

Abstract
Much of our current state of knowledge pertaining to the mechanisms controlling intestinal epithelial homeostasis derives from epidemiological, molecular genetic, cell biological, and biochemical studies of signaling pathways that are dysregulated during the process of colorectal tumorigenesis. Activating mutations in members of the RAS oncoprotein family play an important role in the progression of colorectal cancer (CRC) and, by extension, intestinal epithelial homeostasis. Mutations in K-RAS account for 90% of the RAS mutations found in CRC. As such, the study of RAS protein function in the intestinal epithelium is largely encompassed by the study of K-RAS function in CRC. In this review, we summarize the data available from genetically defined in vitro and in vivo models of CRC that aim to characterize the oncogenic properties of mutationally activated K-RAS. These studies paint a complex picture of a multi-functional oncoprotein that engages an array of downstream signaling pathways to influence cellular behaviors that are both pro- and anti-tumorigenic. While the complexity of K-RAS biology has thus far prevented a comprehensive understanding of its oncogenic properties, the work to date lays a foundation for the development of new therapeutic strategies to treat K-RAS mutant CRC.
Keywords: Intestinal epithelium, Colorectal cancer, K-Ras
Introduction
Among the vast array of mutations present in cancers, activating mutations in the RAS family of small, monomeric GTPases (i.e., K-, N-, and H-RAS) are both common and widespread. K-RAS activation is most common, with an incidence of approximately 15% across all human tumor types [1]. RAS family members normally cycle between active GTP-bound and inactive GDP-bound states and function at cellular membranes to transmit signals originating from extracellular stimuli to influence cell growth, proliferation, differentiation, and survival [2]. Oncogenic missense mutations at codons 12, 13, 61, and 146 strongly attenuate GTPase activity and cause RAS to accumulate in the active GTP-bound state, resulting in sustained activation of downstream signaling pathways [3]. The downstream “effector” pathways engaged by GTP-bound RAS are numerous and include both phosphorylation cascades (e.g. RAF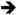 MEK
MEK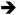 ERK, PI3K
ERK, PI3K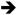 AKT, PLCε
AKT, PLCε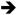 PKC) and secondary GTPase pathways (e.g. RAC and RAL) [4]. In theory, these effectors constitute potential therapeutic targets for RAS-mutant cancers, yet activation of individual pathways is context-dependent, making it difficult to predict which would be an effective target in a given cancer.
PKC) and secondary GTPase pathways (e.g. RAC and RAL) [4]. In theory, these effectors constitute potential therapeutic targets for RAS-mutant cancers, yet activation of individual pathways is context-dependent, making it difficult to predict which would be an effective target in a given cancer.
In the context of colorectal cancer (CRC), K-RAS mutations far outnumber those in N-RAS and H-RAS, arising in approximately 40% of cases [5, 6]. N-RAS mutations occur in 3-5% of cases and H-RAS mutations have not been identified [6-8]. Interestingly, while codon 12/13 mutations account for the majority of activating mutations in K-RAS (G12D is the most common mutation in CRC), weak activating mutations at codon 146 significantly outnumber strong activating mutations at codon 61 [8]. Moreover, codon 146 mutations appear to be specific for CRC, as they are not found in other cancers that commonly express mutant K-RAS (e.g. non-small cell lung cancer) [9]. While molecular and pathologic characterization of pre-neoplastic and neoplastic colonic lesions confirms that mutant K-RAS has an early and broad oncogenic role in the process of colorectal carcinogenesis [6, 10-12], epidemiological data provides a more sinister view of K-RAS with respect to CRC outcomes. For example, multiple studies have identified a connection between K-RAS mutation and poor clinical outcome [8, 13]. More recently, K-RAS activating mutations have been found to be strongly predictive for failure of colorectal cancers to respond to therapies targeting the Epidermal Growth Factor Receptor (EGFR) [14-19].
Epidemiological studies underscore the importance of K-RAS mutations in the progression and treatment of CRC, and therefore the urgent need for therapies targeting the pathway(s), but their retrospective nature makes them largely uninformative at the mechanistic level. In this review we survey data from in vitro and in vivo model systems that provide insight into the mechanisms underlying the contribution of mutant K-RAS to colorectal cancer.
Oncogenic properties of K-RAS in CRC cell lines
Much of what we know about the oncogenic properties of mutationally activated RAS is derived from in vitro transformation assays, which measure an oncogene’s ability to influence cellular properties that are integral to tumorigenesis, for example replicative potential, clonogenic survival, or anchorage independent growth. A large body of work establishes the pro-tumorigenic properties of activated RAS, but the development of a unifying model of RAS function has been hindered by the fact that the phenotypes associated with oncogenic RAS are highly dependent upon which family member is mutated, the level of expression of the mutant protein, and the cellular context in which the oncoprotein is expressed. For K-RAS in particular, over-expression studies are complicated by the fact that the endogenous gene is alternatively spliced to produce two distinct proteins, K-RAS4A and K-RAS4B, which differ only in their extreme C-termini. Endogenous activating point mutations affect both splice forms, but over-expression studies (both in vitro and in vivo) typically measure the transforming activity of a single K-RAS isoform. As a result of these caveats, the molecular mechanisms underlying the contribution of mutationally activated K-RAS to CRC progression are poorly characterized.
One powerful approach that has been utilized to characterize the contribution of oncogenic K-RAS to CRC utilizes somatic gene targeting to remove the endogenous mutant allele of KRAS from CRC cell lines by homologous recombination, creating a derivative cell line that is essentially isogenic with the exception of the K-RAS mutational status [20]. While this system has limitations (e.g. the derivative cell lines carry one wild-type and one null allele of KRAS), its major advantage in comparison to RAS over-expression strategies is that it allows for the study of endogenous mutations affecting both K-RAS4A and 4B in the cell type relevant for CRC. A summary of the phenotypes associated with mutant K-RAS, as revealed by the use of isogenic pairs, is shown in Fig. 1.
Fig 1.
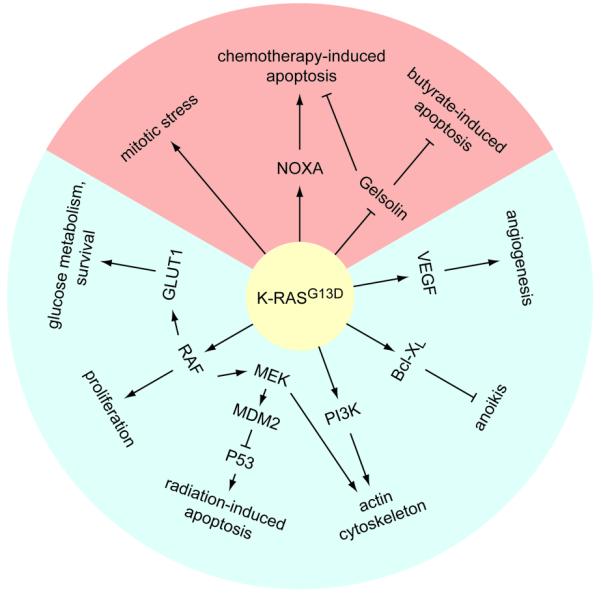
Properties of mutant K-RAS revealed by analysis of isogenic human CRC cell line pairs. Oncogenic K-RAS engages both pro-tumorigenic pathways (shaded in blue) and, paradoxically, anti-tumorigenic pathways (shaded in red). Arrows represent both direct and indirect interactions.
Deletion of the mutant KRAS allele from HCT-116 and DLD-1 CRC cells, which both harbor a single endogenous G13D activating mutation, results in a morphologic change, a decrease in proliferation, abrogation of anchorage-independent growth, and a complete loss of in vivo tumorigenic potential [20]. The signaling pathways connecting K-RASG13D to each of these phenotypes have been studied in more detail. The ability of mutant K-RAS to affect cellular morphology may be related to its effect on cytoskeletal organization; deletion of K-RASG13D from HCT-116 cells restores their ability to assemble stress fibers and focal adhesions/complexes [21]. The capacity of oncogenic K-RAS to disrupt the actin cytoskeleton is mediated by activation of MEK and PI3K, with both pathways shown to be important for uncoupling RHO signaling from actin stress fiber formation [21]. While the effect of mutant K-RAS on the actin cytoskeleton appears to be dependent on canonical downstream effector pathways, the high rate of proliferation due to K-RAS activation in DLD-1 cells is dependent on RAF, but independent of MEK, suggesting that K-RAS can engage non-canonical effector pathways to control cell division [22]. Mutant K-RAS also confers resistance to detachment-induced apoptosis (anoikis), which plays an important role in anchorage independent growth. When the K-RAS wild-type derivative of DLD-1 is grown in suspension, cell death results from down-regulation of the anti-apoptotic protein Bcl-XL, but mutant K-RAS promotes Bcl-XL stability in the parental line, thereby suppressing anoikis [23]. Finally, deletion of K-RASG13D from DLD-1 or HCT-116 leads to a dramatic reduction in the amount of VEGF produced and VEGF is required for the growth of these cells in Nude mice [24, 25]. Since subcutaneous growth of human cancer xenografts in immunocompromised mice requires efficient vascularization, it is likely that the failure of K-RAS wild-type derivatives of DLD-1 and HCT-116 cells to grow subcutaneously is at least partially due to defects in vascularization. Altogether, these studies highlight (1) that K-RAS is such an effective oncoprotein because it influences many aspects of tumorigenesis and (2) that combinatorial therapies will likely be required to inhibit the pathways operating downstream of mutant K-RAS.
In addition to its effects on cellular characteristics associated with traditional measures of transformation, mutant K-RAS regulates other cancer-related properties of CRC cells, such as radiation response and glucose metabolism. For example, the parental lines DLD-1 and HCT-116 are resistant to ionizing radiation when compared to their KRAS wild-type derivatives [26, 27]. In HCT-116 cells, K-RASG13D stabilizes MDM2, resulting in an attenuated P53 response to radiation [26]. It is not clear if a similar or distinct mechanism functions in DLD-1 cells, which are mutant for P53. Since patients with locally advanced rectal cancer are treated with a combination of chemotherapy and radiation, the radiation resistance conferred by mutant K-RAS is significant. Indeed, some clinical studies suggest that patients with K-RAS mutant rectal cancers respond poorly to chemoradiation therapy [28, 29].
Mutant K-RAS also alters the metabolism of CRC cells by allowing them to grow in the present of low levels of glucose [30]. Parental DLD-1 and HCT-116 cells express high levels of glucose transporter 1 (GLUT1) compared to their wild-type derivatives, which leads to enhanced glucose uptake and glycolysis [30]. A preferential use of aerobic glycolysis, termed the Warburg effect, is a hallmark of cancer cells, providing transformed cells with a proliferative and survival advantage in the presence of low oxygen [31].
A paradoxical attribute of mutant K-RAS, which seems to contradict its strong tumor promoting activity, is its ability to potentiate stress-induced apoptosis. Expression of K-RASG13D sensitizes HCT-116 and DLD-1 cells to the chemotherapeutic agents 5-fluorouracil (5-FU) and oxaliplatin [32, 33]. Parental HCT-116 and DLD-1 cells are also more sensitive to sodium butyrate- and staurosporine-induced apoptosis than their wild-type derivatives [34, 35]. At least two independent pro-apoptotic mechanisms function in K-RAS mutant CRC cells. In the case of chemotherapy response, mutant K-RAS up-regulates the pro-apoptotic protein NOXA and NOXA knockdown confers resistance to 5-FU and oxaliplatin in HCT-116 cells [32]. K-RASG13D also regulates the transcription of several genes known to play a role in apoptosis, including the anti-apoptotic protein gelsolin [34]. Down-regulation of gelsolin by mutant K-RAS was shown to be important for the hypersensitivity of HCT-116 cells to both sodium butyrate and 5-FU [34]. While the full extent of the pro-apoptotic program elicited by mutant K-RAS remains to be characterized, therapeutic engagement of this program may constitute a viable therapeutic strategy for K-RAS mutant CRC. In a similar vein, DLD-1 and HCT-116 cells expressing mutant K-RAS exhibit evidence of mitotic stress, such as slow mitotic progression and an increased number of lagging chromosomes in anaphase [36]. Inhibitors of mitosis preferentially induce cell death in the parental lines compared to K-RAS wild-type derivatives, suggesting that targeting this anti-tumorigenic property of K-RAS may also constitute a viable therapeutic strategy [36].
From the comparisons between K-RAS mutant colorectal cancer cells and their isogenic K-RAS wild-type derivatives, it is clear that K-RAS is involved in a wide variety of processes important for cancer (Fig. 1). What these in vitro studies cannot address, however, is how mutant K-RAS functions within the context of an intact tissue and how it contributes to the progression of an autochthonous developing CRC.
Mouse models of K-Ras activation in the intestinal epithelium
The laboratory mouse represents an ideal model system for studying oncogenes in an in vivo setting. Traditionally, loss of function (i.e. knockout) studies have been used to probe the function of specific genes in physiologic processes. Targeted homozygous deletion of K-Ras, for example, results in embryonic lethality due to defective fetal liver hematopoiesis [37]. K-Ras function is dispensable in most adult tissues, including the intestinal epithelium, which is essentially unaffected by loss of K-Ras [37](K. Haigis, unpublished) (Fig. 2A). While this observation may be significant with respect to targeted therapy – it suggests that a therapy that completely and specifically eliminates K-Ras function may not exhibit excessive toxicity – it does not provide much insight into the oncogenic function of mutationally activated K-Ras.
Fig. 2.

Phenotypic effects of Kras mutations in mice. (A) Histology of the small intestinal epithelium from an adult wild-type <-> Kras−/− chimeric animal. The Kras null component is labeled with LacZ. There is no effect of K-Ras deficiency on the development of the intestinal epithelium. (B) Wholemount view of a large ACF (outlined in white) from a KrasLA/+ animal. The tissue is stained with methylene blue to highlight the ACF. (C) Scanning electron microscopy (SEM) of colonic tissue from an Fabpl4X@-132-Cre ; KrasLSL-G12D/+ animal. Because Cre expression is mosaic, both wild-type and K-Ras mutant tissue areas are observed. The wild-type colonic tissue (outlined in red) exhibits a smooth appearance, while the tissue expressing K-RasG12D exhibits a cerebriform morphology. SEM courtesy of Paul Appleton and Inke Näthke, University of Dundee. (D) Fluorescent immunohistochemistry for lysozyme (Lys), which marks Paneth cells at the base of the small intestinal crypt. Wild-type crypts have several Lys-positive cells near the base, but crypts expressing K-RasG12D have no mature Lys-expressing Paneth cells. (E) Colonoscopy of Fabpl4X@-132-Cre ; Apc2lox14/+ and Fabpl4X@-132-Cre ; Apc2lox14/+ ; KrasLSL-G12D/+ animals. Pedunculated tumors (P) are indicated with black arrows. A sessile tumor (S) is indicated with a red arrow. (F) Histology of colonic tumors from Fabpl4X@-132-Cre ; Apc2lox14/+ and Fabpl4X@-132-Cre ; Apc2lox14/+ ; KrasLSL-G12D/+ animals. Tumors expressing wild-type K-Ras (top panel) are well differentiated, but those expressing mutant K-Ras (bottom panel) are poorly differentiated and highly dysplastic.
A major advance in the study of oncogenic K-Ras was the generation of genetically engineered mice expressing the mutated protein in the intestinal epithelium. The first such model employed transgenic over-expression of K-RasG12V from the Villin promoter, which confers expression throughout the small and large intestinal epithelia [38]. Villin-KrasG12V animals develop a range of pre-neoplastic and neoplastic intestinal lesions, including aberrant crypt foci (ACF), adenomas, and invasive adenocarcinomas, suggesting that mutant K-Ras strongly promotes intestinal neoplasia, even in the absence of secondary oncogenic mutations [38]. Similar to cellular models relying on ectopic over-expression, the physiological relevance, and thus the interpretation of the phenotype, of this transgenic model is not entirely clear. Nevertheless, the development of mouse models with mutational activation of endogenously expressed K-Ras has made it possible to address this uncertainty. KrasLA/+ mice carry a duplication of exon 2 and a silent G12D activating mutation in the endogenous Kras locus. In its un-recombined state, this duplication constitutes a null allele (which can be transmitted through the germ line in heterozygous form), but somatic recombination between the duplicated exons leads to restoration of normal gene structure and expression of activated K-Ras [39]. The expression of a single mutant Kras allele is sufficient to induce the formation of hyperplastic ACF in the colon (Fig. 2B), but these animals do not develop colon cancer [39].
Because the activation of K-Ras is dependent upon stochastic somatic recombination in the KrasLA/+ mice, their phenotype is not restricted to the intestinal epithelium. In fact, KrasLA/+ animals typically die from lung cancer [39], which precludes an in-depth analysis of the colonic phenotype. The phenotypic effects of mutant K-Ras in the intestinal epithelium are more effectively studied in animals that are genetically engineered to carry a Cre-dependent activated allele in the endogenous locus. The first such study utilized a Cre-dependent activated allele of Kras (KrasLSLV12) and a P450-inducible Cre transgenic line (Ah-Cre) [40, 41]. Injection of Ah-Cre ; KrasLSLV12/+ mice with β-naphthoflavone (βNF) leads to widespread expression of K-RasG12V throughout the epithelium of the proximal small intestine, but no change in basal homeostasis was seen [42]. Because Ah-Cre is not activated by βNF in the colonic epithelium [41], this study did not reveal how expression of K-RasG12V in the colonic epithelium compared to the sporadic activation seen in KrasLA/+ mice.
Subsequently, three studies revealed a strong phenotype associated with expression of activated K-Ras in the colonic epithelium. These studies utilized a Cre-dependent mutant allele of endogenous Kras (KrasLSL-G12D) [43] crossed with distinct intestinal epithelium-specific Cre transgenic strains, Fabpl4X@-132-Cre and Villin-Cre. Both of these transgenic lines express Cre recombinase in the distal small intestinal and colonic epithelia, while only Villin-Cre expresses in the proximal small intestinal epithelium [44, 45]. Fabpl4X@-132-Cre ; KrasLSL-G12D/+ animals develop widespread epithelial hyperplasia throughout the colon (Fig. 2C), which is associated with an expansion of the progenitor zone in the crypt and hyperproliferation of transit amplifying cells [22]. Villin-Cre ; KrasLSL-G12D/+ animals also exhibit widespread hyperplasia that is associated with increased proliferation in proximal colon, but not in the distal colon [46, 47].
What accounts for the distinct observations made from the Ah-Cre ; KrasLSLV12/+ study, which found no phenotype associated with expression of K-RasG12V, and the subsequent studies in which K-RasG12D induced hyperplasia? Differences in allele (G12V vs. G12D), organ site (small intestine vs. colon), or Cre driver could account for the phenotypic variation, but Fabpl4X@-132-Cre ; KrasLSL-G12D/+ animals develop hyperplasia in the distal small intestine as well as in the colon (K. Haigis, unpublished), suggesting that a difference in organ site does not underlie the differing results. Interestingly, animals expressing mutant K-Ras in the small intestine also fail to develop Paneth cells, suggesting that oncogenic K-Ras impinges upon pathways that control differentiation (Fig. 2D) [48].
Mouse models of endogenous K-Ras activation have also provided insight into the signaling pathways that function downstream of mutant K-Ras to control proliferation. Hyperplastic growth in both Fabpl4X@-132-Cre ; KrasLSL-G12D/+ and Villin-Cre ; KrasLSL-G12D/+ animals was associated with activation of the Raf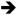 Mek
Mek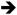 Erk signaling cascade [22, 46, 47]. Interestingly, phosphorylated Mek was detected throughout the intestinal epithelium, but phosphorylated Erk was detected only in the upper part of the crypts, outside of the proliferative zone [22, 46]. Nevertheless, treatment of Fabpl4X@-132-Cre ; KrasLSL-G12D/+ animals with CI-1040, a pharmacologic inhibitor of Mek, suppressed colonic epithelial hyper-proliferation and caused a return to normal histology [22]. These observations suggest that mutationally activated K-Ras promotes proliferation and hyperplasia through Mek and, possibly, independently of Erk.
Erk signaling cascade [22, 46, 47]. Interestingly, phosphorylated Mek was detected throughout the intestinal epithelium, but phosphorylated Erk was detected only in the upper part of the crypts, outside of the proliferative zone [22, 46]. Nevertheless, treatment of Fabpl4X@-132-Cre ; KrasLSL-G12D/+ animals with CI-1040, a pharmacologic inhibitor of Mek, suppressed colonic epithelial hyper-proliferation and caused a return to normal histology [22]. These observations suggest that mutationally activated K-Ras promotes proliferation and hyperplasia through Mek and, possibly, independently of Erk.
Another interesting observation from Fabpl4X@-132-Cre ; KrasLSL-G12D/+ animals was a significant suppression of Akt phosphorylation compared to wild-type, suggesting that mutant K-Ras negatively regulates PI3K signaling in this setting [22]. Although this result contrasts the historical view of PI3K as a direct effector of mutant RAS, epidemiological studies support the idea that PI3K is not a K-RAS effector in the context of CRC. Activating mutations in PIK3CA, the p110 catalytic subunit of PI3Kα, are often coincident with K-RAS mutations in primary human CRC [11].
As discussed above, in cultured CRC cells, oncogenic K-RAS paradoxically promotes stress-induced apoptosis. This phenomenon holds true in vivo as well. For example, Villin-Cre ; KrasLSL-G12D/+ animals exhibit an increased frequency of spontaneous apoptosis in the distal colon and Fabpl4X@-132-Cre ; KrasLSL-G12D/+ mice were highly sensitive to colonic epithelial apoptosis induced by dextran sodium sulfate (DSS) treatment [22, 46]. These data support the notion that the oncogenic effects of K-Ras activation are context-dependent and suggest that mutant K-Ras may not contribute to tumorigenesis in the context of a chronic apoptotic stimulus (e.g. inflammation). Consistent with this notion, KRAS mutations are reported to be less frequent in CRC that arises in a background of ulcerative colitis (UC) compared to CRC from the general population [49, 50].
Taken together, studies using animals expressing endogenous levels of mutant K-Ras in the intestinal epithelium indicate that activated K-Ras promotes hyperplasia. Importantly, the activation of K-Ras on its own is insufficient to induce neoplasia in the mouse intestinal epithelium. This observation is consistent with the paradigm of colorectal cancer as a multistep process that requires cooperating mutations and epigenetic events targeting several oncogenes and tumor suppressor genes.
Cooperation between mutant K-Ras and Apc
Molecular genetic studies of human tumors and phenotypic analyses of animal models indicate that, unlike in pancreatic and lung cancer, K-Ras activation does not act as an initiating event in CRC. Instead, mutation of K-Ras is more commonly a progression event that occurs in the context of an adenoma that has activated the Wnt signaling pathway via inactivation of the Adenomatous polyposis coli (Apc) tumor suppressor protein or activation of the β-catenin oncoprotein [51]. Potential cooperation between activating mutations in K-Ras and inactivating mutations in Apc was first explored in mouse models in a cross between Villin-KrasG12V animals and those carrying a targeted hypomorphic allele of Apc (Apc1638N/+). Villin-KrasG12V ; Apc1638N/+ animals exhibit reduced lifespan due to a significant increase in tumor multiplicity when compared with Villin-KrasG12V or Apc1638N/+ single mutant animals [52]. Moreover, tumors from double mutant animals exhibit higher rates of proliferation and reduced levels of apoptosis when compared with single mutant mice, demonstrating strong cooperativity between over-expressed mutant K-Ras and Apc inactivation in intestinal tumor development [52]. A second study crossed Ah-Cre ; KrasLSLV12/+ animals with those carrying the ApcMin allele and then injected animals with βNF to activate K-Ras in the proximal small intestine. While K-Ras activation on its own had no effect on homeostasis, combined mutation of K-Ras and Apc led to enhanced tumor invasion [42].
Subsequent studies examined cooperativity between K-Ras and Apc mutations in the colonic epithelium by crossing Fabpl4X@-132-Cre ; KrasLSL-G12D/+ animals to a strain carrying a Cre-dependent inactivating allele of Apc (Apc2lox14) [53]. Cre-mediated recombination of the Apc2lox14 allele induces deletion of exon 14 and, as a result, a frame shift leading to truncation after amino acid 580 of the Apc protein. As in the Villin-KrasG12V ; Apc1638N/+ study, activation of K-Ras led to a significant increase in the number of colonic tumors in Fabpl4X@-132-Cre ; Apc2lox14/+ animals (Fig. 2E) [22]. Yet, while colonic tumors from Fabpl4X@-132-Cre ; Apc2lox14/+ animals were benign, those from Fabpl4X@-132-Cre ; Apc2lox14/+ ; KrasLSL-G12D/+ animals were malignant, characterized by the presence of uniform high-grade dysplasia throughout the tumor and invasion into the underlying smooth muscle (Fig. 2F) [22]. In addition, colonic tumors from double mutant animals exhibited distinct morphologic features when compared to tumors from animals expressing wild-type K-Ras. In essence, mutational activation of K-Ras induced a shift from the development of benign, pedunculated adenomas to the development of malignant, sessile adenocarcinomas (Fig. 2E) [22]. These data are all consistent with mutant K-Ras having a major effect on the differentiation of Apc-mutant colonic epithelial cells. Indeed, all of the epithelial cells from double mutant tumors expressed markers of intestinal progenitor cells, suggesting that K-Ras activation locks them into a relatively primitive differentiation state [22].
A limitation of these studies is that the Apc and K-Ras mutations occur very early during colonic development, owing to the embryonic expression of the Cre transgenes. Indeed, the increase in tumor multiplicity seen in Apc/K-Ras double mutant animals, relative to Apc single mutant animals, probably results from an increased likelihood of loss of heterozygosity at the Apc locus due to the widespread hyperplasia caused by K-Ras activation [22, 52]. A more accurate model of colorectal cancer would require the ability to mutate K-Ras and Apc, simultaneously or sequentially, in single crypts of the adult colonic epithelium. This was accomplished by infecting Apc2lox14/2lox14 ; KrasLSL-G12D/+ animals with Adenovirus carrying a transgene for Cre recombinase (Adeno-Cre) [54]. Similarly to the Fabpl4X@-132-Cre ; Apc2lox14/+ ; KrasLSL-G12D/+ experiment, mutant K-Ras strongly promoted tumor progression in this study. But in contrast, the low tumor multiplicity that occurred following Adeno-Cre infection allowed the double mutant animals to live long enough to develop metastatic disease [54]. Currently, the Adeno-Cre infected Apc2lox14/2lox14 ; KrasLSL-G12D/+ model is the only bona fide mouse model of metastatic CRC.
One common feature of the mouse models that combine mutations in K-Ras and Apc is that the double mutant tumors displayed an increase in the levels and nuclear accumulation of β-catenin when compared with tissues from K-Ras or Apc single mutant animals [22, 42, 52, 54]. This is most likely associated with an increased expression of Wnt target genes, for example c-Myc. This observation suggests a synergistic role for K-Ras activation and Apc loss in enhancing the activity of the canonical Wnt/β-catenin signaling pathway, a phenomenon that has been described in human CRC cell lines as well [55].
While mouse models have effectively recapitulated the phenotypic effects of mutating K-Ras in colorectal cancers, they have thus far failed to reveal the molecular mechanisms underlying tumor promotion. In some cases, mutation of K-Ras was associated with constitutively phosphorylated Erk [52, 54]. Nevertheless, one study demonstrated high levels of phosphorylated Mek in cancers from Fabpl4X@-132-Cre ; Apc2lox14/+ ; KrasLSL-G12D/+ mice, but no signs of Erk activation [22]. Inhibition of Mek with CI-1040 did not decrease proliferation in cancers from Fabpl4X@-132-Cre ; Apc2lox14/+ ; KrasLSL-G12D/+ animals, suggesting that, in contrast to what was observed in Fabpl4X@-132-Cre ; KrasLSL-G12D/+ mice, Mek is not required for the oncogenic activity of mutant K-Ras in colon cancers arising in an Apc mutant context [22]. This observation is entirely consistent with the failure of CI-1040 to elicit a clinical response in cancers that commonly have K-RAS mutations, although cancers in this particular Phase II trial were not screened for KRAS genotype [56].
Another therapeutically relevant observation was made in animals in which Adeno-Cre infection was employed to trigger K-Ras activation and/or Apc inactivation. Colonic tumors from Adeno-Cre infected animals were found to have active mTOR signaling, regardless of K-Ras genotype, as ascertained by the levels of phosphorylated S6 protein [54]. The levels of phosphorylated S6 were reduced by treatment with the mTOR inhibitor rapamycin. Interestingly rapamycin treatment reduced the size of tumors expressing wild-type K-Ras, but had no effect on the growth of tumors expressing K-RasG12D [54]. Thus, while the presence of an oncogenic K-Ras allele did not appear to alter signaling through the mTOR pathway, it did confer resistance to inhibition of mTOR signaling. This observation is similar to what is observed in the clinic, where K-RAS mutations correlate with resistance to targeted therapies, such as inhibitors of EGFR [14-19]. This mouse model may constitute a good system for studying the effects of mutant K-Ras on response to targeted therapies.
Cooperation between mutant K-Ras and Ink4a/Arf
In the late 1990’s, over-expression of mutationally activated H-RAS was shown to induce growth arrest and stabilization of p53 and p16INK4A in cultured fibroblasts, a phenomenon now called oncogene-induced senescence (OIS) [57]. Over the ensuing decade, a debate emerged as to whether OIS was a cell culture artifact or whether it was restricted to instances of RAS over-expression, but endogenous activated K-Ras was subsequently shown to induce OIS in a mouse model of lung cancer [40]. The hyperplasic colonic epithelium seen in Villin-Cre ; KrasLSL-G12D/+ animals expresses high levels of Ink4a and is positive for senescence-associated β-galactosidase (SAβgal) activity, a hallmark of OIS [47]. Moreover, when Villin-Cre ; KrasLSL-G12D/+ animals are crossed to Ink4a null mice, they develop adenomas with a serrated morphology [47]. These data suggest that, in an in vivo setting, mutant K-Ras is incapable of initiating neoplasia because it induces a senescence response. It remains unclear how this study integrates with those studying the interaction between Apc and K-Ras, since mutant K-Ras has not been reported to induce OIS in Apc-mutant tumors and activation of Wnt has not been demonstrated to overcome senescence. Perhaps, as the authors suggest, K-Ras/Ink4a represents an alternate route to tumorigenesis in the colon.
Cooperation between mutant K-Ras and Tgfbr2
Mutational inactivation of Transforming Growth Factor beta Receptor II (TGFBR2) occurs in approximately 30% of all CRCs [58]. Owing to a mononucleotide repeat in the coding region, the TGFBR2 gene is highly sensitive to loss of DNA mismatch repair, and thus it is mutated in approximately 80% of CRCs with microsatellite instability (MSI+) [59]. Mutations in TGFBR2 are associated with an increase in cell proliferation and with a higher angiogenic potential [60]. Mice expressing K-RasG12D (Villin-Cre ; KrasLSL-G12D/+) in a Tgfbr2 null background exhibit an increase in the colonic crypt length and an expansion of the proliferative compartment of the crypt when compared with wild-type mice [48]. While these phenotypes are likely attributable simply to the expression of activated K-Ras, the Tgfbr2/K-Ras double mutant animals also develop invasive adenocarcinomas that metastasizes to the regional lymph nodes or lungs [48]. A major difference between this model and those that we described previously is that tumors from Tgfbr2/K-Ras animals appear to arise without any obvious activation of β-catenin, suggesting that mutant K-Ras may act as a modifier of Wnt signaling only in the presence of a primary mutation within the Wnt pathway [48]. Similar to some of the Apc/K-Ras models, tumorigenesis in Tgfbr2/K-Ras double mutant animals is associated with a lack of Erk activation and an apparent down-regulation of PI3K signaling [48]. In addition, increased levels of the EGFR ligand epiregulin were observed in tumors from Tgfbr2/K-Ras double mutant animals, suggesting that mutant K-Ras induces an autocrine feed-forward loop in the absence of Tgfbr2 [48]. This K-RAS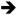 Epiregulin
Epiregulin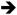 EGFR feed-forward loop was also found to play an important role regulating proliferation in HCT-116 cells, which are mutant for TGFBR2 [61].
EGFR feed-forward loop was also found to play an important role regulating proliferation in HCT-116 cells, which are mutant for TGFBR2 [61].
Conclusion
Mutational activation of K-RAS is among the most significant genetic events in the progression of CRC, both in terms of its prevalence and also because of its effect on the response to cancer therapies. While K-RAS mutations were first identified in CRC over two decades ago, targeted therapies for K-RAS mutant cancer are still lacking. As a result, knowledge of the K-RAS mutational status of a given tumor is currently useful only for its negative predictive value. The development of novel therapies to treat K-RAS mutant CRC relies upon a better understanding of the molecular mechanisms underlying the pleiotropic oncogenic phenotypes associated with activated K-RAS, which will ultimately require the integration of detailed mechanistic studies performed in cell lines and mouse models. In the end, given the multi-dimension nature of its effects, a therapy that targets K-RAS directly or, more likely, combination therapies will be required to counteract the action of this oncoprotein.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Lau KS, Haigis KM. Non-redundancy within the RAS oncogene family: Insights into mutational disparities in cancer. Mol. Cells. 2009;28:315–320. doi: 10.1007/s10059-009-0143-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Hancock JF. Ras proteins: different signals from different locations. Nat. Rev. Mol. Cell. Biol. 2003;4:373–384. doi: 10.1038/nrm1105. [DOI] [PubMed] [Google Scholar]
- [3].Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat. Rev. Cancer. 2003;3:459–465. doi: 10.1038/nrc1097. [DOI] [PubMed] [Google Scholar]
- [4].Yeh JJ, Madigan JP, Campbell PM, Roberts PJ, DeGraffenreid L, Der CJ. Targeting Ras for Anticancer Drug Discovery. In: Bradshaw RA, Dennis EA, editors. Handbook of Cell Signaling. 2nd edition Vol. 3. Academic Press; Oxford: 2009. pp. 2837–2857. [Google Scholar]
- [5].Bos JL, Fearon ER, Hamilton SR, Verlaan-de Vries M, van Boom JH, van der Eb AJ, Vogelstein B. Prevalence of ras gene mutations in human colorectal cancers. Nature. 1987;327:293–297. doi: 10.1038/327293a0. [DOI] [PubMed] [Google Scholar]
- [6].Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. New Engl. J. Med. 1988;319:525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- [7].Irahara N, Baba Y, Nosho K, Shima K, Yan L, Dias-Santagata D, Iafrate AJ, Fuchs CS, Haigis KM, Ogino S. NRAS mutations are rare in colorectal cancer. Diagn. Mol. Pathol. 2010;19:157–163. doi: 10.1097/PDM.0b013e3181c93fd1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Janakiraman M, Vakiani E, Zeng Z, Pratilas CA, Taylor BS, Chitale D, Halilovic E, Wilson M, Huberman K, Filho J.C. Ricarte, Persaud Y, Levine DA, Fagin JA, Jhanwar SC, Mariadason JM, Lash A, Ladanyi M, Saltz LB, Heguy A, Paty PB, Solit DB. Genomic and biological characterization of exon 4 KRAS mutations in human cancer. Cancer Res. 2010;70:5901–5911. doi: 10.1158/0008-5472.CAN-10-0192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Edkins S, O’Meara S, Parker A, Stevens C, Reis M, Jones S, Greenman C, Davies H, Dalgliesh G, Forbes S, Hunter C, Smith R, Stephens P, Goldstraw P, Nicholson A, Chan TL, Velculescu VE, Yuen ST, Leung SY, Stratton MR, Futreal PA. Recurrent KRAS codon 146 mutations in human colorectal cancer. Cancer Biol. Ther. 2006;5:928–932. doi: 10.4161/cbt.5.8.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].O’Brien MJ. Hyperplastic and serrated polyps of the colorectum. Gastroenterol. Clin. North Am. 2007;36:947–968. viii. doi: 10.1016/j.gtc.2007.08.007. [DOI] [PubMed] [Google Scholar]
- [11].Velho S, Moutinho C, Cirnes L, Albuquerque C, Hamelin R, Schmitt F, Carneiro F, Oliveira C, Seruca R. BRAF, KRAS and PIK3CA mutations in colorectal serrated polyps and cancer: primary or secondary genetic events in colorectal carcinogenesis? BMC Cancer. 2008;8:255. doi: 10.1186/1471-2407-8-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ohnishi T, Tomita N, Monden T, Ohue M, Yana I, Takami K, Yamamoto H, Yagyu T, Kikkawa N, Shimano T, Monden M. A detailed analysis of the role of K-ras gene mutation in the progression of colorectal adenoma. Brit. J. cCncer. 1997;75:341–347. doi: 10.1038/bjc.1997.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Andreyev HJ, Norman AR, Cunningham D, Oates J, Dix BR, Iacopetta BJ, Young J, Walsh T, Ward R, Hawkins N, Beranek M, Jandik P, Benamouzig R, Jullian E, Laurent-Puig P, Olschwang S, Muller O, Hoffmann I, Rabes HM, Zietz C, Troungos C, Valavanis C, Yuen ST, Ho JW, Croke CT, O’Donoghue DP, Giaretti W, Rapallo A, Russo A, Bazan V, Tanaka M, Omura K, Azuma T, Ohkusa T, Fujimori T, Ono Y, Pauly M, Faber C, Glaesener R, de Goeij AF, Arends JW, Andersen SN, Lovig T, Breivik J, Gaudernack G, Clausen OP, De Angelis PD, Meling GI, Rognum TO, Smith R, Goh HS, Font A, Rosell R, Sun XF, Zhang H, Benhattar J, Losi L, Lee JQ, Wang ST, Clarke PA, Bell S, Quirke P, Bubb VJ, Piris J, Cruickshank NR, Morton D, Fox JC, Al-Mulla F, Lees N, Hall CN, Snary D, Wilkinson K, Dillon D, Costa J, Pricolo VE, Finkelstein SD, Thebo JS, Senagore AJ, Halter SA, Wadler S, Malik S, Krtolica K, Urosevic N. Kirsten ras mutations in patients with colorectal cancer: the ‘RASCAL II’ study. Brit. J. Cancer. 2001;85:692–696. doi: 10.1054/bjoc.2001.1964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lievre A, Bachet JB, Corre D. Le, Boige V, Landi B, Emile JF, Cote JF, Tomasic G, Penna C, Ducreux M, Rougier P, Penault-Llorca F, Laurent-Puig P. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- [15].Di Fiore F, Blanchard F, Charbonnier F, Pessot F. Le, Lamy A, Galais MP, Bastit L, Killian A, Sesboue R, Tuech JJ, Queuniet AM, Paillot B, Sabourin JC, Michot F, Michel P, Frebourg T. Clinical relevance of KRAS mutation detection in metastatic colorectal cancer treated by Cetuximab plus chemotherapy. Brit. J. Cancer. 2007;96:1166–1169. doi: 10.1038/sj.bjc.6603685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, Humblet Y, Bodoky G, Cunningham D, Jassem J, Rivera F, Kocakova I, Ruff P, Blasinska-Morawiec M, Smakal M, Canon JL, Rother M, Oliner KS, Wolf M, Gansert J. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J. Clin. Oncol. 2010;28:4697–4705. doi: 10.1200/JCO.2009.27.4860. [DOI] [PubMed] [Google Scholar]
- [17].Peeters M, Price TJ, Cervantes A, Sobrero AF, Ducreux M, Hotko Y, Andre T, Chan E, Lordick F, Punt CJ, Strickland AH, Wilson G, Ciuleanu TE, Roman L, Van Cutsem E, Tzekova V, Collins S, Oliner KS, Rong A, Gansert J. Randomized phase III study of panitumumab with fluorouracil, leucovorin, and irinotecan (FOLFIRI) compared with FOLFIRI alone as second-line treatment in patients with metastatic colorectal cancer. J. Clin. Oncol. 2010;28:4706–4713. doi: 10.1200/JCO.2009.27.6055. [DOI] [PubMed] [Google Scholar]
- [18].Van Cutsem E, Kohne CH, Lang I, Folprecht G, Nowacki MP, Cascinu S, Shchepotin I, Maurel J, Cunningham D, Tejpar S, Schlichting M, Zubel A, Celik I, Rougier P, Ciardiello F. Cetuximab Plus Irinotecan, Fluorouracil, and Leucovorin As First-Line Treatment for Metastatic Colorectal Cancer: Updated Analysis of Overall Survival According to Tumor KRAS and BRAF Mutation Status. J. Clin. Oncol. 2011 doi: 10.1200/JCO.2010.33.5091. [DOI] [PubMed] [Google Scholar]
- [19].Bokemeyer C, Bondarenko I, Hartmann JT, de Braud F, Schuch G, Zubel A, Celik I, Schlichting M, Koralewski P. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: the OPUS study. Ann. Oncol. 2011 doi: 10.1093/annonc/mdq632. [DOI] [PubMed] [Google Scholar]
- [20].Shirasawa S, Furuse M, Yokoyama N, Sasazuki T. Altered growth of human colon cancer cell lines disrupted at activated Ki-ras. Science. 1993;260:85–88. doi: 10.1126/science.8465203. [DOI] [PubMed] [Google Scholar]
- [21].Pollock CB, Shirasawa S, Sasazuki T, Kolch W, Dhillon AS. Oncogenic K-RAS is required to maintain changes in cytoskeletal organization, adhesion, and motility in colon cancer cells. Cancer Res. 2005;65:1244–1250. doi: 10.1158/0008-5472.CAN-04-1911. [DOI] [PubMed] [Google Scholar]
- [22].Haigis KM, Kendall KR, Wang Y, Cheung A, Haigis MC, Glickman JN, Niwa-Kawakita M, Sweet-Cordero A, Sebolt-Leopold J, Shannon KM, Settleman J, Giovannini M, Jacks T. Differential effects of oncogenic K-Ras and N-Ras on proliferation, differentiation and tumor progression in the colon. Nat. Genet. 2008;40:600–608. doi: 10.1038/ngXXXX. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Rosen K, Rak J, Leung T, Dean NM, Kerbel RS, Filmus J. Activated Ras prevents downregulation of Bcl-X(L) triggered by detachment from the extracellular matrix. A mechanism of Ras-induced resistance to anoikis in intestinal epithelial cells. J. Cell Biol. 2000;149:447–456. doi: 10.1083/jcb.149.2.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Rak J, Mitsuhashi Y, Bayko L, Filmus J, Shirasawa S, Sasazuki T, Kerbel RS. Mutant ras oncogenes upregulate VEGF/VPF expression: implications for induction and inhibition of tumor angiogenesis. Cancer Res. 1995;55:4575–4580. [PubMed] [Google Scholar]
- [25].Okada F, Rak JW, Croix BS, Lieubeau B, Kaya M, Roncari L, Shirasawa S, Sasazuki T, Kerbel RS. Impact of oncogenes in tumor angiogenesis: mutant K-ras up-regulation of vascular endothelial growth factor/vascular permeability factor is necessary, but not sufficient for tumorigenicity of human colorectal carcinoma cells. Proc.Natl Acad. Sci. U. S. A. 1998;95:3609–3614. doi: 10.1073/pnas.95.7.3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ries S, Biederer C, Woods D, Shifman O, Shirasawa S, Sasazuki T, McMahon M, Oren M, McCormick F. Opposing effects of Ras on p53: transcriptional activation of mdm2 and induction of p19ARF. Cell. 2000;103:321–330. doi: 10.1016/s0092-8674(00)00123-9. [DOI] [PubMed] [Google Scholar]
- [27].Bernhard EJ, Stanbridge EJ, Gupta S, Gupta AK, Soto D, Bakanauskas VJ, Cerniglia GJ, Muschel RJ, McKenna WG. Direct evidence for the contribution of activated N-ras and K-ras oncogenes to increased intrinsic radiation resistance in human tumor cell lines. Cancer Res. 2000;60:6597–6600. [PubMed] [Google Scholar]
- [28].Bengala C, Bettelli S, Bertolini F, Salvi S, Chiara S, Sonaglio C, Losi L, Bigiani N, Sartori G, Dealis C, Malavasi N, D’Amico R, Luppi G, Gatteschi B, Maiorana A, Conte PF. Epidermal growth factor receptor gene copy number, K-ras mutation and pathological response to preoperative cetuximab, 5-FU and radiation therapy in locally advanced rectal cancer. Ann. Oncol. 2009;20:469–474. doi: 10.1093/annonc/mdn647. [DOI] [PubMed] [Google Scholar]
- [29].Zauber NP, Marotta SP, Berman E, Grann A, Rao M, Komati N, Ribiero K, Bishop DT. Molecular genetic changes associated with colorectal carcinogenesis are not prognostic for tumor regression following preoperative chemoradiation of rectal carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2009;74:472–476. doi: 10.1016/j.ijrobp.2008.08.020. [DOI] [PubMed] [Google Scholar]
- [30].Yun J, Rago C, Cheong I, Pagliarini R, Angenendt P, Rajagopalan H, Schmidt K, Willson JK, Markowitz S, Zhou S, Diaz LA, Jr., Velculescu VE, Lengauer C, Kinzler KW, Vogelstein B, Papadopoulos N. Glucose deprivation contributes to the development of KRAS pathway mutations in tumor cells. Science. 2009;325:1555–1559. doi: 10.1126/science.1174229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- [32].de Bruijn MT, Raats DA, Hoogwater FJ, van Houdt WJ, Cameron K, Medema JP, Rinkes I.H. Borel, Kranenburg O. Oncogenic KRAS sensitises colorectal tumour cells to chemotherapy by p53-dependent induction of Noxa. Brit. J. Cancer. 2010;102:1254–1264. doi: 10.1038/sj.bjc.6605633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Klampfer L, Swaby LA, Huang J, Sasazuki T, Shirasawa S, Augenlicht L. Oncogenic Ras increases sensitivity of colon cancer cells to 5-FU-induced apoptosis. Oncogene. 2005;24:3932–3941. doi: 10.1038/sj.onc.1208552. [DOI] [PubMed] [Google Scholar]
- [34].Klampfer L, Huang J, Sasazuki T, Shirasawa S, Augenlicht L. Oncogenic Ras promotes butyrate-induced apoptosis through inhibition of gelsolin expression. The J. Biol. Chem. 2004;279:36680–36688. doi: 10.1074/jbc.M405197200. [DOI] [PubMed] [Google Scholar]
- [35].Keller JW, Haigis KM, Franklin JL, Whitehead RH, Jacks T, Coffey RJ. Oncogenic K-RAS subverts the antiapoptotic role of N-RAS and alters modulation of the N-RAS:gelsolin complex. Oncogene. 2007;26:3051–3059. doi: 10.1038/sj.onc.1210103. [DOI] [PubMed] [Google Scholar]
- [36].Luo J, Emanuele MJ, Li D, Creighton CJ, Schlabach MR, Westbrook TF, Wong KK, Elledge SJ. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell. 2009;137:835–848. doi: 10.1016/j.cell.2009.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Johnson L, Greenbaum D, Cichowski K, Mercer K, Murphy E, Schmitt E, Bronson RT, Umanoff H, Edelmann W, Kucherlapati R, Jacks T. K-ras is an essential gene in the mouse with partial functional overlap with N-ras. Genes Dev. 1997;11:2468–2481. doi: 10.1101/gad.11.19.2468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Janssen KP, el-Marjou F, Pinto D, Sastre X, Rouillard D, Fouquet C, Soussi T, Louvard D, Robine S. Targeted expression of oncogenic K-ras in intestinal epithelium causes spontaneous tumorigenesis in mice. Gastroenterology. 2002;123:492–504. doi: 10.1053/gast.2002.34786. [DOI] [PubMed] [Google Scholar]
- [39].Johnson L, Mercer K, Greenbaum D, Bronson RT, Crowley D, Tuveson DA, Jacks T. Somatic activation of the K-ras oncogene causes early onset lung cancer in mice. Nature. 2001;410:1111–1116. doi: 10.1038/35074129. [DOI] [PubMed] [Google Scholar]
- [40].Guerra C, Mijimolle N, Dhawahir A, Dubus P, Barradas M, Serrano M, Campuzano V, Barbacid M. Tumor induction by an endogenous K-ras oncogene is highly dependent on cellular context. Cancer Cell. 2003;4:111–120. doi: 10.1016/s1535-6108(03)00191-0. [DOI] [PubMed] [Google Scholar]
- [41].Ireland H, Kemp R, Houghton C, Howard L, Clarke AR, Sansom OJ, Winton DJ. Inducible Cre-mediated control of gene expression in the murine gastrointestinal tract: effect of loss of beta-catenin. Gastroenterology. 2004;126:1236–1246. doi: 10.1053/j.gastro.2004.03.020. [DOI] [PubMed] [Google Scholar]
- [42].Sansom OJ, Meniel V, Wilkins JA, Cole AM, Oien KA, Marsh V, Jamieson TJ, Guerra C, Ashton GH, Barbacid M, Clarke AR. Loss of Apc allows phenotypic manifestation of the transforming properties of an endogenous K-ras oncogene in vivo. Proc. Natl Acad. Sci. U. S. A. 2006;103:14122–14127. doi: 10.1073/pnas.0604130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Tuveson DA, Shaw AT, Willis NA, Silver DP, Jackson EL, Chang S, Mercer KL, Grochow R, Hock H, Crowley D, Hingorani SR, Zaks T, King C, Jacobetz MA, Wang L, Bronson RT, Orkin SH, DePinho RA, Jacks T. Endogenous oncogenic K-ras(G12D) stimulates proliferation and widespread neoplastic and developmental defects. Cancer Cell. 2004;5:375–387. doi: 10.1016/s1535-6108(04)00085-6. [DOI] [PubMed] [Google Scholar]
- [44].Wong MH, Saam JR, Stappenbeck TS, Rexer CH, Gordon JI. Genetic mosaic analysis based on Cre recombinase and navigated laser capture microdissection. Proc. Natl Acad. Sci. U. S. A. 2000;97:12601–12606. doi: 10.1073/pnas.230237997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].el Marjou F, Janssen KP, Chang BH, Li M, Hindie V, Chan L, Louvard D, Chambon P, Metzger D, Robine S. Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis. 2004;39:186–193. doi: 10.1002/gene.20042. [DOI] [PubMed] [Google Scholar]
- [46].Calcagno SR, Li S, Colon M, Kreinest PA, Thompson EA, Fields AP, Murray NR. Oncogenic K-ras promotes early carcinogenesis in the mouse proximal colon. International journal of cancer. Int. J. Cancer. 2008;122:2462–2470. doi: 10.1002/ijc.23383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Bennecke M, Kriegl L, Bajbouj M, Retzlaff K, Robine S, Jung A, Arkan MC, Kirchner T, Greten FR. Ink4a/Arf and oncogene-induced senescence prevent tumor progression during alternative colorectal tumorigenesis. Cancer Cell. 2010;18:135–146. doi: 10.1016/j.ccr.2010.06.013. [DOI] [PubMed] [Google Scholar]
- [48].Trobridge P, Knoblaugh S, Washington MK, Munoz NM, Tsuchiya KD, Rojas A, Song X, Ulrich CM, Sasazuki T, Shirasawa S, Grady WM. TGF-beta receptor inactivation and mutant Kras induce intestinal neoplasms in mice via a beta-catenin-independent pathway. Gastroenterology. 2009;136:1680–1688. e1687. doi: 10.1053/j.gastro.2009.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Lyda MH, Noffsinger A, Belli J, Fenoglio-Preiser CM. Microsatellite instability and K-ras mutations in patients with ulcerative colitis. Hum. Pathol. 2000;31:665–671. doi: 10.1053/hupa.2000.7643. [DOI] [PubMed] [Google Scholar]
- [50].Wong NA, Harrison DJ. Colorectal neoplasia in ulcerative colitis-recent advances. Histopathology. 2001;39:221–234. doi: 10.1046/j.1365-2559.2001.01292.x. [DOI] [PubMed] [Google Scholar]
- [51].Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- [52].Janssen KP, Alberici P, Fsihi H, Gaspar C, Breukel C, Franken P, Rosty C, Abal M, El Marjou F, Smits R, Louvard D, Fodde R, Robine S. APC and oncogenic KRAS are synergistic in enhancing Wnt signaling in intestinal tumor formation and progression. Gastroenterology. 2006;131:1096–1109. doi: 10.1053/j.gastro.2006.08.011. [DOI] [PubMed] [Google Scholar]
- [53].Colnot S, Niwa-Kawakita M, Hamard G, Godard C, Plenier S. Le, Houbron C, Romagnolo B, Berrebi D, Giovannini M, Perret C. Colorectal cancers in a new mouse model of familial adenomatous polyposis: influence of genetic and environmental modifiers. Lab. Invest. 2004;84:1619–1630. doi: 10.1038/labinvest.3700180. [DOI] [PubMed] [Google Scholar]
- [54].Hung KE, Maricevich MA, Richard LG, Chen WY, Richardson MP, Kunin A, Bronson RT, Mahmood U, Kucherlapati R. Development of a mouse model for sporadic and metastatic colon tumors and its use in assessing drug treatment. Proc. Natl Acad. Sci. U. S. A. 2010;107:1565–1570. doi: 10.1073/pnas.0908682107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Li J, Mizukami Y, Zhang X, Jo WS, Chung DC. Oncogenic K-ras stimulates Wnt signaling in colon cancer through inhibition of GSK-3beta. Gastroenterology. 2005;128:1907–1918. doi: 10.1053/j.gastro.2005.02.067. [DOI] [PubMed] [Google Scholar]
- [56].Rinehart J, Adjei AA, Lorusso PM, Waterhouse D, Hecht JR, Natale RB, Hamid O, Varterasian M, Asbury P, Kaldjian EP, Gulyas S, Mitchell DY, Herrera R, Sebolt-Leopold JS, Meyer MB. Multicenter phase II study of the oral MEK inhibitor, CI-1040, in patients with advanced non-small-cell lung, breast, colon, and pancreatic cancer. J. Clin. Oncol. 2004;22:4456–4462. doi: 10.1200/JCO.2004.01.185. [DOI] [PubMed] [Google Scholar]
- [57].Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- [58].Biswas S, Trobridge P, Romero-Gallo J, Billheimer D, Myeroff LL, Willson JK, Markowitz SD, Grady WM. Mutational inactivation of TGFBR2 in microsatellite unstable colon cancer arises from the cooperation of genomic instability and the clonal outgrowth of transforming growth factor beta resistant cells. Gene Chromosome Cancer. 2008;47:95–106. doi: 10.1002/gcc.20511. [DOI] [PubMed] [Google Scholar]
- [59].Duval A, Hamelin R. Mutations at coding repeat sequences in mismatch repair-deficient human cancers: toward a new concept of target genes for instability. Cancer Res. 2002;62:2447–2454. [PubMed] [Google Scholar]
- [60].Jakowlew SB. Transforming growth factor-beta in cancer and metastasis. Cancer Metastasis Rev. 2006;25:435–457. doi: 10.1007/s10555-006-9006-2. [DOI] [PubMed] [Google Scholar]
- [61].Baba I, Shirasawa S, Iwamoto R, Okumura K, Tsunoda T, Nishioka M, Fukuyama K, Yamamoto K, Mekada E, Sasazuki T. Involvement of deregulated epiregulin expression in tumorigenesis in vivo through activated Ki-Ras signaling pathway in human colon cancer cells. Cancer Res. 2000;60:6886–6889. [PubMed] [Google Scholar]