

Sod1 is an important antioxidant enzyme that becomes activated by its chaperone, Ccs1. The localization of Ccs1 to mitochondria is controlled by the oxidoreductase Mia40. The formation of a disulfide bond between Cys-27 and Cys-64 in Ccs1 is critical for import and stability but not for Ccs1 activity in the maturation of Sod1.
Abstract
Superoxide dismutase 1 (Sod1) is an important antioxidative enzyme that converts superoxide anions to hydrogen peroxide and water. Active Sod1 is a homodimer containing one zinc ion, one copper ion, and one disulfide bond per subunit. Maturation of Sod1 depends on its copper chaperone (Ccs1). Sod1 and Ccs1 are dually localized proteins that reside in the cytosol and in the intermembrane space of mitochondria. The import of Ccs1 into mitochondria depends on the mitochondrial disulfide relay system. However, the exact mechanism of this import process has been unclear. In this study we detail the import and folding pathway of Ccs1 and characterize its interaction with the oxidoreductase of the mitochondrial disulfide relay Mia40. We identify cysteines at positions 27 and 64 in domain I of Ccs1 as critical for mitochondrial import and interaction with Mia40. On interaction with Mia40, these cysteines form a structural disulfide bond that stabilizes the overall fold of domain I. Although the cysteines are essential for the accumulation of functional Ccs1 in mitochondria, they are dispensable for the enzymatic activity of cytosolic Ccs1. We propose a model in which the Mia40-mediated oxidative folding of domain I controls the cellular distribution of Ccs1 and, consequently, active Sod1.
INTRODUCTION
The respiratory chain is one of the main sources of reactive oxygen species (ROS; Murphy, 2009). Consequently, mitochondria contain numerous ROS-counteracting systems. Key enzymes are two superoxide dismutases (Sods): copper–zinc Sod1, which localizes to the cytosol and the mitochondrial intermembrane space (IMS), and manganese Sod2, which is present in the mitochondrial matrix. Sod1 is a homodimeric enzyme that per subunit contains one zinc ion, one copper ion, and one disulfide bond. The incorporation of the copper ion and the formation of the disulfide bond are mediated by an activating enzyme, the copper chaperone for Sod1 (Ccs1; Culotta et al., 1997; Rae et al., 1999). Although Ccs1 is essential for the activation of Sod1 in yeast (Culotta et al., 1997), Ccs1 knockout mice retain ∼10–20% Sod1 activity (Wong et al., 2000).
Ccs1 is like Sod1 found in the cytosol and in the IMS (Okado-Matsumoto and Fridovich, 2001; Sturtz et al., 2001). Because neither Sod1 nor Ccs1 contains mitochondrial targeting signals, it has been unclear how both proteins are imported into the IMS. Recently, it was shown in yeast and mammalian cells that the import of Ccs1 and Sod1 relies on the mitochondrial disulfide relay (Mesecke et al., 2005; Kawamata and Manfredi, 2008; Reddehase et al., 2009). Thereby, the overexpression or depletion of the oxidoreductase Mia40 influences mitochondrial protein levels of Ccs1 and Sod1. During the import process, Ccs1 directly interacts with Mia40 through an intermolecular disulfide bond, whereas no direct interaction of Mia40 and Sod1 was observed (Reddehase et al., 2009). However, mitochondrial Sod1 amounts increase upon overexpression of mitochondria-targeted Ccs1 in a CCS1-deletion background (Field et al., 2003). Thus Sod1 is presumably enriched in the IMS by specific binding to mitochondrial Ccs1, and Ccs1 in turn is a substrate of the mitochondrial disulfide relay.
Substrates of the mitochondrial disulfide relay usually are small proteins (10–15 kDa) composed of a single domain that is formed by a simple helix–loop–helix structure (Chacinska et al., 2009; Riemer et al., 2009). These proteins contain four conserved cysteines that are arranged in either two CX9C or two CX3C motifs (C, cysteine; X, any other amino acid) and consequently are termed twin CX9C or twin CX3C proteins, respectively. Mia40 substrates can only translocate into the IMS in their reduced and unfolded state. On mitochondrial import the cysteines are oxidized by Mia40 and form disulfide bonds that connect the two helices (Chacinska et al., 2009; Riemer et al., 2009). Thus oxidative folding and import into mitochondria are coupled processes for these small substrates. In addition to its role as thiol oxidase, Mia40 has recently demonstrated to function as a molecular chaperone that induces helix formation in its substrates (Banci et al., 2010).
In contrast with the classic, rather small substrates of Mia40, the Ccs1 monomer has a size of 27 kDa and is composed of three domains: two copper-binding domains I and III at the amino and carboxyl termini, respectively, and a central domain II that is structurally homologous to Sod1 (Figure 1A). Whereas domain III is essential for Sod1 activation, domain I is required only under conditions of copper deprivation (Schmidt et al., 1999). Of the seven cysteines of yeast Ccs1, four localize to domain I (two form the copper-binding CXXC motif), one is found in domain II, and two are in domain III in a CXC motif responsible for copper transfer to Sod1 (Figure 1A). Because its cysteines are not arranged in twin CX9C or twin CX3C motifs, it remains unclear how Ccs1 is imported, which cysteine interacts with Mia40, whether some of its cysteines are present in an oxidized state after import, and whether Mia40 assists in the folding of Ccs1.
FIGURE 1:

Cys-27 and Cys-64 are required for import of Ccs1 into isolated mitochondria. (A) Domain organization of yeast Ccs1. The three domains and the sequence positions of the seven cysteines of Ccs1 are depicted. (B) Import of Ccs1 into isolated mitochondria. In vitro translated radioactive Ccs1 (lanes 1 and 6, 10% control) was incubated with 50 μg of mitochondria isolated from Mia40-overexpressing strains. Subsequently, mitochondria were either analyzed directly (lane 2), treated with 100 μg/μl PK to remove nonimported proteins (lane 3), or turned into mitoplasts (MP) by hypotonic swelling and then treated with PK (lane 4) to remove all IMS proteins. One sample was also completely lysed with Triton X-100 (TX) and incubated with PK to test Ccs1 for protease resistance (lane 5). All samples were analyzed by reducing SDS–PAGE and autoradiography. The treatments with PK on mitochondria and mitoplasts were verified by immunoblotting against Erv1, Tim10 (both IMS), and Mrpl40 (matrix). (C) Glutathione dependence of Ccs1 import. Same as B, except that in vitro translated Ccs1 was incubated with different amounts of reduced glutathione during import. Experiments were quantified with ImageJ. (D) Import of Ccs1 cysteine mutants into isolated mitochondria. Same as B, except that in vitro translated Ccs1 cysteine mutants were incubated with mitochondria. Only whole mitochondria and mitoplasts were analyzed after PK digest. (E) Structure of domain I of Ccs1 (Lamb et al., 1999). This domain encompasses residues 2–74 and contains four cysteines (C17, C20, C27, and C64). Note that the C27–C64 disulfide connects two antiparallel α-helices, resulting in a structure of domain I that is similar to the structure of twin CX3C or twin CX9C substrates. (F) Import of Ccs1–domain I cysteine mutants and DHFR fusion variants into isolated mitochondria. Same as D, except that in vitro translated variants of domain I– or domain I–DHFR fusion proteins were incubated with mitochondria.
In this study, we aimed to understand the process of mitochondrial import and folding of Ccs1 in mechanistic detail. We identified two cysteines (Cys-27 and Cys-64) in domain I that are required for mitochondrial import of Ccs1—but not for its role in Sod1 maturation—by the use of cysteine mutants. We demonstrate that C27 and C64 can be directly oxidized by Mia40 and in vivo form a stable disulfide bond in mitochondria, whereas they are partially reduced in the cytosol. We propose that the formation of the C27–C64 disulfide by Mia40 competes with a slower, nonenzymatic oxidation in the cytosol and thus allows the import of a fraction of Ccs1 into the IMS before folding in the cytosol can take place.
RESULTS
Cys-27 and Cys-64 in domain I are required for mitochondrial import of Ccs1
The import of Ccs1 into mitochondria depends on the disulfide relay of the IMS (Mesecke et al., 2005; Reddehase et al., 2009). For the twin CX9C and twin CX3C substrates of Mia40 the cysteines are critical for efficient import. We therefore decided to identify the cysteine(s) in Ccs1 that influence its localization by analyzing the mitochondrial import of Ccs1 cysteine-to-serine mutants. First, we established a protocol to monitor the mitochondrial import of wild-type Ccs1 (Figure 1B). To this end, we incubated radioactively labeled Ccs1 with isolated mitochondria. At the end of the import reaction the sample was split into four parts and analyzed either directly or after treatment with proteinase K (PK). The latter treatment either was applied to intact mitochondria or was preceded by hypoosmotic swelling of mitochondria to generate mitoplasts (mitochondria lacking their outer membrane) or by mitochondrial lysis with Triton X-100. A band migrating at the expected size of Ccs1 was detected when intact mitochondria were treated with PK, indicating import of Ccs1 into mitochondria (Figure 1B, lane 3). On hypoosmotic swelling and subsequent PK treatment this signal disappeared, implying import of Ccs1 into the IMS (Figure 1B, lane 4). Notably, in some experiments we observed that the band representing Ccs1 migrated slightly faster on SDS–PAGE after PK treatment compared with untreated samples. In these cases a small part of the Ccs1 protein might still be exposed to the exterior after the import reaction. This exposed part would then be truncated upon digestion by PK. However, the main part of the protein was inaccessible to protease. The import of twin CX3C and twin CX9C substrates of Mia40 exhibits a marked dependence on reductants such as reduced glutathione (GSH; Bien et al., 2010). When testing the import of Ccs1 we found a similar dependence of Ccs1 import (Figure 1C). At low concentrations of GSH (5 mM) import of Ccs1 proceeded faster than import in the absence of reductant. However, at higher concentrations of GSH (15 mM) Ccs1 import was strongly decreased.
We next investigated the import of the Ccs1 cysteine mutants (Figure 1D). We generated double mutants of the two cysteine pairs that were previously shown to contribute to copper binding and transfer (C17/C20 and C229/C231; Figure 1A) and single mutants of the remaining three cysteines. The Ccs1 double-cysteine mutants Ccs1C17S/C20S and Ccs1C229S/C231S, as well as Ccs1C159S, were imported with comparable efficiency to that of wild-type Ccs1. However, Ccs1C27S and Ccs1C64S exhibited strongly diminished import, indicating that these cysteines are critical for the mitochondrial localization of Ccs1.
The cysteines at positions 27 and 64 are both situated in domain I of Ccs1 (Figure 1, A and E). A closer analysis of the structures of this domain (Lamb et al., 1999, 2001) revealed that C27 and C64 face each other and, at least in one of the structures, covalently connect two antiparallel α-helices (Figure 1E). Thus the structure of domain I resembles the structures of typical Mia40 substrates. We therefore asked whether domain I (corresponding to residues 2–74 of Ccs1) on its own is imported into mitochondria and whether it can serve as a targeting signal for a nonmitochondrial protein. Applying our import assay, we first demonstrated that domain I can be imported into isolated mitochondria, although with lower efficiency than full-length Ccs1 (Figure 1F, lanes 1–3). Mutation of either C27 or C64 abolished the import of domain I into mitochondria, confirming their relevance for the import process (Figure 1D). Moreover, wild-type domain I fused to the cytosolic protein dihydrofolate reductase (DHFR) allowed the import of this chimeric protein into mitochondria, whereas the C27S and C64S mutants again failed to be transported into mitochondria (Figure 1F, lanes 4–6). In summary, we found that domain I alone is sufficient for C27- and C64-dependent targeting of Ccs1 to mitochondria.
In vitro C27 and C64 of Ccs1 can be oxidized by Mia40
Cellular depletion of Mia40 results in lowered amounts of mitochondrial Ccs1 (Reddehase et al., 2009). We verified this finding by immunoblot analyses of mitochondria isolated from strains with increased or lowered Mia40 levels (Figure 2A). Notably, the mitochondrial levels of Ccs1 did not appear to be as sensitive toward depletion of Mia40 as the levels of the twin CX3C protein Tim10.
FIGURE 2:

A disulfide bond between Cys-27 and Cys-64 of Ccs1 can be formed by Mia40 in vitro. (A) Protein levels in mitochondria isolated from strains with varying Mia40 levels. The yeast strain GalL–Mia40 was used to regulate the protein levels of Mia40. To overexpress Mia40 (Mia40↑), cells were grown in the presence of galactose. To deplete Mia40 (Mia40↓), cells were cultured in lactate medium containing 0.1% glucose for 16 h. Subsequently, mitochondria were isolated, and 10, 30, and 60 μg of isolated mitochondria were analyzed by SDS–PAGE and Western blotting with the indicated antibodies. (B) Interaction of Ccs1 variants with Mia40 during import into isolated mitochondria. Radiolabeled Ccs1 variants or the twin CX3C protein Tim9 were incubated with mitochondria isolated from a strain overexpressing Mia40 for 8 min at 25°C. Thiol–disulfide exchange reactions were stopped by addition of 50 mM NEM, mitochondria were lysed under denaturing conditions and diluted with Triton X-100–containing buffer, and supernatants were subjected to immunoprecipitation (IP) against Mia40. Bound material was analyzed by nonreducing SDS–PAGE and autoradiography. E, elution from beads after IP; T, total supernatant after IP. (C) In vitro oxidation of full-length Ccs1C17S/C20S/C159S/C229S/C231S by Mia40-WT. The in vitro translated radioactive Ccs1 mutant was incubated with 30 μM purified wild-type Mia40 (Mia40-WT) for the indicated times. Subsequently, samples were TCA precipitated, treated with 10 mM mm-PEG24 for 1 h, subjected to nonreducing (lanes 1–8) or reducing (lanes 9–16) SDS–PAGE, and analyzed by autoradiography. Arrowhead, disulfide-linked complex of Mia40 and Ccs1; asterisk, background band—potentially a disulfide-linked Ccs1 dimer. (D) As in C, except that the in vitro translated radioactive Ccs1 mutant was incubated with buffer (lanes 3–8) or 30 μM purified redox-inactive Mia40-SPS (lanes 11–16) for the indicated times. The samples were subjected to nonreducing SDS–PAGE. Asterisk, background band—potentially a disulfide-linked Ccs1 dimer. (E) Quantification of C and D. The autoradiographs of three different experiments were quantified using ImageJ.
During mitochondrial import Ccs1 also forms an intermediate with Mia40 that is connected by a disulfide bond (Reddehase et al., 2009). Because C27 and C64 are critical for Ccs1 import, we next tested whether these cysteines are involved in intermolecular disulfide bond formation with Mia40 (Figure 2B). We imported radioactively labeled Ccs1 into mitochondria, stopped thiol–disulfide exchange reactions by treatment with N-ethylmaleimide (NEM), and then subjected Mia40 to immunoprecipitation. When analyzing the precipitate from the experiment with wild-type Ccs1 by nonreducing SDS–PAGE and autoradiography we indeed identified a band at a size of ∼90 kDa corresponding to the combined size of Mia40 and Ccs1 (Figure 2B, lane 3). When analyzing the C27S and C64S mutants for an interaction with Mia40, we found that the C27S variant still interacted with Mia40, although to a lesser extent than the wild-type protein (Figure 2B, lane 6). However, the interactions between Mia40 and the C64S mutant, as well as those between Mia40 and the C27S/C64S mutant, were completely abolished, indicating that Mia40 presumably interacts with Cys-64 of Ccs1 during import into mitochondria (Figure 2B, lanes 9 and 12). These results are in agreement with a similar experiment performed by Groß et al., (2011, in this issue of MBoC). Notably, the amounts of the Mia40–Ccs1 complex detected by this method were significantly lower than the amounts of the Mia40–Tim9 complex observed in a control experiment (Figure 2B, compare lanes 3 and 15).
The interactions of twin CX3C and twin CX9C substrates with Mia40 result in the formation of intramolecular disulfides in these substrates. We therefore assessed whether Mia40 also can introduce a disulfide between C27 and C64 (Figure 2, C–E). To this end, we applied an in vitro assay that we had previously established to monitor the Mia40-mediated oxidation of small twin CX9C proteins (Bien et al., 2010). In this assay a reduced and radioactively labeled protein is incubated with either buffer, purified oxidized wild-type Mia40 (Mia40-WT), or a purified Mia40 active-site mutant (Mia40-SPS). At certain time points the reaction is stopped by trichloroacetic acid (TCA) precipitation, which prevents all further thiol–disulfide exchange reactions. Subsequently, free (reduced) thiols but not oxidized disulfides are modified with the alkylating agent mm-PEG24. This derivatization results in an increase in the molecular weight of the protein, which can be detected by a changed migration behavior on SDS–PAGE. Thus these alkylation shift experiments enable us to determine the redox state of a given protein. When applying the assay to a Ccs1 variant in which all cysteines except C27 and C64 were mutated (Ccs1C17S/C20S/C159S/C229S/C231S) we found that the C27–C64 disulfide was formed in the presence of Mia40-WT, although with significantly slower kinetics than with the in vitro oxidation of twin CX9C proteins (Bien et al., 2010). We also found that during the reaction Mia40-WT and Ccs1 formed an intermolecular disulfide bond that was absent when we analyzed the reaction on a reducing gel (Figure 2C, compare lanes 3–8 with lanes 11–16). Instead, a semioxidized form of Ccs1 appeared (i.e., a form in which only one cysteine can be modified with mm-PEG24). Of importance, in the absence of functional Mia40 no oxidation of the two cysteines occurred (Figure 2D). Taken together, the results demonstrate that Ccs1 interacts with Mia40 most likely through C64 and that Mia40 can oxidize Ccs1 in vitro.
In vivo mitochondrial Ccs1 contains a structural disulfide bond between C27 and C64
Next we asked whether C27 and C64 form a disulfide bond in vivo. To address this question, we relied on in vivo alkylation shift experiments (Figure 3A). First, we rapidly lysed cells by treatment with TCA, thus “freezing” the in vivo redox state of Ccs1. Then we modified thiols with the alkylating agent 4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid (AMS) to induce changes in migration on SDS–PAGE comparable to those depicted in Figure 2C and analyzed the proteins by SDS–PAGE and immunoblotting against Ccs1.
FIGURE 3:

In vivo Cys-27 and Cys-64 form a structural disulfide bond in IMS-localized Ccs1. (A) Scheme depicting the setup of the in vivo redox state measurements. (B) Migration standard for AMS alkylation experiments. Cells expressing different cysteine-to-serine mutants of Ccs1 were lysed with Triton X-100 (TX) and incubated with 20 mM DTT at 96°C for 10 min. Subsequently, proteins were precipitated with TCA and modified with 15 mM AMS, except for the sample in lane 8, which was left unmodified. This protocol ensures complete reduction of all cysteines and thus their complete AMS modification. The number of AMS moieties added to the proteins is indicated. The samples were analyzed by Western blotting against Ccs1. (C) In vivo redox state of wild-type (WT) Ccs1 and Ccs1C27S/C64S expressed under the control of the endogenous promoter of Ccs1 from a cen plasmid (pRS). Cells were treated as indicated according to the protocol described in A. Redox states at steady state are depicted in lanes 2 (WT) and 9 (Ccs1C27S/C64S). Note that wild-type Ccs1 can be fully reduced (i.e., modified with seven AMS) only upon boiling with 20 mM DTT at 96°C. (D) As in C, except that the in vivo redox state of overexpressed mitochondrial Ccs1 (pYX/b2–CCS1) was determined.
For a detailed analysis of the Ccs1 redox state in vivo we relied on a series of yeast strains expressing different Ccs1 cysteine mutants in a CCS1-deletion background. To this end, we generated constructs that encode CCS1 gene variants under the control of its endogenous promoter and terminator (pRS), as well as under the control of an overexpression promoter (pYX; see Supplemental Figure S1 for expression levels).
We used these strains first to generate an AMS-migration standard for Ccs1 (Figure 3B). We lysed cells from the different Ccs1 mutant strains, incubated the protein samples at 96°C with the reductant dithiothreitol (DTT) to open all disulfide bonds, and, after TCA precipitation to remove excess DTT, modified cysteine residues with AMS. The individual size shifts of AMS-treated Ccs1 variants correlated with the number of cysteine residues (Figure 3B; see added AMS molecules, and compare with lane 8).
Next we assessed the in vivo redox state of the wild-type Ccs1 (Figure 3C). At steady state Ccs1 was present in two fractions: a completely reduced form (modified with seven AMS molecules) and one that migrated at an apparent size that corresponds to a modification with five AMS molecules, indicating the presence of one disulfide bond in this form (Figure 3C, lane 2). The disulfide bond in this semioxidized form of Ccs1 appears to be very stable (i.e., structural) because it could be opened only upon DTT treatment at 96°C but not at 20°C or by DTT treatment of intact cells (Figure 3C, compare lanes 3–5). Because C17/C20 and C229/C231 are localized to CXXC and CXC motifs, respectively, which usually form unstable disulfides, we reasoned that C27 and C64 might form this structural disulfide bond. We therefore determined the redox state of Ccs1C27S/C64S in vivo (Figure 3C, lanes 6–10). Ccs1C27S/C64S was completely reduced (Figure 3C, lane 9), indicating that the stable disulfide in the wild type was indeed formed between C27 and C64. Notably, our results demonstrate that in vivo all other cysteines of Ccs1 (C17, C20, C159, C229, C231) are in the reduced state, which for C17/C20 and C229/C231 is in accordance with their role in copper binding and transfer.
Because wild-type Ccs1 exists at steady state in two different redox forms we wondered whether mitochondrial Ccs1 might have a redox state different from that of cytosolic Ccs1. To perform this experiment in vivo, we generated yeast cells in which Ccs1 exclusively localizes to the IMS. To this end, the CCS1-deletion strain was transformed with plasmids encoding IMS-targeted Ccs1 variants. These plasmids encoded an extended bipartite mitochondrial targeting signal of cytochrome b2 fused to Ccs1 (b2–Ccs1). This signal consists of a matrix-targeting signal followed by a hydrophobic sorting sequence ensuring transfer into the inner membrane, and an additional IMS-localized domain that prevents accidental matrix import. After import into mitochondria a part of the bipartite targeting signal is removed, and the mature b2–Ccs1 protein localizes to the IMS. Using the alkylation shift assay we found that all wild-type b2–Ccs1 molecules contained the stable C27–C64 disulfide bond (Figure 3D, lane 2). b2–Ccs1C27S/C64S migrated at the same position (Figure 3D, lane 9), confirming that in wild-type, IMS-localized Ccs1, C27 and C64 are oxidized (because they could not be modified with AMS). Thus our results indicate that at least a fraction of the cytosolic Ccs1 does not contain the C27–C64 disulfide (Figure 3C, lane 2).
We repeated this redox state determination multiple times and observed that the ratio between the completely reduced and the semioxidized fraction varied significantly in whole cells but not in mitochondria (Supplemental Figure S2, A-C). Such redox state changes at steady state are frequently observed also in other redox systems and might be due, for example, to differing growth states, cell densities, nutrient supplies, and oxygen tensions (Appenzeller-Herzog and Ellgaard, 2008; Appenzeller-Herzog et al., 2008). Indeed, we observed differences in the redox state of Ccs1 by changes in its expression level and by varying the oxygen tension (Supplemental Figure S2, A, B, and D). When comparing the redox state of Ccs1 at endogenous and overexpressed levels (Figure 3C and Supplemental Figure S2, A and B) we found that in both cases the protein was in part semioxidized, but Ccs1 present at endogenous levels appeared to be overall in a more oxidized state. This finding might indicate that upon overexpression of Ccs1 the oxidation system in the cytosol becomes limiting. Similarly, in cells exposed to 20% oxygen a larger portion of C27 and C64 was present in the oxidized state than in cells cultured under hypoxic conditions (Supplemental Figure S2D). Taken together, the results demonstrate that C27 and C64 form a disulfide bond in the mitochondrial fraction of Ccs1, whereas a varying fraction of cytosolic Ccs1 is completely reduced.
Cysteine mutations in domain I affect the cellular distribution of Ccs1
Next we assessed whether the cysteine residues at positions 27 and 64 are also required for import under in vivo conditions by analyzing their cellular distribution. First, we compared the expression levels of the Ccs1 cysteine mutants expressed from the endogenous CCS1 promoter in whole cells (Figure 4A). All cells contained similar amounts of Ccs1 except for the strains expressing Ccs1C27S, Ccs1C64S, and Ccs1C27S/C64S. We assume that these three mutants are less stable compared with the wild-type Ccs1 and thus become rapidly degraded. This is also supported by circular dichroism spectra of the purified domain I of both the wild type and the C27S/C64S mutant of Ccs1, as the latter variant exhibits less secondary structure compared with the wild type (Supplemental Figure S3).
FIGURE 4:

Mutations of Cys-27 and Cys-64 result in decreased mitochondrial levels of Ccs1. (A) Ccs1 protein levels in Δccs1 cells expressing different Ccs1 variants under the control of the endogenous promoter (pRS). Cells were grown to mid-log phase in S(Lac)-Trp medium, lysed, and analyzed by SDS–PAGE and Western blotting against the indicated proteins. (B) Ccs1 protein levels in mitochondria isolated from Δccs1 cells expressing different Ccs1 variants under the control of the endogenous promoter from a cen plasmid (pRS). Cells were grown to mid-log phase in S(Lac)-Trp medium and lysed, and mitochondria were isolated. To remove proteins attached to the outside of mitochondria, the mitochondrial fraction was treated with 100 μg/ml PK. Mitochondrial fractions were analyzed by SDS–PAGE and immunoblotting using antibodies directed against Ccs1, Mrpl40 (mitochondrial matrix), and Mia40 (IMS). (C) Comparison of the cellular distribution of wild-type Ccs1 and Ccs1C27S/C64S in cells with endogenous levels of Ccs1. Experiments were performed as in A and B. Amounts of 5, 10, and 20 μg of mitochondria and cells at OD600 values of 0.05, 0.1, and 0.2 were analyzed. (D) Ccs1 protein levels in Δccs1 cells expressing different Ccs1 variants under the control of the triosephosphate isomerase promoter from a 2μ plasmid (pYX). As in A, except that Ccs1 was overexpressed in Δccs1 cells. (E) Ccs1 protein levels in mitochondria isolated from Δccs1 cells expressing different Ccs1 variants under the control of the triosephosphate isomerase promoter (pYX). As in B, except that Ccs1 was overexpressed in Δccs1 cells. (F) Comparison of the cellular distribution of wild-type Ccs1 in cells with endogenous levels of Ccs1 and cells overexpressing Ccs1. Experiments were performed as in A and B on Δccs1 cells expressing wild-type Ccs1 under the control of either the endogenous promoter (pRS) or the triosephosphate isomerase promoter (pYX). Totals of 5, 10, and 20 μg mitochondria and cells at OD600 values of 0.05, 0.1, and 0.2 were analyzed.
We then isolated mitochondria from all strains and treated them with low amounts of PK to remove any residual proteins still attached to the outside of the mitochondria. We confirmed with control Western blots against the IMS protein Mia40 that mitochondria remained intact during the procedure (Figure 4B, bottom). Notably, we found that mitochondria isolated from the cells expressing Ccs1C27S, Ccs1C64S, and Ccs1C27S/C64S contained only minute amounts of Ccs1 (Figure 4B). To exclude that these minute amounts of mitochondrial Ccs1C27S, Ccs1C64S, and Ccs1C27S/C64S result only from the low cellular levels of Ccs1, we compared strains expressing the wild type and Ccs1C27S/C64S in more detail (Figure 4C). We found that the strain containing Ccs1C27S/C64S harbored only ∼10% of the Ccs1 found in the wild type. However, the mitochondrial Ccs1 levels in the former strain were well below 2% compared with those in the wild type, indicating that the cellular distribution of Ccs1C27S/C64S to mitochondria is indeed impaired.
To further strengthen this notion, we repeated the experiment using cells expressing the different Ccs1 variants from an overexpression plasmid. The protein levels in these cells were ∼10–20 times higher than with wild-type cells (Supplemental Figure S1). Moreover, the amounts of Ccs1C27S, Ccs1C64S, and Ccs1C27S/C64S were also significantly higher in these strains than in the strains expressing Ccs1 under the control of the endogenous promoter (Supplemental Figures S1 and S4, A and D). When comparing total Ccs1 levels with mitochondrial Ccs1 amounts (Figure 4, D and E) we clearly confirmed that Ccs1C27S, Ccs1C64S, and Ccs1C27S/C64S were lacking from the mitochondrial fraction. Moreover, the distribution of Ccs1 between mitochondria and the remainder of the cell was not substantially altered compared with the distribution in cells containing endogenous levels of Ccs1 (Figure 4F; also compare Figure 4, D and E, with A and B). An exception was the C159S mutant of Ccs1, which appeared to become enriched in the mitochondrial fraction upon overexpression. Taken together, these results support that also in vivo C27 and C64 contribute to the accumulation of Ccs1 in mitochondria.
Cysteine mutations in domain I do not affect the enzyme activity of Ccs1
We were also interested in the effect of cysteine mutations in Ccs1 on its activity. In yeast Ccs1 is essential for the activation of Sod1, and thus Sod1 activity can be used as a measure for the enzymatic activity of Ccs1. We applied a gel-based Sod activity assay on lysates of cells and of isolated mitochondria from yeast strains expressing different Ccs1 cysteine mutants in a CCS1-deletion background (Figure 5). When analyzing whole cells we confirmed previous findings (Schmidt et al., 1999) that the mutation of C229 and C231 in Ccs1 results in the complete loss of Sod1 activity (Figure 5A). Deletion of the remaining cysteines, however, did not affect Sod1 activity in whole-cell lysates (Figure 5A). This was in strong contrast with the situation in mitochondria, in which C27 and C64 were essential for the accumulation of Sod1 activity (Figure 5B). This was due to the import defect of these Ccs1 mutants into mitochondria because fusion of the variants to a b2-targeting sequence restored mitochondrial Sod1 activity (Figures 5C).
FIGURE 5:
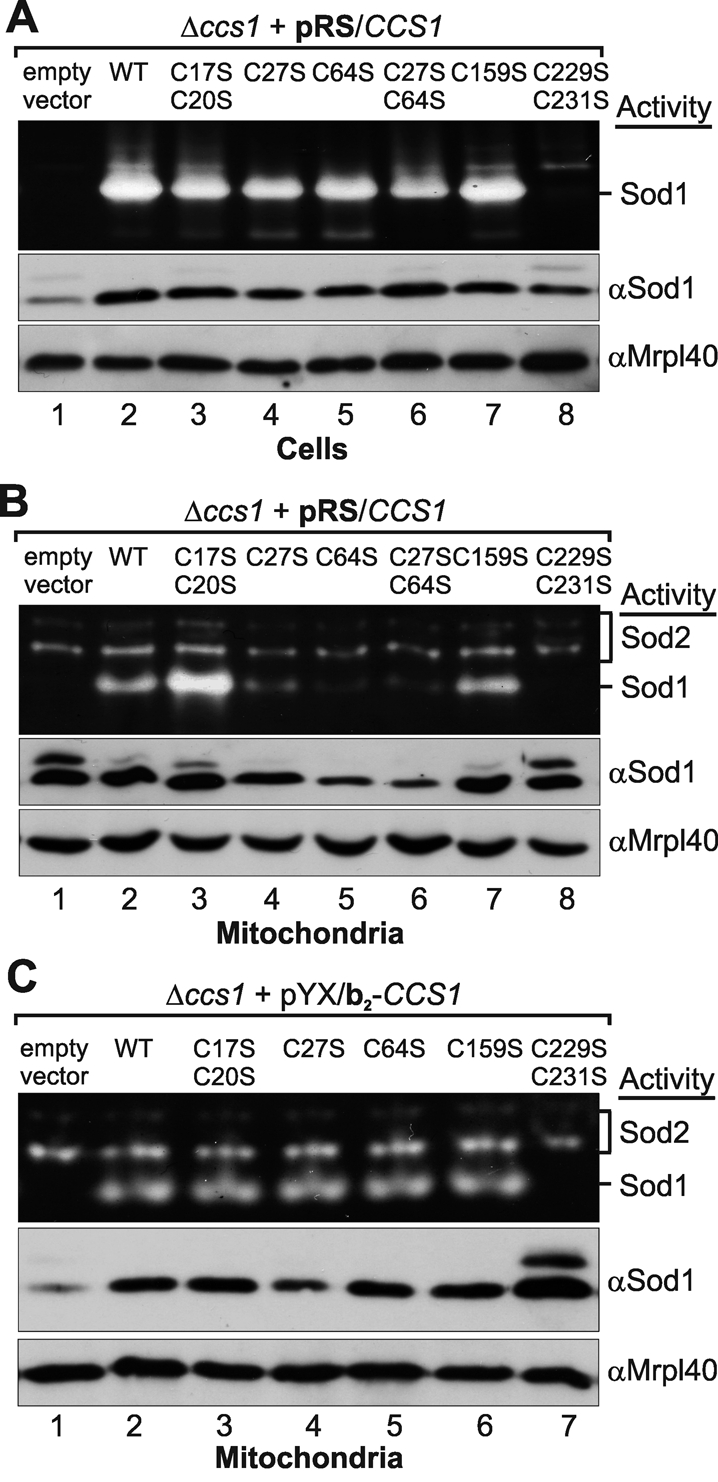
Levels of active mitochondrial Sod1 are lower in strains expressing the C27S and C64S cysteine mutants of Ccs1. (A) Sod1 protein and activity levels in Δccs1 cells expressing different Ccs1 variants under the control of the endogenous promoter (pRS). Cells were grown to mid-log phase in S(Lac)-Trp medium. Subsequently, cells were either lysed in native lysis buffer and analyzed by Sod activity gels (top) or lysed in SDS lysis buffer and analyzed by SDS–PAGE and Western blotting against the indicated proteins (middle and bottom). (B) Sod1 protein and activity levels in mitochondria isolated from Δccs1 cells expressing different Ccs1 variants under the control of the endogenous promoter from a cen plasmid (pRS). Cells were grown to mid-log phase in S(Lac)-Trp medium and lysed, and mitochondria were isolated. To remove proteins attached to the outside of mitochondria, the mitochondrial fraction was treated with 100 μg/ml PK. Mitochondrial fractions were then analyzed by Sod activity gels or by SDS–PAGE and immunoblotting using antibodies directed against Sod1, Mrpl40 (mitochondrial matrix), and Mia40 (IMS). (C) Sod1 activity and protein level in mitochondria isolated from cells expressing different b2–Ccs1 cysteine mutants. As in B, except that Ccs1 variants were fused to a b2-targeting sequence and expressed under the control of the triosephosphate isomerase promoter from a 2μ plasmid (pYX).
Mutations in amino acids forming the potential IMS-targeting signal result in the same phenotype as the C27S and C64S mutants of Ccs1
The small twin CX3C and twin CX9C substrates of Mia40 contain an internal mitochondrial targeting signal, a so-called mitochondria IMS–sorting signal (MISS) or intermembrane space–targeting signal (ITS; Milenkovic et al., 2009; Sideris et al., 2009). Such a signal has not been defined for Ccs1. Closer analysis of the Ccs1 sequence and structure revealed that amino acids I24, I57, and L61 might potentially form part of such a sequence, as they are hydrophobic residues localized to the α-helix presumably interacting with Mia40 (Figure 6A). To investigate the influence of these residues on the cellular distribution, mitochondrial import, and redox state of Ccs1, we generated and analyzed I24E and L61E mutants (Figure 6, B–D). We found that these Ccs1 variants were not imported into isolated mitochondria (Figure 6B) and that they were present only at low levels in whole cells and in minute amounts in mitochondria (Figure 6C). Although they were in principle capable of activating Sod1 in whole cells, mitochondria contained lower levels of active Sod1 (Figure 6D). Thus both Ccs1 variants behaved like Ccs1C27S and CcsC64S and might indeed constitute a part of the MISS/ITS of Ccs1.
FIGURE 6:

MISS/ITS mutants in Ccs1 behave like the C27S and C64S mutants of Ccs1. (A) Scheme of domain I of Ccs1 and location of the amino acids forming the putative MISS/ITS. Locations of amino acids forming the potential MISS/ITS are indicated in the structure of Ccs1 domain I, as well as in the helical-wheel representation of the two helices forming domain I. The black and gray labels of the wheel indicate the hydrophobic and hydrophilic faces of the helices, respectively. (B) Import of MISS/ITS mutants of Ccs1 into isolated mitochondria. The experiment was performed as indicated in Figure 1D. (C) Cellular distribution of the MISS/ITS mutants of Ccs1. The experiment was performed as indicated in Figure 4. (D) Sod activity in cells and mitochondria from CCS1 deletion strains expressing MISS/ITS mutants of Ccs1. The experiment was performed as indicated in Figure 5. (E) Model for the mechanism of Ccs1 distribution between the cytosol and the IMS. See Discussion for details.
DISCUSSION
Ccs1 is dually localized to the cytosol and mitochondria. Mitochondrial import is facilitated by Mia40 (Reddehase et al., 2009). However, the mechanistic details of this import process remained unclear. In this study we provide in-depth analyses of Ccs1 cellular distribution, mitochondrial import, oxidative folding by Mia40, and physiological consequences of Ccs1 mutations.
The oxidation of the Cys-27 and Cys-64 by Mia40 is necessary for efficient import of Ccs1 into mitochondria
We demonstrate that C27 and C64 in domain I of Ccs1 play a critical role in controlling the distribution of Ccs1 and thus the distribution of active Sod1 within the cell. Our findings indicate the following order of events (Figure 6E): After cytosolic synthesis Ccs1 can be either folded in the cytosol or translocated into mitochondria. The latter process is driven by the covalent interaction of Mia40 with immature Ccs1, which results in the formation of a stable intramolecular disulfide bond between C27 and C64 (Figures 2 and 3). The interaction with Mia40 most likely takes place through C64 (Figure 2B). IMS import signals also encompass sequence features that facilitate noncovalent binding to Mia40; in the case of Ccs1 these include the hydrophobic residues I24 and L61 (Figure 6). Without the two cysteines or the two hydrophobic residues Ccs1 is not imported into mitochondria (Figures 1, 4, and 6), and consequently IMS-localized Sod1 is not activated (Figures 5 and 6). The C27–C64 disulfide is also formed in the cytosolic form of Ccs1 even though it appears that a significant fraction of Ccs1 remains completely reduced, indicating that oxidation commences more slowly (Figure 3). It therefore seems likely that cytosolic Ccs1 oxidation occurs without the help of a dedicated oxidoreductase. On the basis of these findings we propose that the mitochondrial import, which is accompanied by enzyme-catalyzed oxidative folding in the IMS, competes with a slow and non–enzyme-catalyzed oxidative folding reaction of Ccs1 in the cytosol. This would ensure that a significant amount of unfolded and reduced Ccs1 remains in the import-competent state after cytosolic translation long enough to be imported into the IMS.
The import and oxidative folding of Ccs1 by Mia40 clearly show differences compared with the twin CX9C and twin CX3C substrates, such as slower folding kinetics, lower import efficiency, and the formation of only one disulfide in a domain with a more complex structure (αββα). The lower efficiency of mitochondrial import of Ccs1 into isolated mitochondria compared with other Mia40 substrates (Figure 1B) might stem from its slower folding kinetics (Figure 2C). Slower folding could result from a longer noncovalent interaction with Mia40. Consequently, less Mia40 is unoccupied and can serve as import receptor for Ccs1. In addition, for Ccs1 import both cysteines in domain I appear to be critical. This is in contrast to twin CX3C and twin CX9C proteins, which are imported into isolated mitochondria if only the cysteine that initially interacts with Mia40 is present (Milenkovic et al., 2007, 2009; Sideris and Tokatlidis, 2007; Sideris et al., 2009). Although we also observed a Mia40–Ccs1C27S complex, we could not detect import of this mutant into mitochondria. This might be explained by the fact that we did not include the reductant β-mercaptoethanol in the import buffer to detect covalent interactions between Mia40 and its substrates (Figure 2B). However, β-mercaptoethanol was added during the import reactions (Figure 1, except C) because reductants normally serve to accelerate the import process of Mia40 substrates (Bien et al., 2010). Furthermore, in vivo both Ccs1C27S and Ccs1C64S are almost absent from the mitochondrial fractions. To our knowledge no such in vivo analyses have been performed for cysteine mutants of twin CX3C and twin CX9C proteins. It thus remains unclear whether these proteins are depleted in mitochondria in intact cells.
Other aspects of Mia40-dependent oxidative folding are conserved, such as the lack of import of a completely cysteine-less mutant, the formation of a stable structural disulfide connecting two antiparallel helices, and internal targeting signals (MISS/ITS) that facilitate recognition by Mia40. It was recently shown that Mia40 also can serve as a chaperone that induces the folding of one α-helix in a twin CX9C substrate and subsequently forms a disulfide between two cysteines in juxtaposed helices (Banci et al., 2010). Although the overall structure of Ccs1 differs from those of the twin CX3C and twin CX9C proteins, domain I resembles these classic Mia40 substrates. This raises the possibility that Mia40, unlike chaperones of the cytosol and the endoplasmic reticulum, might be primed to induce the folding of only one specific fold in a limited subset of substrates.
The disulfide between C27 and C64 is not required for Ccs1 activity but seems to increase the stability of Ccs1
Our results suggest that the structural disulfide between C27 and C64 increases the stability of Ccs1. This is reflected by lower protein levels of Ccs1C27S/C64S at steady state and the lowered secondary structure content of domain I bearing the C27S/C64S mutations compared with the wild type. Ccs1 activity toward Sod1 is not affected, however, by lack of the C27–C64 disulfide. Therefore, the reduced cytosolic form of Ccs1 is still capable of activating Sod1. These results are in agreement with experiments in which a Ccs1 mutant lacking the complete domain I still was capable of activating Sod1 in whole cells (in light of our results, likely the ∼95% of cytosolic Sod1; Schmidt et al., 1999). Notably, an IMS-targeted Ccs1 mutant in which C27 and C64 had been replaced by serines could activate Sod1 in mitochondria, indicating that both cysteines influence mainly the import of Ccs1 into mitochondria.
Consequences of the dual localization of Ccs1 for the cellular antioxidative response
The dual localization of Ccs1 and Sod1 also raised the questions of whether the distribution is variable and can be regulated. Indeed, the expression of a mitochondria-targeted variant of Ccs1 in a CCS1-deletion background results in increased protein and activity levels of mitochondrial Sod1 in yeast (Field et al., 2003). Likewise, growth of mammalian tissue culture cells under hypoxic conditions leads to increased amounts of Sod1 and Ccs1 in mitochondria (Kawamata and Manfredi, 2008). In yeast and in a mouse model, higher levels of mitochondrial Sod1 activity improve the capacity to withstand mitochondria-derived oxidative stress (Klöppel et al., 2010; Fischer et al., 2011).
We thus aimed to identify conditions under which the cellular distribution of Ccs1 changed. To this end, we determined the levels of Ccs1 in mitochondria and whole cells in a number of deletion strains (e.g., Δsod1, Δctt1, Δgrx1Δgrx2Δgrx8, and Δglr1), as well as under varying conditions of oxidative stress (e.g., treatment with paraquat, tetramethylethylenediamine; unpublished data). However, under the tested conditions the cellular Ccs1 distribution remained unchanged. Nevertheless, the lowered stability of reduced Ccs1 could constitute a way to decrease the levels of cellular Ccs1 in the absence of oxidative stress because under these conditions a higher portion of Ccs1 was reduced.
It is important to note that our findings do not exclude the possibility that regulation occurs through changes in distribution in higher eukaryotes. It has already been shown in mammalian cells that the levels of mitochondrial Ccs1 increase during hypoxia (Kawamata and Manfredi, 2008). This also raises the question of how exactly Ccs1 is imported into mammalian mitochondria. We expect that the cellular distribution of human Ccs1 is also controlled by a cysteine-dependent mechanism because overexpression of human Mia40 strongly influences mitochondrial Ccs1 amounts (Kawamata and Manfredi, 2008). However, whereas human Ccs1 domain I shares the same overall structure as its yeast counterpart, it lacks the cysteines corresponding to C27 and C64. Instead, a serine and a threonine are found at the equivalent positions and form a hydrogen bond connecting the two antiparallel α-helices. It will be exciting to test which of the nine cysteines in human Ccs1 contribute to mitochondrial import of the protein.
MATERIALS AND METHODS
Strains and plasmids
For primers and plasmids see Supplemental Table S1. The Δccs1 strain was derived from the wild-type strain YPH499 (MATa ura3-52 lys2-801_amber ade2-101_ochre trp1-Δ63 his3-Δ200 leu2-Δ1) by replacement of the CCS1 open reading frame with a URA3 cassette. Homologous recombination was verified by PCR and controlled by immunoblotting against Ccs1. The Δccs1 strains expressing Ccs1 variants were generated by transformation with the respective plasmids. The GalL–Mia40 strain was introduced before (Terziyska et al., 2005). Strains were grown in S(Lac) medium (0.85% yeast nitrogen base, 2.5% (NH4)2SO4, 9.9% lactate, 20 mg/l adenine sulfate, 20 mg/l uracil, 100 mg/l l-leucine, 30 mg/l l-lysine, 20 mg/l l-histidine, pH 5.5) with 0.2% galactose at 30°C.
Antibodies
The following antibodies were used: anti-Mia40 (Mesecke et al., 2005), anti-Mrpl40 (Gruschke et al., 2010), anti-Pgk1 (Invitrogen, Carlsbad, CA), anti-Sod1 (Klöppel et al., 2010), anti-Tim10, and anti-Tom70 (Mesecke et al., 2005). To generate the Ccs1 antibody, full-length yeast Ccs1 was purified and used for immunization of rabbits.
Isolation of mitochondria, cell fractionation, and Sod activity assays
These methods were performed as described (Klöppel et al., 2010), except that for the analyses of the cellular Ccs1 distribution intact isolated mitochondria were treated with 100 μg/ml PK to remove proteins attached to the outside of mitochondria. The intactness of the outer membrane was verified by immunoblotting against the IMS protein Mia40.
Import of radioactively labeled proteins into isolated mitochondria and in vitro disulfide bond formation assay
These experiments were performed as described (Bien et al., 2010). The import reaction was performed either in the presence of 2 mM β-mercaptoethanol or in the presence of varying amounts of reduced glutathione and in the absence of β-mercaptoethanol.
Analyses of Mia40–Ccs1 interaction after import of radioactively labeled proteins into isolated mitochondria
These experiments were performed as described (Mesecke et al., 2005). During import no reductant was added.
In vivo redox state analyses
To determine the redox state of Ccs1 in whole cells, cells were grown in selective medium to an OD600 of 1.0. Cells were either left untreated or, for the in situ DTT control, incubated for 10 min at 30°C with 100 mM DTT. Then cells were harvested by centrifugation, and cell pellets were resuspended in 10% TCA. For the in vitro DTT controls, cell pellets were resuspended in 20 mM DTT and 1% Triton X-100. Cells were broken by glass bead homogenization, and the lysate was incubated for 10 min at either 20 or 96°C. At 20°C proteins are in their native conformation; however, in lysed cells the accessibility of DTT is improved compared with in situ DTT incubation. TCA was added to all samples, which then were subjected to sonification and glass bead homogenization. TCA pellets were obtained by centrifugation and were resuspended in 40 μl AMS buffer (58 mM Tris, pH 7.0, 7% glycerol, 1.5% SDS, 0.1% bromocresol purple) containing 15 mM AMS in the case of AMS modification. Modification took place for 1 h at 20°C in the dark. Subsequently, samples were analyzed by SDS–PAGE and Western blot against Ccs1.
Supplementary Material
Acknowledgments
We thank Vera Nehr for technical assistance and Martin Ott (University of Kaiserslautern) for critical reading of the manuscript. We are grateful to Johannes Herrmann (University of Kaiserslautern) for many stimulating discussions, critical reading of the manuscript, and continuous support. Funding was obtained from the Fritz-Thyssen-Stiftung. J.R. is a European Molecular Biology Organization Long Term Fellow, and C.P. is a fellow of the Boehringer Ingelheim Fonds.
Abbreviations used:
- AMS
4-acetamido-4′-maleimidylstilbene-2,2′-disulfonic acid
- Ccs1
copper chaperone for Sod1
- IMS
intermembrane space
- ITS
intermembrane space–targeting signal
- MISS
mitochondrial IMS–sorting signal
- NEM
N-ethylmaleimide
- PK
proteinase K
- ROS
reactive oxygen species
- Sod1
copper–zinc superoxide dismutase
- Sod2
manganese superoxide dismutase
- TCA
trichloroacetic acid
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-04-0293) on August 24, 2011.
*These authors contributed equally to this work.
REFERENCES
- Appenzeller-Herzog C, Ellgaard L. In vivo reduction-oxidation state of protein disulfide isomerase: the two active sites independently occur in the reduced and oxidized forms. Antioxid Redox Signal. 2008;10:55–64. doi: 10.1089/ars.2007.1837. [DOI] [PubMed] [Google Scholar]
- Appenzeller-Herzog C, Riemer J, Christensen B, Sorensen ES, Ellgaard L. A novel disulphide switch mechanism in Ero1alpha balances ER oxidation in human cells. EMBO J. 2008;27:2977–2987. doi: 10.1038/emboj.2008.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banci L, et al. Molecular chaperone function of Mia40 triggers consecutive induced folding steps of the substrate in mitochondrial protein import. Proc Natl Acad Sci USA. 2010;107:20190–20195. doi: 10.1073/pnas.1010095107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bien M, Longen S, Wagener N, Chwalla I, Herrmann JM, Riemer J. Mitochondrial disulfide bond formation is driven by intersubunit electron transfer in Erv1 and proofread by glutathione. Mol Cell. 2010;37:516–528. doi: 10.1016/j.molcel.2010.01.017. [DOI] [PubMed] [Google Scholar]
- Chacinska A, Koehler CM, Milenkovic D, Lithgow T, Pfanner N. Importing mitochondrial proteins: machineries and mechanisms. Cell. 2009;138:628–644. doi: 10.1016/j.cell.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culotta VC, Klomp LW, Strain J, Casareno RL, Krems B, Gitlin JD. The copper chaperone for superoxide dismutase. J Biol Chem. 1997;272:23469–23472. doi: 10.1074/jbc.272.38.23469. [DOI] [PubMed] [Google Scholar]
- Field LS, Furukawa Y, O'Halloran TV, Culotta VC. Factors controlling the uptake of yeast copper/zinc superoxide dismutase into mitochondria. J Biol Chem. 2003;278:28052–28059. doi: 10.1074/jbc.M304296200. [DOI] [PubMed] [Google Scholar]
- Fischer LR, Igoudjil A, Magrane J, Li Y, Hansen JM, Manfredi G, Glass JD. SOD1 targeted to the mitochondrial intermembrane space prevents motor neuropathy in the Sod1 knockout mouse. Brain. 2011;134:196–209. doi: 10.1093/brain/awq314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groß DP, Burgard CA, Reddehase S, Leitch JM, Culotta VC, Hell K. Mitochondrial Ccs1 contains a structural disulfide bond crucial for the import of this unconventional substrate by the disulfide relay system. Mol Biol Cell. 2011;22:3758–3767. doi: 10.1091/mbc.E11-04-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruschke S, Grone K, Heublein M, Holz S, Israel L, Imhof A, Herrmann JM, Ott M. Proteins at the polypeptide tunnel exit of the yeast mitochondrial ribosome. J Biol Chem. 2010;285:19022–19028. doi: 10.1074/jbc.M110.113837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata H, Manfredi G. Different regulation of wild-type and mutant Cu,Zn superoxide dismutase localization in mammalian mitochondria. Hum Mol Genet. 2008;17:3303–3317. doi: 10.1093/hmg/ddn226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klöppel C, Michels C, Zimmer J, Herrmann JM, Riemer J. In yeast redistribution of Sod1 to the mitochondrial intermembrane space provides protection against respiration derived oxidative stress. Biochem Biophys Res Commun. 2010;403:114–119. doi: 10.1016/j.bbrc.2010.10.129. [DOI] [PubMed] [Google Scholar]
- Lamb AL, Torres AS, O'Halloran TV, Rosenzweig AC. Heterodimeric structure of superoxide dismutase in complex with its metallochaperone. Nat Struct Biol. 2001;8:751–755. doi: 10.1038/nsb0901-751. [DOI] [PubMed] [Google Scholar]
- Lamb AL, Wernimont AK, Pufahl RA, Culotta VC, O'Halloran TV, Rosenzweig AC. Crystal structure of the copper chaperone for superoxide dismutase. Nat Struct Biol. 1999;6:724–729. doi: 10.1038/11489. [DOI] [PubMed] [Google Scholar]
- Mesecke N, Terziyska N, Kozany C, Baumann F, Neupert W, Hell K, Herrmann JM. A disulfide relay system in the intermembrane space of mitochondria that mediates protein import. Cell. 2005;121:1059–1069. doi: 10.1016/j.cell.2005.04.011. [DOI] [PubMed] [Google Scholar]
- Milenkovic D, Gabriel K, Guiard B, Schulze-Specking A, Pfanner N, Chacinska A. Biogenesis of the essential Tim9-Tim10 chaperone complex of mitochondria: site-specific recognition of cysteine residues by the intermembrane space receptor Mia40. J Biol Chem. 2007;282:22472–22480. doi: 10.1074/jbc.M703294200. [DOI] [PubMed] [Google Scholar]
- Milenkovic D, Ramming T, Muller JM, Wenz LS, Gebert N, Schulze-Specking A, Stojanovski D, Rospert S, Chacinska A. Identification of the signal directing Tim9 and Tim10 into the intermembrane space of mitochondria. Mol Biol Cell. 2009;20:2530–2539. doi: 10.1091/mbc.E08-11-1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J Biol Chem. 2001;276:38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- Rae TD, Schmidt PJ, Pufahl RA, Culotta VC, O'Halloran TV. Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science. 1999;284:805–808. doi: 10.1126/science.284.5415.805. [DOI] [PubMed] [Google Scholar]
- Reddehase S, Grumbt B, Neupert W, Hell K. The disulfide relay system of mitochondria is required for the biogenesis of mitochondrial Ccs1 and Sod1. J Mol Biol. 2009;385:331–338. doi: 10.1016/j.jmb.2008.10.088. [DOI] [PubMed] [Google Scholar]
- Riemer J, Bulleid N, Herrmann JM. Disulfide formation in the ER and mitochondria: two solutions to a common process. Science. 2009;324:1284–1287. doi: 10.1126/science.1170653. [DOI] [PubMed] [Google Scholar]
- Schmidt PJ, Rae TD, Pufahl RA, Hamma T, Strain J, O'Halloran TV, Culotta VC. Multiple protein domains contribute to the action of the copper chaperone for superoxide dismutase. J Biol Chem. 1999;274:23719–23725. doi: 10.1074/jbc.274.34.23719. [DOI] [PubMed] [Google Scholar]
- Sideris DP, Petrakis N, Katrakili N, Mikropoulou D, Gallo A, Ciofi-Baffoni S, Banci L, Bertini I, Tokatlidis K. A novel intermembrane space-targeting signal docks cysteines onto Mia40 during mitochondrial oxidative folding. J Cell Biol. 2009;187:1007–1022. doi: 10.1083/jcb.200905134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sideris DP, Tokatlidis K. Oxidative folding of small Tims is mediated by site-specific docking onto Mia40 in the mitochondrial intermembrane space. Mol Microbiol. 2007;65:1360–1373. doi: 10.1111/j.1365-2958.2007.05880.x. [DOI] [PubMed] [Google Scholar]
- Sturtz LA, Diekert K, Jensen LT, Lill R, Culotta VC. A fraction of yeast Cu,Zn-superoxide dismutase and its metallochaperone, CCS, localize to the intermembrane space of mitochondria. A physiological role for SOD1 in guarding against mitochondrial oxidative damage. J Biol Chem. 2001;276:38084–38089. doi: 10.1074/jbc.M105296200. [DOI] [PubMed] [Google Scholar]
- Terziyska N, Lutz T, Kozany C, Mokranjac D, Mesecke N, Neupert W, Herrmann JM, Hell K. Mia40, a novel factor for protein import into the intermembrane space of mitochondria is able to bind metal ions. FEBS Lett. 2005;579:179–184. doi: 10.1016/j.febslet.2004.11.072. [DOI] [PubMed] [Google Scholar]
- Wong PC, Waggoner D, Subramaniam JR, Tessarollo L, Bartnikas TB, Culotta VC, Price DL, Rothstein J, Gitlin JD. Copper chaperone for superoxide dismutase is essential to activate mammalian Cu/Zn superoxide dismutase. Proc Natl Acad Sci USA. 2000;97:2886–2891. doi: 10.1073/pnas.040461197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.