

Glucagon- and salbutamol-derived cAMP prevents estrogen-induced alteration of canalicular transporter localization and function via different pathways. Glucagon-derived protection depends on PKA activation, whereas salbutamol protection is exerted through a pathway that depends on Epac/MEK and microtubules.
Abstract
In estradiol 17β-d-glucuronide (E17G)–induced cholestasis, the canalicular hepatocellular transporters bile salt export pump (Abcb11) and multidrug-resistance associated protein 2 (Abcc2) undergo endocytic internalization. cAMP stimulates the trafficking of transporter-containing vesicles to the apical membrane and is able to prevent internalization of these transporters in estrogen-induced cholestasis. Hepatocyte levels of cAMP are regulated by hormones such as glucagon and adrenaline (via the β2 receptor). We analyzed the effects of glucagon and salbutamol (a β2 adrenergic agonist) on function and localization of Abcb11 and Abcc2 in isolated rat hepatocyte couplets exposed to E17G and compared the mechanistic bases of their effects. Glucagon and salbutamol partially prevented the impairment in Abcb11 and Abcc2 transport capacity. E17G also induced endocytic internalization of Abcb11 and Abcc2, which partially colocalized with the endosomal marker Rab11a. This effect was completely prevented by salbutamol, whereas some transporter-containing vesicles remained internalized and mainly colocalizing with Rab11a in the perinuclear region after incubation with glucagon. Glucagon prevention was dependent on cAMP-dependent protein kinase (PKA) and independent of exchange proteins activated directly by cAMP (Epac) and microtubules. In contrast, salbutamol prevention was PKA independent and Epac/MEK and microtubule dependent. Anticholestatic effects of glucagon and salbutamol were additive in nature. Our results show that increases in cAMP could activate different anticholestatic signaling pathways, depending on the hormonal mediator involved.
INTRODUCTION
Bile secretion depends on the normal activity of ATP-dependent transporters belonging to the ABC superfamily located in the canalicular pole of the hepatocyte (Gatmaitan and Arias, 1995; Borst and Elferink, 2002). Hence alterations in their activity, localization, and/or expression lead to secretory failure and cholestasis (Trauner et al., 1999; Crocenzi et al., 2003a). Among the most relevant transporters involved in bile formation are the bile salt export pump (Abcb11, also named Bsep), which transports monoanionic bile salts, and the multidrug-resistance associated protein 2 (Abcc2, also named Mrp2), which transports glutathione and glutathione conjugates, as well as a wide variety of anionic compounds, including bipolar, sulfated, or glucuronidated bile salts and bilirubin monoglucuronides and diglucuronides (Gatmaitan and Arias, 1995; Borst and Elferink, 2002). Bile salts and glutathione are chief determinants of the so-called bile salt-dependent and bile salt-independent fractions of the bile flow, respectively (Esteller, 2008).
Studies in different models of experimental cholestasis of clinical relevance, including estrogen-induced cholestasis, revealed a series of characteristic alterations in the localization of canalicular transporters (Dombrowski et al., 2000; Mottino et al., 2002; Crocenzi et al., 2003a). These works demonstrated that, in cholestatic conditions, Abcb11 and Abcc2 left the canalicular membrane, undergoing endocytic internalization into vesicular compartments. This phenomenon was systematically associated with a failure in the secretion of their specific substrates, pointing to a key role of this pathomechanism in the cholestatic process. The endocytic internalization of transporters in cholestasis can be counterbalanced, at least conceptually, by signaling molecules that stimulate routes of exocytic insertion, therefore modifying the internalization/insertion balance toward the latter process. In addition, the exocytic reinsertion of the endocytosed transporters that spontaneously occurs after the initial cholestatic insult with estradiol 17β-d-glucuronide (E17G; Crocenzi et al., 2003a) can be accelerated by these signaling molecules, thus providing a means to speed up recovery from cholestasis. A signaling mediator with these stimulatory properties is cAMP. This signaling messenger stimulates, in the liver, the traffic of hepatocellular transporters from its places of synthesis to the canalicular membrane in a microtubule-dependent manner, as well as the fusion of transporter-containing vesicles from the subapical compartment with the apical membrane (Roelofsen et al., 1998; Kipp and Arias, 2002). Our group demonstrated that cAMP prevents the internalization and accelerates the reinsertion of ABC transporters in E17G-induced cholestasis (Mottino et al., 2002; Crocenzi et al., 2003a).
cAMP is the second messenger of a number of hormones that act at the hepatic level, such as glucagon (Glu), vasoactive intestinal peptide, and catecholamines (Steinberg et al., 2003). The potential therapeutic use of such hormones is, in addition, rationally justified by the fact that, in cholestasis, the hormonal mechanisms that lead to cAMP synthesis are diminished due to the altered expression of subunit α of small stimulatory G proteins (Bouscarel et al., 1998). Among the hormones with potential anticholestatic properties, Glu and adrenaline could represent the most appropriate ones. Glu is recognized for its capacity to promote bile flow (Branum et al., 1991) and to elevate cAMP levels in humans (Pecker et al., 1979). In addition, Glu is produced in the pancreas and delivered to portal circulation, thus arriving at the liver in high levels before reaching systemic circulation. Adrenaline increases cAMP levels in liver by means of the β2 adrenergic receptor (Morgan et al., 1983) and shares with Glu some cAMP-dependent hormonal effects in this organ such as glycogenolysis (Bollen et al., 1998). Consequently, any modification of the synthesis or release rate of these hormones is expected to have an important impact in the liver in terms of signaling.
Within the cell, adenylyl-cyclases, phosphodiesterases, and pathway effectors are closely associated. Therefore, cAMP generation and degradation is compartmentalized, leading to the existence of localized pools of the messenger. cAMP signaling can be regulated by the integration of all stimuli and cellular processes implied in this generation/degradation (Tasken and Aandahl, 2004). Experimental data confirm that passive diffusion of cAMP in the cytosol is substantially restricted, as shown by monitoring the spatial distribution of cAMP in single living cells and by more indirect in vitro approaches (Zaccolo et al., 2006). The local elevation of cAMP can trigger different pathways. Among them, two are known as effectors of cAMP in exocytosis processes: cAMP-dependent protein kinase (PKA) and exchange proteins activated directly by cAMP (Epac; de Rooij et al., 1998; Seino and Shibasaki, 2005). The aim of the present work is to evaluate the capability of the cAMP-elevating hormones Glu and adrenaline to prevent E17G-induced cholestasis, using isolated rat hepatocyte couplets, and to characterize the eventual mechanism of protection in term of the signaling pathway involved (PKA or Epac) and cytoskeletal requirements, including a systematic comparison of their effects.
RESULTS
Salbutamol and glucagon partially prevent E17G-induced impairment of canalicular secretory function in a concentration-dependent manner
To assess which concentrations of Glu and salbutamol (Sal; β2 adrenergic agonist) produce a maximal protective effect, we performed concentration–response studies with various concentrations of Glu (0.001–10 μM) and Sal (0.1–1000 μM). Both Glu and Sal partially prevented the effect of 50 μM E17G on canalicular vacuolar accumulation (cVA) of cholyl-lysylfluorescein (CLF) and glutathione methylfluorescein (GS-MF) throughout the range of concentrations evaluated. Glu concentrations higher than 0.1 μM did not produce further protection; likewise, Sal preventive effect was maximal at the dose of 1 μM (Supplemental Figure S1). Hence the remaining experiments were performed using these concentrations.
To further characterize the preventive effect of these compounds, we carried out different concentration–response studies in which the concentration of E17G was modified in the presence of fixed concentrations of Sal or Glu (Figure 1). Curves were adjusted assuming that the parameter minimal effect (bottom) was equal to 0 and that the Hill slope coefficient was 1. As shown in Table 1, the IC50 of CLF and GS-MF accumulation induced by E17G was significantly increased in the presence of Glu by 104 and 67%, respectively. Moreover, Glu prevented the maximal inhibition of GS-MF accumulation induced by E17G. Similarly, the hormone tended to decrease the maximal effect of the estrogen glucuronide on CLF accumulation, although this difference did not reach statistical significance. Sal also increased the IC50 of E17G in CLF and GS-MF accumulation (90 and 100%, respectively). However, Sal did not alter the maximal effect of the estrogen metabolite.
FIGURE 1:

Prevention by Glu (top) and Sal (bottom) of E17G-induced impairment of cVA of CLF (left) and GS-MF (right). IRHCs were preincubated with Glu (0.1 μM) or Sal (1 μM) for 15 min and then exposed to E17G (12.5–800 μM) for an additional 20 min. cVAs of CLF and GS-MF were calculated as the percentage of couplets displaying visible fluorescence in their canalicular vacuoles from a total of at least 200 couplets per preparation, referred to control cVA values. cVA control values were 75 ± 2% for CLF and 76 ± 2% for GS-MF. Data are expressed as mean ± SEM (n = 3).
TABLE 1:
Estimates of the concentration–response curve parameters of E17G-induced impairment of canalicular secretory function in the presence of Glu (0.1 μM) and Sal (1 μM) depicted in Figure 1.
| CLF | GS-MF | |||
|---|---|---|---|---|
| E17G alone | E17G + agent | E17G alone | E17G + agent | |
| Glu | ||||
| Maximal effect (%) | 0 ± 2 | 8 ± 4 | 2 ± 2 | 22 ± 4a |
| IC50 (μM) | 46 ± 1 | 94 ± 2a | 51 ± 1 | 85 ± 2a |
| Sal | ||||
| Maximal effect (%) | 4 ± 3 | 6 ± 3 | 7 ± 4 | 6 ± 3 |
| IC50 (μM) | 69 ± 1 | 131 ± 1a | 44 ± 2 | 88 ± 2a |
Data were analyzed with GraphPad Prism software, assuming the minimal effect of E17G (100% value) to be the cVA in control IRHCs.
aSignificantly different from the E17G-alone curve (p < 0.05).
Salbutamol and glucagon increase intracellular cAMP in isolated rat hepatocyte couplets
To determinate whether Sal and Glu increase cAMP levels, we incubated isolated rat hepatocyte couplets (IRHCs) with these agonists in the presence of the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine (IBMX) and compared their effects with IRHC exposed to IBMX alone (control). Glu and Sal significantly increased the intracellular levels of cAMP compared with control (control, 2.62 ± 0.40; Glu, 25.79 ± 2.85*; Sal, 25.67 ± 0.91* pmol/105 IRHC; *different from control, p < 0.05, n = 3).
Glucagon but not salbutamol requires PKA activation for its preventive effect on E17G-induced impairment of canalicular secretory function
To study whether Sal or Glu protection is mediated by PKA, IRHCs were preincubated with two different PKA inhibitors. As shown in Figure 2, the ability of Glu to prevent the decrease in cVA of both CLF and GS-MF was blocked by the PKA inhibitors H89 and KT5720, indicating that this prevention requires PKA activation. It was surprising that when we analyzed Sal prevention, its preventive capability was not blocked by these inhibitors. These findings indicate that Sal preventive effect does not involve PKA activation. To confirm glucagon activation of PKA, kinase activity was evaluated by Western blot of the phosphorylated forms of PKA substrates (Figure 3). As previously reported, these immunoblots present several bands that could represent phosphorylated PKA substrates (Brennan et al., 2006; Lei et al., 2007). On the basis of their response to the PKA activator DBcAMP and PKA inhibitors, we analyzed two bands, of ∼25 and 110 kDa. Glucagon increased the phosphorylation of these two PKA substrates, and these increases were blocked by PKA inhibitors. These experiments also revealed that salbutamol activates PKA, as previously demonstrated (Meja et al., 2004). 8-(4-Chlorophenylthio)-2´-O-methyladenosine 3´,5´-cyclicmonophosphate (8-CPT-2´-O-Me-cAMP), a specific Epac activator, did not affect PKA phosphorylation.
FIGURE 2:
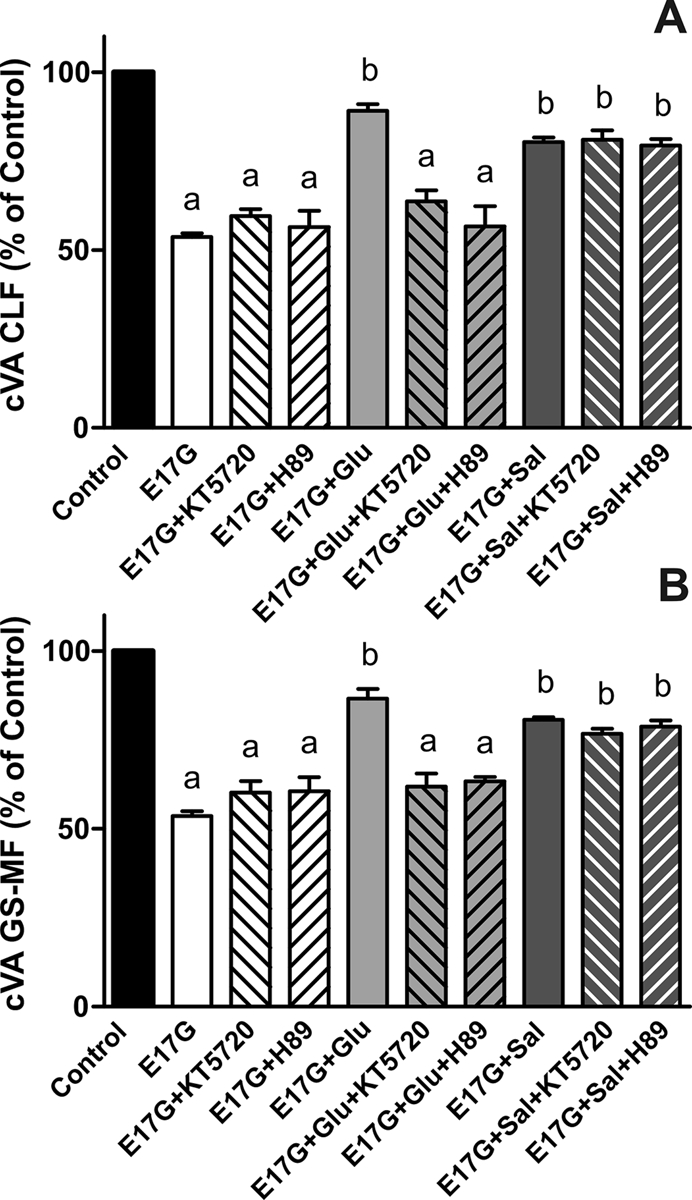
Effect of the PKA inhibitors H-89 and KT5720 on the prevention by Glu and Sal of E17G-induced impairment of cVA of CLF (A) and GS-MF (B). Couplets pretreated for 15 min with H-89 (200 nM), KT5720 (50 nM), or vehicle (DMSO) in controls were subsequently exposed to Glu or Sal for a further 15-min period and then exposed to E17G (50 μM) for an additional 20-min period. cVAs of CLF and GS-MF were calculated as the percentage of couplets displaying visible fluorescence in their canalicular vacuoles from a total of at least 200 couplets per preparation, referred to control cVA values. Control cVA values were 76 ± 1% for CLF and 76 ± 2% for GS-MF. Data are expressed as mean ± SEM (n = 3). aSignificantly different from control (p < 0.05). bSignificantly different from E17G and control (p < 0.05).
FIGURE 3:

Estimation of PKA activation by Glu and Sal. Isolated rat hepatocytes were incubated with DBcAMP (10 μM, positive control), Glu (0.1 μM), Sal (1 μM), and 8-CPT-2´-O-Me-cAMP (8-CPT-cAMP, 50 μM, negative control) in the presence or absence of PKA inhibitors (50 nM KT5720 or 200 nM H-89). PKA activity was determined by Western blot, using an antibody against phosphorylated PKA substrates. Two bands of 25 and 110 kDa were analyzed based on their response to DBcAMP and PKA inhibitors. Two exposure times were necessary to reveal these bands, a short exposure for the 25-kDa band and a long exposure for the 110-kDa band. Differences in sample loading were corrected by the densitometric signal of the corresponding actin band. The ratio of each phosphorylated substrate/actin band density was compared with that of control bands (100%). Data are expressed as mean ± SEM (n = 3). aSignificantly different from control (p < 0.05). bSignificantly different from the agonist alone (p < 0.05).
Salbutamol but not glucagon requires microtubule integrity for its preventive effect on E17G-induced impairment of canalicular secretory function
To evaluate whether the protection afforded by these compounds occurs by a microtubule-dependent mechanism, we pretreated IRHC with colchicine, an inhibitor of microtubule polymerization. As shown in Figure 4, the preincubation with colchicine blocked the preventive effect of Sal on E17G impairment of Abcc2 and Abcb11 function. On the other hand, colchicine did not cause inhibition in the protective effect of Glu. Therefore, unlike Sal, Glu exerts its protective effect by a microtubule-independent mechanism.
FIGURE 4:
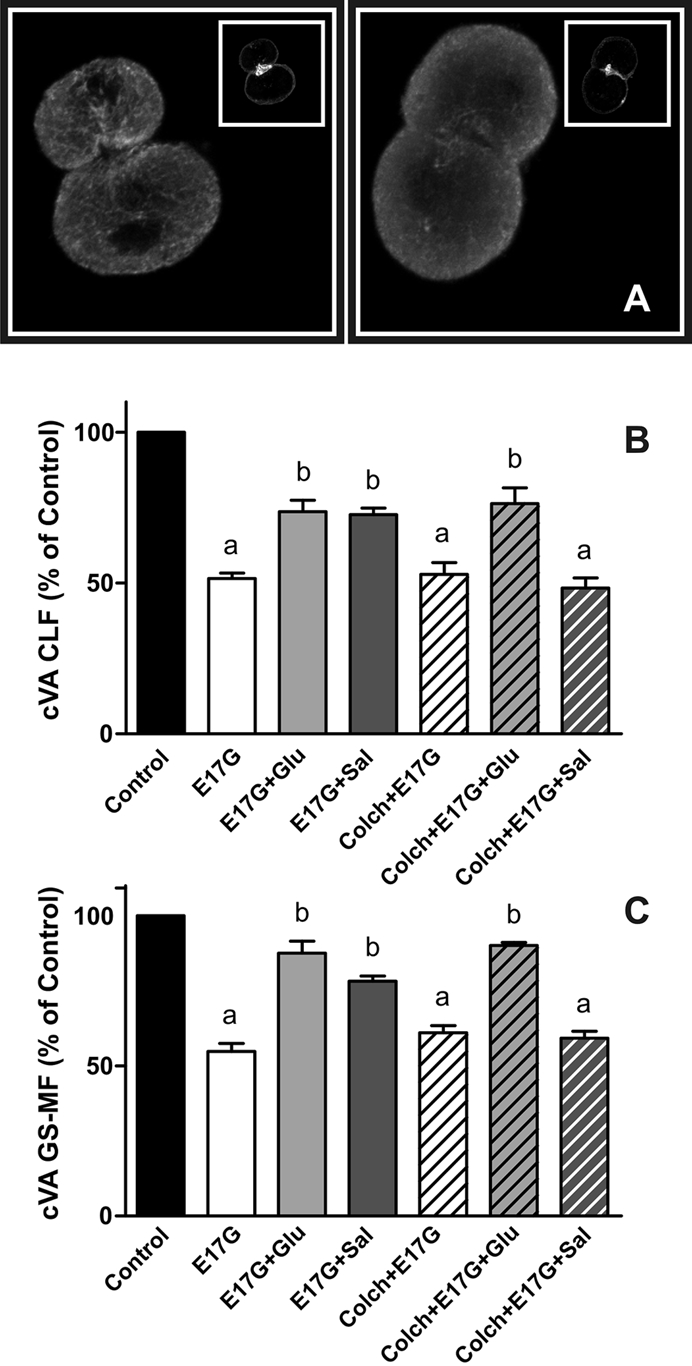
(A) Representative confocal images of control-treated (left) and colchicine-treated (right) IRHCs stained for β-tubulin and actin (inset). (B, C) Effect of colchicine on Glu and Sal prevention of E17G-induced impairment of cVAs of CLF (B) and GS-MF (C). Couplets pretreated for 30 min with colchicine (Colch, 1 μM) or vehicle (DMSO) were subsequently exposed to Glu (0.1 μM) or Sal (1 μM) for 15 min and then exposed to E17G (50 μM) for an additional 20 min. cVAs of CLF and GS-MF were calculated as the percentage of couplets displaying visible fluorescence in their canalicular vacuoles from a total of at least 200 couplets per preparation, referred to control cVA values. Control cVA values were 75 ± 3% for CLF and 76 ± 1% for GS-MF. Data are expressed as mean ± SEM (n = 3). aSignificantly different from control (p < 0.05). bSignificantly different from E17G and control (p < 0.05).
Glucagon and salbutamol have an additive protective effect in E17G-induced impairment of canalicular secretory function
To corroborate that Glu and Sal present different preventive mechanisms, we coincubated IRHC with these compounds and evaluated cVA of CLF and GS-MF. Figure 5 shows that cells cotreated with Glu and Sal present an additive preventive effect on the impairment of the canalicular secretory function induced by E17G. Moreover, preincubation with PKA inhibitors (KT5720 and H89) led to a partial inhibition of these additive effects, in accordance with the results shown in Figure 2.
FIGURE 5:
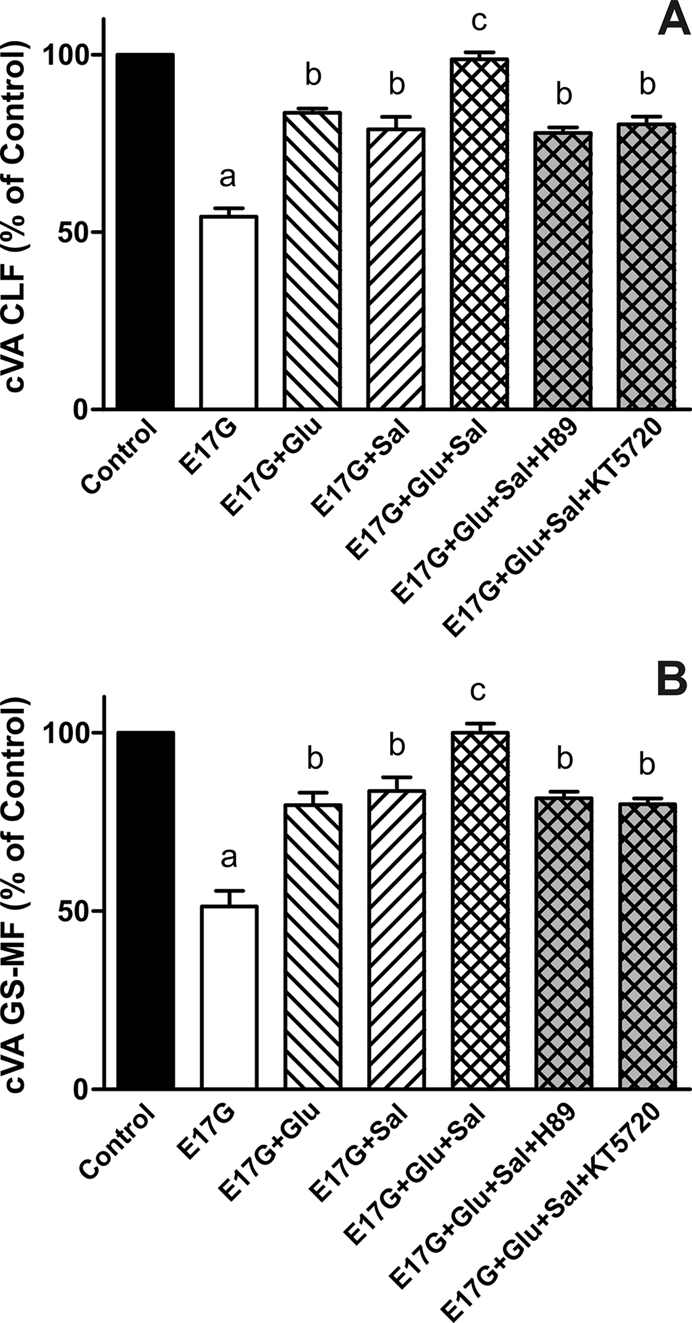
Effect of coincubation with Glu and Sal on E17G-induced impairment of canalicular vacuolar accumulations (cVAs) of CLF (A) and GS-MF (B). IRHCs were incubated with Glu (0.1 μM) and Sal (1 μM), either alone or together, for 15 min and then exposed to E17G (50 μM) for an additional 20 min. Finally cVAs of CLF and GS-MF were calculated as the percentage of couplets displaying visible fluorescence in their canalicular vacuoles from a total of at least 200 couplets per preparation, referred to control cVA values. Control cVA values were 76 ± 3% for CLF and 75 ± 1% for GS-MF. Data are expressed as mean ± SEM (n = 3). aSignificantly different from control (p < 0.05). bSignificantly different from E17G and control (p < 0.05).cSignificantly different from E17G, E17G + Glu, E17G + Sal, E17G + Glu + Sal + H-89, and E17G + Glu + Sal + KT5720 (p < 0.05).
Salbutamol activation of Epac pathway is necessary to prevent E17G-induced impairment in biliary secretory function
To evaluate whether Epac is implied in the protection by Sal of E17G-induced cholestatic effects, IRHC were incubated with 8-CPT-2´-O-Me-cAMP, a specific Epac agonist. Figure 6 shows that treatment with 8-CPT-2´-O-Me-cAMP partially prevented E17G-induced alteration of Abcb11 and Abcc2 transport function in a microtubule-dependent manner. In addition, western blots of pMEK, an indicator of mitogen-activated protein kinase kinase (MEK) activation (Epac downstream; Bos et al., 2001), revealed that Sal and 8-CPT-2´-O-Me-cAMP increased the amount of pMEK (Figure 7). The same figure shows that Glu does not affect pMEK levels with respect to control. To confirm the possible involvement of the Epac pathway in the beneficial effects of Sal, we used the specific MEK inhibitor PD98059. As shown in Figure 8, the ability of Sal and 8-CPT-2´-O-Me-cAMP to prevent the decrease in cVA of both CLF and GS-MF induced by E17G was blocked by the MEK inhibitor, linking Sal prevention with Epac activation. When we analyzed the ability of Glu to prevent E17G effects, we found that its preventive capability was not blocked by this inhibitor. Furthermore, IRHC coincubated with 8-CPT-2´-O-Me-cAMP together with Sal or Glu (Figure 9) showed an additive protective effect only in 8-CPT-2´-O-Me-cAMP + Glu group but not in 8-CPT-2´-O-Me-cAMP + Sal group, corroborating that Sal, but not Glu, shares with 8-CPT-2´-O-Me-cAMP the protective mechanism triggered by activation of the Epac pathway.
FIGURE 6:
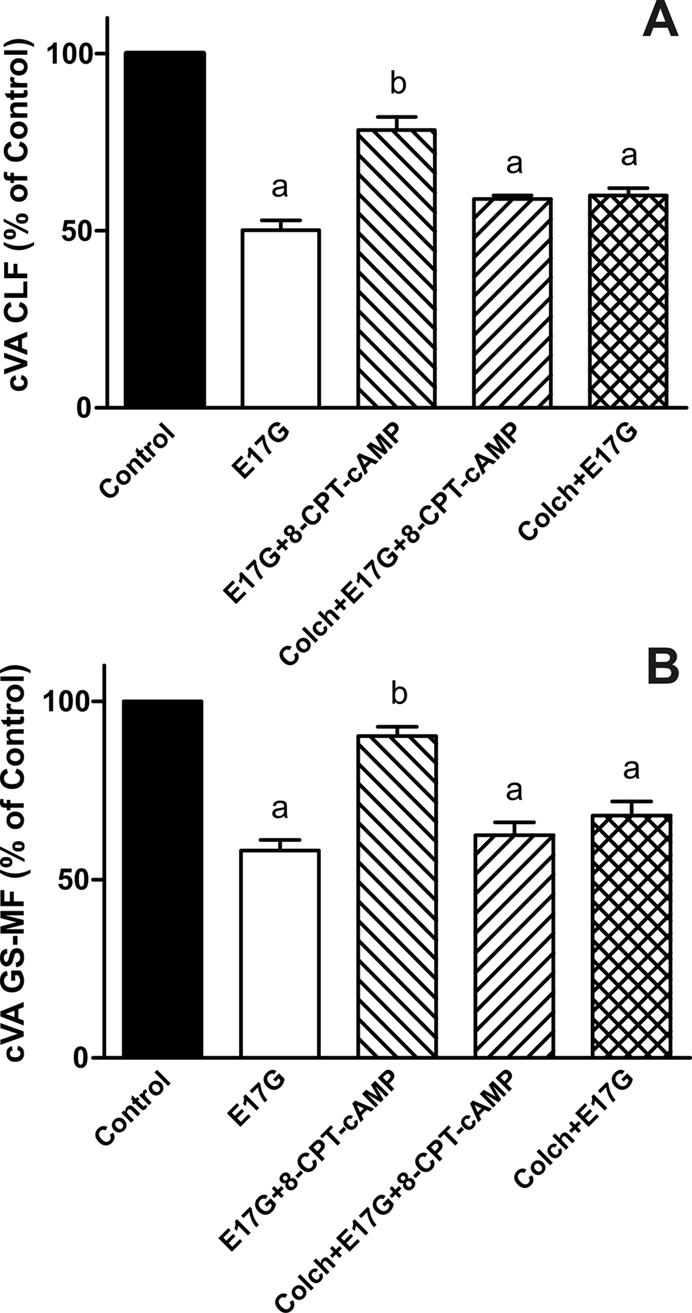
Effect of colchicine on the prevention of E17G-induced impairment in the canalicular vacuolar accumulation (cVA) of CLF (A) and GS-MF (B) afforded by activation of the Epac pathway with 8-CPT-2´-O-Me-cAMP. IRHCs were preincubated with colchicine for 30 min and then with 8-CPT-2´-O-Me-cAMP (8-CPT-cAMP, 50 μM) for a further 15 min and subsequently exposed to E17G (50 μM) for an additional 20 min. Finally, cVAs of CLF and GS-MF were calculated as the percentage of couplets displaying visible fluorescence in their canalicular vacuoles from a total of at least 200 couplets per preparation, referred to control cVA values. Control cVA values were 74 ± 1% for CLF and 75 ± 2% for GS-MF. Data are expressed as mean ± SEM (n = 3). aSignificantly different from control (p < 0.05). bSignificantly different from E17G and control (p < 0.05).
FIGURE 7:
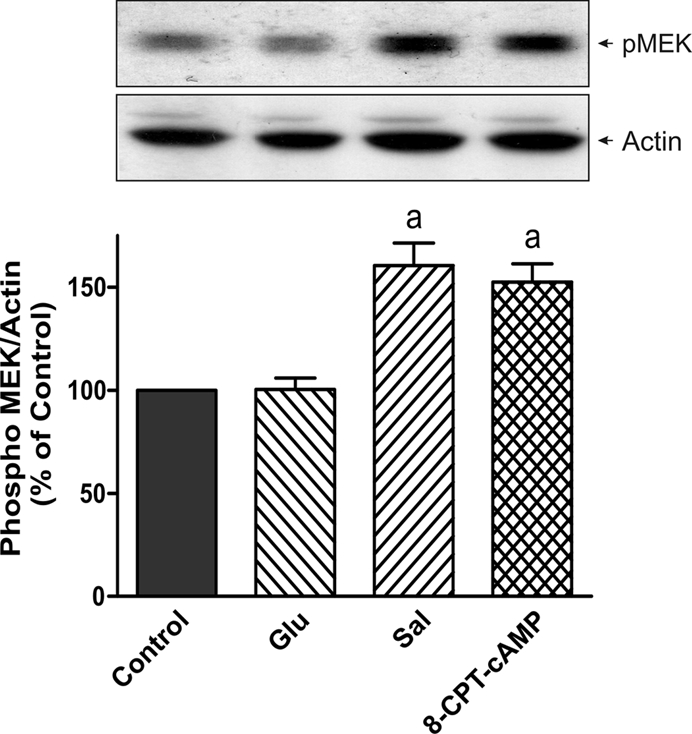
Estimation of MEK activation by salbutamol and 8-CPT-2´-O-Me-cAMP. Isolated rat hepatocytes were incubated with Glu (0.1 μM), Sal (1 μM), and 8-CPT-2´-O-Me-cAMP (8-CPT-cAMP, 50 μM, positive control). MEK activity was determined by immunoblots, using an antibody against phosphorylated MEK1/2. A band at 45 kDa was detected. Differences in sample loading were corrected by the densitometric signal of the corresponding actin band. The ratio of each pMEK/actin band density was compared with the ratio of control bands (100%). Data are expressed as mean ± SEM (n = 3). aSignificantly different from control (p < 0.05).
FIGURE 8:
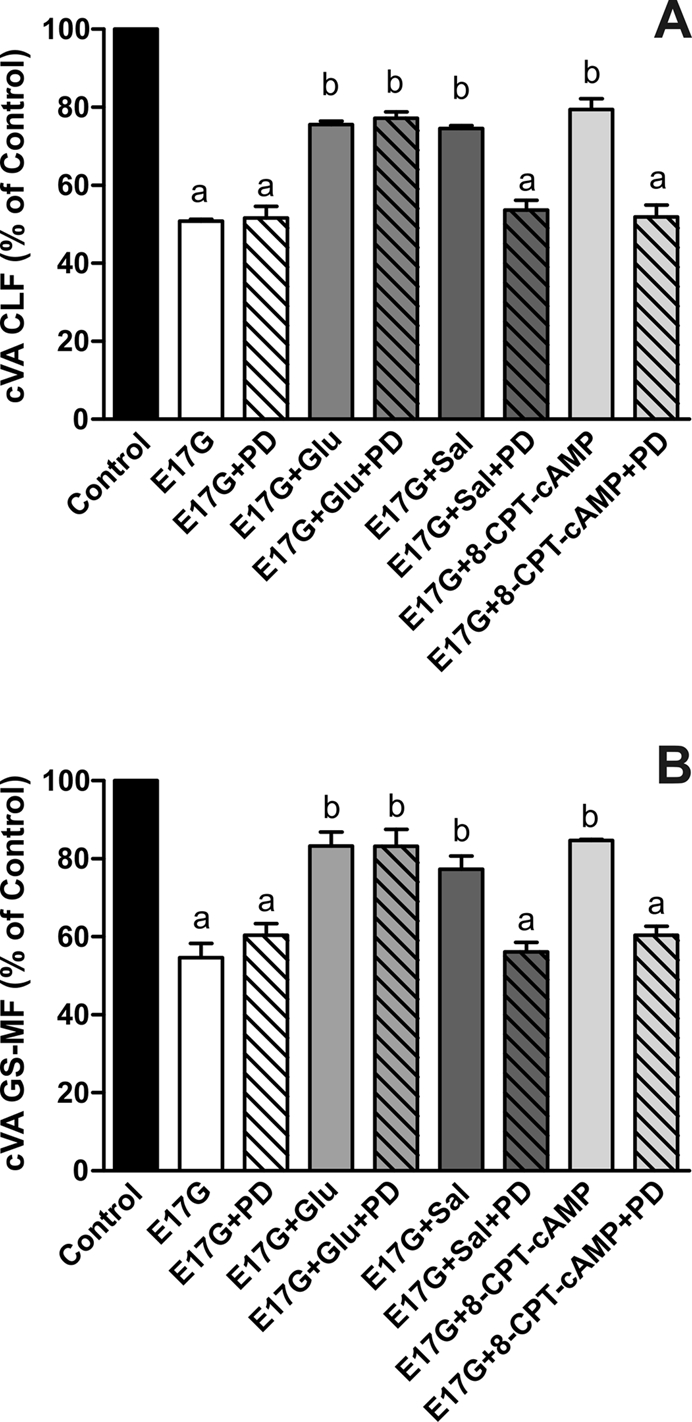
Effect of MEK1/2 inhibitor PD98059 on Glu, Sal, and 8-CPT-2´-O-Me-cAMP prevention of E17G-induced impairment of canalicular vacuolar accumulations (cVAs) of CLF (A) and GS-MF (B). Couplets pretreated for 15 min with PD98059 (PD, 1 μM) or vehicle (DMSO) were subsequently exposed to Glu (0.1 μM), Sal (1 μM), or 8-CPT-2´-O-Me-cAMP (8-CPT-cAMP, 50 μM) for 15 min and then exposed to E17G (50 μM) for an additional 20 min. cVAs of CLF and GS-MF were calculated as the percentage of couplets displaying visible fluorescence in their canalicular vacuoles from a total of at least 200 couplets per preparation, referred to control cVA values. Control cVA values were 75 ± 1% for CLF and 76 ± 2% for GS-MF. Data are expressed as mean ± SEM (n = 3). aSignificantly different from control (p < 0.05). bSignificantly different from E17G and control (p < 0.05).
FIGURE 9:
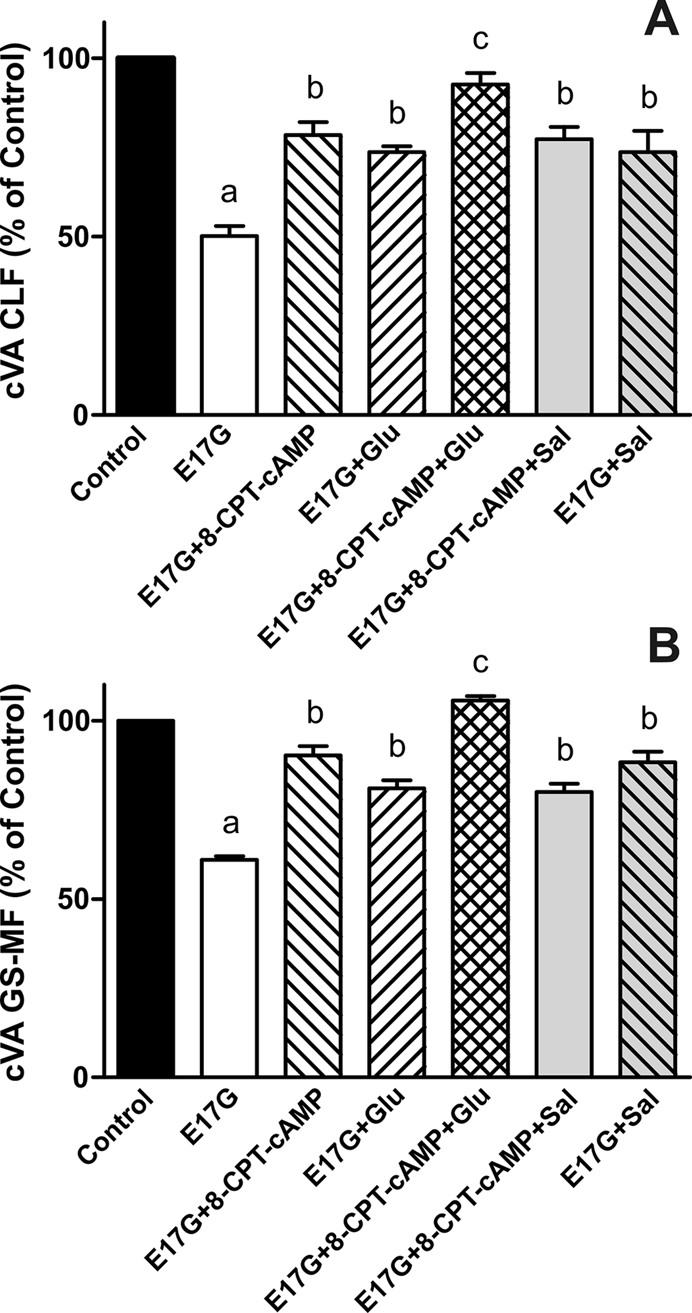
Effect of coincubation with Glu + 8-CPT-2´-O-Me-cAMP and Sal + 8-CPT-2´-O-Me-cAMP on E17G-induced impairment of cVA of CLF (A) and GS-MF (B). IRHCs were coincubated with Glu (0.1 μM) and 8-CPT-2´-O-Me-cAMP (8-CPT-cAMP, 50 μM), or Sal (1 μM) and 8-CPT-2´-O-Me-cAMP, for 15 min and then exposed to E17G (50 μM) for an additional 20 min. Finally cVAs of CLF and GS-MF were calculated as the percentage of couplets displaying visible fluorescence in their canalicular vacuoles from a total of at least 200 couplets per preparation, referred to control cVA values. Control cVA values were 77 ± 3% for CLF and 74 ± 1% for GS-MF. Data are expressed as mean ± SEM (n = 3). aSignificantly different from control (p < 0.05). bSignificantly different from E17G and control (p < 0.05).cSignificantly different from E17G, E17G + Glu, and E17G + 8-CPT-cAMP (p < 0.05).
Glucagon and salbutamol prevent E17G-induced retrieval of Abcb11 and Abcc2 by different mechanisms
Figures 10 and 11 illustrate the effect of the different treatments on Abcb11 and Abcc2 cellular localization in IRHC, respectively. F-Actin was stained and used to demarcate the limits of the canalicular membrane since actin network exhibits a predominant ring-shaped pericanalicular distribution (Roma et al., 1998). Confocal images show that, in control cells, Abcb11 and Abcc2 were mainly localized within the actin pericanalicular ring space. IRHC treated with Glu, Sal, or 8-CPT-2´-O-Me-cAMP alone presented the same distribution pattern (unpublished data). E17G induced redistribution of both Abcb11 and Abcc2 over a greater distance from the center of the canalicular vacuoles. Glu, Sal, and 8-CPT-2´-O-Me-cAMP prevented E17G-induced retrieval of Abcb11 and Abcc2. However, some remnant transporter-containing vesicles remain visible in a deep compartment after preincubation with Glu of E17G-treated IRHC, which is not evident under Sal and 8-CPT-2´-O-Me-cAMP treatment (Figures 10 and 11A, arrowheads). Protection of canalicular transporters internalization by Glu, Sal, and 8-CPT-2´-O-Me-cAMP was confirmed by densitometric analysis of the distribution of the transporters in the vicinity of the canalicular vacuole (Supplemental Figures S3 and S4). The mechanisms involved in their preventive effects were the same as those involved in the protection against the functional impairment of these transporters induced by E17G, that is, Glu-mediated prevention of canalicular transporter delocalization was blocked when cells were pretreated with the PKA inhibitors H89 and KT5720 (Figures 10 and 11B), whereas Sal- and 8-CPT-2´-O-Me-cAMP–mediated protection was abolished by colchicine, suggesting a microtubule-dependent protective effect and PD98059, suggesting MEK1/2 involvement (Figures 10 and 11, C and D). Thus, these findings support the participation of the Epac–MEK1/2 pathway in the anticholestatic effects of Sal.
FIGURE 10:

Representative confocal images showing cellular distribution of Abcb11, actin, and merged images (with actin in red and Abcb11 in green) in IRHCs under different treatments. (A) Prevention by Glu (0.1 μM) and Sal (1 μM) of E17G (200 μM)-induced retrieval of Abcb11. Note that under control conditions Abcb11-associated fluorescence is mainly localized at the canalicular membrane in the area delimited by the pericanalicular actin ring. E17G induced a clear internalization of Abcb11-containing vesicles beyond the limits of the pericanalicular actin ring, a phenomenon significantly prevented by Glu and Sal. The arrowhead in the E17G + Glu group shows some transporter-containing vesicles that remain internalized in a deep intracellular compartment. (B) Preincubation with the PKA inhibitor H-89 (200 nM) or KT5720 (KT, 50 nM) significantly inhibited the preventive effect of Glu on E17G-induced internalization of Abcb11. (C) Pretreatment of IRHC with the microtubule-disrupting agent colchicine (Colch, 1 μM), or the MEK1/2 inhibitor PD98059 (PD, 1 μM) abolished the preventive effect of Sal on E17G-induced internalization of Abcb11. (D) Pretreatment of IRHC with 8-CPT-2´-O-Me-cAMP (8-CPT-cAMP, 50 μM), an Epac agonist, also prevented delocalization of Abcb11 induced by E17G, a phenomenon also abolished by preincubation with Colch and PD. None of the treatments affected the normal distribution of actin, which showed a predominant pericanalicular distribution.
FIGURE 11:

Representative confocal images showing cellular distribution of Abcc2, actin, and merged images (with actin in red and Abcc2 in green) in IRHCs under different treatments. (A) Prevention by Glu (0.1 μM) and Sal (1 μM) of E17G (200 μM)-induced retrieval of Abcc2. Note that under control conditions Abcc2-associated fluorescence is mainly localized at the canalicular membrane in the area delimited by the pericanalicular actin ring. E17G induced a clear internalization of Abcc2-containing vesicles beyond the limits of the pericanalicular actin ring, a phenomenon significantly prevented by Glu and Sal. The arrowhead in the E17G + Glu group shows some transporter-containing vesicles that remain internalized in a deep intracellular compartment. (B) Preincubation with the PKA inhibitor H-89 (200 nM) or KT5720 (KT, 50 nM) significantly inhibited the preventive effect of Glu on E17G-induced internalization of Abcc2. (C) Pretreatment of IRHCs with the microtubule-disrupting agent colchicine (Colch, 1 μM) or the MEK1/2 inhibitor PD98059 (PD, 1 μM) abolished the preventive effect of Sal on E17G-induced internalization of Abcc2. (D) Pretreatment of IRHCs with 8-CPT-2´-O-Me-cAMP (8-CPT-cAMP, 50 μM), an Epac agonist, also prevented delocalization of Abcc2 induced by E17G, a phenomenon also abolished by preincubation with Colch and PD. None of the treatments affected the normal distribution of actin, which showed a predominant pericanalicular distribution.
E17G-induced colocalization of canalicular transporters with endosomal marker Rab11a is prevented by Sal and only partially prevented by Glu
It was reported that canalicular Abcb11 and Abcc2, during their normal recycling between the canalicular membrane and the subapical compartment or under an endocytic stimulus, colocalize intracellularly with the endosomal marker Rab11a (Wakabayashi et al., 2004; Wang et al., 2006). In line with this, Figure 12 shows that E17G induced delocalization of Abcb11 (top) and Abcc2 (bottom) to the inner part of the cell. Part of transporter-associated fluorescence colocalized with Rab11a in the perinuclear region (arrowheads in amplified images). Sal treatment led all transporters back to the canalicular domain. In turn, Glu treatment partially prevented the effects of E17G on canalicular transporters since part of those transporter-containing vesicles internalized by the estrogen remained colocalized with Rab11a in the perinuclear region after Glu treatment (arrowheads in amplified images).
FIGURE 12:

Representative confocal images showing cellular distribution of canalicular transporters, endosomal marker Rab11a, and merged images (with endosomal marker in red, canalicular transporter in green, and nuclei in blue) in IRHCs under the different treatments. (Top) Prevention by Glu (0.1 μM) and Sal (1 μM) of E17G (200 μM)-induced retrieval of Abcb11. (Bottom) Prevention by Glu (0.1 μM) and Sal (1 μM) of E17G (200 μM)-induced retrieval of Abcc2. Square demarcated zones are shown amplified below the corresponding microphotographs. E17G induced a clear internalization of transporter-containing vesicles beyond the canalicular region, reaching the perinuclear zone and partially colocalizing with Rab11a (arrowheads), a phenomenon significantly prevented by Glu and Sal. The E17G + Glu group shows some transporter-containing vesicles that remained mainly colocalizing with the endosomal marker Rab11a in a deep intracellular compartment (arrowheads).
DISCUSSION
This study gives insight into the mechanism of prevention of E17G-induced cholestasis afforded by cAMP and the role of different downstream pathways in the series of events leading to this prevention.
Both cAMP-elevating compounds analyzed—Glu and Sal—displaced the E17G concentration–response curve of cVA of both Abcc2 and Abcb11 substrates to the right, indicating a partial protective effect of these compounds on the cholestatic failure induced by E17G. However, Glu presented a clear tendency toward impeding the complete blockage of the canalicular transport function that occurs at high estrogen concentrations, although this was significant only for the transport of GS-MF. This could indicate that part of the transporters endocytosed by E17G are readily available to be reinserted by Glu (Figure 1, top). In turn, Sal does not prevent the E17G maximal effect (Figure 1, bottom), suggesting a different mechanism of action. Confocal images also suggest different mechanisms of action since Sal fully prevented transporter delocalization induced by E17G, whereas Glu relocalized a great part of delocalized transporters but could not avoid that part of the transporters that remained in the perinuclear zone (Figures 10 and 11), mainly colocalizing with the endosomal marker Rab11a (Figure 12).
A detailed analysis of the results showed that Glu protection is carried out by PKA activation not requiring microtubule integrity, whereas Sal protection, surprisingly, did not require PKA activation but was dependent on microtubule integrity. Accordingly, protection afforded by Sal and Glu together presented an additive effect, corroborating the existence of independent mechanisms of protection. Moreover, we observed that activation of Epac pathway prevented E17G-induced impairment of canalicular secretory function. Similar to Sal action, this prevention was microtubule dependent. Finally, inhibition of MEK, a kinase downstream of Epac, blocked Sal protective effect. All of these facts indicate that Sal mediates its beneficial effects by activating a cAMP-Epac signaling pathway, probably via MEK1 or 2. Further supporting this, coincubation with Sal and 8-CPT-2´-O-Me-cAMP, an Epac agonist, did not produce an additive protective effect, which points to a shared intracellular pathway. In contrast, Glu and 8-CPT-2´-O-Me-cAMP induced additive effects, giving additional evidence for the existence of different mechanisms of protection. It is worth noting that these results represent the first evidence that Epac activation could play a role in protection against E17G-induced cholestasis.
Previously, our group demonstrated that microtubule integrity is necessary for the recovery of bile secretion and the retargeting of the endocytosed transporters back to the canalicular membrane that spontaneously occurs after a single E17G insult (Mottino et al., 2005). In line with this, Roelofsen et al. (1998) showed that cAMP participates in the three steps of the reinsertion of Abcc2 following the redistribution that occurs after IRHC isolation, that is, the endocytosis from the basolateral plasma membrane where Abcc2 is initially redistributed, its transcytosis to the apical pole in a microtubule-dependent manner, and, finally, the fusion of transporter-containing vesicles with the apical membrane in a microtubule-independent manner. Although this approach differs from our cholestatic model in the cause of the transporter redistribution process and in the extent at which this redistribution occurs, the two last steps can, in principle, be applied to the spontaneous reinsertion of transporters that occurs after E17G cholestasis. Our approach using different hormones that increase intracellular levels of cAMP allowed us to discriminate different actions of this second messenger, depending on its different origins within the cell. cAMP intracellular distribution following Glu/Sal–induced synthesis is compartmentalized in spatially restricted zones underneath the plasma membrane (Garcia et al., 2001; Tasken and Aandahl, 2004), and Glu-associated cAMP seems to act only in the last step of fusion of subapical vesicles with the apical membrane, which is independent of microtubules but dependent on PKA activity. In contrast, Sal seems to increase cAMP in the microdomain responsible for the microtubule-dependent trafficking of transporter-containing vesicles from a deep vesicular domain to the immediacies of the apical membrane, without the final intervention of the PKA-dependent fusion process. Lack of action of Glu on microtubule-dependent reinsertion of transporters may be the cause of the persistence of part of Abcb11 and Abcc2 colocalizing with Rab11a in the perinuclear zone.
The protection induced by Sal indicates that transporter-containing vesicles not only are transferred to the vicinity of the apical membrane, but also are fused to the membrane. This final fusion is, unlike that induced by Glu, PKA independent. This could indicate the existence of two pools of transporters ready to be inserted in the canalicular membrane with differential fusion mechanisms in terms of PKA requirements. In line with this, Kipp et al. (2001) postulated the existence of a pool of Abcb11 that depends on cAMP and another pool that depends on bile salts. Our results suggest that both Abcb11 and Abcc2 endocytosed from the membrane can be readily reinserted by a PKA-dependent mechanism and that transporters derived from microtubular traffic are spontaneously fused to the membrane in a PKA-independent manner. Because Kipp et al. did not study the mechanism of vesicle fusion, it is impossible to associate PKA-dependent and PKA-independent pools with cAMP- and bile salt–dependent pools with certainty.
Regarding E17G-induced cholestasis, the present data demonstrate that E17G administration leads to transporter relocalization at two different levels, one next to the apical membrane, which can be reverted by Glu, and another one to a deeper compartment, which needs microtubule integrity to be reverted. This reversion could depend on reinsertion of previously deinserted transporters and/or on the transfer of newly synthesized transporters from the Golgi apparatus (Kipp and Arias, 2000). Experimental data support the former explanation since transporters that were delocalized by E17G colocalized with Rab11a, and those transporters that failed to be relocalized by glucagon also mainly colocalized with Rab11a. A tentative model to explain the differential actions of Sal and Glu is depicted in Figure 13.
FIGURE 13:

Proposed model for cAMP-dependent transporter reinsertion after their endocytic internalization induced by E17G. The cholestatic estrogen glucuronide produces deinsertion of canalicular transporters to a subapical vesicle pool and to a deeper endocytic compartment. Glu mediates the reinsertion of subapical transporters via a PKA-dependent mechanism. On the other hand, Sal, via Epac–MEK activation, promotes the microtubule-dependent trafficking of transporter-containing vesicles toward the canalicular pole from a deep endocytic pool, the final fusion of subapical vesicles with the apical membrane being independent of PKA.
Potential targets of activated PKA include the actin cytoskeleton and the transporters themselves. Transporter phosphorylation cannot be ruled out, but no evidence for such a process exists. Some pieces of evidence point to actin reorganization as a potential target. cAMP greatly accelerated the reorganization of F-actin in IRHCs after the disarrangement induced by cell isolation, and this closely paralleled the reinsertion of canalicular transporters back to the canalicular domain; conversely, maneuvers retarding actin reorganization (e.g., protein kinase C activation, Ca2+ sequestration) delayed transporter retargeting (Roma et al., 2000). In line with this, cAMP protects against oxidative stress–induced alteration in actin cytoskeleton via a PKA-dependent mechanism (Perez et al., 2006), which suggests a role for the kinase in actin organization in hepatocytes. cAMP also stimulated the microfilament-mediated fusion of the basolateral bile salt transporter Na+–taurocholate cotransporting polypeptide with its membrane domain (Dranoff et al., 1999).
On the other hand, Epac actions could be involved in microtubule assembly. Sehrawat et al. (2008) found that Epac1 activation is involved in increasing the length of microtubules from human umbilical vein endothelial cells. Mei and Cheng (2005) found similar results, and they concluded that Epac binds to tubulin and its activation is necessary for microtubule formation. In view of this evidence, our results suggest that cAMP activation of Epac prevents cholestasis by acting at the level of microtubules, probably increasing microtubule assembly necessary for the traffic of transporter-containing vesicles that were deeply endocytosed by E17G action. Epac involvement in the final PKA-independent vesicular fusion could not be ruled out based on our results.
Summarizing, we can say that Glu favors the reinsertion of subapical transporters that are endocytosed by E17G through a mechanism that involves both microtubule-independent trafficking and activation of PKA. In contrast, Sal stimulates selectively a microtubule-dependent pathway, which is PKA independent and depends on Epac activation. On the basis of evidence on trafficking mechanisms, we propose that cAMP derived from Sal modulates the long-range trafficking of transporter-containing vesicles, whereas cAMP derived from Glu participates in the microfilament-mediated fusion of these vesicles with the apical membrane. Because endocytic internalization of canalicular transporters relevant to bile formation is a common feature of most forms of cholestasis (Roma et al., 2008) and probably a main pathomechanism, a better understanding of the cAMP-dependent beneficial signaling pathways, such as that provided by this work, may help to develop new therapeutic strategies aimed at affording appropriate transporter localization after a cholestatic insult.
MATERIALS AND METHODS
Materials
E17G, Leibovitz-15 (L-15) culture medium, collagenase type A from Clostridium histolyticum, colchicine, albumin, paraformaldehyde, Triton X-100, 8-CPT-2´-O-Me-cAMP, adrenaline, isoproterenol, salbutamol, fentolamine, butoxamine, propranolol, atenolol, PD98059, N6,2´-O-dibutyryladenosine 3′:5′ cyclic monophosphate (DBcAMP), and dimethyl sulfoxide (DMSO) were from Sigma-Aldrich (St. Louis, MO). Glucagon was from Eli Lilly (Indianapolis, IN). 5-Chloromethylfluorescein diacetate (CMFDA) was from Life Technologies Corporation (Carlsbad, CA). Cholyl-lysylfluorescein (CLF) was kindly provided by Charles O. Mills (Birmingham, United Kingdom). KT5720 and N-[2-(methylamino) ethyl]-5-isoquinolinesulfonamide (H-89) were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). All other chemicals were of the highest grade available.
Isolation and culture of rat hepatocyte couplets
To obtain a preparation enriched in IRHCs, livers from adult, female Wistar rats weighing 250–300 g and bred in our animal house as described (Crocenzi et al., 2003b), were perfused under urethane anesthesia (1 g/kg intraperitoneally) according to the two-step collagenase perfusion procedure and were further enriched by centrifugal elutriation (Gautam et al., 1987; Wilton et al., 1991). The final preparation contained 70–80% of IRHCs with viability >95%, as assessed by the trypan blue exclusion test. After isolation, IRHCs were plated onto 24-well plastic plates at a density of 0.5 × 105 U/ml in L-15 culture medium, and they were cultured for 5 h to allow the restoration of couplet polarity.
IRHC treatments
To determine which adrenergic receptor is involved in the preventive effect of adrenaline, in preliminary studies cells were incubated with different specific adrenergic agonists (1 μM adrenaline, 1 μM isoproterenol; 10 μM Sal and 30 μM fentolamine) for 15 min before addition of E17G (50 μM), and thus maintained throughout the period of exposure to E17G (20 min). To discriminate which subtype of receptor β is involved in the prevention, IRHCs were preincubated with different specific β-receptor antagonists (1 μM propranolol; 1 μM atenolol and 5 μM butoxamine) before the addition of the adrenergic agonists. Results from these ancillary studies showed protective effect of adrenaline, isoproterenol, and Sal and no protection of fentolamine for Abcb11 and Abcc2 function. This protection was reverted by butoxamine and propranolol but not by atenolol, indicating that protective effect was mediated by β2 receptors (Supplemental Figure S3). Hence further studies concerning adrenergic protection were carried out using Sal, a β2 agonist.
To evaluate the protective effects of Glu and Sal, IRHCs were incubated for 15 min with Glu (0.001–10 μM) or Sal (0.1–1000 μM) and then exposed to DMSO (control group) or various concentrations of E17G (12.5–800 μM) for 20 min in the presence of Glu or Sal.
The possible involvement of PKA in the protective effect of Glu and Sal against E17G-induced secretory failure was evaluated by incubating the cells for 15 min with the PKA inhibitors KT5720 (50 nM) or H-89 (200 nM) (Kimura and Ogihara, 1997), followed by incubation with 0.1 μM Glu or 1 μM Sal 15 min before the addition of E17G (50 μM, 20 min) to the culture medium.
The possible participation of the Epac pathway in the preventive effect of the studied compounds was evaluated by treating IRHCs for 15 min with a specific Epac agonist (50 μM 8-CPT-2´-O-Me-cAMP) in the presence or absence of 0.1 μM Glu and 1 μM Sal 15 min before addition of E17G (50 μM, 20 min). To confirm Epac participation in Sal effects, we incubated IRHC for 15 min with a specific MEK (Epac downstream) inhibitor PD98059 (1 μM) in the presence of Glu, Sal, and 8-CPT-2´-O-Me-cAMP 15 min before addition of E17G (50 μM, 20 min).
To assess the possible involvement of microtubules in the actions of Sal, Glu, or 8-CPT-2´-O-Me-cAMP, IRHCs were preincubated for 30 min with the inhibitor of microtubule polymerization colchicine (1 μM) (Mottino et al., 2005). Then IRHCs were preincubated with 8-CPT-2´-O-Me-cAMP, Glu, or Sal before the addition of E17G, as described earlier.
Assessment of Abcb11 and Abcc2 secretory function and localization in IRHCs
The transport function of Abcb11 and Abcc2 was evaluated by analyzing the canalicular vacuolar accumulation of the fluorescent substrates CLF and GS-MF, respectively (Roma et al., 2000; Wang et al., 2002). CLF is a bile salt analogue transported selectively by Abcb11 (Mills et al., 1999), whereas CMFDA is a lipophilic compound taken up by passive diffusion across the basolateral membrane and converted into GS-MF by the sequential action of intracellular esterases and glutathione S-transferases. For transport studies, cells were washed twice with L-15 and exposed to 2 μM CLF (Crocenzi et al., 2003b; Roma et al., 2003) or 2.5 μM CMFDA (Roelofsen et al., 1998; Roma et al., 2000) for 15 min. Finally, cells were washed twice with L-15, and canalicular transport activity for both substrates was assessed by fluorescence microscopy (Roma et al., 2000) under an inverted microscope (Axiovert 25, Zeiss, Göttingen, Germany). Images were captured with a digital camera (Q-Color5; Olympus America, Center Valley, PA), and the cVA of the fluorescent substrates was determined as the percentage of IRHCs in the images displaying visible green fluorescence in their canalicular vacuoles from a total analysis of at least 200 couplets per preparation.
To evaluate the intracellular distribution of Abcb11 and Abcc2, IRHCs were fixed and stained as previously reported (Roma et al., 2000). The E17G concentration used in these experiments (200 μM) was higher than that used in functional experiments to render transporter retrieval more evident. After treatments, IRHCs were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) and permeabilized with PBS–Triton X-100 2%–bovine serum albumin (BSA). Then cells were incubated with a polyclonal antibody against mouse Abcb11 (1:100, 2 h) (Kamiya Biomedical, Seattle, WA) or a monoclonal antibody against human ABCC2 (1:100, 2 h; M2III-6; Alexis Biochemicals, San Diego, CA), followed by incubation with Cy2-conjugated donkey anti–immunoglobulin G (IgG; 1:200) for 40 min or fluorescein isothiocyanate (FITC)–labeled goat anti–mouse IgG (1:200, 1 h; Zymed, San Francisco, CA). To delimit the canaliculi, F-actin was stained by coincubating cells with Alexa Fluor 568 phalloidin (Invitrogen, Carlsbad CA) (1:100, 1 h). In colocalization studies of Abcc2 and Abcb11 with Rab11a, after fixation, transporters were stained using the primary and secondary antibodies just described. Rab11a was detected by using two different specific antibodies: a mouse monoclonal to Rab11a antibody (1:100, 2 h; Abcam, Cambridge, MA) for colabeling with Abcb11 and a rabbit polyclonal to Rab11a antibody (1:100, 2 h; Invitrogen) for colabeling with Abcc2, followed by incubation with a Cy3-conjugated donkey anti–mouse IgG and a Cy3-conjugated goat anti–rabbit IgG (1:200 1 h; Jackson ImmunoResearch Laboratory, West Grove, PA), respectively. Cellular nuclei were stained by incubating during 10 min with 1.5 μM 4,6-diamidino-2-phenylindol (Invitrogen). Finally, cells were mounted and examined with a Nikon C1 Plus confocal laser scanning microscope attached to a Nikon TE-2000 inverted microscope (Nikon, Tokyo, Japan).
Assessment of microtubule status in IRHCs
Colchicine specificity on microtubule integrity was confirmed by immunofluorescence after treatment of cells with DMSO (control) or colchicine for 30 min. Then cells were washed three times with PBS, fixed (4% paraformaldehyde, 0.1% Triton X-100, 80 mM potassium 1,4- piperazinediethanesulfonate, pH 7.2, 1 mM ethylene glycol tetraacetic acid, 1 mM MgSO4, 30% glycerol) for 20 min at room temperature (Larocca et al., 2004) and permeabilized with PBS–Triton X-100 2%–BSA, and incubated with a monoclonal antibody against mouse β-tubulin (Sigma-Aldrich; 1:100, 2 h). To delimit the canaliculi, actin network was stained by coincubating cells with Alexa Fluor 568 phalloidin (Invitrogen; 1:100). Finally, cells were incubated for 40 min with FITC-labeled goat anti–mouse IgG (1:100, 1 h; Zymed). Cells were then mounted and examined with a Nikon C1 Plus confocal laser scanning microscope, attached to a Nikon TE-2000 inverted microscope.
Assessment of intracellular cAMP levels in IRHCs
In the IRHC model, couplets were plated in 35-mm plastic plates covered with collagen (0.5 × 105 U/ml) and cultured for 5 h at 37°C to allow restoration of couplet polarity. Subsequently, IRHCs were preincubated with the phosphodiesterase inhibitor IBMX (0.8 mM) for 5 min and then incubated with DMSO (control), Glu (0.1 μM), or Sal (1 μM) for 15 min. Reaction was stopped by the addition of ice-cold ethanol for 20 min. Then ethanol was evaporated, and the residue was resuspended for cAMP determination, which was carried out by competition of [3H]cAMP for carbon/dextran–immobilized PKA, as previously described (Davio et al., 1995; Sabbatini et al., 2003). Results are expressed as pmol/105 IRHC.
Immunoblot analysis of PKA substrate phosphorylation status
The phosphorylation status of PKA substrates was analyzed to evaluate PKA activation by hormonal modulators, as well as to test the efficacy of the PKA inhibitors used in this study. Briefly, isolated hepatocytes were obtained by collagenase perfusion as previously described (Garcia et al., 2001) and cultured in 3-cm, collagen-coated Petri dishes at a density of 2 × 106 cells/ml. After a 24-h culture period, cells were preincubated for 5 min with IBMX (0.8 mM) and then exposed to DBcAMP (10 μM, positive control), Glu (0.1 μM), Sal (1 μM), and 8-CPT-2´-O-Me-cAMP (50 μM) in the presence or absence of the PKA inhibitors KT5720 (50 nM) or H-89 (200 nM). After treatments, cells were washed with cold PBS and finally resuspended in a cellular lysis buffer containing Protease Inhibitor Cocktail (Sigma-Aldrich) and phosphatase inhibitors (Phosphatase Inhibitor Cocktail 3 [Sigma-Aldrich], 1 mM NaF, and 1 mM Na3VO4). Aliquots containing an equivalent total protein content, as determined by the Lowry procedure with BSA as the standard (Lowry et al., 1951), were subjected to SDS/12% PAGE. Separated proteins were electrotransferred to Immobilon-P membranes and probed with an anti–Phospho-(Ser/Thr) PKA Substrate Antibody (Cell Signaling Technology, Danvers, MA; 1:1000) overnight. After the use of a donkey anti–rabbit IgG secondary antibody (1:5000), membranes were revealed using standard chemiluminescence protocols. Bands were quantified by densitometry with ImageJ 1.44p software (National Institutes of Health, Bethesda, MD). The membranes were then stripped and reprobed with an anti-actin antibody (Invitrogen; 1:5000).
Immunoblot analysis of MEK phosphorylation
Activation of Epac was confirmed by Western blot analysis of the phosphorylation status of MEK, a downstream Epac effector, in primary cultured hepatocytes. Briefly, after a 24-h culture period, cells were exposed to DMSO (control), Glu (0.1 μM), Sal (1 μM), and 8-CPT-2′-O-Me-cAMP (50 μM) for 15 min, then washed with cold PBS, and finally resuspended in a cellular lysis buffer containing Protease Inhibitor Cocktail (Sigma-Aldrich) and phosphatase inhibitors (Phosphatase Inhibitor Cocktail 3 [Sigma-Aldrich], 1 mM NaF, and 1 mM Na3VO4). Aliquots containing an equivalent total protein content were subjected to SDS/12% PAGE. Separated proteins were electrotransferred to Immobilon-P membranes and probed with an anti-Phospho-MEK1/2 (Ser-217/221) antibody (Cell Signaling Technology; 1:1000) overnight. After the use of a donkey anti–mouse IgG secondary antibody (1:5000), membranes were revealed using standard chemiluminescence protocols. Bands were quantified by densitometry with ImageJ 1.44p software. The membranes were then stripped and reprobed with an anti-actin antibody (1:5000).
Statistical analysis
Results are expressed as mean ± SE of the mean (SEM). One-way analysis of variance, followed by a Newman–Keuls test, was used for multiple comparisons. Statistical analysis of cAMP levels was performed by using the paired Student's t test. The four-parameter dose–response curves were compared using GraphPad Prism software (GraphPad Software, La Jolla, CA). Values of p < 0.05 were considered to be statistically significant.
Supplementary Material
Acknowledgments
This work was supported by grants from the Agencia Nacional de Promoción Científica y Tecnológica (PICTs 2006-02012 and 05-26115), the Consejo Nacional de Investigaciones Científicas y Técnicas (PIP 6442 and 0691), and the Fundación Allende. We deeply apprecitate the collaboration of Carlos Davio (Cátedra de Química Medicinal, Facultad de Farmacia y Bioquímica, Universidad de Buenos Aires) for performing cAMP measurements, María Cecilia Larocca (Instituto de Fisiología Experimental, Consejo Nacional de Investigaciones Científicas y Técnicas, Universidad Nacional de Rosario) for help with tubulin confocal images, and Rodrigo Vena (Instituto de Biología Celular y Molecular de Rosario, CONICET-UNR, Consejo Nacional de Investigaciones Científicas y Técnicas, Universidad Nacional de Rosario) for his help with the use the confocal microscope.
Abbreviations used:
- Abcb11
bile salt export pump
- Abcc2
multidrug-resistance associated protein 2
- CLF
cholyl-lysylfluorescein
- CMFDA
5-chloromethylfluorescein diacetate
- 8-CPT-2´-O-Me-cAMP
8-(4-chlorophenylthio)-2´-O-methyladenosine 3´,5´-cyclicmonophosphate
- cVA
canalicular vacuolar accumulation
- DMSO
dimethyl sulfoxide
- E17G
estradiol 17β-d-glucuronide
- Glu
glucagon
- GS-MF
glutathione methylfluorescein
- H-89
N-[2- (methylamino) ethyl]-5- isoquinolinesulfonamide
- IRHC
isolated rat hepatocyte couplets
- Sal
salbutamol.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-01-0047) on August 24, 2011.
REFERENCES
- Bollen M, Keppens S, Stalmans W. Specific features of glycogen metabolism in the liver. Biochem J. 1998;336:19–31. doi: 10.1042/bj3360019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borst P, Elferink RO. Mammalian ABC transporters in health and disease. Annu Rev Biochem. 2002;71:537–592. doi: 10.1146/annurev.biochem.71.102301.093055. [DOI] [PubMed] [Google Scholar]
- Bos JL, de Rooij J, Reedquist KA. Rap1 signalling: adhering to new models. Nat Rev Mol Cell Biol. 2001;2:369–377. doi: 10.1038/35073073. [DOI] [PubMed] [Google Scholar]
- Bouscarel B, Matsuzaki Y, Le M, Gettys TW, Fromm H. Changes in G protein expression account for impaired modulation of hepatic cAMP formation after BDL. Am J Physiol. 1998;274:G1151–G1159. doi: 10.1152/ajpgi.1998.274.6.G1151. [DOI] [PubMed] [Google Scholar]
- Branum GD, Bowers BA, Watters CR, Haebig J, Cucchiaro G, Farouk M, Meyers WC. Biliary response to glucagon in humans. Ann Surg. 1991;213:335–340. doi: 10.1097/00000658-199104000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan JP, Bardswell SC, Burgoyne JR, Fuller W, Schroder E, Wait R, Begum S, Kentish JC, Eaton P. Oxidant-induced activation of type I protein kinase A is mediated by RI subunit interprotein disulfide bond formation. J Biol Chem. 2006;281:21827–21836. doi: 10.1074/jbc.M603952200. [DOI] [PubMed] [Google Scholar]
- Crocenzi FA, Mottino AD, Cao J, Veggi LM, Sanchez Pozzi EJ, Vore M, Coleman R, Roma MG. Estradiol-17-βd-glucuronide induces endocytic internalization of Bsep in rats. Am J Physiol Gastrointest Liver Physiol. 2003a;285:G449–G459. doi: 10.1152/ajpgi.00508.2002. [DOI] [PubMed] [Google Scholar]
- Crocenzi FA, Mottino AD, Sanchez Pozzi EJ, Pellegrino JM, Rodríguez Garay EA, Milkiewicz P, Vore M, Coleman R, Roma MG. Impaired localisation and transport function of canalicular Bsep in taurolithocholate-induced cholestasis in the rat. Gut. 2003b;52:1170–1177. doi: 10.1136/gut.52.8.1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davio CA, Cricco GP, Bergoc RM, Rivera ES. H1 and H2 histamine receptors in N-nitroso-N-methylurea (NMU)-induced carcinomas with atypical coupling to signal transducers. Biochem Pharmacol. 1995;50:91–96. doi: 10.1016/0006-2952(95)00108-c. [DOI] [PubMed] [Google Scholar]
- de Rooij J, Zwartkruis FJ, Verheijen MH, Cool RH, Nijman SM, Wittinghofer A, Bos JL. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature. 1998;396:474–477. doi: 10.1038/24884. [DOI] [PubMed] [Google Scholar]
- Dombrowski F, Kubitz R, Chittattu A, Wettstein M, Saha N, Haussinger D. Electromicroscopic demonstration of multidrug resistance protein 2 (Mrp2) retrieval from the canalicular membrane in response to hyperosmolarity and lipopolysaccharide. Biochem J. 2000;348:183–188. [PMC free article] [PubMed] [Google Scholar]
- Dranoff JA, McClure M, Burgstahler AD, Denson LA, Crawford AR, Crawford JM, Karpen SJ, Nathanson MH. Short-term regulation of bile acid uptake by microfilament-dependent translocation of rat NTCP to the plasma membrane. Hepatology. 1999;30:223–229. doi: 10.1002/hep.510300136. [DOI] [PubMed] [Google Scholar]
- Esteller A. Physiology of bile secretion. World J Gastroenterol. 2008;14:5641–5649. doi: 10.3748/wjg.14.5641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia F, Kierbel A, Larocca MC, Gradilone SA, Splinter P, LaRusso NF, Marinelli RA. The water channel aquaporin-8 is mainly intracellular in rat hepatocytes, and its plasma membrane insertion is stimulated by cyclic AMP. J Biol Chem. 2001;276:12147–12152. doi: 10.1074/jbc.M009403200. [DOI] [PubMed] [Google Scholar]
- Gatmaitan ZC, Arias IM. ATP-dependent transport systems in the canalicular membrane of the hepatocyte. Physiol Rev. 1995;75:261–275. doi: 10.1152/physrev.1995.75.2.261. [DOI] [PubMed] [Google Scholar]
- Gautam A, Ng OC, Boyer JL. Isolated rat hepatocyte couplets in short-term culture: structural characteristics and plasma membrane reorganization. Hepatology. 1987;7:216–223. doi: 10.1002/hep.1840070203. [DOI] [PubMed] [Google Scholar]
- Kimura M, Ogihara M. Density-dependent proliferation of adult rat hepatocytes in primary culture induced by epidermal growth factor is potentiated by cAMP-elevating agents. Eur J Pharmacol. 1997;324:267–276. doi: 10.1016/s0014-2999(97)00078-2. [DOI] [PubMed] [Google Scholar]
- Kipp H, Arias IM. Newly synthesized canalicular ABC transporters are directly targeted from the Golgi to the hepatocyte apical domain in rat liver. J Biol Chem. 2000;275:15917–15925. doi: 10.1074/jbc.M909875199. [DOI] [PubMed] [Google Scholar]
- Kipp H, Arias IM. Trafficking of canalicular ABC transporters in hepatocytes. Annu Rev Physiol. 2002;64:595–608. doi: 10.1146/annurev.physiol.64.081501.155793. [DOI] [PubMed] [Google Scholar]
- Kipp H, Pichetshote N, Arias IM. Transporters on demand: intrahepatic pools of canalicular ATP binding cassette transporters in rat liver. J Biol Chem. 2001;276:7218–7224. doi: 10.1074/jbc.M007794200. [DOI] [PubMed] [Google Scholar]
- Larocca MC, Shanks RA, Tian L, Nelson DL, Stewart DM, Goldenring JR. AKAP350 interaction with cdc42 interacting protein 4 at the Golgi apparatus. Mol Biol Cell. 2004;15:2771–2781. doi: 10.1091/mbc.E03-10-0757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei H, Venkatakrishnan A, Yu S, Kazlauskas A. Protein kinase A-dependent translocation of Hsp90 alpha impairs endothelial nitric-oxide synthase activity in high glucose and diabetes. J Biol Chem. 2007;282:9364–9371. doi: 10.1074/jbc.M608985200. [DOI] [PubMed] [Google Scholar]
- Lowry OH, Rosembrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- Mei FC, Cheng X. Interplay between exchange protein directly activated by cAMP (Epac) and microtubule cytoskeleton. Mol Biosyst. 2005;1:325–331. doi: 10.1039/b511267b. [DOI] [PubMed] [Google Scholar]
- Meja KK, Catley MC, Cambridge LM, Barnes PJ, Lum H, Newton R, Giembycz MA. Adenovirus-mediated delivery and expression of a cAMP-dependent protein kinase inhibitor gene to BEAS-2B epithelial cells abolishes the anti-inflammatory effects of rolipram, salbutamol, and prostaglandin E2: a comparison with H-89. J Pharmacol Exp Ther. 2004;309:833–844. doi: 10.1124/jpet.103.060020. [DOI] [PubMed] [Google Scholar]
- Mills CO, Milkiewicz P, Muller M, Roma MG, Havinga R, Coleman R, Kuipers F, Jansen PL, Elias E. Different pathways of canalicular secretion of sulfated and non-sulfated fluorescent bile acids: a study in isolated hepatocyte couplets and TR− rats. J Hepatol. 1999;31:678–684. doi: 10.1016/s0168-8278(99)80348-1. [DOI] [PubMed] [Google Scholar]
- Morgan NG, Blackmore PF, Exton JH. Age-related changes in the control of hepatic cyclic AMP levels by alpha 1- and beta 2-adrenergic receptors in male rats. J Biol Chem. 1983;258:5103–5109. [PubMed] [Google Scholar]
- Mottino AD, Cao J, Veggi LM, Crocenzi FA, Roma MG, Vore M. Altered localization and activity of canalicular Mrp2 in estradiol-17β-d-glucuronide-induced cholestasis. Hepatology. 2002;35:1409–1419. doi: 10.1053/jhep.2002.33327. [DOI] [PubMed] [Google Scholar]
- Mottino AD, Crocenzi FA, Pozzi EJ, Veggi LM, Roma MG, Vore M. Role of microtubules in estradiol-17beta-D-glucuronide-induced alteration of canalicular Mrp2 localization and activity. Am J Physiol Gastrointest Liver Physiol. 2005;288:G327–G336. doi: 10.1152/ajpgi.00227.2004. [DOI] [PubMed] [Google Scholar]
- Pecker F, Duvaldestin P, Berthelot P, Hanoune J. The adenylate cyclase system in human liver: characterization, subcellular distribution and hormonal sensitivity in normal or cirrhotic adult, and in foetal liver. Clin Sci (Lond) 1979;57:313–325. doi: 10.1042/cs0570313. [DOI] [PubMed] [Google Scholar]
- Perez LM, Milkiewicz P, Ahmed-Choudhury J, Elias E, Ochoa JE, Sanchez Pozzi EJ, Coleman R, Roma MG. Oxidative stress induces actin-cytoskeletal and tight-junctional alterations in hepatocytes by a Ca2+-dependent, PKC-mediated mechanism: protective effect of PKA. Free Radic Biol Med. 2006;40:2005–2017. doi: 10.1016/j.freeradbiomed.2006.01.034. [DOI] [PubMed] [Google Scholar]
- Roelofsen H, Soroka CJ, Keppler D, Boyer JL. Cyclic AMP stimulates sorting of the canalicular organic anion transporter (Mrp2/cMoat) to the apical domain in hepatocyte couplets. J Cell Sci. 1998;111:1137–1145. doi: 10.1242/jcs.111.8.1137. [DOI] [PubMed] [Google Scholar]
- Roma MG, Crocenzi FA, Mottino AD. Dynamic localization of hepatocellular transporters in health and disease. World J Gastroenterol. 2008;14:6786–6801. doi: 10.3748/wjg.14.6786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roma MG, Milkiewicz P, Ahmed-Choudhury J, Elias E, Coleman R. Oxidative stress induces F-actin and Bsep relocalisation in isolated rat hepatocyte couplet model: Possible involvement of PKC and protective effect of PKA. J Hepatol. 2003;38(Suppl 1):81. [Google Scholar]
- Roma MG, Milkiewicz P, Elias E, Coleman R. Control by signaling modulators of the sorting of canalicular transporters in rat hepatocyte couplets: role of the cytoskeleton. Hepatology. 2000;32:1342–1356. doi: 10.1053/jhep.2000.20519. [DOI] [PubMed] [Google Scholar]
- Roma MG, Stone V, Shaw R, Coleman R. Vasopressin-induced disruption of actin cytoskeletal organization and canalicular function in isolated rat hepatocyte couplets: possible involvement of protein kinase C. Hepatology. 1998;28:1031–1041. doi: 10.1002/hep.510280418. [DOI] [PubMed] [Google Scholar]
- Sabbatini ME, Villagra A, Davio CA, Vatta MS, Fernandez BE, Bianciotti LG. Atrial natriuretic factor stimulates exocrine pancreatic secretion in the rat through NPR-C receptors. Am J Physiol Gastrointest Liver Physiol. 2003;285:G929–G937. doi: 10.1152/ajpgi.00010.2003. [DOI] [PubMed] [Google Scholar]
- Sehrawat S, Cullere X, Patel S, Italiano J Jr, Mayadas TN. Role of Epac1, an exchange factor for Rap GTPases, in endothelial microtubule dynamics and barrier function. Mol Biol Cell. 2008;19:1261–1270. doi: 10.1091/mbc.E06-10-0972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seino S, Shibasaki T. PKA-dependent and PKA-independent pathways for cAMP-regulated exocytosis. Physiol Rev. 2005;85:1303–1342. doi: 10.1152/physrev.00001.2005. [DOI] [PubMed] [Google Scholar]
- Steinberg SF, Chow YK, Bilezikian JP. Cyclic AMP and guanine nucleotide regulatory proteins. In: Arias IM, Jakoby WB, Popper H, editors. In: The Liver: Biology and Pathobiology. New York: Raven Press; 2003. pp. 769–776. [Google Scholar]
- Tasken K, Aandahl EM. Localized effects of cAMP mediated by distinct routes of protein kinase A. Physiol Rev. 2004;84:137–167. doi: 10.1152/physrev.00021.2003. [DOI] [PubMed] [Google Scholar]
- Trauner M, Meier PJ, Boyer JL. Molecular regulation of hepatocellular transport systems in cholestasis. J Hepatol. 1999;31:165–178. doi: 10.1016/s0168-8278(99)80179-2. [DOI] [PubMed] [Google Scholar]
- Wang L, Soroka CJ, Boyer JL. The role of bile salt export pump mutations in progressive familial intrahepatic cholestasis type II. J Clin Invest. 2002;110:965–972. doi: 10.1172/JCI15968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Soroka CJ, Mennone A, Rahner C, Harry K, Pypaert M, Boyer JL. Radixin is required to maintain apical canalicular membrane structure and function in rat hepatocytes. Gastroenterology. 2006;131:878–884. doi: 10.1053/j.gastro.2006.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi Y, Lippincott-Schwartz J, Arias IM. Intracellular trafficking of bile salt export pump (ABCB11) in polarized hepatic cells: constitutive cycling between the canalicular membrane and rab11-positive endosomes. Mol Biol Cell. 2004;15:3485–3496. doi: 10.1091/mbc.E03-10-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilton JC, Williams DE, Strain AJ, Parslow RA, Chipman JK, Coleman R. Purification of hepatocyte couplets by centrifugal elutriation. Hepatology. 1991;14:180–183. doi: 10.1002/hep.1840140129. [DOI] [PubMed] [Google Scholar]
- Zaccolo M, Di BG, Lissandron V, Mancuso L, Terrin A, Zamparo I. Restricted diffusion of a freely diffusible second messenger: mechanisms underlying compartmentalized cAMP signalling. Biochem Soc Trans. 2006;34:495–497. doi: 10.1042/BST0340495. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.