH2-terminal alternatively spliced variants of cTnT also showed different degrees of phosphorylation, in which the embryonic cTnT transgenically expressed in adult mouse hearts had the highest level of phosphorylation. However, the difference in phosphorylation did not correlate to the production of cTnT-ND and, therefore, the NH2-terminal amino acid sequence may play a predominant role in modulating the overall conformation of cTnT and the sensitivity to restrictive μ-calpain cleavage over the effect, if any, of NH2-terminal phosphorylation. Supporting a role of substrate structure in the profound effect of exon 7 deletion on cTnT-ND production, we previously showed that exon 7 deletion alters the overall molecular conformation and function of cTnT, resulting in increased Ca2+-sensitivity in actomyosin ATPase assay [10].
The mechanistic link between calcium overload and the μ-calpain-sensitive conformation in cTnT remains to be better understood. Future studies investigating the effect of changes in myofibril contractile state due to calcium overload on the molecular conformation of cTnT and its sensitivity to μ-calpain modification would help to further understand the pathophysiological regulation of cTnT-ND.
The NH2-terminal variable region of cardiac troponin T is a regulatory structure
Its removal has a compensatory effect on heart function in energetic crisis
Calcium overload conditions induced this restrictive proteolysis
The NH2-terminal structure of cardiac troponin forms a basis of this cleavage
Molecular conformation of the cleavage site may play a determining role
Acknowledgments
We thank Dr. M. Moazzem Hossain and Ms. Hui Wang for maintaining the transgenic mouse lines. This study was supported by grants from the National Institutes of Health (HL078773, HL098945 and AR048816 to J.-P.J). ZZ was supported in part by a postdoctoral fellowship from the American Heart Association Great Midwest Affiliate.
Abbreviations used
- TnT
troponinT
- BDM
2,3-butanedione monoxime
- BSA
bovine serum albumin
- cTnI
cardiac troponin I
- cTnT
cardiac troponin T
- cTnT-ND
NH2-terminal truncated cTnT
- mAb
monoclonal antibody
- PKC
protein kinase C
- TBS
Tris-buffered saline
- TnC
troponin C
- TnI
troponin I
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gordon AM, Homsher E, Regnier M. Physiol Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 2.Perry SV. J Muscle Res Cell Motil. 1998;19:575–602. doi: 10.1023/a:1005397501968. [DOI] [PubMed] [Google Scholar]
- 3.Jin J-P, Zhang Z, Bautista JA. Crit Rev Eukaryot Gene Expr. 2008;18:93–124. doi: 10.1615/critreveukargeneexpr.v18.i2.10. [DOI] [PubMed] [Google Scholar]
- 4.Wei B, Jin J-P. Arch Biochem Biophys. 2011;505:144–154. doi: 10.1016/j.abb.2010.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jin J-P, Lin JJ. J Biol Chem. 1988;263:7309–7315. [PubMed] [Google Scholar]
- 6.Wang J, Jin J-P. Gene. 1997;193:105–114. doi: 10.1016/s0378-1119(97)00100-5. [DOI] [PubMed] [Google Scholar]
- 7.Yu ZB, Gao F, Feng HZ, Jin J-P. Am J Physiol Cell Physiol. 2007;292:C1192–C1203. doi: 10.1152/ajpcell.00462.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Larsson L, Wang X, Yu F, Hook P, Borg K, Chong SM, Jin J-P. Am J Physiol Cell Physiol. 2008;295:C722–C731. doi: 10.1152/ajpcell.00110.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anderson PA, Greig A, Mark TM, Malouf NN, Oakeley AE, Ungerleider RM, Allen PD, Kay BK. Circ Res. 1995;76:681–686. doi: 10.1161/01.res.76.4.681. [DOI] [PubMed] [Google Scholar]
- 10.Biesiadecki BJ, Jin J-P. J Biol Chem. 2002;277:18459–18468. doi: 10.1074/jbc.M200788200. [DOI] [PubMed] [Google Scholar]
- 11.Biesiadecki BJ, Elder BD, Yu ZB, Jin J-P. J Biol Chem. 2002;277:50275–50285. doi: 10.1074/jbc.M206369200. [DOI] [PubMed] [Google Scholar]
- 12.Pearlstone JR, Smillie LB. J Biol Chem. 1982;257:10587–10592. [PubMed] [Google Scholar]
- 13.Pearlstone JR, Smillie LB. Can J Biochem Cell Biol. 1985;63:212–218. doi: 10.1139/o85-030. [DOI] [PubMed] [Google Scholar]
- 14.Hinkle A, Goranson A, Butters CA, Tobacman LS. J Biol Chem. 1999;274:7157–7164. doi: 10.1074/jbc.274.11.7157. [DOI] [PubMed] [Google Scholar]
- 15.Pan BS, Gordon AM, Potter JD. J Biol Chem. 1991;266:12432–12438. [PubMed] [Google Scholar]
- 16.Fujita S, Maeda K, Maeda Y. J Biochem (Tokyo) 1992;112:306–308. doi: 10.1093/oxfordjournals.jbchem.a123896. [DOI] [PubMed] [Google Scholar]
- 17.Chandra M, Montgomery DE, Kim JJ, Solaro RJ. J Mol Cell Cardiol. 1999;31:867–880. doi: 10.1006/jmcc.1999.0928. [DOI] [PubMed] [Google Scholar]
- 18.Biesiadecki BJ, Chong SM, Nosek TM, Jin J-P. Biochemistry. 2007;46:1368–1379. doi: 10.1021/bi061949m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tobacman LS. J Biol Chem. 1988;263:2668–2672. [PubMed] [Google Scholar]
- 20.Gomes AV, Guzman G, Zhao J, Potter JD. J Biol Chem. 2002;277:35341–35349. doi: 10.1074/jbc.M204118200. [DOI] [PubMed] [Google Scholar]
- 21.Wang J, Jin J-P. Biochemistry. 1998;37:14519–14528. doi: 10.1021/bi9812322. [DOI] [PubMed] [Google Scholar]
- 22.Jin J-P, Root DD. Biochemistry. 2000;39:11702–11713. doi: 10.1021/bi9927437. [DOI] [PubMed] [Google Scholar]
- 23.Jin J-P, Chen A, Ogut O, Huang Q-Q. Am J Physiol Cell Physiol. 2000;279:C1067–C1077. doi: 10.1152/ajpcell.2000.279.4.C1067. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Z, Biesiadecki BJ, Jin J-P. Biochemistry. 2006;45:11681–11694. doi: 10.1021/bi060273s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feng HZ, Biesiadecki BJ, Yu ZB, Hossain MM, Jin J-P. J Physiol (London) 2008;586:3537–3550. doi: 10.1113/jphysiol.2008.153577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murphy E, Steenbergen C. Physiol Rev. 2008;88:581–609. doi: 10.1152/physrev.00024.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Steinberg SF, Robinson RB, Lieberman HB, Stern DM, Rosen MR. Circ Res. 1991;68:1216–1229. doi: 10.1161/01.res.68.5.1216. [DOI] [PubMed] [Google Scholar]
- 28.Mirabet M, Garcia-Dorado D, Ruiz-Meana M, Barrabes JA, Soler-Soler J. J Mol Cell Cardiol. 2005;39:277–283. doi: 10.1016/j.yjmcc.2005.03.005. [DOI] [PubMed] [Google Scholar]
- 29.Zhou YY, Wang SQ, Zhu WZ, Chruscinski A, Kobilka BK, Ziman B, Wang S, Lakatta EG, Cheng H, Xiao RP. Am J Physiol Heart Circ Physiol. 2000;279:H429–H436. doi: 10.1152/ajpheart.2000.279.1.H429. [DOI] [PubMed] [Google Scholar]
- 30.McDonough AA, Velotta JB, Schwinger RH, Philipson KD, Farley RA. Basic Res Cardiol. 2002;97:I19–I24. doi: 10.1007/s003950200024. [DOI] [PubMed] [Google Scholar]
- 31.Kometiani P, Askari A, Liu J, Xie Z, Askari FK. Am J Physiol Heart Circ Physiol. 2001;280:H1415–H1421. doi: 10.1152/ajpheart.2001.280.3.H1415. [DOI] [PubMed] [Google Scholar]
- 32.Feng H-Z, Jin J-P. Am J Physiol Heart Circ Physiol. 2010;299:H97–H105. doi: 10.1152/ajpheart.01105.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin J-P, Chong SM. Archiv Biochem Biophys. 2010;500:144–150. doi: 10.1016/j.abb.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jin J-P, Yang FW, Yu ZB, Ruse CI, Bond M, Chen A. Biochemistry. 2001;40:2623–2631. doi: 10.1021/bi002423j. [DOI] [PubMed] [Google Scholar]
- 35.Jin J-P, Malik ML, Lin JJ. Hybridoma. 1990;9:597–608. doi: 10.1089/hyb.1990.9.597. [DOI] [PubMed] [Google Scholar]
- 36.Jin J-P, Wang J, Zhang J. Gene. 1996;168:217–221. doi: 10.1016/0378-1119(95)00803-9. [DOI] [PubMed] [Google Scholar]
- 37.Jin J-P. Biochem Biophys Res Commun. 1996;225:883–889. doi: 10.1006/bbrc.1996.1267. [DOI] [PubMed] [Google Scholar]
- 38.Martin AF. J Biol Chem. 1981;256:964–968. [PubMed] [Google Scholar]
- 39.Michele DE, Albayya FP, Metzger JM. J Cell Biol. 1999;145:1483–1495. doi: 10.1083/jcb.145.7.1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sumandea MP, Pyle WG, Kobayashi T, de Tombe PP, Solaro RJ. J Biol Chem. 2003;278:35135–35144. doi: 10.1074/jbc.M306325200. [DOI] [PubMed] [Google Scholar]
- 41.He X, Liu Y, Sharma V, Dirksen RT, Waugh R, Sheu SS, Min W. Am J Pathol. 2003;163:243–251. doi: 10.1016/S0002-9440(10)63647-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vahebi S, Kobayashi T, Warren CM, de Tombe PP, Solaro RJ. Circ Res. 2005;96:740–747. doi: 10.1161/01.RES.0000162457.56568.7d. [DOI] [PubMed] [Google Scholar]
- 43.Mellor H, Parker PJ. Biochem J. 1998;332:281–292. doi: 10.1042/bj3320281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Di Lisa F, De Tullio R, Salamino F, Barbato R, Melloni E, Siliprandi N, Schiaffino S, Pontremoli S. Biochem J. 1995;308:57–61. doi: 10.1042/bj3080057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kihara Y, Grossman W, Morgan JP. Circ Res. 1989;65:1029–1044. doi: 10.1161/01.res.65.4.1029. [DOI] [PubMed] [Google Scholar]
- 46.Kusuoka H, Porterfield JK, Weisman HF, Weisfeldt ML, Marban E. J Clin Invest. 1987;79:950–961. doi: 10.1172/JCI112906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Eckardt L, Kirchhof P, Breithardt G, Haverkamp W. Basic Res Cardiol. 2001;96:369–380. doi: 10.1007/s003950170045. [DOI] [PubMed] [Google Scholar]
- 48.Pinet C, Algalarrondo V, Sablayrolles S, Le Grand B, Pignier C, Cussac D, Perez M, Hatem SN, Coulombe A. Mol Pharmacol. 2008;73:1622–16231. doi: 10.1124/mol.107.043182. [DOI] [PubMed] [Google Scholar]
- 49.Jideama NM, Noland TA, Jr, Raynor RL, Blobe GC, Fabbro D, Kazanietz MG, Blumberg PM, Hannun AY, Kuo JF. J Biol Chem. 1996;271:23277–23283. doi: 10.1074/jbc.271.38.23277. [DOI] [PubMed] [Google Scholar]
- 50.Liu Y, Ytrehus K, Downey JM. J Mol Cell Cardiol. 1994;26:661–668. doi: 10.1006/jmcc.1994.1078. [DOI] [PubMed] [Google Scholar]
- 51.Wei B, Gao J, Huang X-P, Jin J-P. J Biol Chem. 2010;285:27806–27816. doi: 10.1074/jbc.M110.137844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sancho Solis R, Ge Y, Walker JW. J Muscle Res Cell Motil. 2008;29:203–212. doi: 10.1007/s10974-009-9168-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Marston SB, Walker JW. J Muscle Res Cell Motil. 2009;30:93–95. doi: 10.1007/s10974-009-9184-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barbato JC, Huang QQ, Hossain MM, Bond M, Jin JP. J Biol Chem. 2005;280:6602–6609. doi: 10.1074/jbc.M408525200. [DOI] [PubMed] [Google Scholar]